
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric one-pot reactions using heterogeneous chemical catalysis: recent steps towards sustainable processes
György
Szőllősi

MTA-SZTE Stereochemistry Research Group, University of Szeged, H-6720 Szeged, Dóm tér 8, Hungary. E-mail: szollosi@chem.u-szeged.hu; Fax: +36 62 544200
First published on 20th November 2017
Abstract
The preparation of optically pure fine chemicals is among the most important and challenging tasks met by organic chemists. Recently, significant efforts have been focused on the development of green and sustainable procedures for the synthesis of these high value-added compounds. Asymmetric heterogeneous catalysis has provided efficient solutions to these challenges. The application of heterogeneous chiral catalysts in one-pot processes combines the advantages of use of these materials with time, material, and energy savings associated with cascade or sequential procedures. This review surveys these asymmetric one-pot reactions reported until July 2017, in which a heterogeneous chemical catalyst has been applied either as a single multifunctional catalyst or in combination with a second catalytically active material. These processes include one-pot procedures catalysed by carefully designed solids obtained by the immobilization of chiral metal complexes, by anchoring chiral organocatalysts, or by modifying catalytic surfaces with optically pure compounds, which may also incorporate uncatalyzed and homogeneously catalysed steps. Methods applying achiral heterogeneous catalysts in combination with soluble chiral chemical catalysts or biocatalysts are also presented. Sophisticated, finely tuned materials have been applied in most of these reactions, which have been discussed along with the main requirements necessary to perform these transformations in a one-pot manner.
 György Szőllősi | György Szőllősi was born in Targu-Mures in 1967, and graduated from the Technical University of Timisoara, Romania, as a Chemical Engineer in 1993. In 1994, he joined the Organic Catalysis Research group of the Hungarian Academy of Sciences, Szeged, Hungary, where he obtained a PhD in Chemistry in 2002. Following a two year postdoctoral fellowship at the AIST Tohoku, Sendai, Japan, he was appointed Senior Research Associate in the Stereochemistry Research Group of HAS. His research focuses on metal and organocatalysed asymmetric reactions, mostly involving the development of novel heterogeneous asymmetric catalytic systems for the preparation of chiral organic intermediates. |
Introduction
Presently, the most important challenge in organic chemistry is to develop procedures having low environmental impact, which are able to produce target compounds in the desired quantities. Recent synthetic procedures have focused on green and sustainable alternatives for synthesizing complex organic compounds by innovative approaches. The widespread application of catalysis by metal complexes over the last few decades, the remarkable development of organocatalysis in this century, the introduction of alternative reaction media (water or supercritical fluids), the utilization of novel activation methods (ultrasound, microwave, and mechano-chemistry), and exploitation of the special properties of materials developed by material scientists (ordered micro- or mesoporous materials, layered materials, inorganic–organic hybrids, nanomaterials, and carbon allotropes, such as nanotubes or graphene) have contributed to the improvement of chemical processes, which tend to satisfy the increasingly strict environmental regulations.1,2Most of the modern methods of preparing organic fine chemicals incorporate catalytic reactions as key synthetic steps. In the last few decades, development of novel catalysts or finding new catalytic applications for materials reported previously attracted significant attention.3 The trends of replacing the widely applied soluble catalysts with insoluble, heterogeneous materials, were motivated by the simplicity of removal, regeneration and reuse of the latter.4 Nowadays, sustainability is hardly imagined without applying heterogeneous catalysts, which also allow process intensification in flow systems, accompanied by space, time and energy savings.5,6
The increased demand for optically pure chiral intermediates of the pharmaceutical industry and the introduction of green and sustainable chemistry concepts in the preparation of high value-added fine chemicals have accelerated the development of asymmetric catalytic procedures.7–16 Several types of chiral catalysts allowing high stereocontrol have been reported to date. Besides biocatalysts such as enzymes,8,12 metal complexes bearing chiral ligands and organic molecules used as asymmetric organocatalysts have found numerous applications in organic synthesis. However, in many cases, the cumbersome and costly preparation of these catalysts have motivated studies aimed at preparing similarly efficient and selective heterogeneous catalytically active chiral solids.17–19
A simple method for obtaining chiral solid catalysts that were found to be surprisingly efficient in a limited number of reactions, is the surface modification of heterogeneous catalysts such as metals or metal oxides, by optically pure, preferably cheap, natural compounds.17–24 In the asymmetric hydrogenation of certain unsaturated compounds, exceptionally high enantioselectivities were attained over chirally modified metal catalysts. However, widespread application of these catalysts in the fine chemical industry was hampered by their narrow substrate scope.
Another general approach to preparing chiral, catalytically active solid materials is the immobilization of efficient homogeneous catalysts, whether chiral complexes, organocatalysts or enzymes, on solid support by various methods.18,19,25–31 The large variety of highly efficient chiral homogeneous catalysts warrants the possibility of developing solid catalysts for numerous applications. Furthermore, the selection of an appropriate anchoring method and choosing supports with special properties designed for specific purposes have often led to enhanced results, as compared to those obtained using the corresponding soluble catalytic species. These types of immobilized chiral catalysts usually possess all the favourable properties of the heterogeneous catalysts. Still, their application is obstructed by their tedious preparation procedures and by preserving some drawbacks of the corresponding homogeneous catalysts, which are often expensive materials; many of them are highly sensitive to manipulations and require high purity reaction components. However, oftentimes by appropriate material design, robust and stable chiral catalysts have been obtained, which allow preserved performances upon recycling, and the possibility of application in flow systems.32–34
Further increase in the sustainability of the catalytic reactions may result from carrying out several reactions in a single vessel, without isolation of the intermediate products in a so-called one-pot procedure.35–38 One-pot reactions that allow two or more bond-forming transformations under the same conditions without intervention and with subsequent transformations occurring at the functionalities resulting from a previous step, are designated domino or cascade reactions.39,40 Contrary to the stop-and-go methods, during these reactions, all components are introduced from the onset and only the purification of the final product is necessary, as shown in Fig. 1. Difficulties in designing cascade processes are attributable to requirements including the following: all reagents and catalysts needed in different steps must tolerate each other's presence; all reactions must occur under the same conditions; functional groups present in the molecules should participate only in the desired steps. However, due to their operational simplicity and enormous advantages provided by the material, energy, space and time savings, as compared with the classical stop-and-go methods, extensive efforts are devoted to the development of such synthetic procedures.41–45
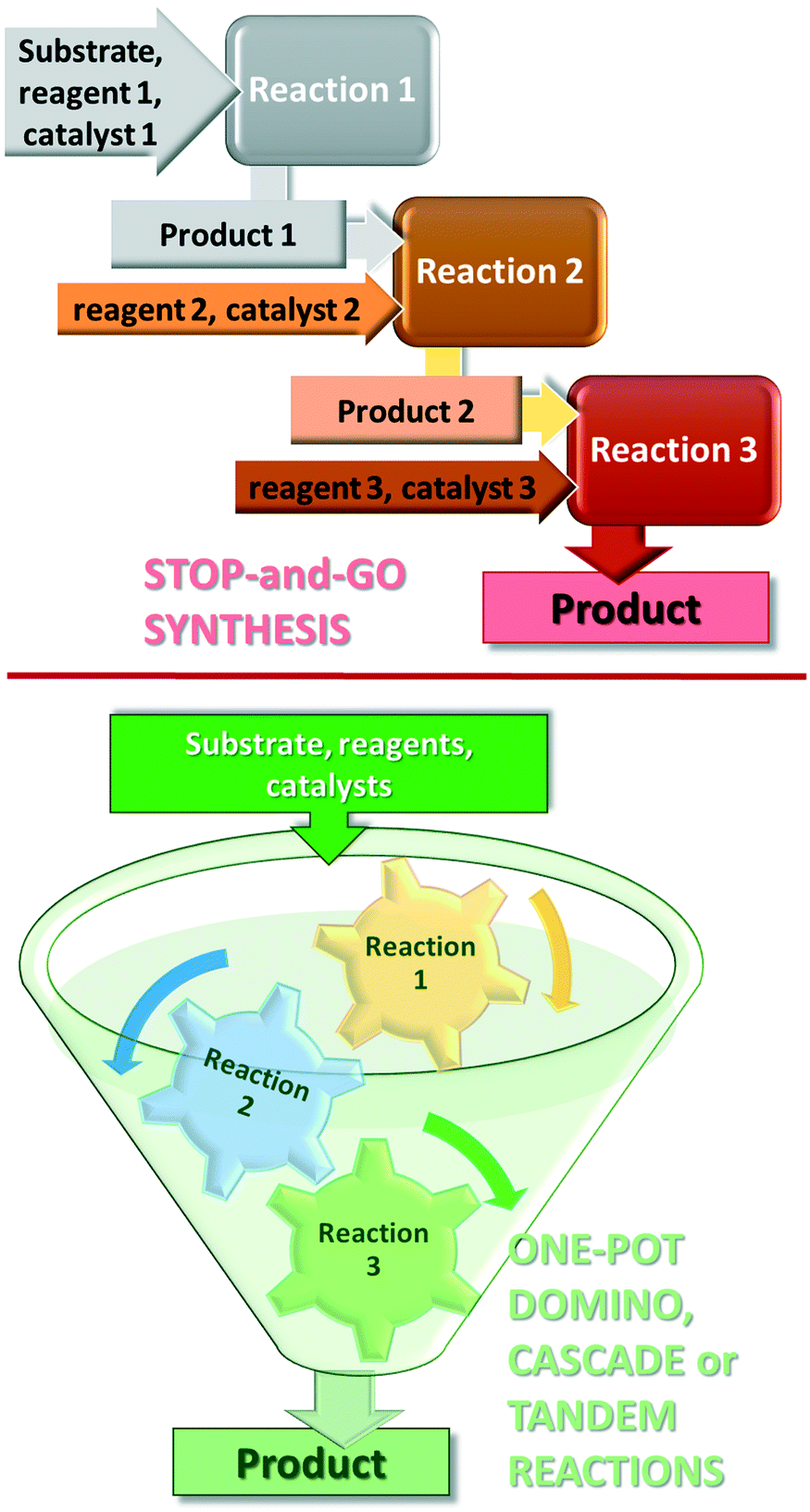 | ||
| Fig. 1 Schemes of the stop-and-go and one-pot domino, cascade or tandem synthesis procedures. | ||
During the last decade, the availability of a large variety of chiral catalysts7–19 has been the driving force for the development of asymmetric catalytic one-pot procedures.46–56 Such reactions could be applied in the synthesis of optically pure building blocks and active pharmaceuticals.36–39 Enzymes, chiral metal complexes and organocatalysts, either as single catalysts or their various combinations with achiral or a second chiral catalyst, have been used in these reactions, allowing the preparation of complex organic molecules such as natural products and other bioactive compounds by significantly simplified procedures.57–59
Although extraordinary complexity may be easily achieved in these reactions, the recovery of the expensive chiral catalysts remain a major task. Accordingly, asymmetric catalytic one-pot reactions using recyclable heterogeneous catalysts have also been investigated. Among these were reactions catalysed by heterogeneous achiral materials in combination with chiral soluble catalysts and procedures in which the stereoselective step itself was promoted by a chiral solid catalyst and was mostly combined with another catalyst or with a spontaneous, uncatalysed reaction step. Nowadays, the number of reported asymmetric heterogeneous catalytic one-pot reactions utilizing chemical catalysis is growing rapidly. However, an overview of these reactions is still lacking. Some of these have been incorporated in monographs or reviews having broader scope, such as asymmetric reactions catalysed by transition-metal functionalized supports, domino or cascade reactions catalysed by heterogeneous catalysts31,45,60–62 or recently, in surveys covering a narrower area or a different segment of such reactions, i.e. processes catalysed by combinations of enzymes and inorganic heterogeneous catalysts, by bio-nanocatalysts, or aminocatalysts combined with metals.56,63,64
The aim of the present review is to give a comprehensive survey of the asymmetric one-pot reactions in which heterogeneous chemical catalysts have been applied. Those procedures are included in which in a catalysed step stereoselective formation of at least one new chiral centre occurs. Reactions are classified according to the nature of the chiral catalyst used in the enantioselective step, i.e. homogeneous, heterogeneous and biocatalytic. Biocatalysts, especially enzymes, are among the most often used stereoselective catalysts,8,12,39,65 which have also been applied in asymmetric one-pot processes,66–68 often in combination with chemical catalysts.39,63,64,69–71 The scope of the present review is limited to asymmetric one-pot processes utilizing heterogeneous chemical catalysis; accordingly, only those reactions will be included in which biocatalysts are used along with such materials. Dynamic kinetic resolutions (DKR) unite the kinetic resolution (KR) of racemic substrates, often catalysed by enzymes, with racemization of the unreacted enantiomers, and are therefore considered biocatalytic cyclic cascade reactions.67 Although the racemization step may be carried out using homogeneous or heterogeneous chemical catalysts,72–82 unless other heterogeneous catalytic steps are included in the process, these will be omitted, as only a part (ideally half) of the chiral compounds are involved in both steps. Procedures utilizing heterogeneous chiral chemical catalysts will be further divided according to the catalyst type, i.e. immobilized metal complexes, heterogeneous organocatalysts and chirally modified catalytic surfaces. The reaction pathways making possible these processes will be briefly discussed.
Before reviewing the asymmetric heterogeneous catalytic one-pot transformations, definitions of terms and classification of the one-pot processes is necessary. Without debate, one-pot reactions are defined as processes that allow more than one reaction to occur in a single flask, without isolation of the intermediate products. Introduction of reagents or catalysts between steps, or changes in the reaction conditions, may be necessary during these processes. Domino or cascade reactions are considered one-pot transformations in which all components, such as substrates, reagents, catalysts, auxiliaries, solvents etc., are introduced in the system at the onset and the reaction conditions are not altered during the process. Accordingly, two or more bond forming transformations occur without any intervention.40 In most of the catalytic cascade reactions reported recently, more than one catalyst or multifunctional catalysts are used. These reactions are defined as tandem catalytic processes: auto-tandem, assisted tandem (when a single catalyst is applied) or orthogonal catalytic processes (using more than one catalysts).41 According to a different approach, domino or cascade reactions are those one-pot transformations in which the intermediates cannot be isolated and the individual steps cannot be performed separately. One-pot reactions performed sequentially and with intervention between steps were denoted as tandem reactions.83 However, this latter definition diverges from the most often used terminology. Recently a novel classification of the catalytic one-pot reactions was proposed to suit better discoveries in this field.84 The so-called tandem reactions were divided into cooperative and relay catalytic processes, whether the catalysts share the same catalytic cycles or not (Fig. 2). Relay catalysis was further divided into self-relay and orthogonal relay reactions, depending on the types and number of catalysts. Unlike sequential catalytic processes during which intervention between steps is necessary, all catalysts are present at the onset of the cooperative and relay catalytic procedures.84
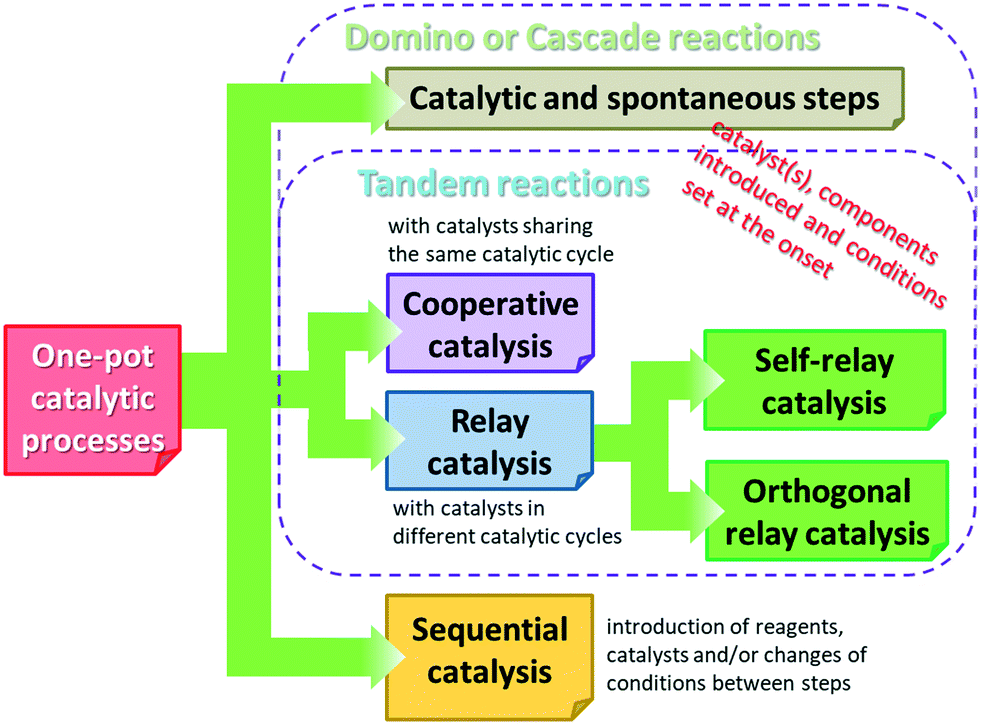 | ||
| Fig. 2 Scheme of classification of the catalytic one-pot processes. | ||
In spite of these classifications and denominations, often the general term “cascade reaction” is used for one-pot catalytic processes. In order to avoid confusion, in this review denominations met in the original reports are used, even if they are not in agreement with the above taxonomies. For clarity, intermediates that could not be or were not isolated are included in square brackets in schemes.
1. Heterogeneous catalytic one-pot reactions including homogeneous asymmetric steps
The last two decades of developments in organic chemistry resulted in the discovery of a large structural variety of highly efficient soluble chiral catalysts. Still, only few asymmetric catalytic one-pot reactions are known, which apply heterogeneous achiral solids combined with chiral soluble catalysts. A possible explanation of the scarcity in reports could be the often observed undesired interaction of the two types of catalysts leading to diminished performances of at least one of the active species, which compromises the formation of the target compounds. Besides, another, more practical reason is that the chiral catalysts are usually the more expensive, which should be recovered; thus the use of an achiral reusable heterogeneous catalysts is less attractive or important, than applying a chiral solid. Consequently, such reactions are by far less studied, as one would expect based on the vast number of efficient chiral homogeneous catalysts.In their pioneering studies Hénin, Muzart and co-workers, described the hydrogenation of unsaturated benzyl carbonates or benzyl β-keto esters over Pd/C in the presence of chiral amino alcohols, which afforded optically enriched α-substituted ketones (Scheme 1).85–87 In the early reports, 2-methyl-1-indanone (3) and 2-methyl-1-tetralone (4) were obtained in up to 52% and 50% enantiomeric excesses (ee) from the corresponding β-keto esters 1 or 2,86 whereas the benzyl carbonate 5 gave the optically enriched ketone 4 in 64% ee.87 Several chiral diamines and amino alcohols were tested,88,89 and the best results were obtained with (+)-endo-2-hydroxy-endo-3-aminobornane (Scheme 1, CA2*). Optimization of the reaction conditions using this chiral amine afforded 3 in up to 99% ee in the cascade reaction of 1.88 Later, the scope of the reaction was extended to various α-substituted-β-keto esters and enol carbonates. Representative substrate structures along with the best results obtained are summarized in Fig. 3.88–95 In most of these reactions, high yields were reached accompanied by good ees (70 ± 5%). Prominent ee was reported only in the reaction of 1.88 It is noteworthy that with alterations in the structure of the substrate, the most efficient chiral amino alcohol was also varied, and thus in some reactions cinchona alkaloids CA4* or CA5* outperformed CA2* with respect to the attained ee. Therefore, the most efficient chiral base is also given next to the results (Fig. 3).
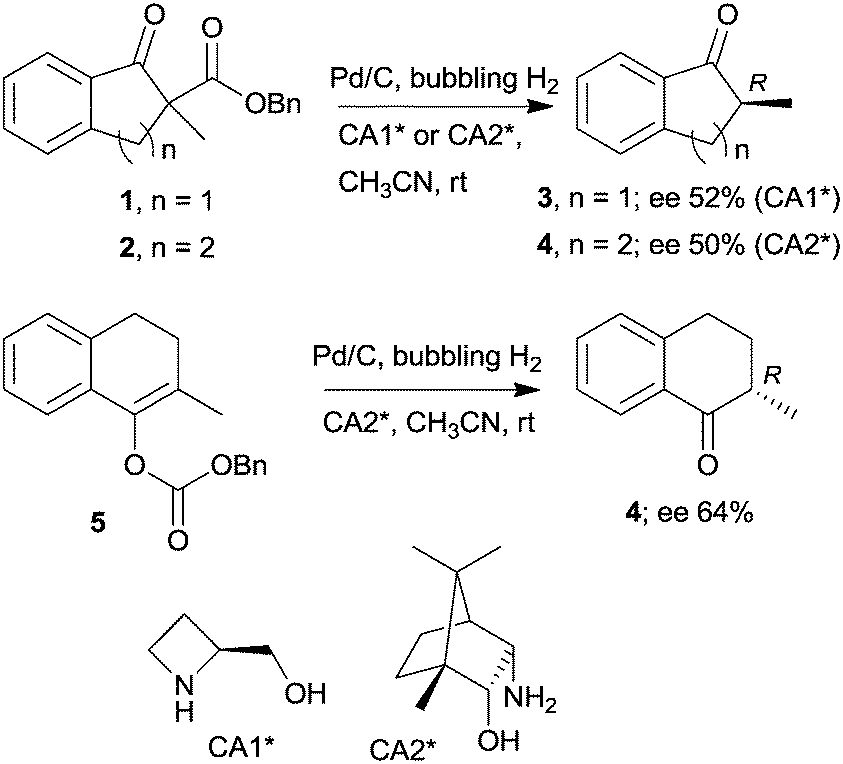 | ||
| Scheme 1 Enantioselective cascade reactions of β-keto esters or benzyl carbonates initiated by Pd/C leading to optically enriched 2-alkyl ketones; Bn: benzyl.85–88 | ||
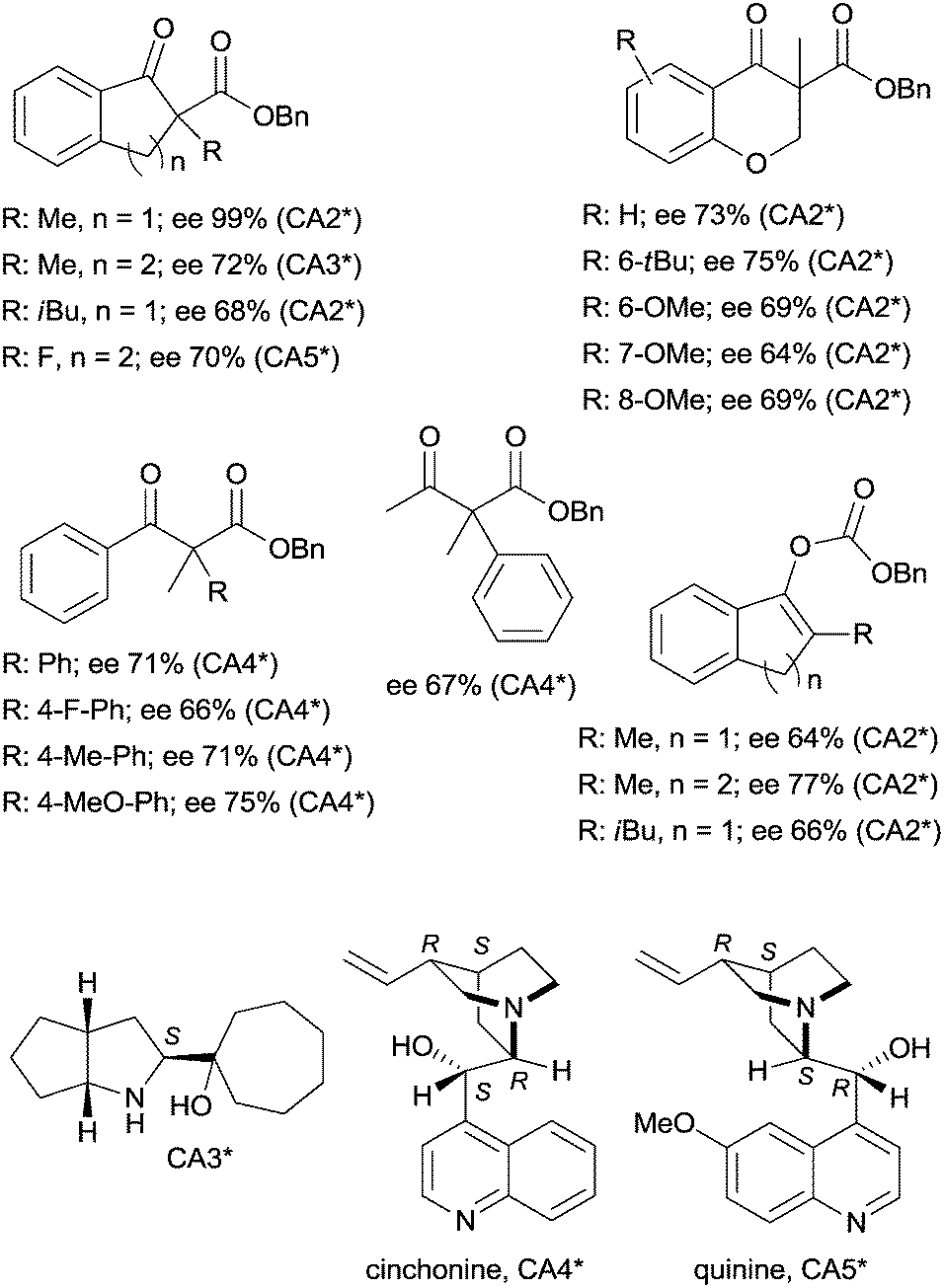 | ||
| Fig. 3 Structure of benzyl β-keto esters and benzyl carbonates transformed in the Pd initiated heterogeneous enantioselective cascade reaction to α-substituted ketones and chiral amino alcohols used in these reactions.88–93 | ||
The heterogeneous Pd catalyst initiates the above cascade reactions through hydrogenolysis of the benzyl esters or carbonates. This step is followed by decarboxylation and enantioselective protonation, with both steps possibly assisted by the chiral amine. Thus, the first step of the cascade takes place on the Pd surface, leading to the formation of the corresponding β-keto acids as was indicated by UV spectroscopy.94 For a long time, it was uncertain whether the second and third steps occur in solution as organocatalysed processes or as a surface reaction over the Pd catalyst modified by the chiral amine.22 Later, based on results obtained using cinchona alkaloids and other chiral amino alcohols of different adsorption strengths, it was suggested that the decarboxylation is catalysed by the chiral base in solution, followed by the stereoselective protonation also directed by the amino alcohol.95 Kinetic studies of decarboxylation by NMR, IR and UV spectroscopy using cinchona alkaloids confirmed that following hydrogenolysis the dominant reaction route was catalysed by the chiral compound in the liquid phase as a homogeneous asymmetric organocatalytic reaction (Scheme 2).96
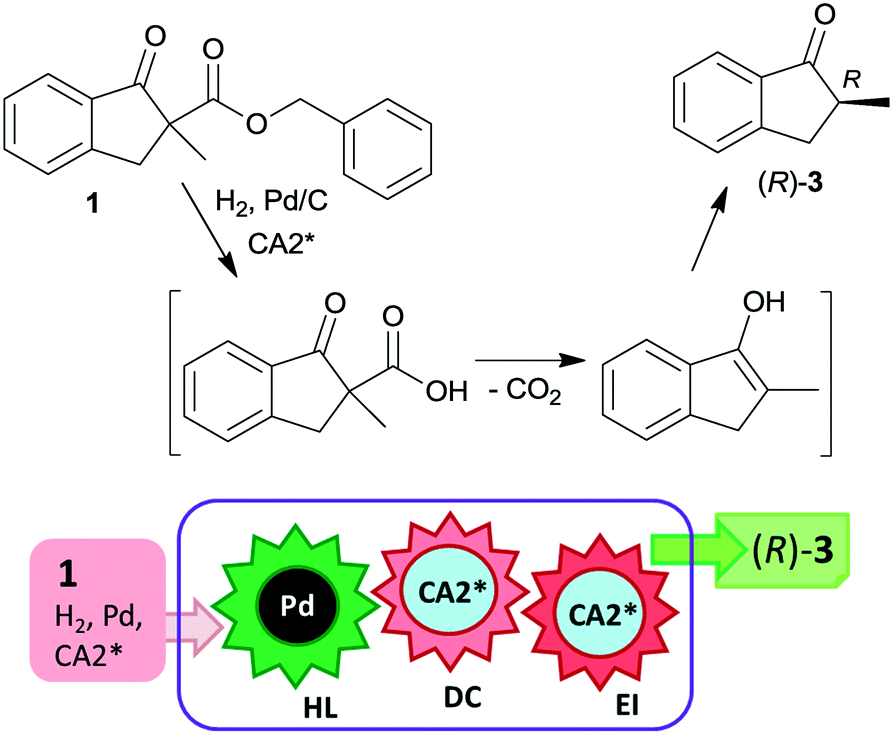 | ||
| Scheme 2 Asymmetric catalytic cascade transformation of 1 using heterogeneous Pd catalyst and chiral amine organocatalyst; HL: hydrogenolysis, DC: decarboxylation and EI: enantioselective isomerization; the catalysts that promote the steps are given in the circles. | ||
Recently, asymmetric cascade reactions using combinations of a soluble chiral organocatalyst and supported Pd catalysts were developed for the preparation of chiral five membered unsaturated cyclic compounds.97 Good yields and high stereoselectivities were obtained in reactions of cinnamaldehyde derivatives (6) with propargylic C-, O- or N-nucleophiles (7, 9, 11), leading to the formation of cyclopentene, dihydrofuran or pyrroline derivatives (Scheme 3). Combinations of two catalysts were used in these reactions. One is a soluble pyrrolidine derivative chiral organocatalyst (CA6*) reported initially by Jørgensen and co-workers98 and Hayashi and co-authors,99 efficient in several organocatalysed reactions, including cascade reactions.100–103 The second achiral heterogeneous catalyst was home-made, with either Pd(II) or Pd(0) nanoparticles (NPs) anchored on aminopropyl-functionalized silica-based mesocellular foam (the latter material being prepared by mild reduction of the former). Interestingly, the Pd NPs/support usually provided slightly better diastereomeric ratio (dr) and ee than the immobilized Pd complex Pd(II)/support. By recycling of the immobilized Pd(II) catalyst, improved activity and stereoselectivity were obtained, as a consequence of the adsorption of CA6* on the solid material, which increased the organocatalyst amount in the successive runs (additionally to the freshly added amount).
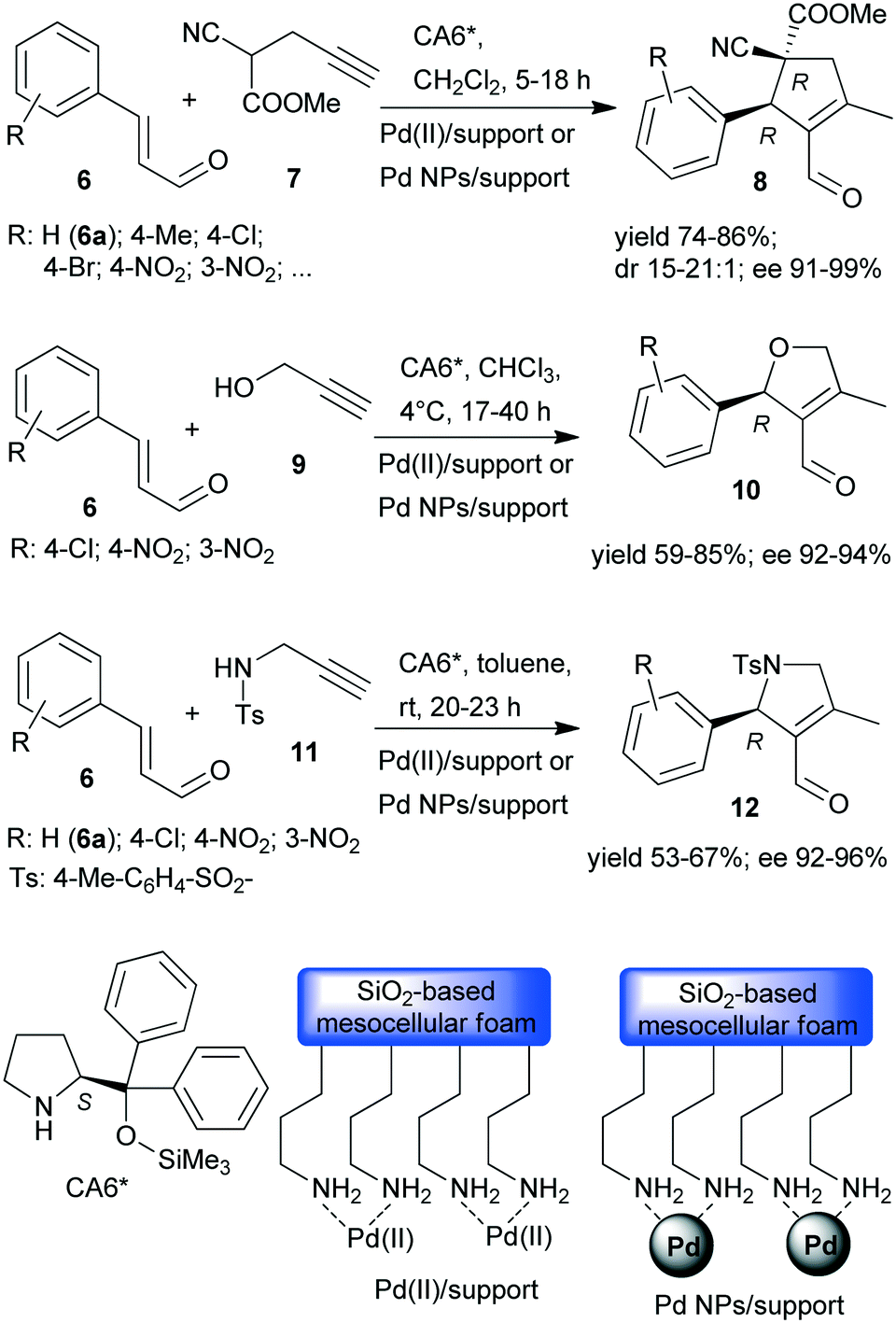 | ||
| Scheme 3 Asymmetric cascade reactions of unsaturated aldehydes and nucleophiles bearing the propargylic group, catalysed by chiral amine and heterogeneous Pd catalysts.97 | ||
The cascade reaction included a homogeneous enantioselective organocatalysed Michael addition and a supported Pd(II) or Pd(0) NPs catalysed intramolecular carbocyclization, as shown in Scheme 4. The authors demonstrated that the second cyclization step proceeds via a heterogeneous pathway. Accordingly, the reaction is a heterogeneous variant of the earlier reported homogeneous one-pot dynamic kinetic asymmetric transformation.104,105 The initial amine-catalysed reversible asymmetric Michael addition is followed by Pd catalysed intramolecular stereoselective irreversible cyclization of the two diastereomeric enamine intermediates. The different cyclization rates of the stereoisomers determined the stereochemical course of the reaction.
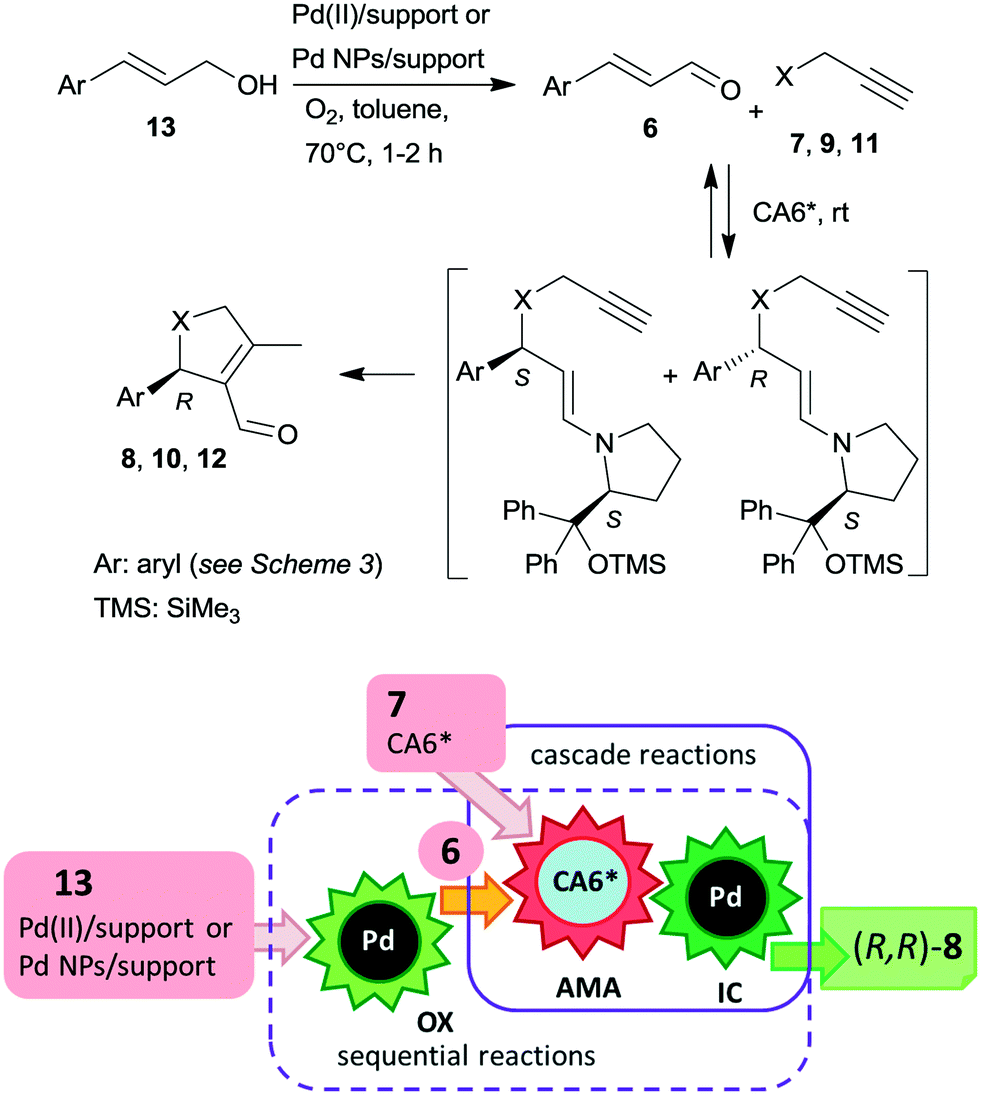 | ||
| Scheme 4 Mechanism of the asymmetric cascade reaction of unsaturated aldehydes and alkyne nucleophiles using chiral organocatalyst CA6* and Pd catalyst coupled with sequential preparation of the aldehydes by oxidation (OX); AMA: asymmetric Michael addition, IC: intramolecular cyclization. | ||
The scope of the above reaction was also extended to the preparation of saturated carbocyclic compounds by the reaction of olefin nucleophiles containing leaving groups in allylic position.106 Later, the cascade reaction was complemented by the in situ preparation of the α,β-unsaturated aldehyde in a one-pot manner by oxidation of the corresponding allylic alcohols (13) with O2.107–109 Although the supported Pd catalysed both the oxidation and the final carbocyclization steps, the reaction temperature was modified and 7 and CA6* was introduced following the oxidation of the allylic alcohol, leading to sequential one-pot reactions coupled with the above cascade process, as illustrated in Scheme 4. Heterogeneous catalysts prepared by immobilization of Pd(II) species over polyimine or azolinked porous polymer supports were also successfully used.110,111
The same chiral organocatalyst (CA6*) was also applied in the tandem asymmetric Michael addition, followed by the asymmetric photocatalytic oxamination process. In the latter visible light-induced step, a heterogeneous TiO2-bound Ru photocatalyst (N719/TiO2) was employed in order to increase the catalytic activity of the photoredox material.112 By this unprecedented tandem iminium/heterogeneous photoinduced catalytic reaction, α,β-substituted aldehydes (16) were obtained in reactions of aromatic unsaturated aldehydes 6, diethyl malonate (14) and 2,2,6,6-tetramethyl-1-piperidinyloxy free radical (15) in good yields, high diastereomeric excesses (de) and excellent ees, as shown in Scheme 5. Recycling of the solid photocatalyst resulted in a gradual decrease in the yield of 16. The role of the heterogeneous photocatalyst in this process was to generate the radical cation from the enamine of the Michael adduct, which reacts with 15 in the second step.
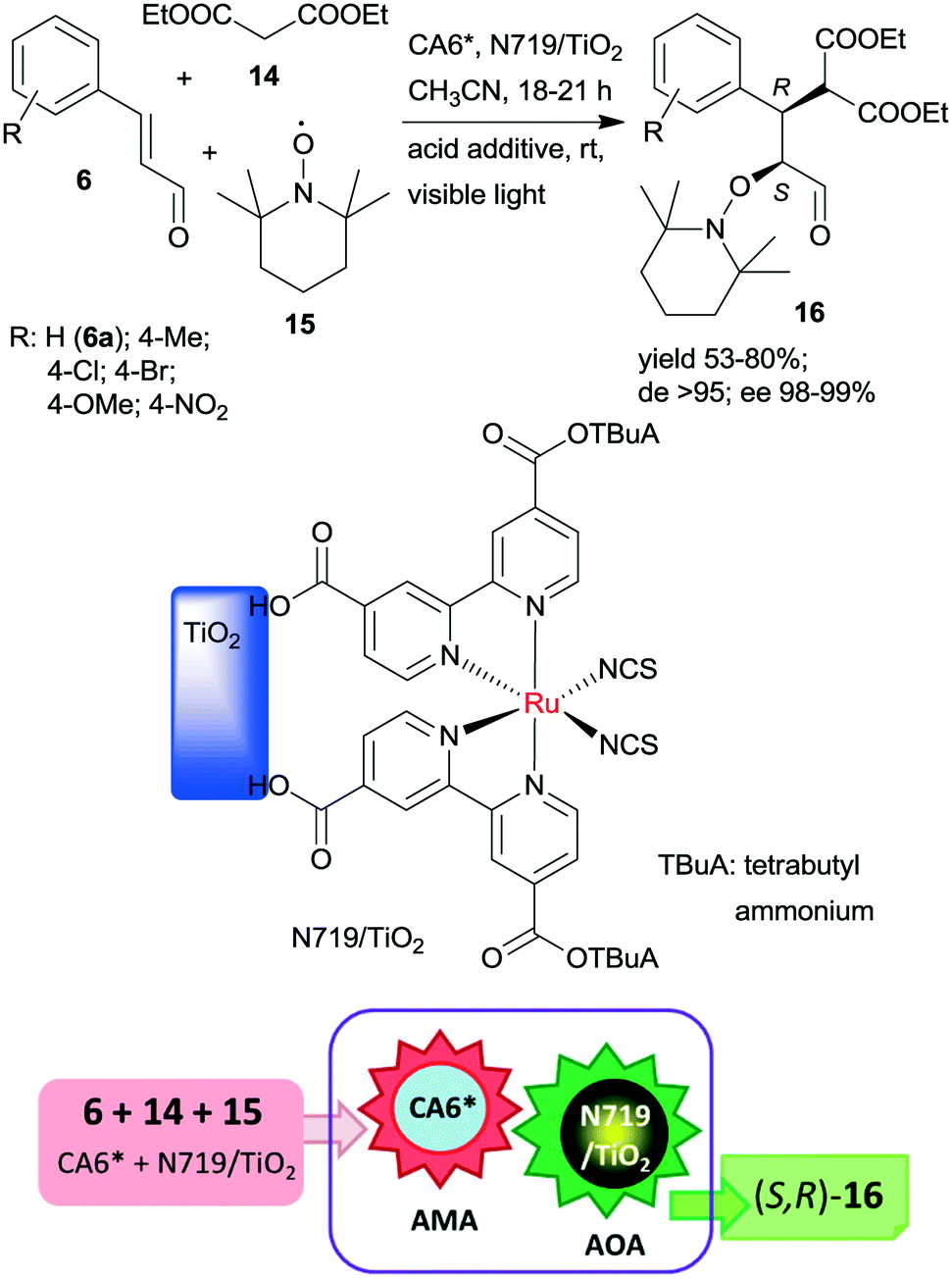 | ||
| Scheme 5 Asymmetric tandem AMA and asymmetric oxamination (AOA) of unsaturated aldehydes using CA6* and heterogeneous photocatalyst N719/TiO2.112 | ||
Besides the above cascade reactions, sequential one-pot transformations using combinations of homogeneous chiral and heterogeneous achiral chemical catalysts have also been described. Asymmetric allylic alkylation and the Pauson–Khand reaction sequence was catalysed by the successive addition of the homogeneous Pd complex formed with the chiral ligand CL1* and the Co/C heterogeneous catalyst (Scheme 6a). The second step of this one-pot reaction also required changes in the conditions, such as increase of the temperature and introduction of CO. Interestingly, the use of Co2(CO)8 for catalysing the second step was inefficient.113 Similarly, during the one-pot asymmetric epoxidation followed by reductive ring opening of α,β-unsaturated amides (20), catalysed by chiral Sm-(S)-BINOL–Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O complex (BINOL: 1,1′-bi-2-naphthol) and Pd/C catalyst, the addition of the latter catalyst and introduction of H2 and MeOH were necessary between the two steps (Scheme 6b).114
O complex (BINOL: 1,1′-bi-2-naphthol) and Pd/C catalyst, the addition of the latter catalyst and introduction of H2 and MeOH were necessary between the two steps (Scheme 6b).114
 | ||
| Scheme 6 Asymmetric one-pot sequential processes using achiral heterogeneous and soluble chiral catalysts.113,114 | ||
Interestingly, until now only the above discussed heterogeneous asymmetric catalytic cascade or sequential one-pot reactions have been reported with the stereoselective steps catalysed by soluble chiral catalysts. The most important advantage of using the heterogeneous catalysts, i.e. their easy separation from the product, was the main reason for developing these systems. In one-pot reactions including the reductive step application of heterogeneous catalysts such as supported Pd was a convenient choice; thus, comparison with homogeneous catalysts was omitted. Still, the rarity of these one-pot reactions may be due to more efforts being dedicated to the recycling of the chiral catalysts and less elegance of methods in which a chiral catalyst is sacrificed, whereas the achiral heterogeneous catalyst, even if is a noble metal, is recovered and reused.
2. One-pot reactions including a heterogeneous catalytic asymmetric step
In contrast to the above reactions, much effort has been devoted lately to developing efficient one-pot processes using heterogeneous chiral catalysts. Chiral modification of catalytic solid surfaces would be the simplest approach to preparing such catalysts, however, the limited applicability of such systems has directed the attention of the researchers to using more arduously prepared homogeneous chiral catalysts anchored on solid supports. The latter, due to the large variety of available soluble chiral catalysts, solid supports and immobilization methods, may warrant efficient solutions to many challenges. However, the undesired interactions of reagents, catalysts needed in different steps and the often-necessary changes in reaction conditions have many times resulted in the development of sequential one-pot catalytic methods.2.1. One-pot reactions with asymmetric steps catalysed by anchored chiral metal complexes
Widespread application of metal complexes formed with chiral ligands in asymmetric catalytic processes have made their immobilization over insoluble supports an attractive way to obtain efficient heterogeneous enantioselective catalysts.26–29,31 However, in most instances, following immobilization, many of the unfavourable properties of the parent complexes were kept. Most importantly, their sensitivity to the impurities of the reactants and solvents and to the presence of other reagents and catalysts was the main obstacle to their application in one-pot reactions. Accordingly, only a few one-pot catalytic processes were reported using surface bonded chiral metal complexes, and part of these were applied in sequential procedures. With few exceptions, these materials were prepared by covalent bonding of the chiral ligand to insoluble supports followed by ex or in situ preparation of the anchored chiral complex employing an appropriate metal precursor.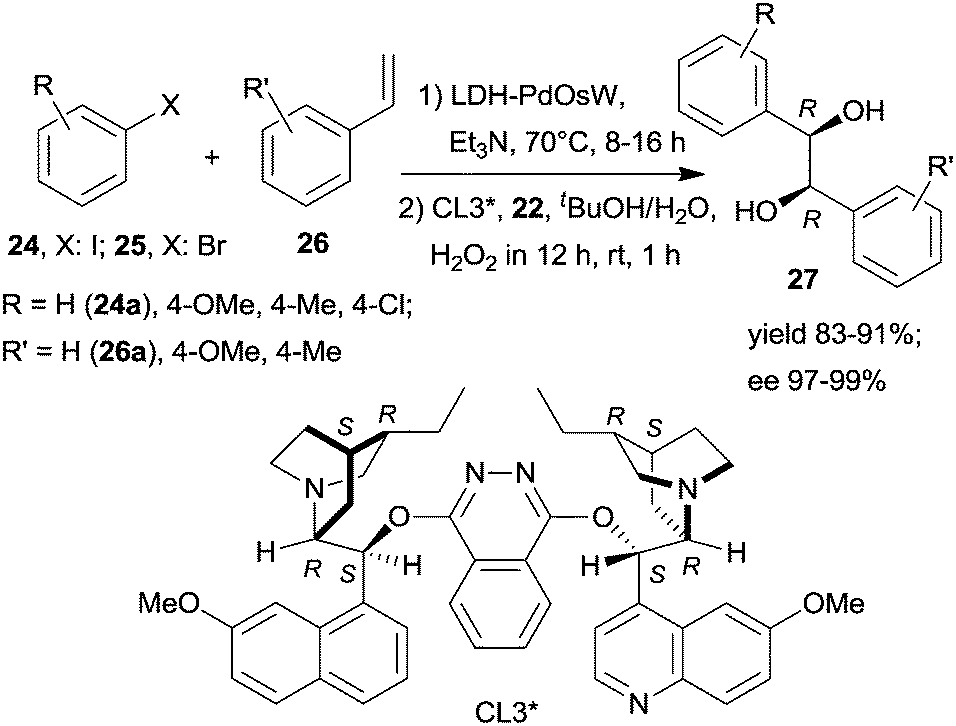 | ||
| Scheme 7 Heterogeneous sequential Heck reaction and asymmetric dihydroxylation coupled with tandem generation and consumption of the oxidant.115 | ||
Substituents on both the aryl halide 24 or 25 and the olefin 26 had only limited influence on results; moreover, acrylic esters also afforded optically pure diols. Steps of the reaction are illustrated in Scheme 8. The catalytic system was applied for the preparation of the dithiazem intermediate ethyl (2R,3S)-2,3-dihydroxy-3-(4-methoxyphenyl)propionate.118 Other supports, such as nanocrystalline MgO or a quaternary ammonium cation functionalized resin were also successfully applied for preparing bifunctional catalysts, such as MgO–OsW, resin–OsW and MgO–PdOs, resin–PdOs. The former two were used in the oxidation-asymmetric dihydroxylation tandem process, whereas the latter two in the Heck reaction-asymmetric dihydroxylation sequence in combination with 23 or K3Fe(CN)6 co-oxidants used in over stoichiometric amounts.119,120
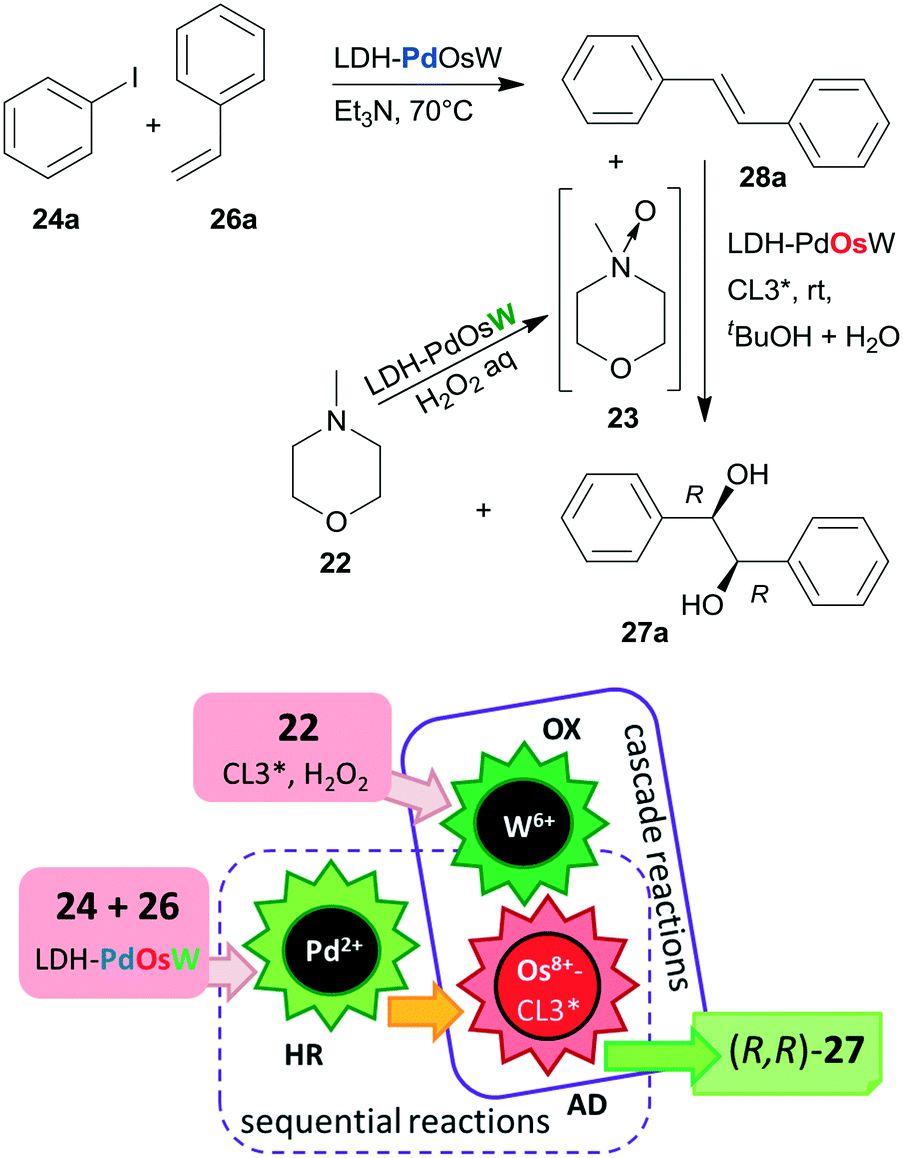 | ||
| Scheme 8 Heck reaction and asymmetric dihydroxylation using 23, obtained in situ from 22 over a trifunctional catalyst containing immobilized Os–CL3* chiral complex; HR: Heck reaction, AD: asymmetric dihydroxylation. | ||
The same authors immobilized the ligand CL3* on SiO2 surface by –On(OMe)mSi–(CH2)3–S– linker and used the chiral solid for the in situ preparation of the SiO2–CL3*–OsO4 complex. This material was efficient in the asymmetric dihydroxylation of olefins in a relay catalytic process, in which H2O2 was used as the terminal oxidant, and the oxidation of 22 to 23 was assisted by titanium silicalite heterogeneous catalyst.121 Furthermore, deposition of PdCl2 on unreacted OnSi–(CH2)3–SH surface groups of the functionalized support and reduction of the metal ions led to a catalyst containing both the anchored chiral ligand and Pd NPs. The material was used in the one-pot Heck reaction followed by the asymmetric dihydroxylation sequential process.122
Polymer supported chiral dirhodium(II)-complex [resin*–Rh2–(CL4*)3] was prepared from tetrakis[N-tetrachlorophthaloyl-(S)-tert-leucinate and N-phthaloyl-(S)-tert-leucine functional groups containing monomer copolymerized with achiral monomers (Scheme 9).123 This heterogeneous Rh complex provided similar results to its soluble dirhodium counterpart in the tandem carbonyl ylide formation and intramolecular cycloaddition reaction of 2-diazo-3,6-diketo esters (29) with 26a or phenylacetylene (30a). The reactions were highly stereocontrolled, both in batch and in flow systems.
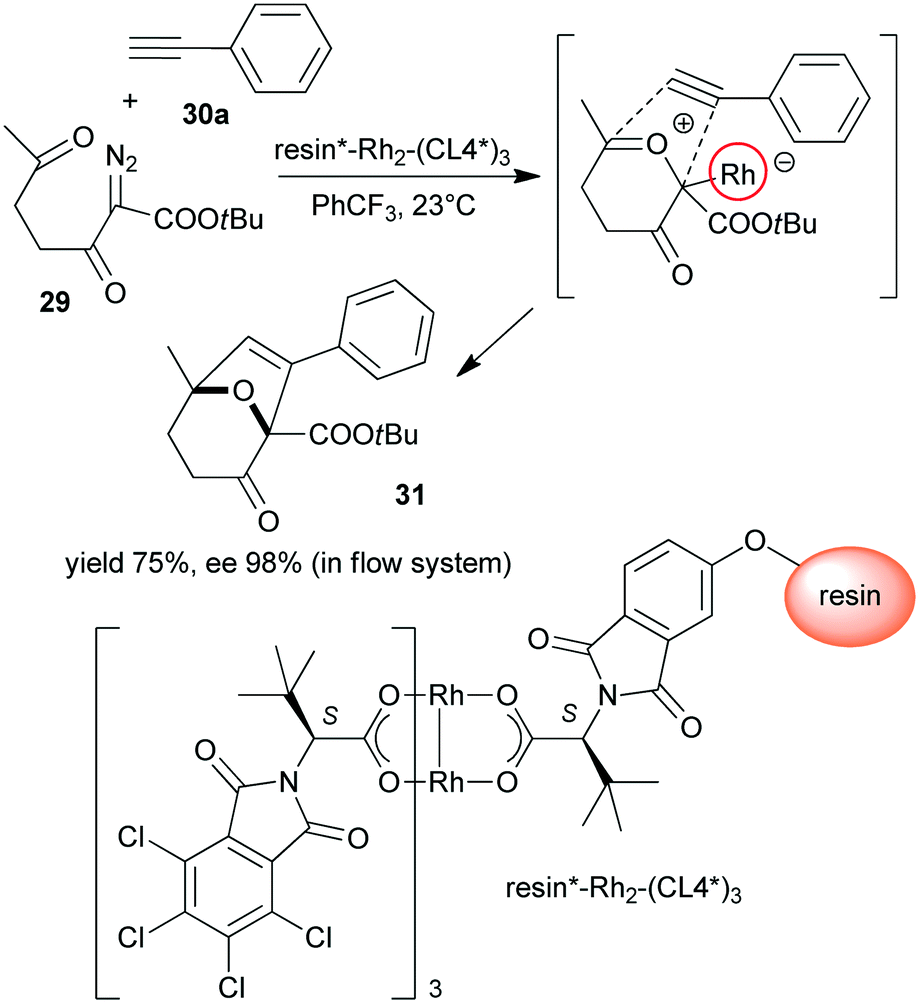 | ||
| Scheme 9 Heterogeneous tandem carbonyl ylide formation and cycloaddition reactions using immobilized dirhodium complex bearing tert-leucine derived chiral ligand.123 | ||
Periodic mesoporous organosilica (PMO) functionalized with (R,R)-1,2-diphenyl-1,2-ethylenediamine (R,R-32) through sulphonamide linker (CL5*) was used as a chiral insoluble ligand to immobilize a Rh complex formed from (CpRhCl2)2 precursor (Cp: pentamethylcyclopentadiene). This heterogeneous catalyst in combination with FeCl3 catalysed the hydration of 30a and the enantioselective transfer hydrogenation of the intermediate acetophenone (33a) in a cascade reaction providing similar results to the corresponding homogeneous Rh complex (Scheme 10).124 The heterogeneous chiral catalyst was recycled four times without significant loss in the activity or enantioselectivity.
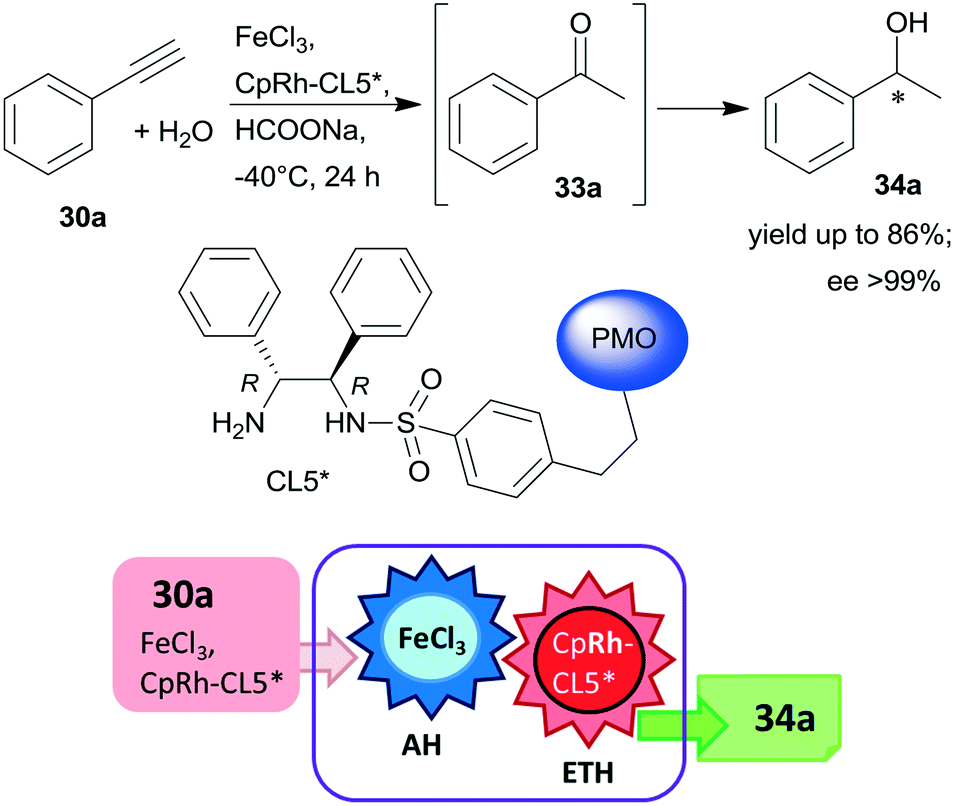 | ||
| Scheme 10 Preparation of chiral alcohol 34a from 30a by a heterogeneous catalytic cascade reaction using FeCl3 and chiral Rh complex catalyst immobilized on PMO; AH: alkyne hydration, ETH: enantioselective transfer hydrogenation.124 | ||
Liu and co-workers, prepared hollow-shell-structured (HSS) PMO nanospheres-bonded S,S-32 heterogeneous chiral ligand (CL6*), which was used for immobilization of Rh, Ir or Ru complexes.125 These materials were applied as catalysts in the tandem enantioselective transfer hydrogenation and lactonization of 2-acylarylcarboxylates 35 to chiral phthalides 37 using HCOONa hydrogen donor and water as solvent. The best results were obtained with the immobilized Rh catalyst (Scheme 11). During this process, the heterogeneous catalytic asymmetric transfer hydrogenation step preceded the intramolecular spontaneous cyclization of the reduced chiral intermediates 36. Remarkably, the solid catalyst was more active and enantioselective, than the corresponding soluble CpRh-S,S-Ts-32 complex (Ts: para-toluenesulfonyl), which was explained by the high hydrophobicity of the hollow-shell-structured nanospheres and the increased activity of the confined, uniformly dispersed chiral Rh species. Similarly high enantioselectivities were obtained in this tandem reaction by using a silica-supported catalyst prepared by sequential grafting of S,S-32 through benzenesulfonamide linker and acrylamide-acrylonitrile copolymer on SiO2 NPs, followed by complexation using (CpRhCl2)2 precursor. The resulting surface-bonded thermoresponsive polymer formed a closed shell, which locked the catalytically active sites at low temperature (15 °C), whereas at 40 °C the polymer turned into an extended form, making the chiral complex accessible.126 The above two heterogeneous catalysts were recycled without decrease in activity or enantioselectivity after 10 and 8 uses, respectively.
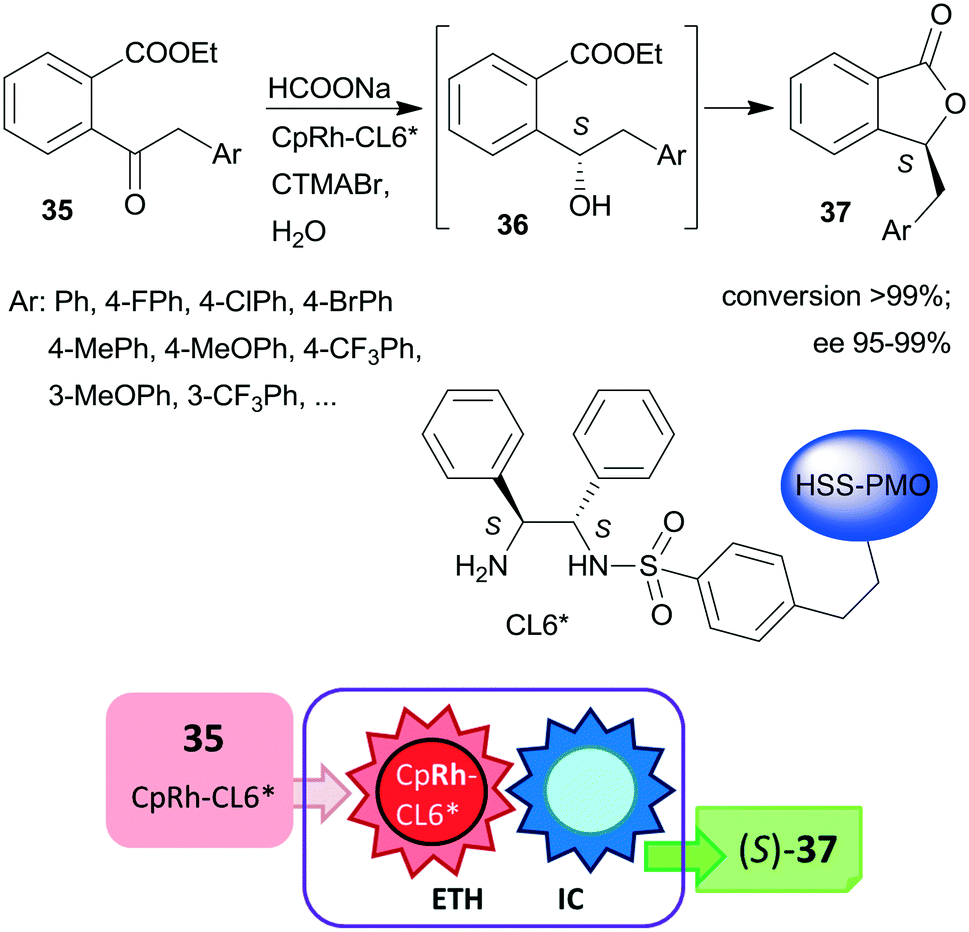 | ||
| Scheme 11 Heterogeneous catalytic preparation of phthalides by tandem ETH and spontaneous intramolecular cyclization (IC) using HSS-PMO immobilized chiral Rh complex; CTMABr: cetyltrimethylammonium bromide.125 | ||
Heterogeneous bifunctional PMO-supported catalysts were obtained by co-condensation of appropriately functionalized Pd and Ru complexes and were used in tandem catalytic processes, such as in ETH-Suzuki coupling, in Heck reaction-ETH or in Sonogashira coupling-ETH transformations (Scheme 12).127,128 The order of the steps in these one-pot cascade reactions was deduced based on the product distribution determined during the transformations, or in the case of the Heck reaction based on the enantiomeric ratios obtained with three iodine substituted acetophenone derivatives. All these processes afforded the corresponding chiral alcohols (40, 41, 42) in high yields and excellent enantioselectivities; moreover, the catalyst used in the ETH-Suzuki coupling kept its efficiency following 8 runs.127 The excellent performances of these catalysts were attributed to the presence of site-isolated, uniformly distributed Pd and Ru catalytic species within a single mesoporous support.
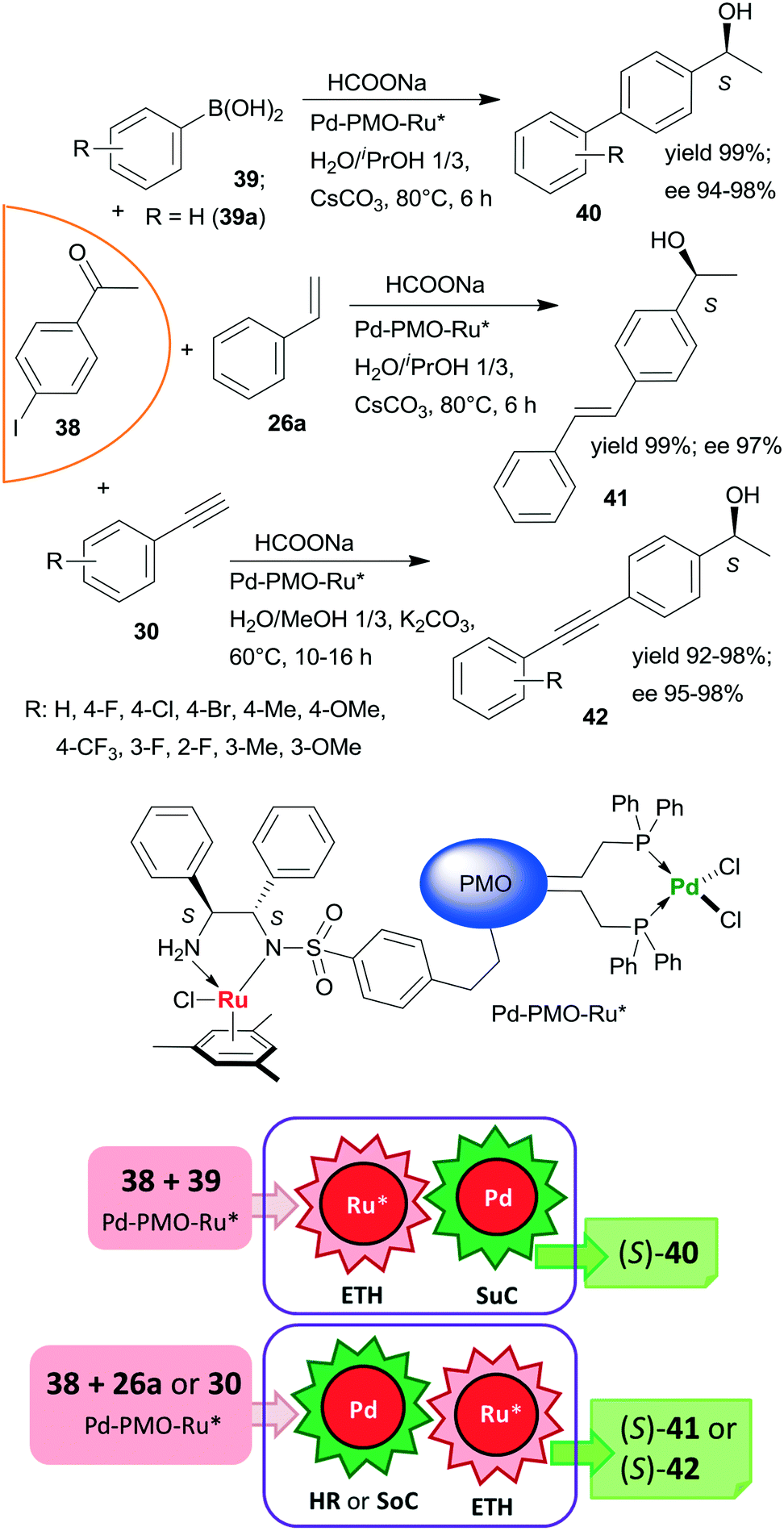 | ||
| Scheme 12 Bimetallic catalyst promoted tandem ETH – C–C coupling reactions of iodine substituted acetophenone 38; SuC: Suzuki coupling, SoC: Sonogashira coupling.127,128 | ||
Large-pore mesoporous silica (FDU-12) was recently used as support for preparing chiral Ru and Au-carbene complexes containing bifunctional heterogeneous catalyst (Scheme 13). Initially, the chiral ligand functionalized mesoporous silica was synthetized using the appropriate S,S-32 derivative, which by complexation with (MesRuCl2)2 (Mes: mesitylene) provided the anchored chiral Ru complex. Hydrogen bonding of Au(carbene)BF4 complex to the silanol groups of this material resulted in the bifunctional catalyst Au-FDU-12-Ru*, which was highly efficient in the tandem AH-ETH process of haloalkynes 43 to chiral halohydrins 45 through haloketone intermediates 44.129 The high yields and enantioselectivities attained using a series of both bromo- and chloroalkynes proved the wide applicability of this catalytic material, which was reused several times without significant loss in its performance. Mass transport limitations were prevented by using mesoporous material with large pore size, as indicated by similar results obtained with this material as compared with homogeneous counterparts applied as dual catalysts. Besides the uniformly distributed two metal complexes, the assistance of the surface silanol-functionalities also contributed to promoting this tandem reaction.129
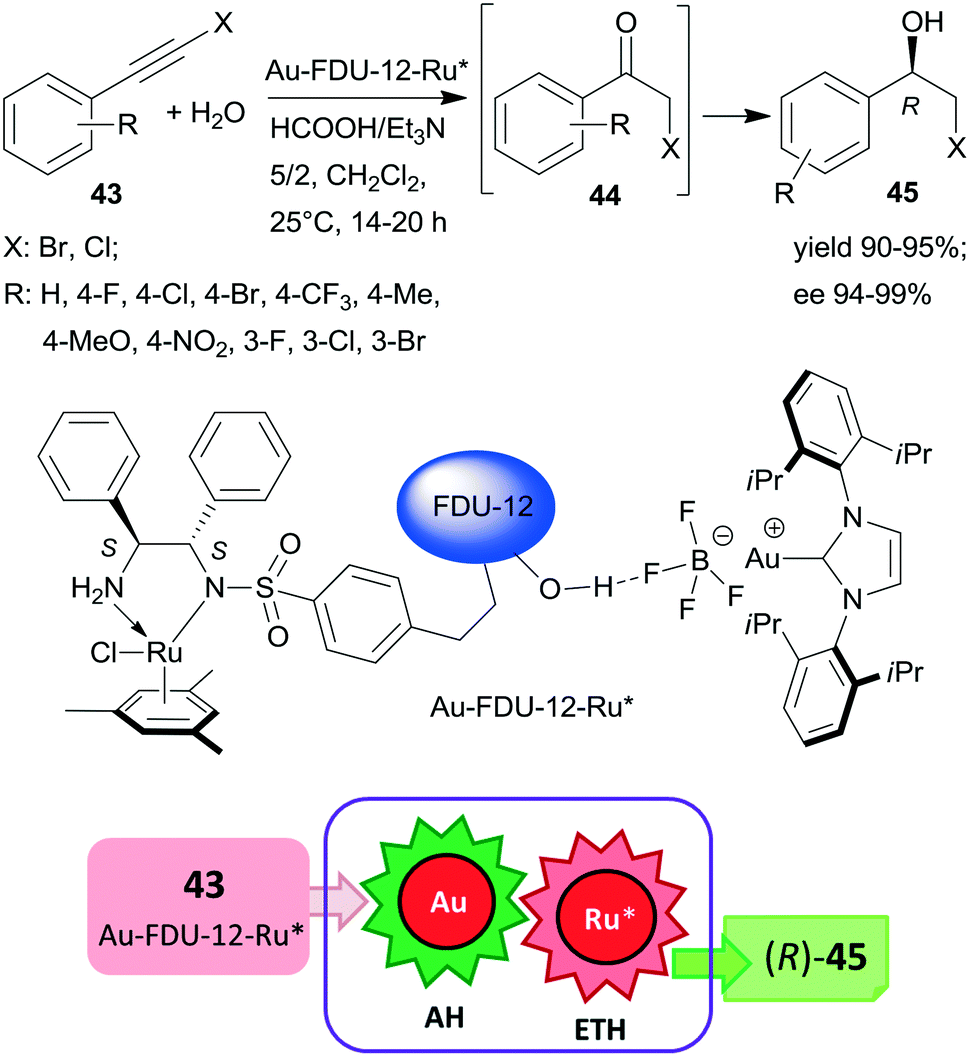 | ||
| Scheme 13 Preparation of chiral halohydrins 45 from haloalkynes 43 by tandem catalysis using large-pore mesoporous silica (FDU-12) immobilized Au-carbene and chiral Ru complexes.129 | ||
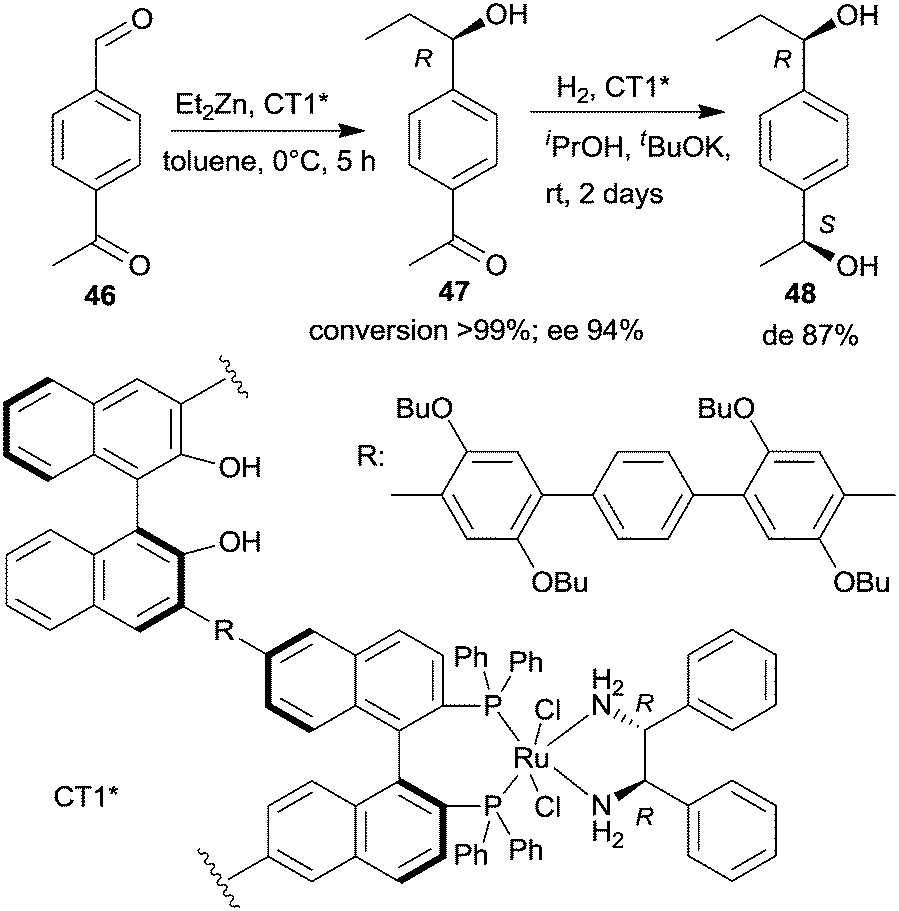 | ||
| Scheme 14 Sequential enantioselective Et2Zn addition – enantioselective hydrogenation by a chiral copolymer immobilized R,R-32-Ru-complex.130 | ||
Hydrogenation was the enantioselective step in a one-pot sequential preparation of L-alanine (51) starting from N-acetyl-dehydroalanine methyl ester 49 (Scheme 15).131 A previously developed heterogeneous chiral Rh-(S)-BINOL complex immobilized by simple ion-exchange on mesoporous Brønsted acidic aluminosilicate (CT2*)132,133 was used in the enantioselective hydrogenation of 49 to 50 under mild conditions and in water as the sole solvent. Thus, the enzymatic hydrolysis of the acetamido and ester groups could be carried out sequentially by the addition of an appropriate biocatalyst.
 | ||
| Scheme 15 Sequential enantioselective hydrogenation – enzymatic hydrolysis using a chiral Rh complex anchored by ion-exchange on mesoporous aluminosilicate.131 | ||
Sequential Suzuki couplings and enantioselective transfer hydrogenations were carried out by Liu and co-workers using a combination of two heterogeneous catalysts, one containing Pd immobilized on organic–inorganic hybrid silica (CT3), and the other carrying chiral Ru complexes formed within ethylene-coated Fe3O4 magnetic nanoparticles (MNPs) using surface bonded benzenesulfonamido-S,S-32 as chiral ligand (CT3*).134 The solvent, reactants and additives needed in both steps were introduced at the onset of the reactions, however, CT3* was added following the first step, when the reaction temperature was also decreased (Scheme 16). Close to complete conversion to biarylalcohols (40, 52) and high ees were obtained using various halogenated acetophenones (38, and others) and arylboronic acids (39). The scope of this system was evidenced using other iodo- and bromo-arylketones, acetylboronic acids and their application in HR-ETR sequences. The MNPs eased the separation of the two heterogeneous catalysts; CT3 could be recovered by centrifugation following removal of CT3* by an external magnet. The catalysts maintained their activity even in their ninth use.
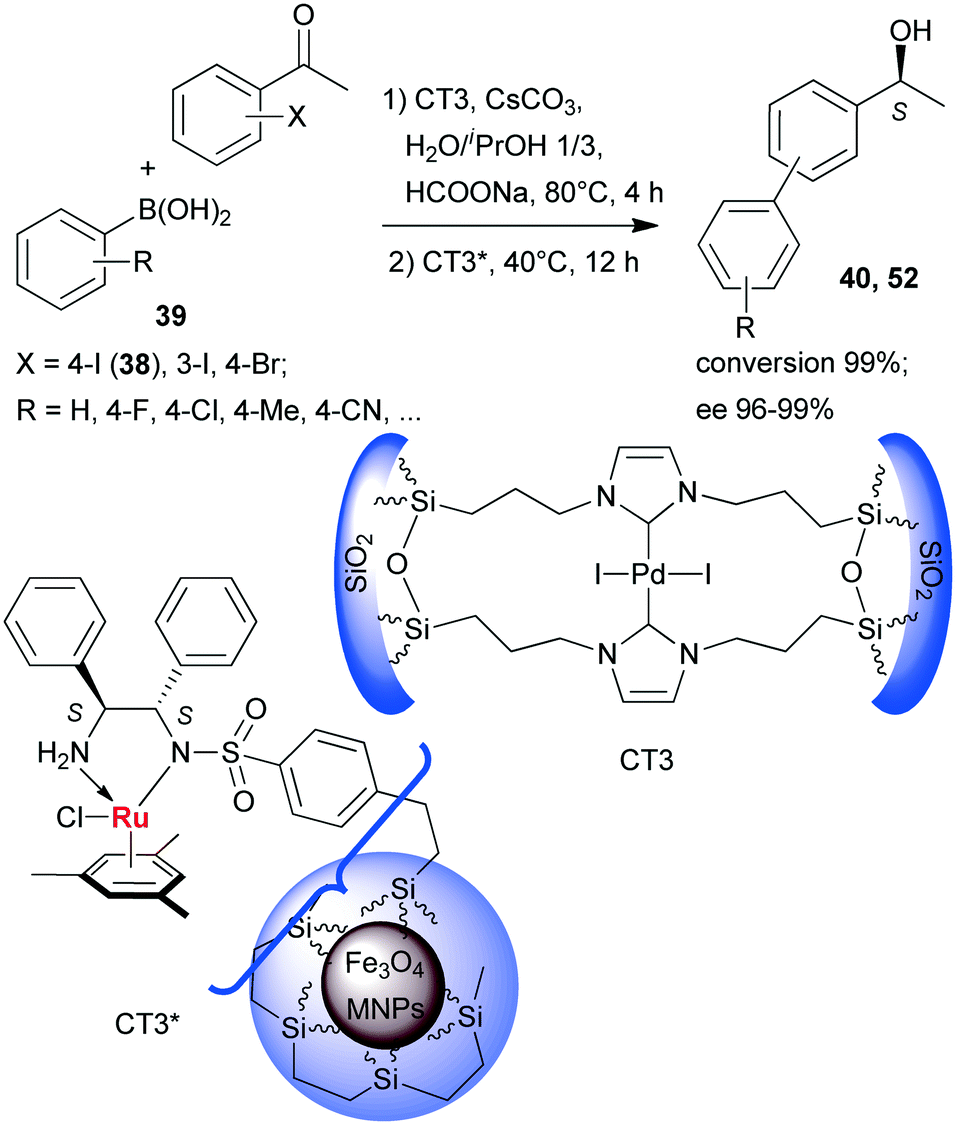 | ||
| Scheme 16 Sequential SuC–ETH reactions catalysed by two heterogeneous catalysts containing anchored Pd and chiral Ru complexes.134 | ||
The same research group prepared a catalyst using CL5*-like PMO-supported ligand (CL5’*) and (CyRuCl2)2 precursor (Cy: para-cymene). This material was used in the sequential enantioselective transfer hydrogenation of β-trifluoromethyl-α,β-unsaturated ketones 53 in water at room temperature and isomerization catalysed by RuCl(PPh3)3 at 70 °C by microwave irradiation.135 Several unsaturated ketones were transformed, obtaining the corresponding ketones 54 in high yields and excellent enantioselectivities (Scheme 17). The stability of the chiral heterogeneous catalyst was examined only in the ETH reaction, i.e. the first step of the sequence, thus, it is unknown whether following the second step, which was carried out under harsher conditions, the surface chiral sites were still preserved.
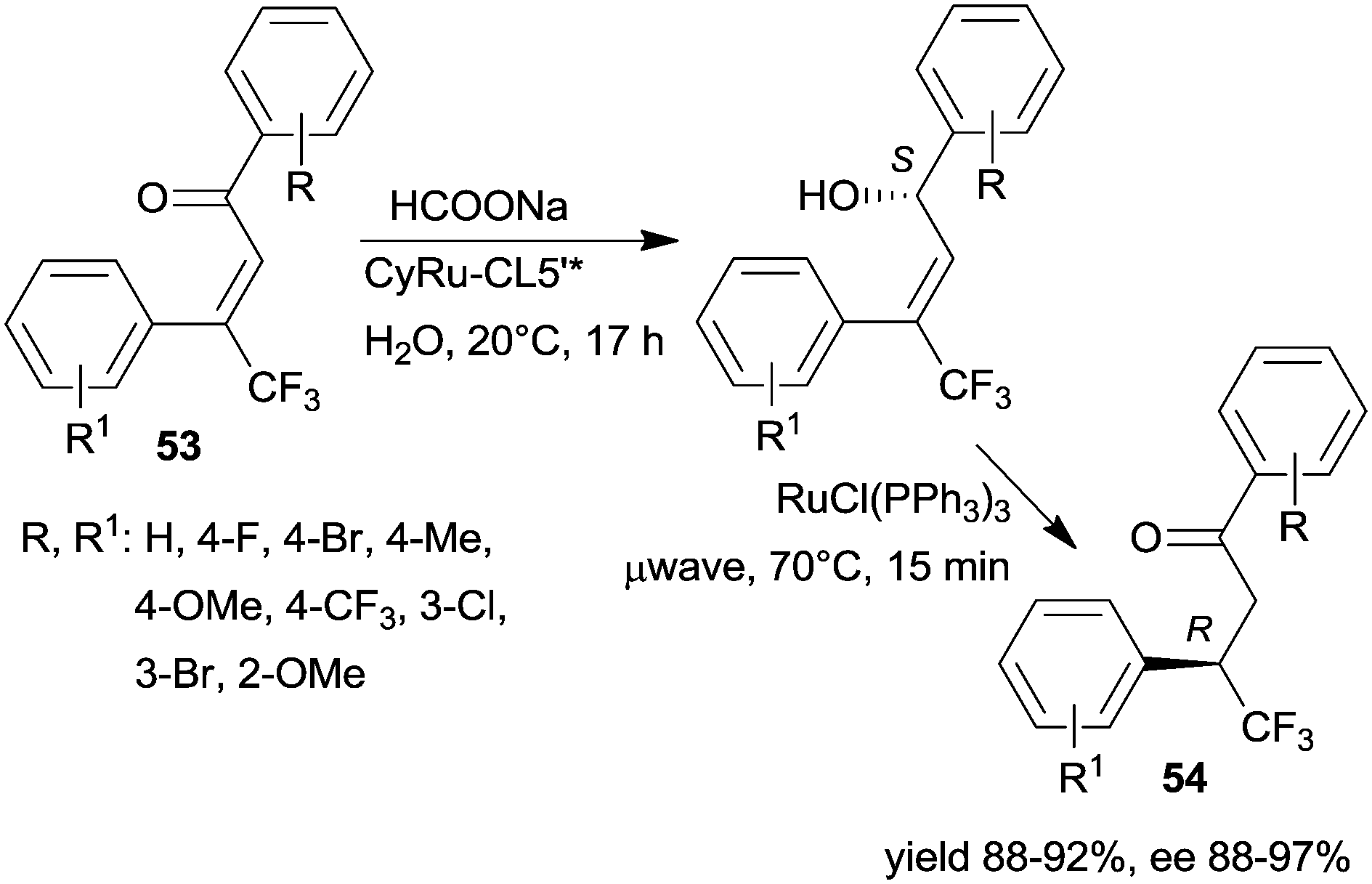 | ||
| Scheme 17 Enantioselective preparation of β-trifluoromethyl ketones by sequential heterogeneous ETH and homogeneous catalytic asymmetric isomerization (AI).135 | ||
Sequential epoxidations of olefins followed by asymmetric opening of the rings using heterogenized chiral complexes were also developed. A metal–organic framework (MOF) containing chiral Mn-Salen units (CT4*) was efficient in the preparation of α-hydroxyazide 56 from the unsaturated cyclic compound 55 (Scheme 18a). The asymmetric epoxidation catalysed by the chiral Mn unit was followed by ring opening catalysed by the Zn species found in the secondary building unit of the MOF, with preservation of the molecule chirality.136 Chiral titanium metal–organic assemblies were prepared from BINOL/Salalen or BINOL/Salan heteroditopic ligands. These materials were used in the sequential epoxidation of a cyclic olefin 57 with H2O2, followed by the stereoselective ring-opening of the resulting meso-epoxide to hydroxylamine 59 using benzylamine (58) (Scheme 18b).137 Among the most efficient chiral solids was the salan derived material CT5* shown in Scheme 18. Remarkably, in the latter reaction the use of the corresponding homogeneous catalysts either in a one-pot process or in a step-wise manner did not result in product formation or provide lower yield and enantioselectivity.137
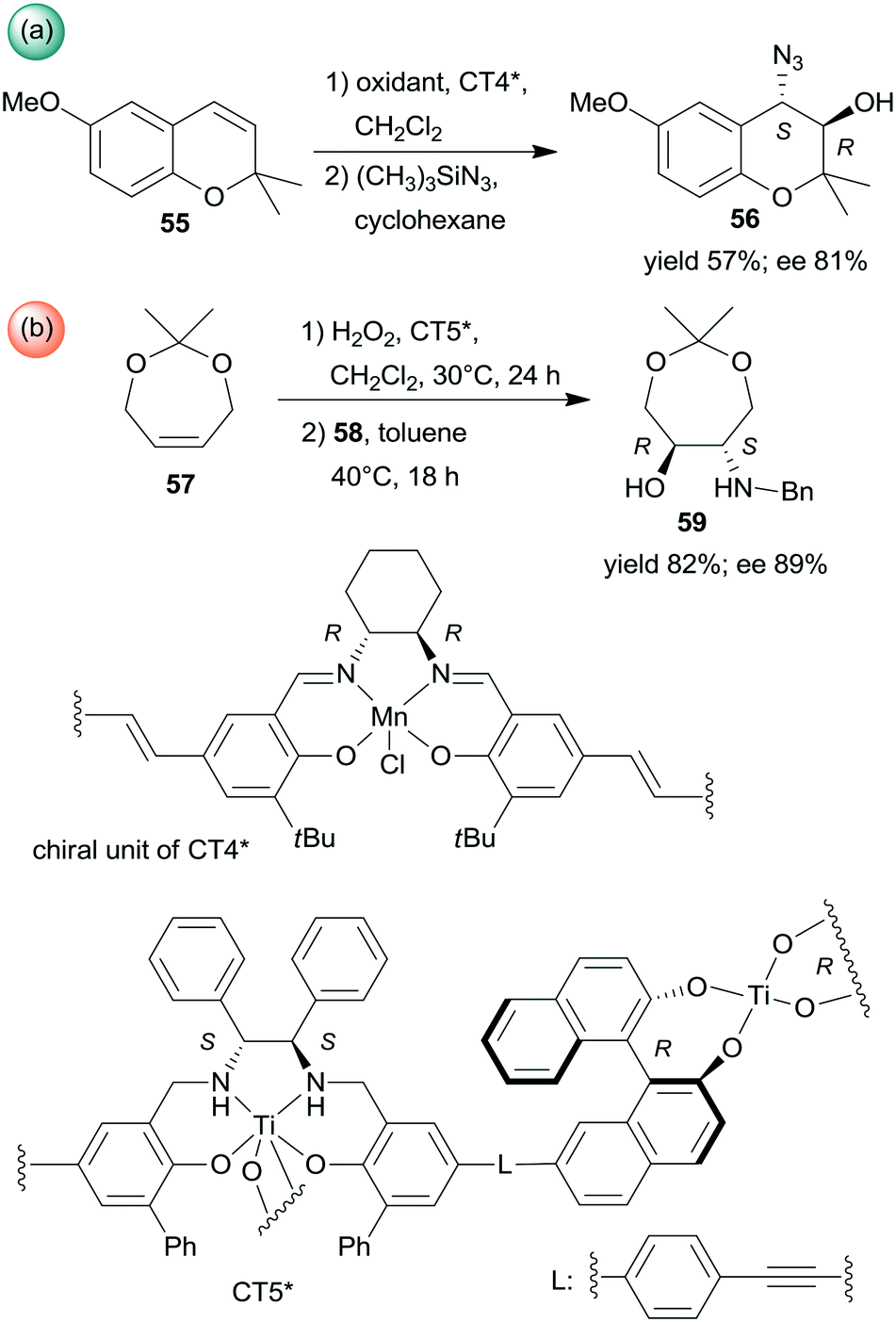 | ||
| Scheme 18 Sequential processes combining asymmetric epoxidation and ring opening (a) or epoxidation and stereoselective ring opening (b).136,137 | ||
Epoxidation was the second step of a sequence initiated by asymmetric allylation of the unsaturated cyclic ketone 60 with in situ formed resin-supported titanium-R-BINOL complex using the anchored chiral ligand CL7* (Scheme 19a).138 The same catalyst was also used in a one-pot sequential asymmetric allylation and enantioselective intramolecular Pauson–Khand reaction (Scheme 19b).138 Recycling of the chiral solid catalyst following the first asymmetric allylation step required remetallation of the anchored ligand with Ti(OiPr)4, due to leaching of the titanium from the resin. Self-supported chiral titanium clusters were obtained by hydrolysis of chiral amino alcohol titanate complexes (CT6*, Scheme 20).139 These materials were efficient in the preparation of α-aminonitriles (66) from benzaldehyde derivatives 64 by sequential imine formation with benzhydrylamine followed by asymmetric cyanation in a flow system. Recycling of the catalyst was examined in the cyanation of the 64a derived imine. The material kept its activity even in the 10th run, and the ee value remained constantly high.139
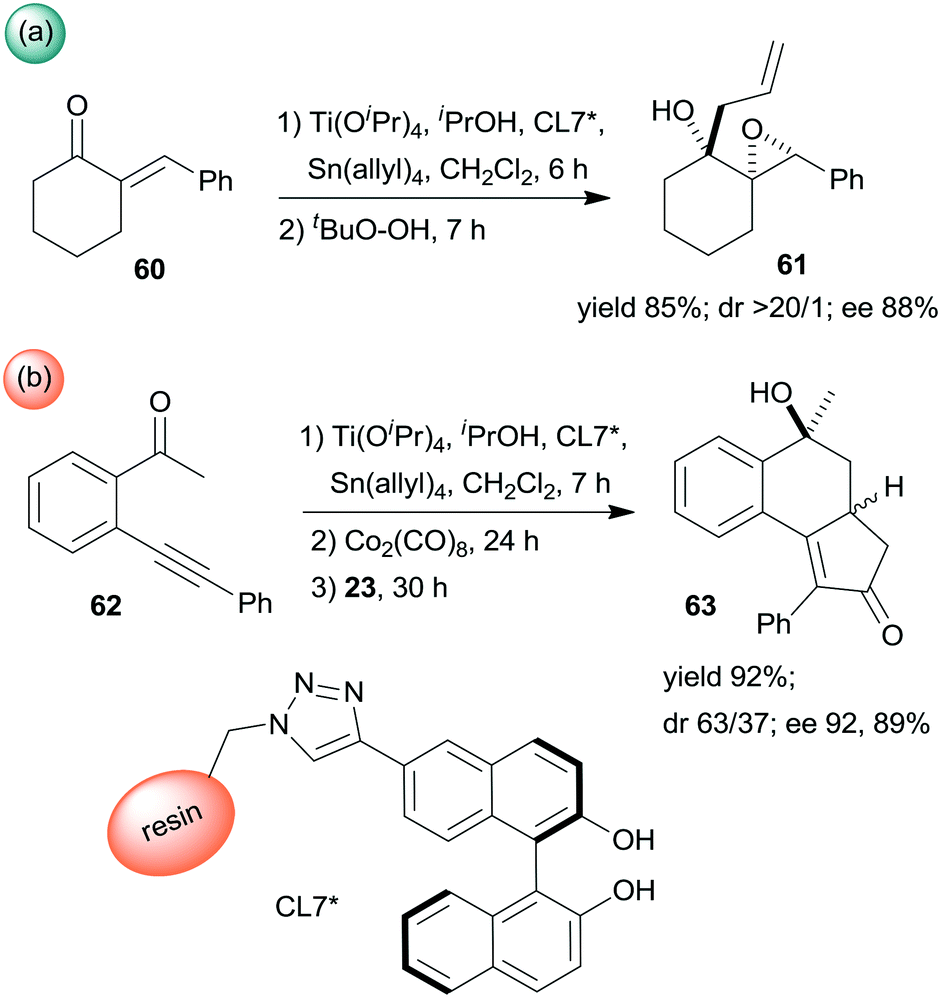 | ||
| Scheme 19 Enantioselective allylation and epoxidation (a) and allylation and Pauson–Khand sequential reactions (b) using in situ formed resin immobilized titanium R-BINOLate complex.138 | ||
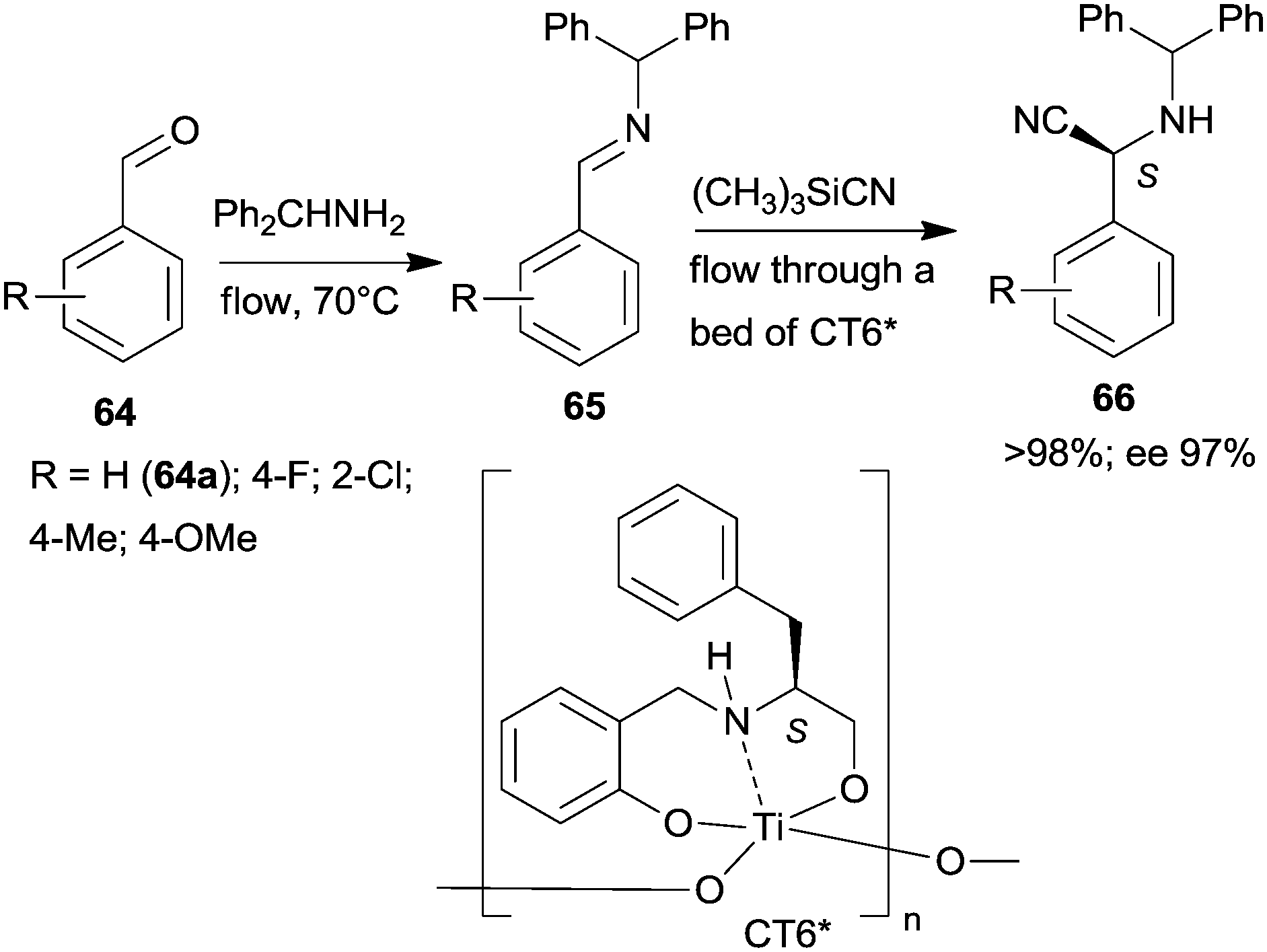 | ||
| Scheme 20 Sequential imine formation and asymmetric cyanation catalysed by self-supported chiral titanium cluster CT6*.139 | ||
As the above presented examples showed, heterogenized chiral metal complexes proved their utility in asymmetric one-pot sequential and cascade reactions. Special care is required in designing such systems, due to possible deterioration of the catalytically active chiral sites of the heterogeneous catalysts in the presence of additives, reactants, other catalysts, or by harmful reaction conditions needed in other steps of these processes. However, as several examples indicated, it is possible to design such recyclable chiral materials for application in combination with homogeneous catalysts. Moreover, for application in some one-pot, especially cascade reactions, materials containing two distinct catalytic species immobilized on the same solid, were also reported. Due to site-isolation effects, these could outperform the corresponding soluble dual catalysts. Additionally, some examples were found in which the support was also involved in activation of the substrates. However, taking into account the sensitivity of most of the metal complexes, the development of such catalytic systems is a particularly difficult and demanding task, as illustrated by the limited number of the systems reported until now.
2.2. One-pot reactions using heterogenized chiral organocatalysts in the asymmetric step
Since the beginning of this century, the use of relatively simple chiral organic molecules as catalysts in the synthesis of optically pure products has become an increasingly attractive alternative to the reactions catalysed by metal complexes. The previously known asymmetric organocatalytic transformations,140–143 have been complemented by reports demonstrating the versatility of these reactions,144–147 leading to explosive growth in the application of such catalysts.9,10,13–15 Although initially inexpensive, natural compounds or their easily prepared derivatives have been used as organocatalysts, which were usually less sensitive to the reaction conditions than the metal complexes; later, as the structure of the catalyst was tuned for defined applications, the complexity and value of the applied catalysts increased. Consequently, the recovery and reuse of these also became of paramount importance.Accordingly, during the last fifteen years, the development of heterogeneous recyclable organocatalysts became a significant task.16,32,148–152 Moreover, several efficient organocatalysts were able to catalyse various reactions or could tolerate the presence of other catalytic species. Thus, their application in one-pot reactions also evolved rapidly.51–56 As a consequence, one-pot processes using heterogeneous organocatalyts, either in combination with soluble catalysts or using multifunctional catalytic materials became one of the preferred ways of increasing the sustainability of preparation of chiral fine chemicals. Due to the larger numbers of such materials, it was expedient to subdivide this section according to the type of immobilized organocatalyst.
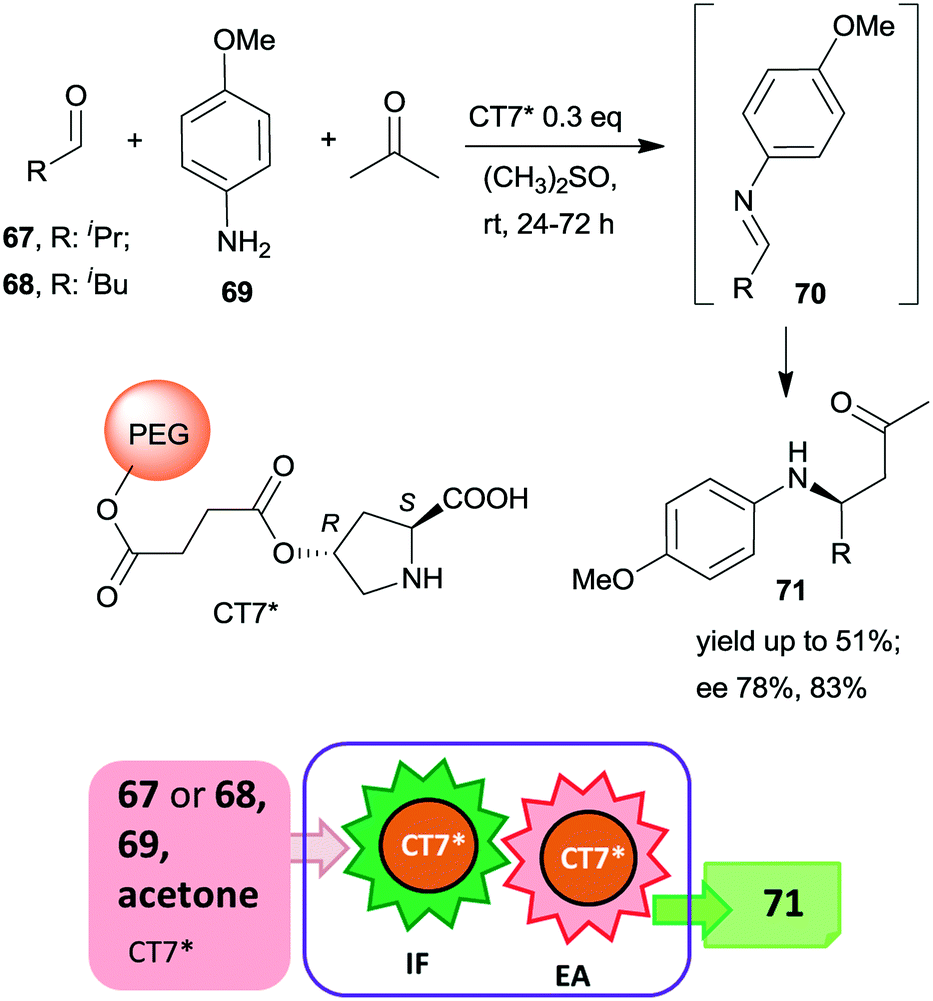 | ||
| Scheme 21 Tricomponent asymmetric tandem Mannich-reaction using poly(ethylene glycol)-bonded chiral catalyst CT7*; IF: imine formation, EA: asymmetric addition.153 | ||
One-pot tricomponent Mannich-reactions using catalysts prepared by anchoring L-Pro over Fe3O4 MNPs (CT8*) were also studied.154 Although good yields and high anti/syn isomer ratios were obtained, the enantioselectivity values were not reported. The same catalyst was used in a tricomponent cascade reaction between N-arylhydroxylamines 72, aromatic aldehydes 64 and crotonaldehyde (73), leading to isoxazolidines 75 by catalytic dipolar cycloaddition between the nitrone 74 and the iminium intermediate formed from the catalyst and 73 (Scheme 22).155 High yields and ees were obtained. The scope of the reaction was extended using few isatin derivatives instead of 64, leading to the formation of spiroisoxazolidines 76, also in high yields and good optical purities. Easy recovery of the catalyst was facilitated by using MNPs as support and recycling four times showed no activity decrease.
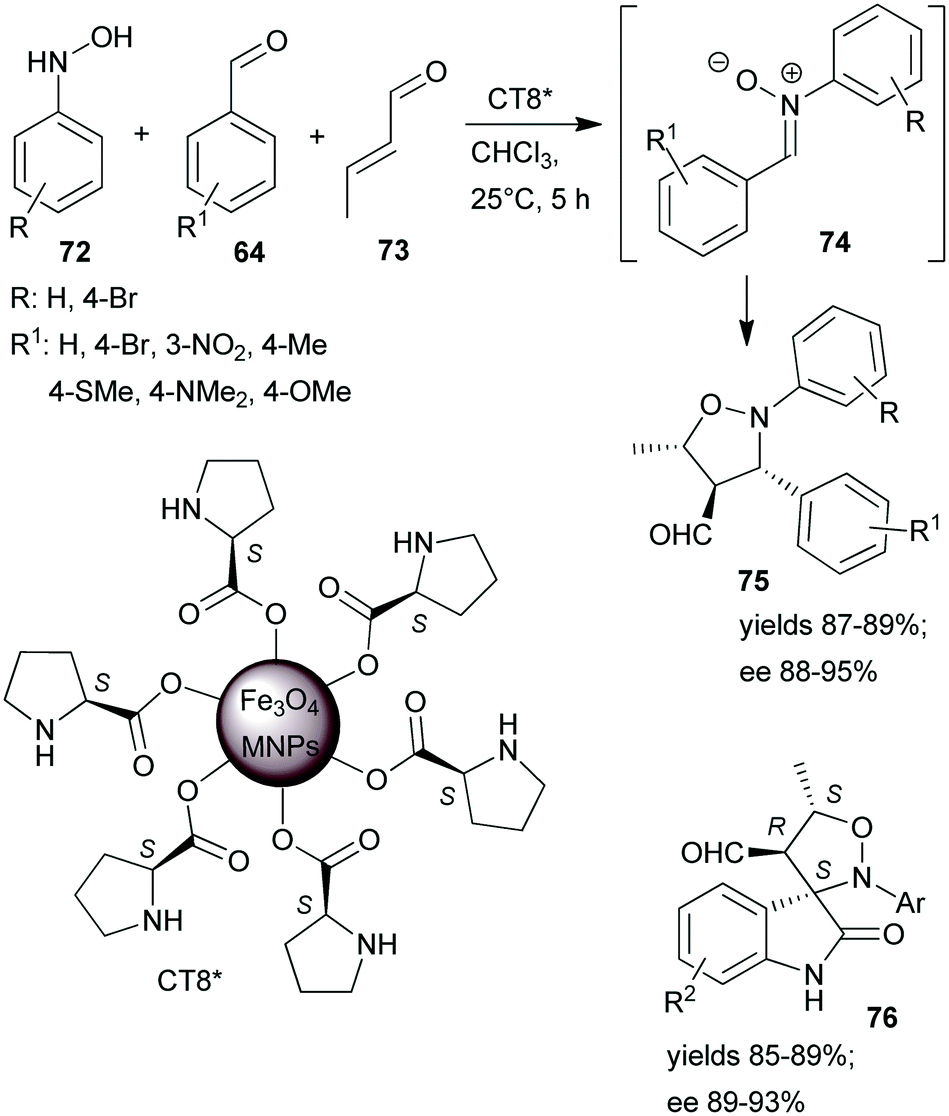 | ||
| Scheme 22 Tricomponent asymmetric cascade reactions catalysed by L-proline immobilized on the surface of Fe3O4 MNPs.155 | ||
Heterogeneous bifunctional catalysts were prepared by anchoring proline derivatives on SBA-15 mesoporous silica and were used in the one-pot sequential nitroaldol reaction followed by asymmetric Michael addition (Scheme 23).156 The synergistic catalytic effect of the surface achiral hydroxyl groups and the immobilized chiral pyrrolidine moiety assured high yields and excellent enantioselectivities in the one-pot reaction of 64a, nitromethane and cyclohexanone (78). The surface hydroxyl groups contributed to the activation of 64a and nitromethane, and thus accelerated the first step of the one-pot reaction and played a role in the orientation of the intermediate 77a in the second step. The sequential addition of reactants, i.e. introduction of 78 following the first step, and decrease of the reaction temperature to 25 °C was necessary in order to obtain good stereoselectivities in the second step. The best results were obtained using CT9* catalyst, containing L-Hyp thioester derivative bonded by the 4-hydroxyl group to SBA-15 functionalized with pendant mercaptopropyl surface groups. The corresponding proline derivative was not able to catalyse the Henry reaction, which showed the probable involvement of the surface achiral groups in this reaction. The CT9* catalyst was reused three times, with only slight decrease in the activity and stereoselectivities.
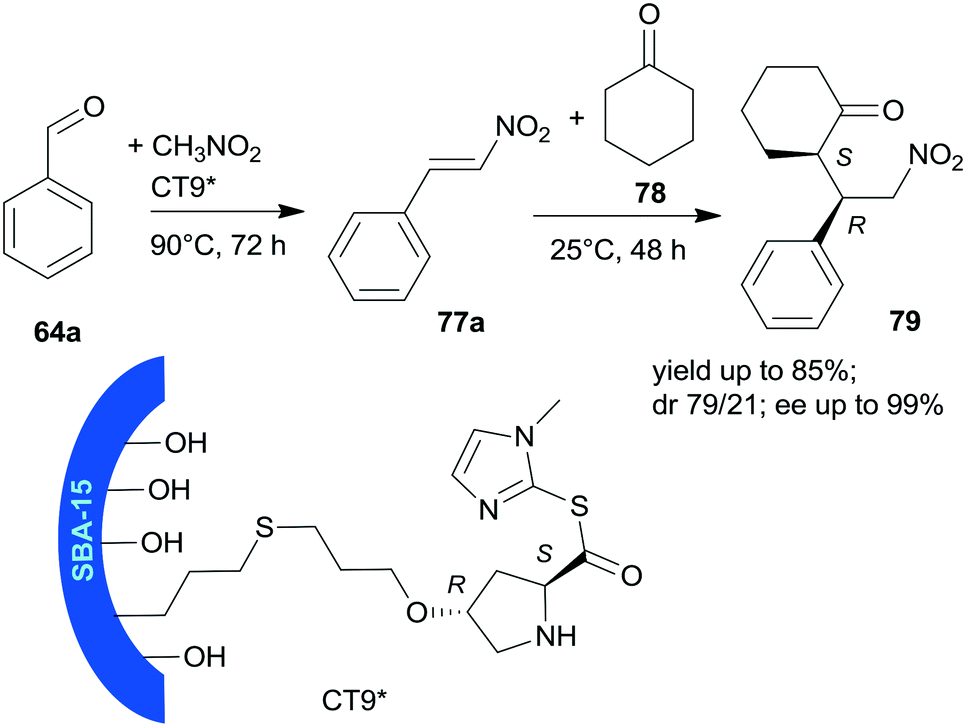 | ||
| Scheme 23 Sequential one-pot nitroaldol reaction and AMA using SBA-15 mesoporous silica anchored L-Pro derivative.156 | ||
Later, the same research group prepared mesoporous silicas with either hydrophobic (HbMs) pores, hydrophilic pores or containing both hydrophobic and hydrophilic alternating blocks (HHMs) in the pore walls. These tailored supports were used to anchor L-Hyp, leading to materials containing surface immobilized L-Pro.157 The heterogeneous organocatalysts were tested in the Knoevenagel condensation, followed by the asymmetric Michael addition cascade reaction of isatin (80), malononitrile (81) and acetone in biphasic organic-aqueous solvent mixtures (Scheme 24). The best results were achieved when the surface was either completely or partially hydrophobic (CT10*), attributed to the more effective organic–water interface developed in the channels of these materials, where the surface hydroxyl groups and/or the water molecules at the interface were also involved in the catalytic processes. The possibility of carrying out the two steps in cascade was ensured by the much faster condensation step as compared with the AMA. The catalyst having alternating surface pore properties (HHMs) was reused three times.
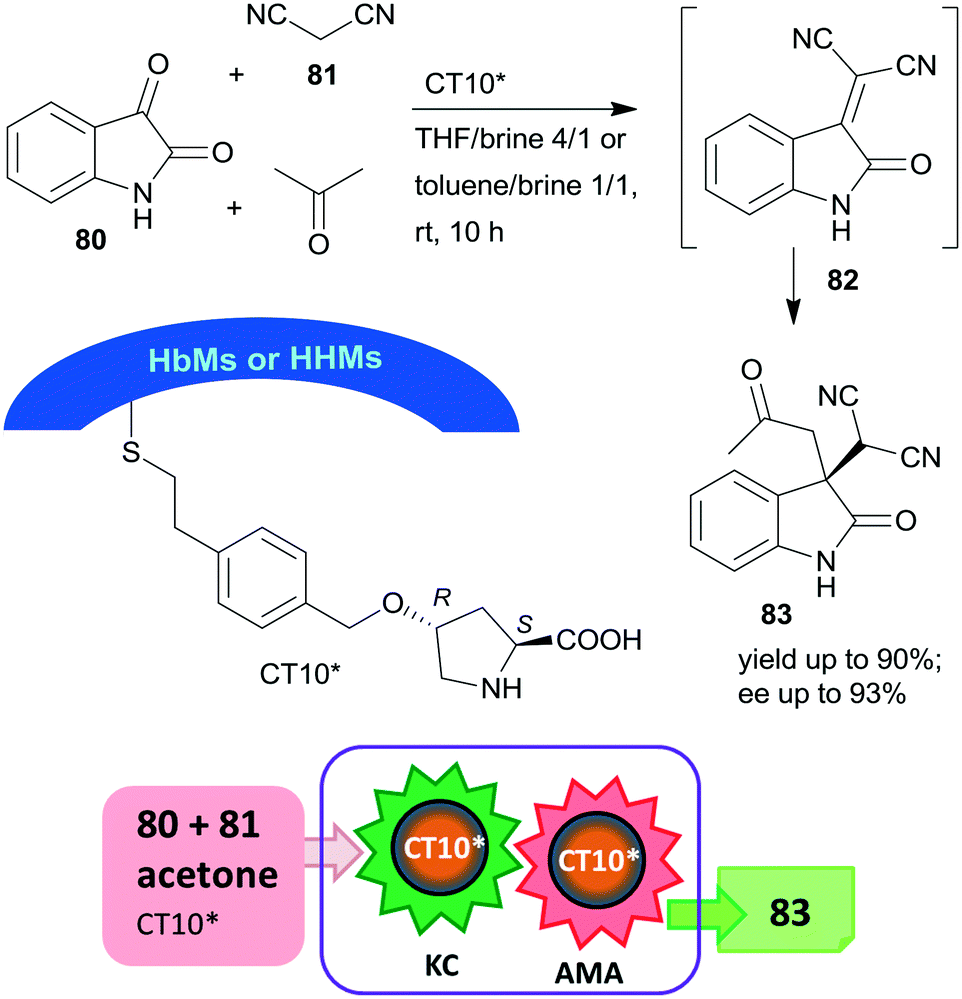 | ||
| Scheme 24 Knoevenagel condensation (KC) and AMA cascade reaction catalysed by L-Pro grafted on hydrophobic and hydrophilic blocks containing mesoporous silica.157 | ||
In a so-called “one-pot like” reaction, the initial asymmetric direct aldol addition of 3-chlorobenzaldehyde (84) and acetone was catalysed in high yield and excellent enantioselectivity158 by a resin immobilized L-prolineamide derivative (CT11*) developed for this purpose (Scheme 25).159 The subsequent reduction to chiral diol 87 was catalysed by alcohol dehydrogenase (ADH) immobilized on a super-adsorbent polymer together with its cofactor NAD+. The process could be run in organic solvent, i.e. cyclohexane, which, following the removal of the volatile components such as acetone after the first step, allowed direct addition of the co-immobilized enzyme (S)-ADH and its cofactor and iPrOH co-substrate. Fortunately, neither the unreacted 84 nor 85, or CT11* found in the system, showed inhibition in the biocatalytic step, and diol 87 was obtained in excellent overall yield and optical purity.158 The stereochemical outcome of the second reduction step was fully controlled by the biocatalyst, overriding the influence of the chiral centre formed in the first step, which implies that all stereoisomers are selectively accessible by using appropriate catalysts.
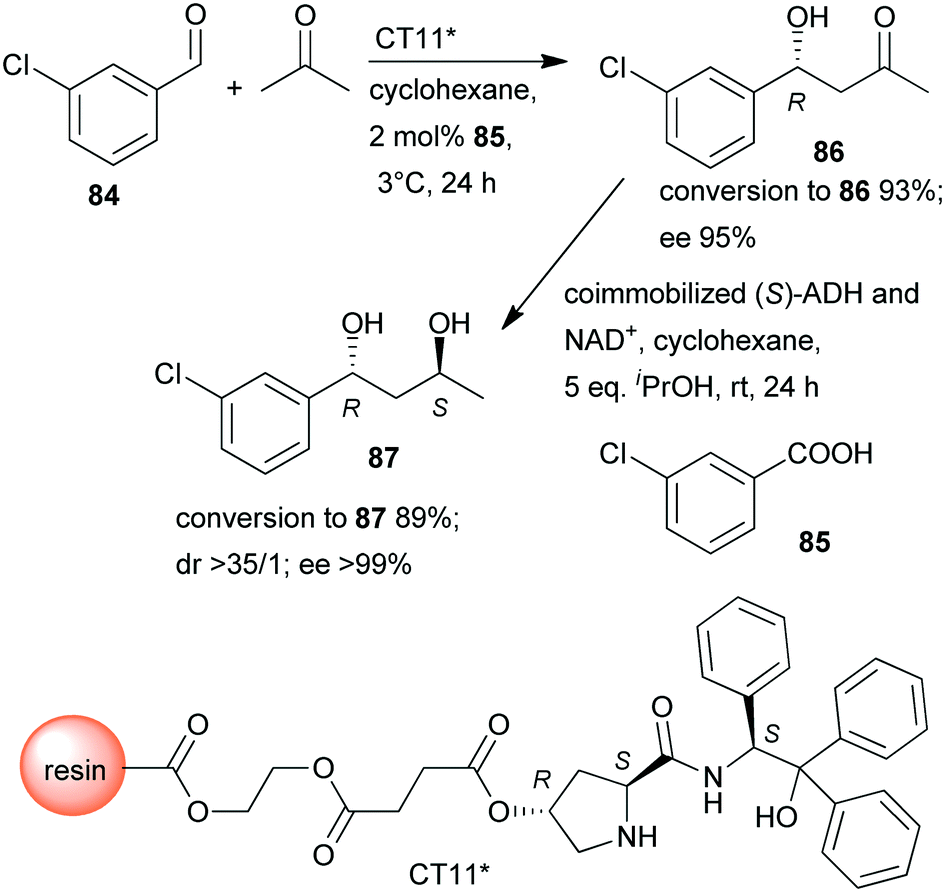 | ||
| Scheme 25 Sequential one-pot asymmetric direct aldol addition and enzymatic reduction using immobilized chemical and biocatalysts.158 | ||
This organocatalyst was bonded by azide-alkyne cycloaddition of the corresponding 4-propargyloxy derivative to Merrifield resin containing azidomethyl groups (CT12*).160 The chiral polymer was used as catalyst in the asymmetric Michael addition followed by the Knoevenagel condensation domino reaction of unsaturated aldehydes 6 and dimethyl 3-oxoglutarate (88) (Scheme 26). These reactions resulted in the formation of cyclohex-2-en-1-ones 90 in good yields and excellent enantioselectivities. In a subsequent step, 90 was reduced to cyclohexane derivatives 91 containing four chiral centres, obtained as single enantiomers. The activity of the catalyst slightly decreased upon reuse, thus, increasingly longer reaction times were necessary during recycling. The solid catalyst charged in a fixed-bed reactor was also able to provide the cyclic compound 90 with similar enantioselectivity to that obtained in the batch reactor.
 | ||
| Scheme 26 AMA and KC cascade reactions catalysed by immobilized CA6*.160 | ||
An ingeniously prepared heterogeneous layered bifunctional catalyst was reported by Kobayashi and co-workers.161 A chiral copolymer containing immobilized CA6* was placed in the inner layer, whereas achiral polymer incarcerated Au/Pd alloy NPs constituted the outer shell of the catalyst beds (CT13*). This layering and separation of the active components assured the isolation of the catalytic sites and resulted in an active and enantioselective catalyst for the tandem catalytic aerobic oxidation of allylic alcohols 13, followed by asymmetric Michael addition of dibenzyl malonate 92 to the resulting unsaturated aldehyde intermediates 6 (Scheme 27). When the metal alloy NPs were placed in the inner part and the chiral copolymer was in the outer layer of the composite, the tandem reaction did not proceed, showing the importance of the layering order of the two active phases. Moreover, recycling of CT13* was obstructed by deactivation as a consequence of transformation of the pyrrolidine secondary amino group.
 | ||
| Scheme 27 Cascade aerobic oxidation followed by AMA catalysed by bifunctional layered catalyst containing anchored CA6*; PI: polymer incarcerated.161 | ||
A chiral heterogeneous catalyst containing both an achiral metal complex and CA6* immobilized on SiO2 support (CT14*, Scheme 28) was developed by Córdova and co-workers162 for heterogeneous synergistic catalysis of the asymmetric Michael addition and intramolecular carbocyclization cascade reaction described initially using soluble organocatalyst (see Scheme 3 (ref. 97)). Interestingly, this heterogeneous catalyst provided a slightly better enantiomeric ratio than was obtained in reaction with the homogeneous CA6*. The scope of the heterogeneous relay catalytic process was investigated using 7 or the oxindole derivative 94,162 the latter resulting in spirolactams 95 with various unsaturated aldehydes 6.108–110 However, recycling of the catalyst was not successful, due to deactivation of the surface bonded chiral amine as a consequence of the inhibition by the aldehyde or by the product.
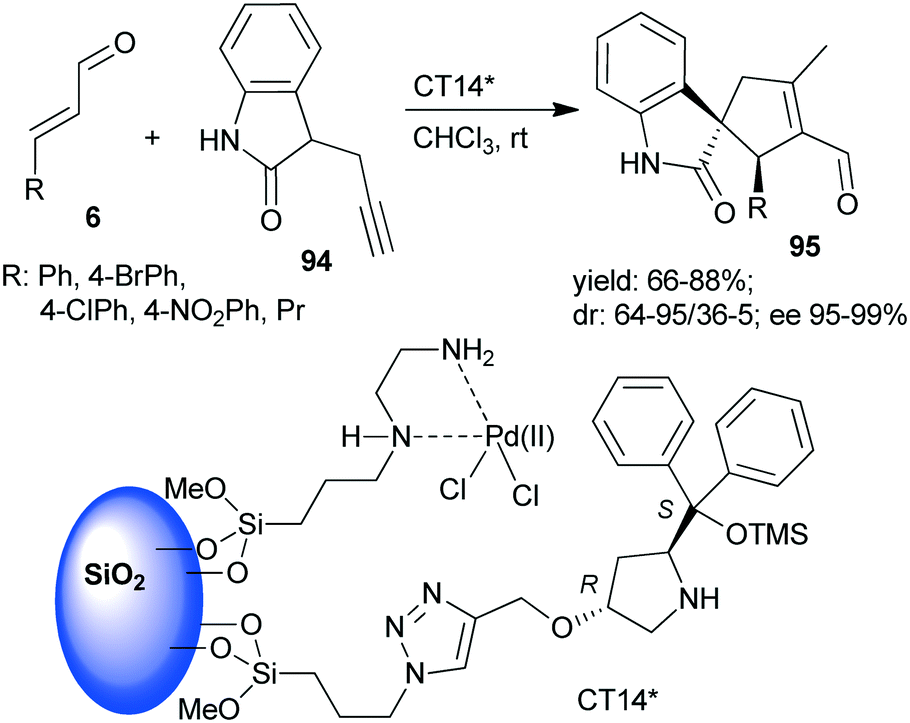 | ||
| Scheme 28 The asymmetric cascade reaction of unsaturated aldehydes and alkyne nucleophiles using the heterogeneous catalyst CT14* (see Schemes 3 and 4).162 | ||
Recently, multi-hollow organic microspheres containing anchored CA6* organocatalyst were prepared by solvent etching the poly(styrene/acrylic acid) core of microparticles having a chiral copolymer shell. This shell was obtained by copolymerization of acrylamide, styrene and CA6* derived monomer crosslinked with para-divinylbenzene and ethylene glycol dimethacrylate (CT15*).163 The material was used in the tricomponent triple cascade process reported earlier,101,164,165 which resulted in the formation of four chiral centres in a stereoselective manner in the first two asymmetric Michael additions (Scheme 29). The initial addition of the aliphatic aldehyde 96 to β-nitrostyrene derivatives 77 by enamine catalysis was followed by addition of the resulting intermediate 97 to the activated unsaturated aldehyde 6avia iminium catalysis. Intramolecular cyclization of the enamine intermediate 98 by aldol condensation eventually led to the cyclohexene derivative 99. Results obtained using the heterogeneous catalyst CT15* were similar to those reported using the homogeneous counterpart CA6*. The stereoselectivities were maintained following five uses of CT15*; however yields slightly decreased, which was ascribed to several reasons, among which the partial collapse of the hollow structure may have a significant contribution to this phenomenon.
 | ||
| Scheme 29 Tricomponent asymmetric triple cascade reaction catalysed by multi-hollow copolymer microparticles containing immobilized CA6*; AC: aldol condensation.163 | ||
Non-interpenetrating star polymers immobilized chiral organocatalysts were used in the sequential asymmetric Friedel–Crafts reaction taking place by iminium catalysis followed by asymmetric Michael addition through enamine catalysis.166 The first step was catalysed by MacMillan's catalyst167 bonded by ionic interactions to pendant benzenesulfonic acid groups of a star polymer (CT16*), whereas the second was with CA6* anchored on star polymer by copolymerization (CT17*). Activation of the Michael acceptor by hydrogen-bonding using 103 increased the yield. CT17*, 103 and the unsaturated ketone 104 were added to the system following the completion of the first step (Scheme 30). Application of star polymers assured the catalytic site isolation necessary in this sequential reaction; the use of either TsOH acid or CA6* instead of the corresponding chiral copolymers or using similar linear copolymers did not provide the desired product 105.
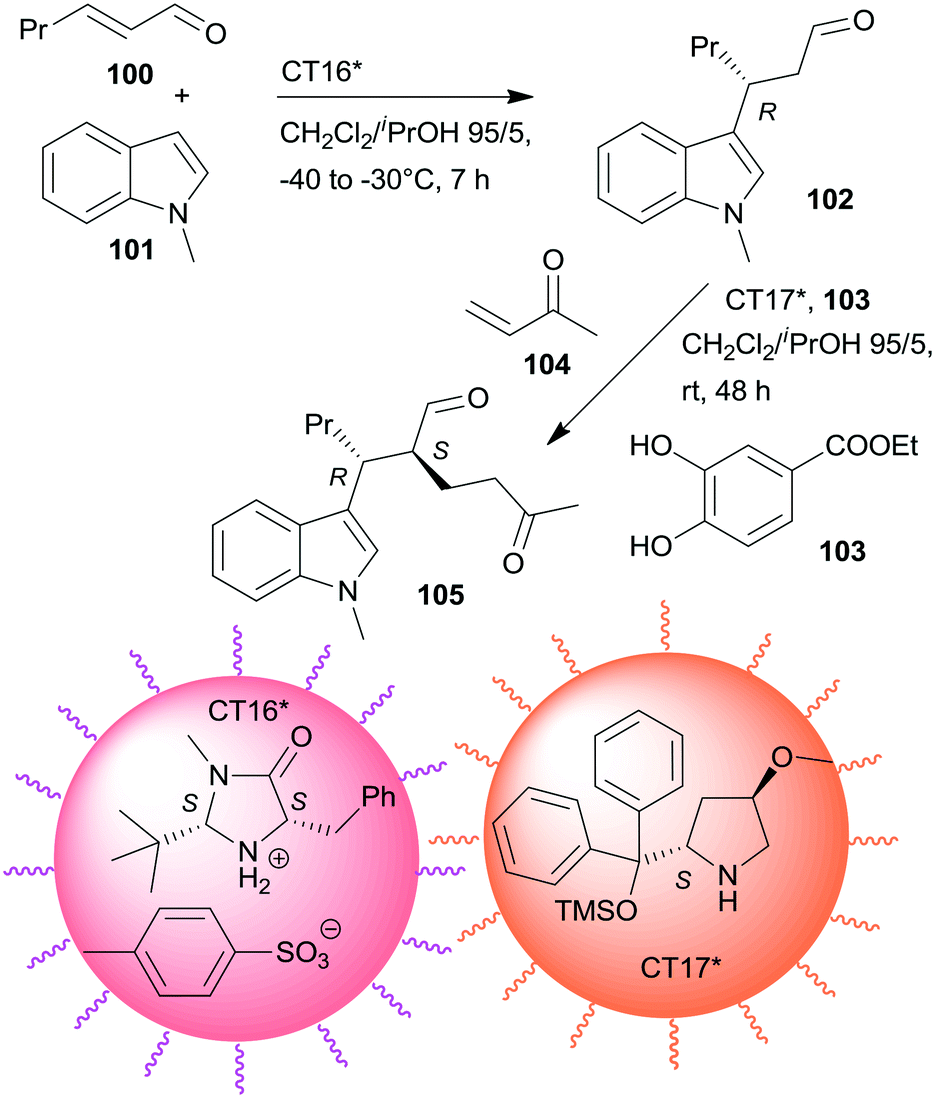 | ||
| Scheme 30 One-pot sequential asymmetric Friedel–Crafts reaction and AMA catalysed by chiral catalysts immobilized on non-interpenetrating star polymers.166 | ||
Finally, a highly efficient example of chiral heterogeneous organocatalyst in which diphenyl prolinol was anchored by the hydroxyl group through ether bond and used in asymmetric cascade reactions was recently reported (CT18*).168 Similar to the previous report from the same research group,156 the use of SBA-15 mesoporous silica as support resulted in a synergistic catalytic effect of the surface acidic achiral hydroxyl groups and the immobilized chiral secondary amine moiety, complemented by a presumed geometrical constraint of the mesopores. These interactions were supposed to be responsible for the high yields and excellent enantioselectivities in the oxa-Michael reaction coupled with asymmetric Michael addition cascades of 106 with 107 or 109 (Scheme 31a). Catalyst CT18* was also successfully used in the asymmetric Michael addition and acetalization cascade of 106 and butanal (not shown); moreover, high enantioselectivity was obtained in the asymmetric aza-Michael addition followed by nitro-aldol condensation of 111 and 77a leading to 1,2-dihydroquinoline derivative 112 (Scheme 31b). The activity of this heterogeneous organocatalyst and the resulting stereoselectivity hardly decreased following five uses in the oxa-Michael reaction and asymmetric Michael addition cascade.168
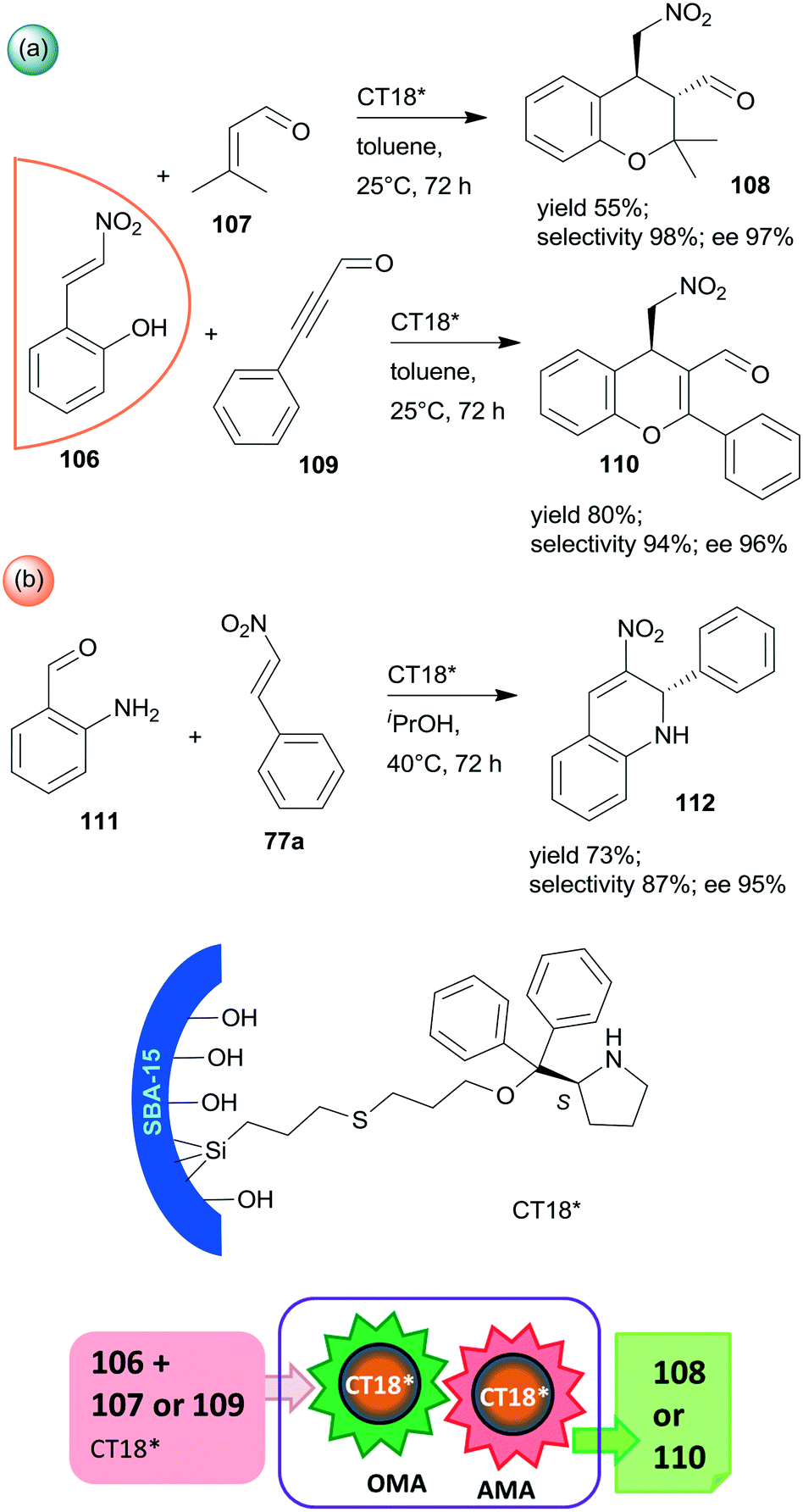 | ||
| Scheme 31 Cascade oxa-Michael reaction (OMA) and AMA (a) and asymmetric aza-Michael reaction and nitro-aldol condensation (b) catalysed by SBA-15 anchored diphenyl prolinol.168 | ||
A polymer-supported oligopeptide catalyst (CT19*) was used as a heterogeneous organocatalyst in the tandem oxidation of primary alcohols 113 and asymmetric α-oxamination of the obtained aldehydes 114 with 15 and CuCl (Scheme 32).169 The asymmetric oxamination reported a few years previous,170 takes place by the enamine activation mechanism in homogeneous systems.171 This oxamination was catalysed by the resin-supported oligopeptide CT19* catalyst using FeCl2 and NaNO2 in aqueous media under aerobic conditions.172 The novel tandem reaction occurred under similar conditions, except that 15 used as reactant in the second step also served as oxidant in the Cu(I)/15 catalysed and 1,1′-bipyridine additive assisted first homogeneous oxidation step of the process. Good yields and high enantioselectivities were reached, determined following an additional reduction of the cascade products 115 to alcohols 116.169 The immobilized peptide could be reused following the tandem reaction and similar results were obtained even in the seventh run.
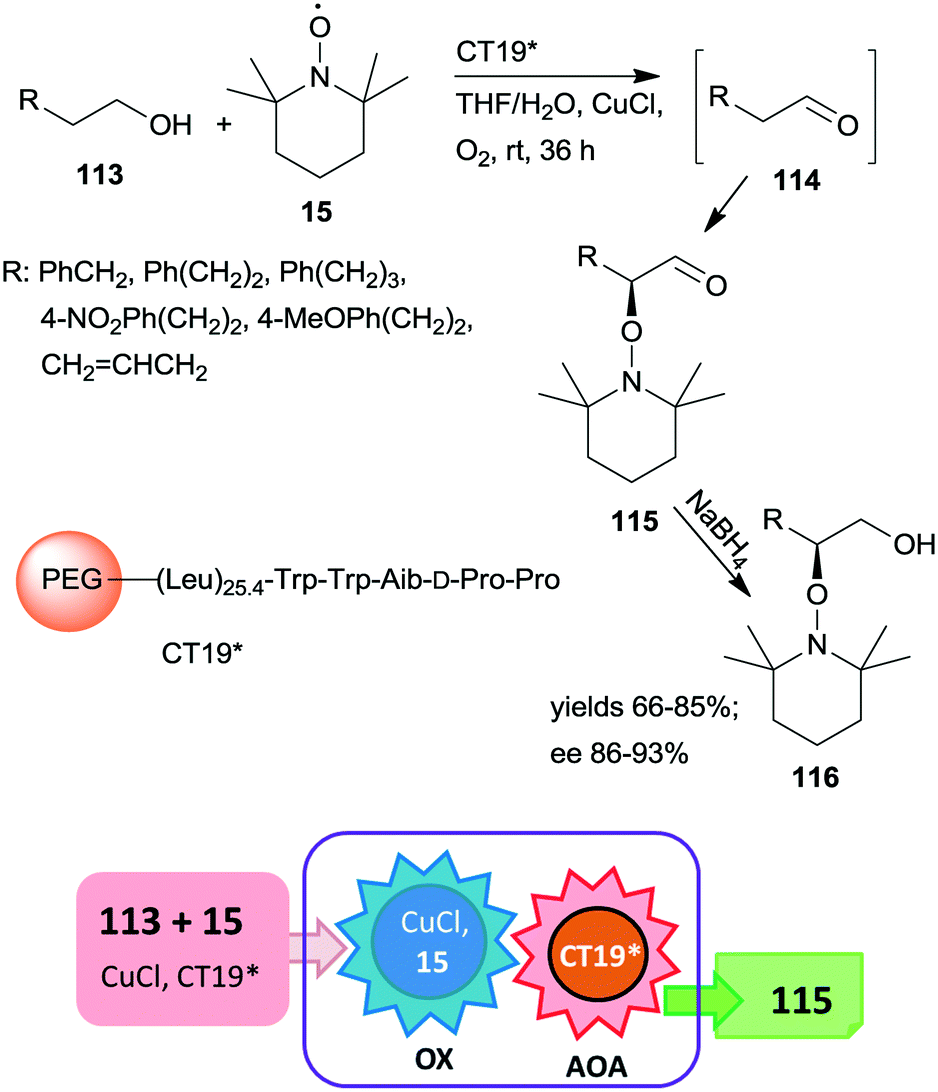 | ||
| Scheme 32 Tandem oxidation and asymmetric oxamination catalysed by resin-supported oligopeptide organocatalyst; AOA: asymmetric oxamination.169 | ||
Kudo and co-workers also developed one-pot sequential prodesses in which asymmetric Friedel–Crafts reactions between N-methylindole derivatives 117 and aromatic unsaturated aldehydes 6 were the first steps (Scheme 33).173,174 Several supported oligopeptide catalysts were used, such as the previously developed CT19* (see Scheme 32). The best results were obtained using (Leu–Leu–Aib)2 linker between the terminal penta-peptide and the polymer (CT20*) in water or a mixture of water and THF. The reaction sequences were continued either by one-pot reduction or by asymmetric α-oxamination of the resulting aldehydes 118 with 15. The latter reaction was carried out in the presence of the oxidative enzyme laccase. In this sequence, the stereochemistry of the α-oxamination step was determined by the immobilized peptide, demonstrated by effects observed upon changing the configurations of the chiral amino acids in the terminal penta-peptide unit. Insufficient water in the solvent mixture inactivated the laccase, thus inhibiting the second peptide/enzyme-catalysed oxidation step.
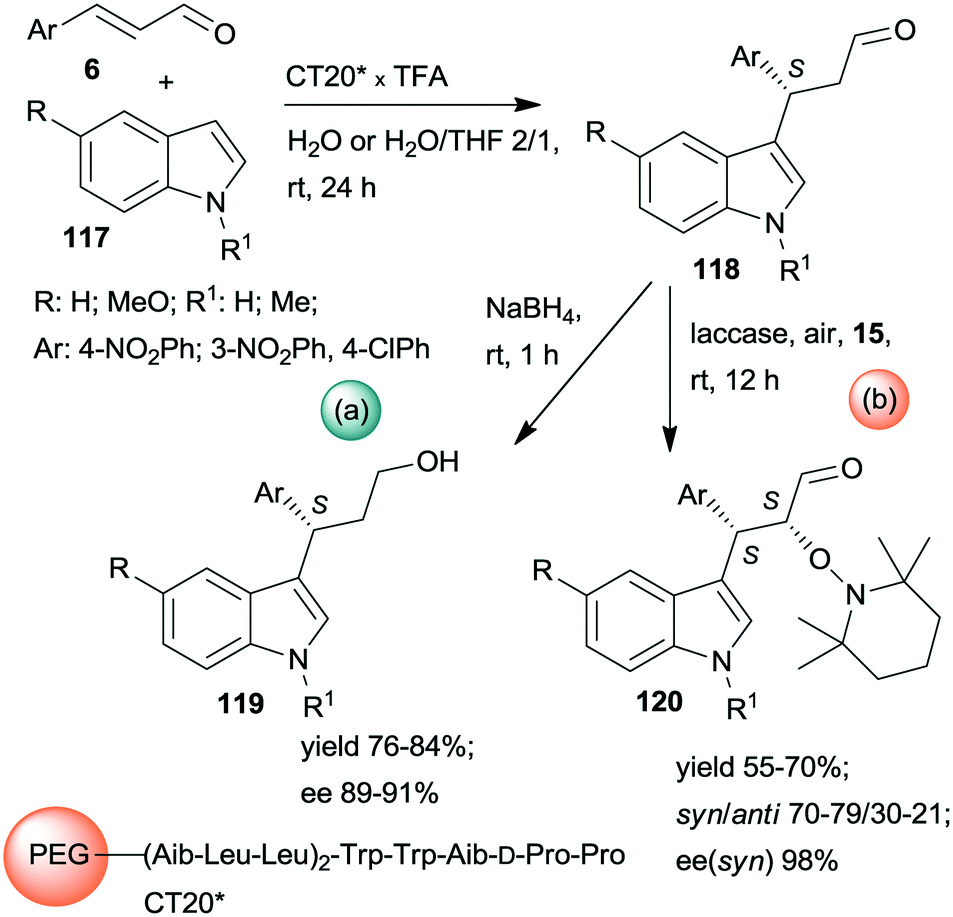 | ||
| Scheme 33 One-pot sequential heterogeneous catalytic asymmetric Friedel–Crafts reaction followed by homogeneous reduction (a) or enzymatic AOA (b) using polymer-bonded oligopeptides; TFA: trifluoroacetic acid.173,174 | ||
Inspired by the Juliá–Colonna asymmetric epoxidation using polyamino acid catalysts,175 poly-L-leucine was used as a recyclable organocatalyst in the second step of the sequential Claisen–Schmidt condensation, followed by an asymmetric epoxidation one-pot process, whereas the initial step was carried out using KOH aqueous solution.176 The scope of the reaction was demonstrated using various benzaldehyde and acetophenone derivatives. The recycled chiral catalyst gave a slightly decreased yield and enantioselectivity in the tenth run. Recently, rehydrated hydrotalcite was used as a support to immobilize poly-L-leucine by simple adsorption.177 The resulting inorganic–organic hybrid material (CT21*) was applied as catalyst in the above sequential Claisen–Schmidt condensation – asymmetric Juliá–Colonna epoxidation one-pot process (Scheme 34). The first step catalysed by basic sites of the inorganic material proceeded most efficiently under neat conditions leading to the unsaturated ketones 121, whereas between steps, toluene, NaOH, H2O2 and Bu4N+Br− were added. The catalyst was stable, providing high yields of 122 and slightly decreased ee during the fourth use. Interestingly, the ortho-nitro-substituted benzaldehyde gave the opposite enantiomer in excess (2S,3R), as compared with the unsubstituted or para-substituted derivatives (excess of 2R,3S enantiomer), attributed to the steric effect of the ortho substituent.177
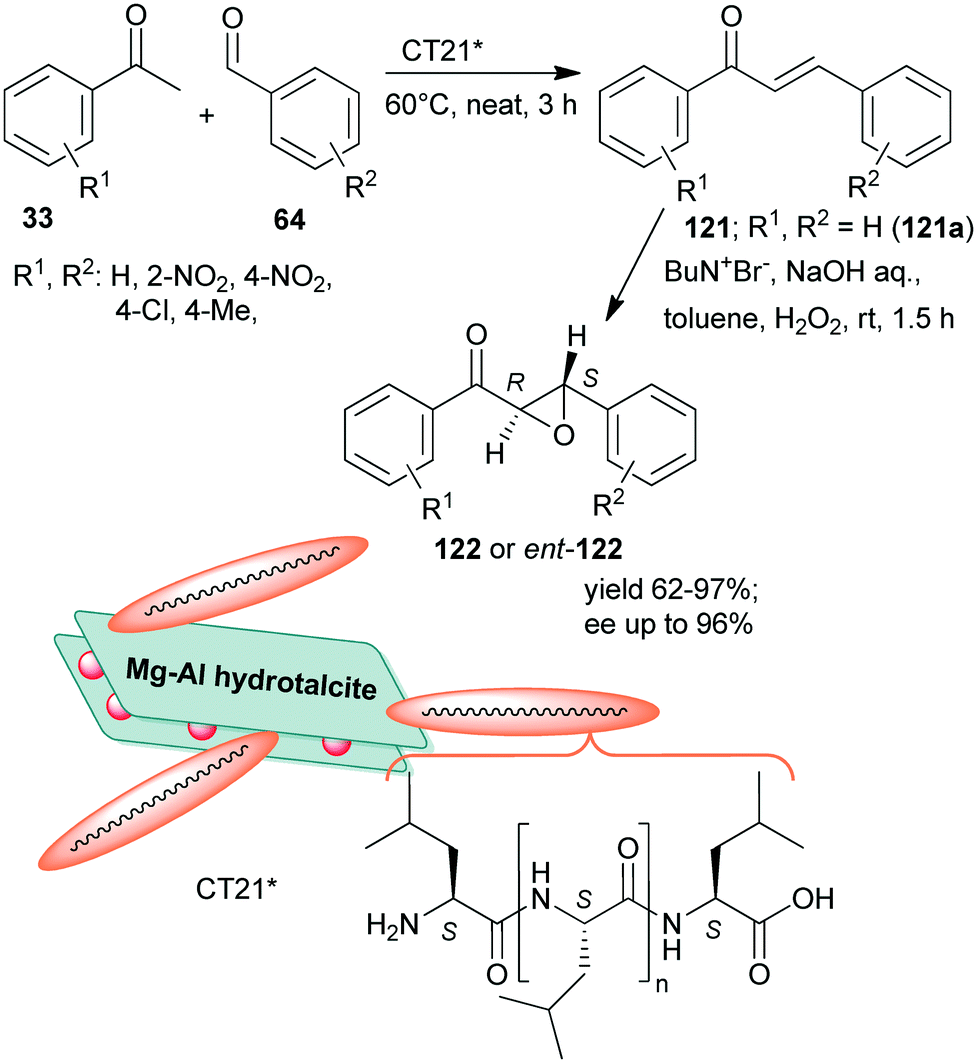 | ||
| Scheme 34 Sequential one-pot Claisen–Schmidt condensation and asymmetric Juliá–Colonna epoxidation catalysed by hydrotalcite-supported poly-L-leucine; ent: enantiomer.177 | ||
Among the first attempts, an epi-cinchona alkaloid thiourea derivative was immobilized on a carrier containing silica supported Fe3O4 MNPs.179 This chiral solid (CT22*) was found efficient in asymmetric inverse-electron-demand Diels–Alder reactions in which the dienophiles were generated by the activation of azlactones 124 (Scheme 35). The resulting intermediates 125 underwent regioselective intramolecular C–O bond cleavage leading to chiral cyclohex-3-en-1-one derivatives 126 in high yields and excellent enantioselectivities (Scheme 35a). The second step of the cascade occurred spontaneously. The heterogeneous catalyst provided similar results to the corresponding homogeneous thiourea derivative. When 123 was replaced with unsaturated N-Ts imines 127, the inverse-electron-demand Diels–Alder reaction was followed by retro-hemiaminalization, resulting in unsaturated amino acid precursors 128 in similarly high enantioselectivities, as obtained in the previous cascade (Scheme 35b).179 It must be mentioned that the role of the MNPs is solely to ease the separation of the catalyst from the reaction mixture; moreover, using Fe3O4 as support led to poor activity and an almost racemic product, indicating the paramount role of the silica in anchoring the cinchona alkaloid. Recycling of CT22* resulted in a slight decrease in the yield and ee following ten uses.
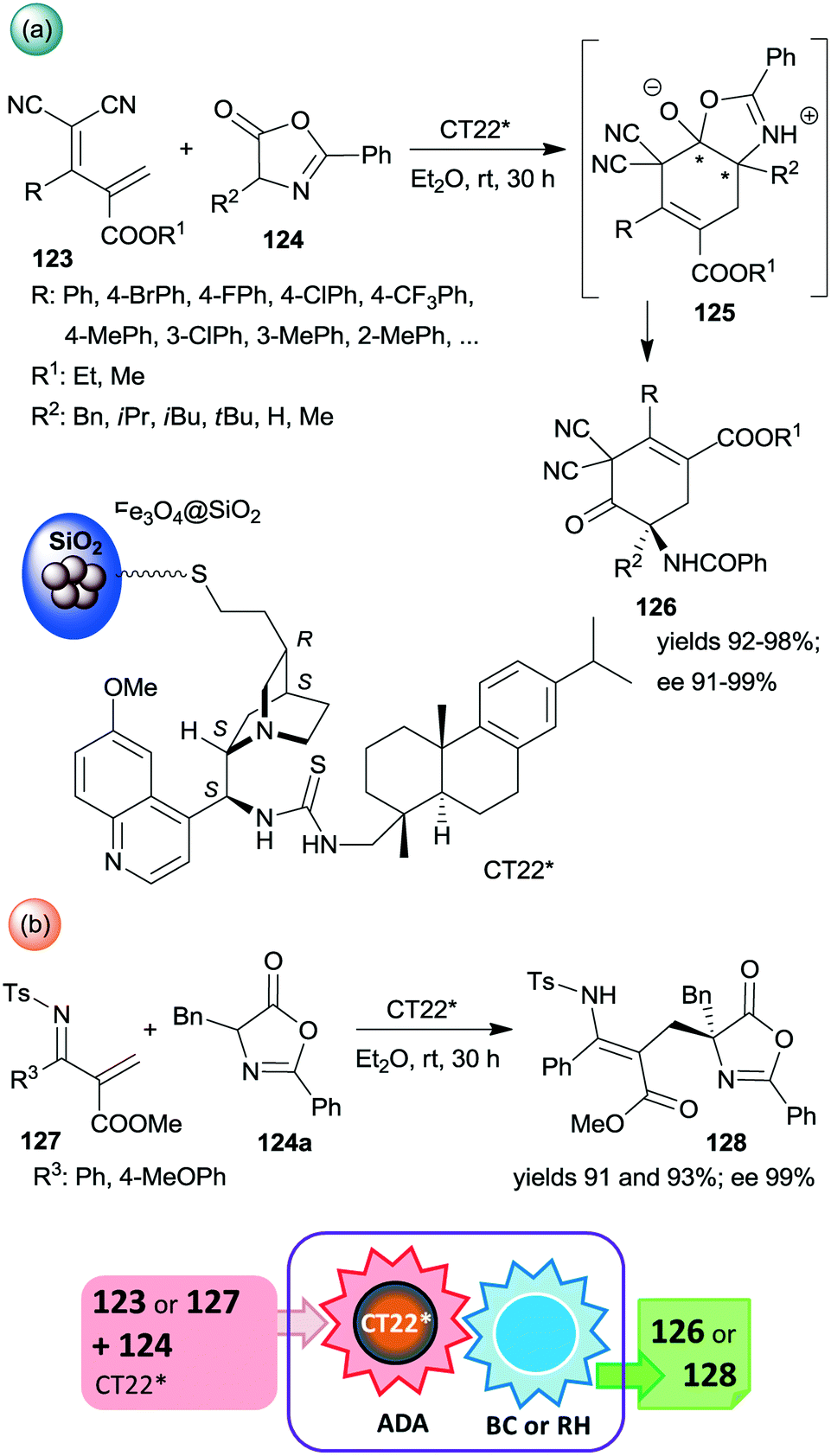 | ||
| Scheme 35 Inverse-electron-demand Diels–Alder reaction (ADA) and intramolecular C–O bond cleavage (BC) cascade catalysed by cinchona alkaloid derivative bonded on the surface of MNPs decorated SiO2 (a) and the cascade occurring by reacting unsaturated N-Ts-imines followed by retro-hemiaminalization (RH) (b).179 | ||
At the same time, the preparation of a hybrid catalyst containing epi-cinchona alkaloid thiourea derivative and aminopropyl units incorporated in the structure of a mesoporous silica support (CT23*) was reported.180 The multifunctional chiral material was obtained by co-condensation of tetramethylorthosilicate with the silylated cinchona alkaloid precursor prepared from quinine functionalized by carbamate groups. Due to partial hydrolysis of the carbamate moiety, the material also contained free aminopropyl pendant groups. This hybrid catalyst afforded high yields and good enantiomeric ratios in the tricomponent reaction of aldehydes, nitromethane and malonates via nitroaldol condensation and asymmetric Michael addition one-pot cascade (Scheme 36).180 It must be noted that the generation of the primary amino groups by partial hydrolysis of the chiral precursor produced the surface sites necessary to catalyse the first step of the cascade reaction. The recovered catalyst was reused two times with a small decrease in its performance; however, the activity of the material decreased with further recycling. This heterogeneous asymmetric cascade procedure was later combined with an ulterior transformation of the products 129 through another one-pot domino process over Pd/C, incorporating the reduction of the nitro group, intramolecular cyclization, ester hydrolysis and decarboxylation reactions in order to prepare γ-aminobutyric acid derivatives 130, which are pharmaceutically relevant chiral building blocks.181 New chiral centres in the final product were not generated during this second domino reaction over Pd/C; however, the chirality of the intermediates 129 was preserved in the final products 130.
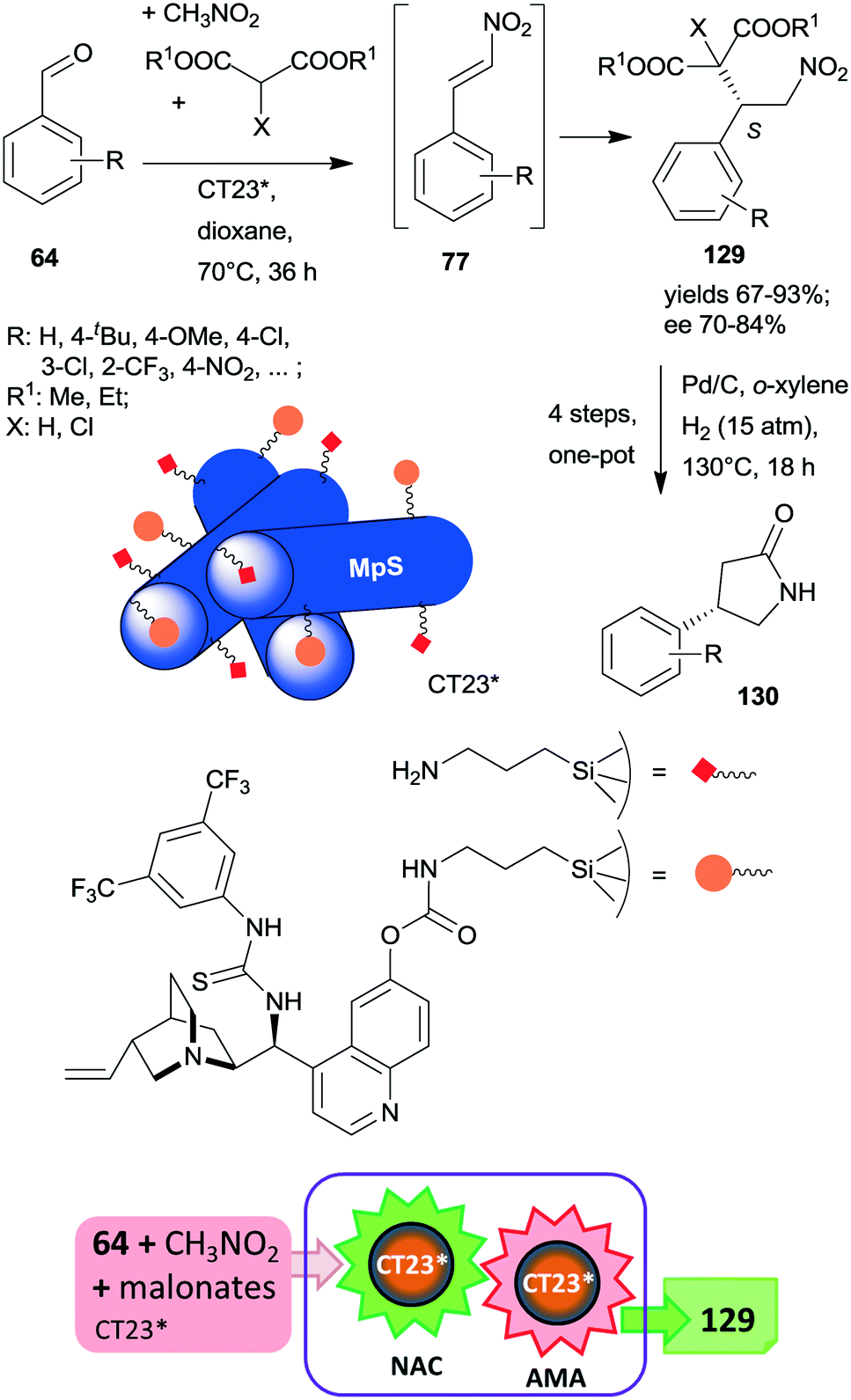 | ||
| Scheme 36 One-pot cascade nitroaldol condensation (NAC) and AMA catalysed by the multifunctional chiral catalyst and transformation of the products in a consecutive one-pot four step reaction over Pd/C; MpS: mesoporous silica.180,181 | ||
A similar one-pot reaction carried out sequentially was developed by Liu and co-workers using bifunctional yolk–shell structured mesoporous silica nanospheres (CT24*).182 The nitroaldol reaction of 64 and nitromethane, affording 77, was catalysed by the basic amino groups bonded to the yolk of the particles, whereas the asymmetric Michael addition of acetylacetone to 77 occurred in the mesoporous shell, where a cinchona-based squaramide derivative was anchored (Scheme 37). Products 131 were obtained in high yields and enantioselectivities using various substituted benzaldehydes; however, between the two steps, the addition of solvent, acetylacetone, and a change in the reaction temperature were necessary. It was shown that the cinchona squaramide hardly affected the first step of this sequential reaction, whereas the surface silanol groups accelerated it by contributing to the activation of the reactants, similar to what was suggested in the nitroaldol or oxa-Michael reactions followed by AMA one-pot transformations catalysed by SBA-15 anchored chiral pyrrolidine derivatives (Schemes 23 and 31).156,168 Recycling of the heterogeneous catalyst showed that the material was active in four consecutive runs. The same research group prepared mesostructured silica anchored N-(3,5-bistrifluoromethylbenzyl)quininium bromide (CT25*) and used this solid as the heterogeneous chiral catalyst in the asymmetric epoxidation of 53 to 132 (Scheme 38).183 The addition of Zn/NH4Cl and ethanol to the slurry afforded in sequential one-pot manner the corresponding β-hydroxy ketones 133 by reductive ring opening of the chiral intermediates in high yields and excellent ees. The reusability of the heterogeneous catalyst was examined in the first step of the sequential process, i.e. the epoxidation of the unsaturated ketones, leading to high yields and preserved ee values following seven recycling processes.
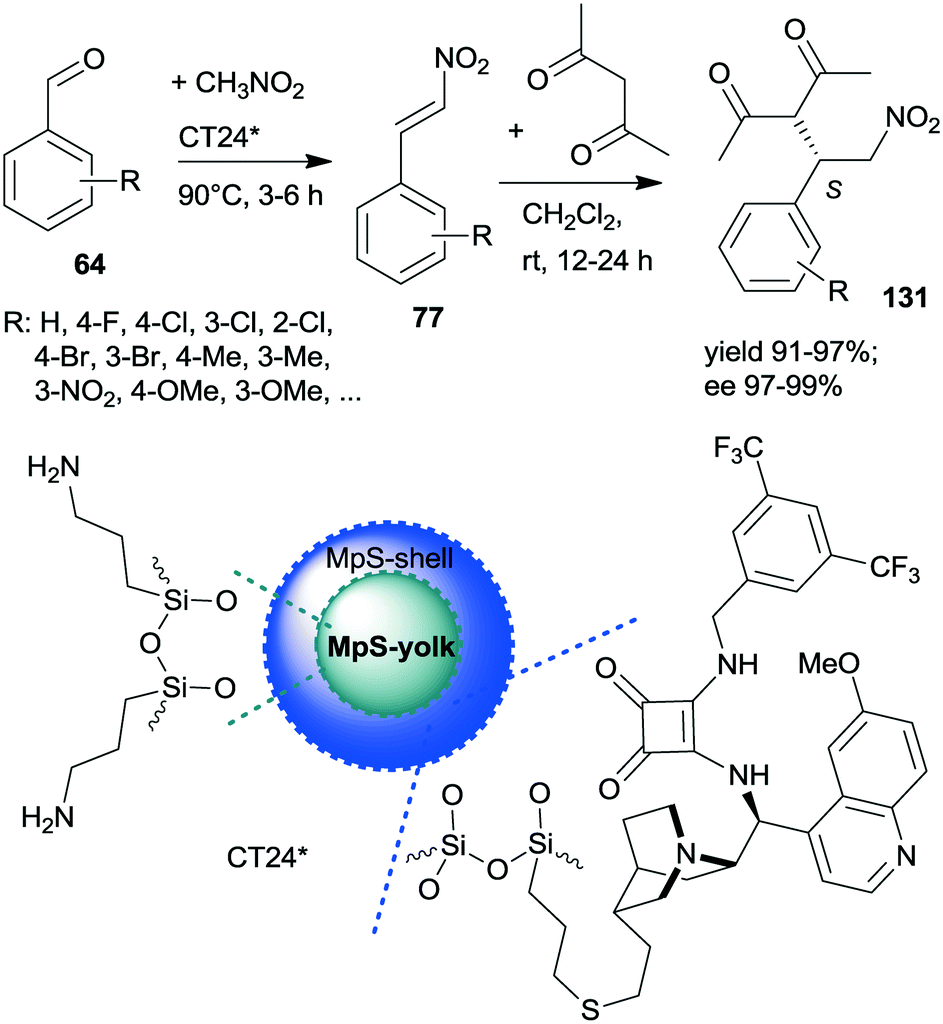 | ||
| Scheme 37 Sequential one-pot NAC and AMA using anchored cinchona alkaloid derivative and pendant primary amine containing catalyst.182 | ||
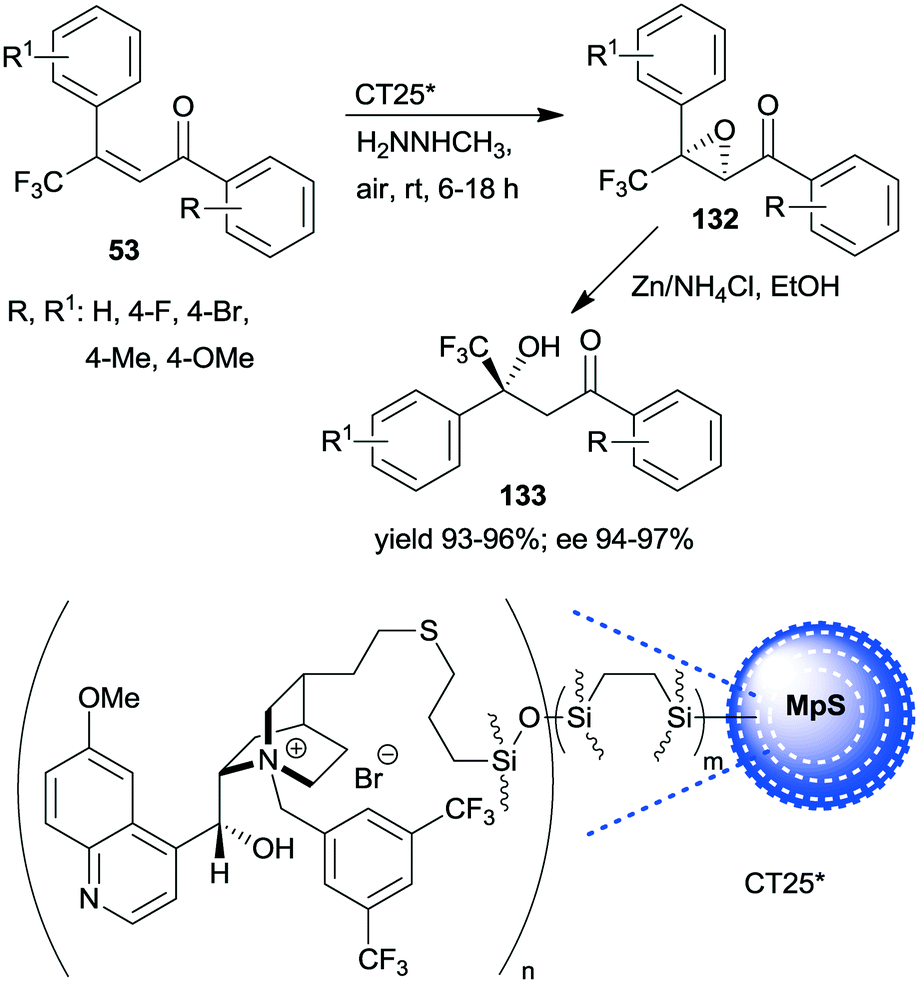 | ||
| Scheme 38 One-pot sequential heterogeneous catalytic asymmetric epoxidation followed by homogeneous reductive ring opening.183 | ||
Very recently, chiral solid organocatalysts were prepared by direct precipitation of cinchona alkaloids derived phosphonic acids with sodium aluminate.184 The materials containing 9-amino(9-deoxy)epi-quinine tethered aluminum phosphonates along with defective P–OH acid sites (CT26*) were used in the double asymmetric Michael addition cascade occurring between unsaturated ketones 134 and nitroolefins 77 through enamine and iminium catalysis (Scheme 39). The reaction afforded cyclohexanone derivatives 135 in good yields and diastereoselectivities and excellent enantiomeric purities. Compared with the corresponding functionalized cinchona derivative, the chiral solid material provided significantly increased yield and better diastereomeric ratio, explained by the acid strength of the free –PO3H2 groups. Furthermore, the amount and acidity of the P–OH defects of the solid material also had significant influence on the catalytic performance of the heterogeneous catalyst. The catalyst CT26* was recycled four times with only slight loss in its performance.184
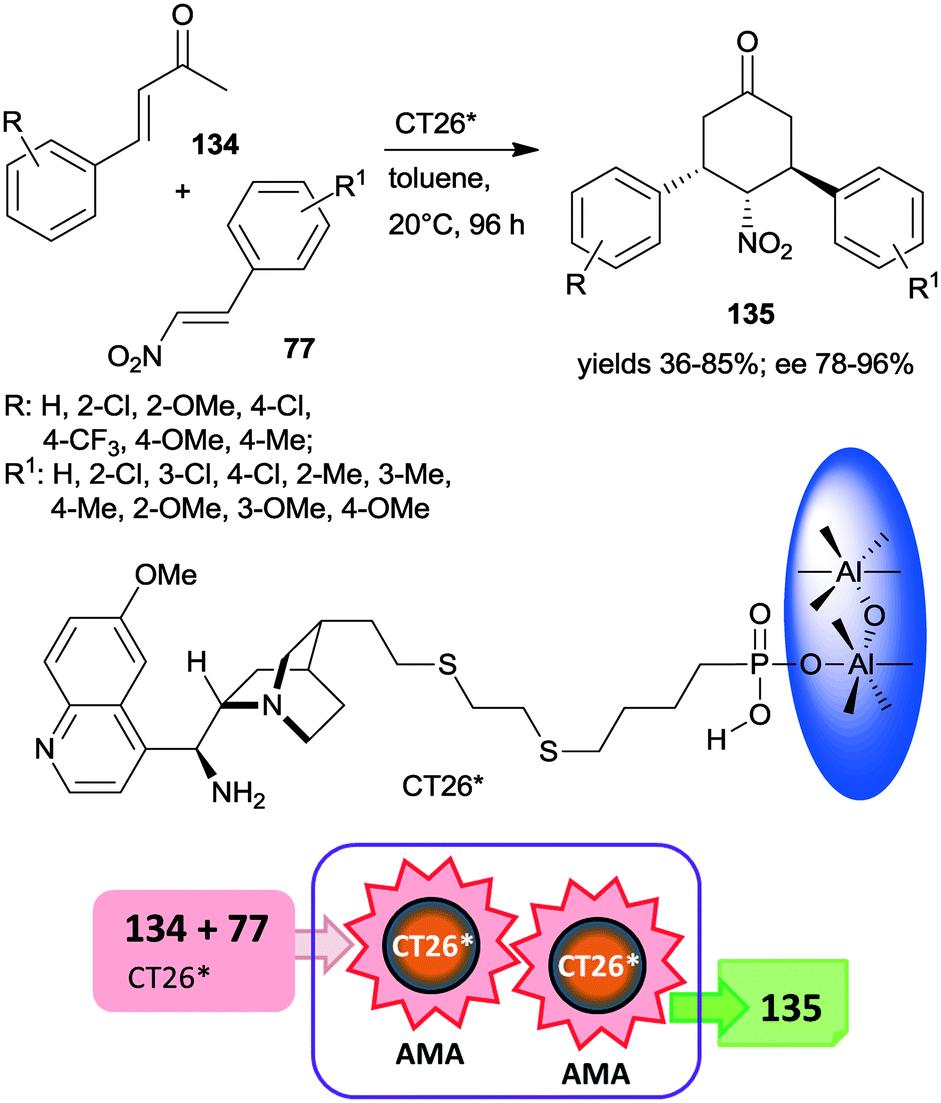 | ||
| Scheme 39 Double AMA cascade reaction of unsaturated ketones and nitroolefins using epi-cinchona alkaloid derived chiral aluminum phosphonate.184 | ||
Heterogeneous asymmetric ketene dimerization was accomplished using cinchona alkaloid derivatives immobilized either on polystyrene resin or on cross-linked poly(methylhydrosiloxane) elastomeric films.185,186 Ketenes 137 were generated in situ from acid chlorides 136 using EtNiPr2. The chiral dimers 138 were further transformed into the corresponding Weinreb amides 140 by ring opening with HN(OMe)Me using 2-pyridone 139 as a homogeneous catalyst.
The latter reaction was not carried out via a one-pot procedure, in order to prevent the contamination of the heterogeneous organocatalyst by reactants used in the final step. Thus, before the ring opening reaction, the heterogeneous catalyst was separated from the reaction mixture (Scheme 40). In these studies both polymer anchored (dihydro)quinidine dimers (CT27*)185 or dihydroquinidine immobilized by functionalization of the C–OH group (CT28*)186 were found to be similarly efficient to their homogeneous counterparts, both providing high enantioselectivities. The immobilized dimeric cinchona alkaloid CT27* was recycled 19 times with only small decreases in the yields and enantioselectivities in the final runs.
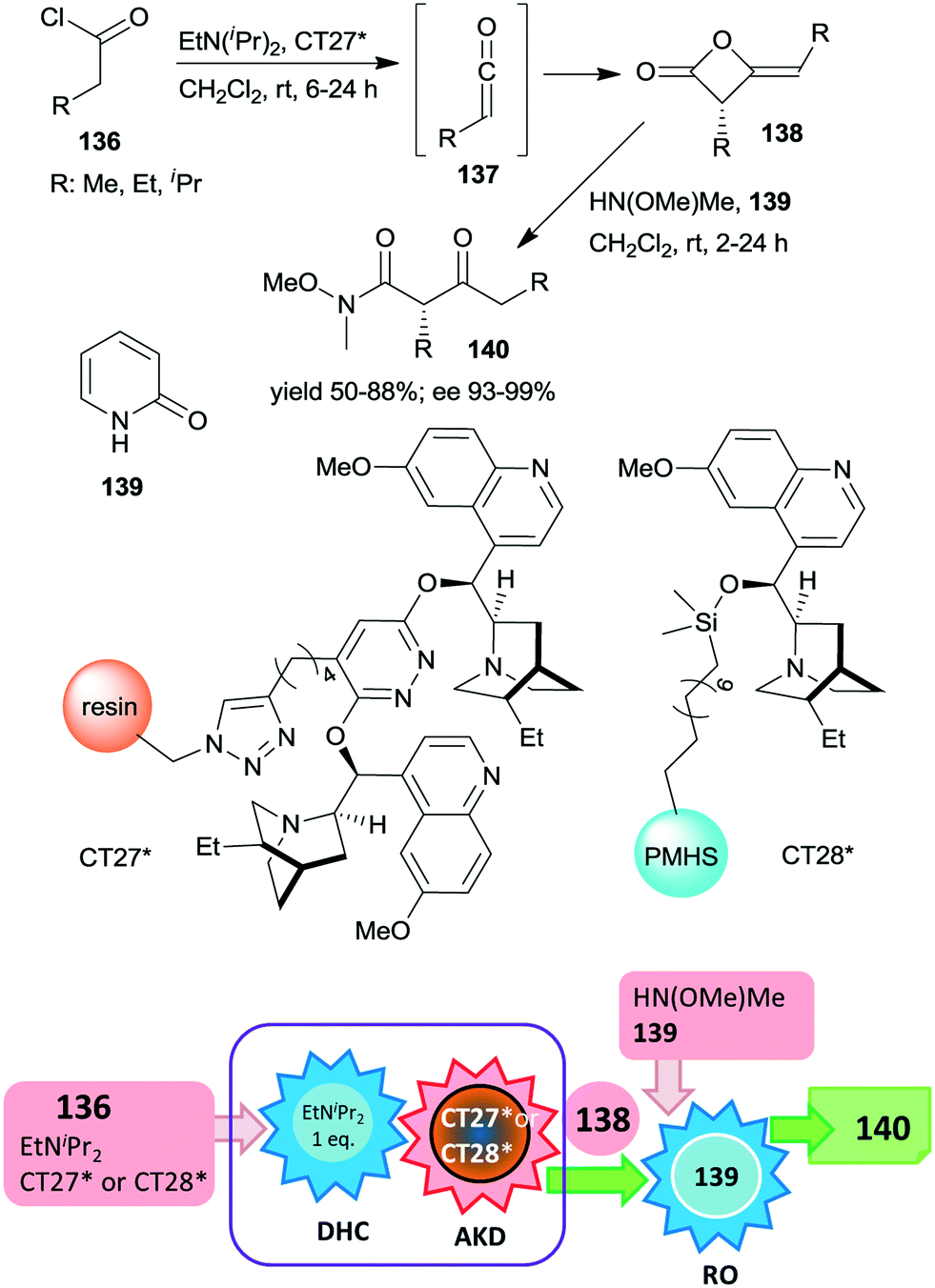 | ||
| Scheme 40 One-pot acid chloride dehydrochlorination (DHC) and asymmetric ketene dimerization (AKD) using heterogeneous cinchona alkaloid catalyst followed by ring opening (RO) to Weinreb amides; PMHS: poly(methylhydrosiloxane).185,186 | ||
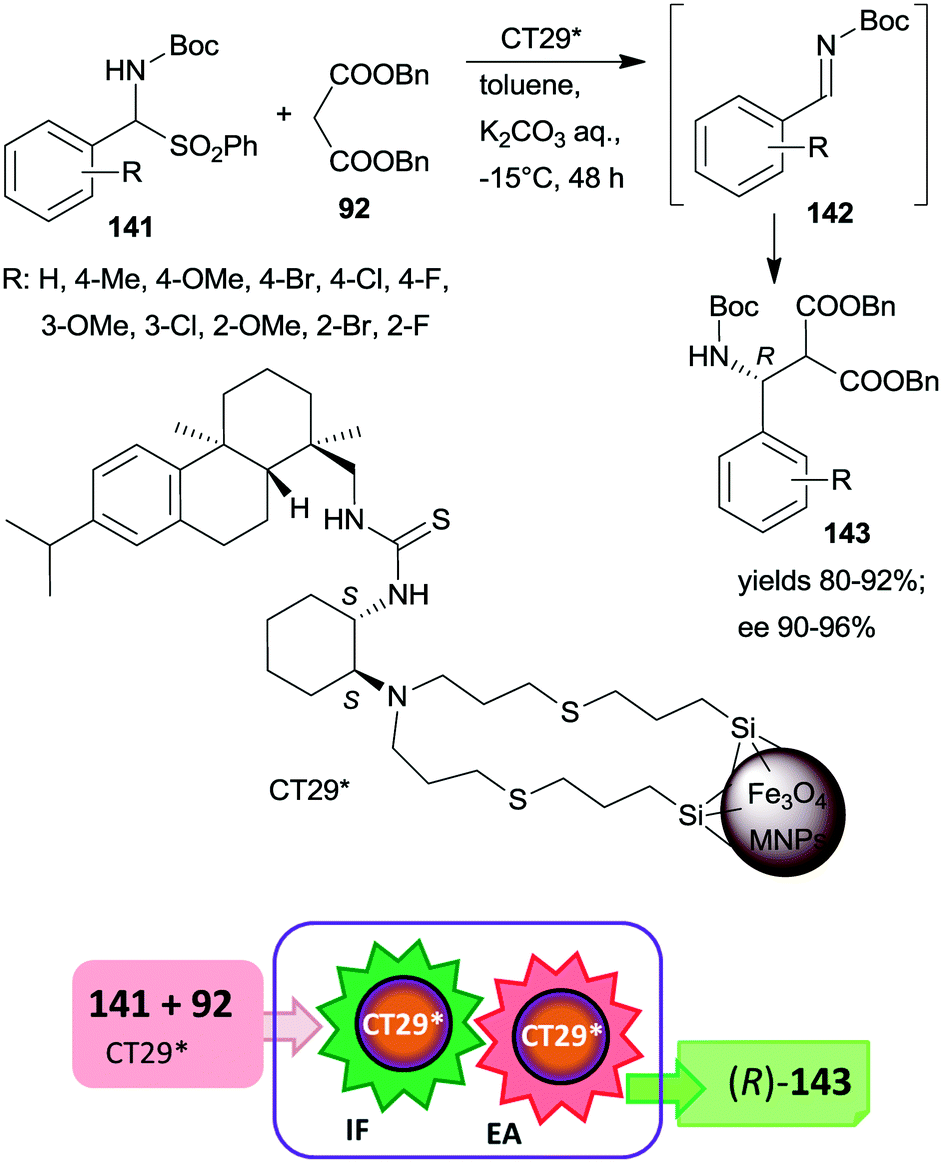 | ||
| Scheme 41 Tandem IF and EA starting from Boc-protected α-amino sulfones catalysed by 1,2-cyclohexanediamine derived chiral catalyst anchored on MNPs.187 | ||
Continuing their efforts at designing bifunctional chiral heterogeneous catalysts for one-pot reactions (Scheme 27),161 Kobayashi and co-workers prepared layered bifunctional catalysts that had an achiral polymer incarcerated carbon black-supported Au/Pd or Au/Pt NPs core and a copolymer shell containing chiral phosphoric acid units (CT30* or CT30*’).188 Previously, chiral phosphoric acids proved their efficiency in homogeneous asymmetric organocatalytic reactions.189 The novel chiral materials were used in the sequential aerobic oxidation of benzyl alcohols 144 followed by organocatalytic asymmetric aza-Friedel–Crafts reaction of the aldehydes 64 and N-aminoethylpyrrole (145), leading to piperazine derivatives 146 (Scheme 42). Although the one-pot reaction did not proceed in a cascade manner, due to the deactivation of the NPs by the primary amine, sequential elaboration by addition of 145 and additives following the oxidation step provided the desired products in high yields and ees. Reverse placement of the metal alloy and organocatalyst in the layered material resulted in a similarly efficient catalyst as CT30*. Substituents on the phenyl ring of 144 had a significant effect on the results when Au/Pd NPs were applied, whereas the Au/Pt NPs were efficient catalysts even in the presence of electron withdrawing substituents. The catalysts could be reused, unlike CT13*, which was deactivated under the reaction conditions.161
 | ||
| Scheme 42 Sequential oxidation – enantioselective aza-Friedel–Crafts reaction catalysed by bifunctional layered catalysts; PI: polymer incarcerated.188 | ||
The relatively numerous examples of applications of heterogeneous chiral organocatalysts in cascade or sequential one-pot asymmetric processes, the majority being developed in the last decade, are due to the insensitive nature and ease of anchoring these catalytically highly efficient chiral compounds. Furthermore, the successful recycling of these heterogeneous catalysts has promoted their application in continuous-flow systems.190,191 Sequential asymmetric reactions using immobilized reagents, chiral organocatalysts and scavengers placed in successive fixed-bed reactors provided the opportunity for the convenient preparation of optically pure fine chemicals. Similar to the one-pot reactions, during these processes, several purification steps may be eliminated; moreover, reactions occurring through sensitive, unstable, hazardous intermediates are easily carried out. Lectka and co-workers described an elegant early example using immobilized quinine catalyst. A chiral β-lactam was prepared from ketenes and iminoesters, both obtained in preceding columns.192 The chiral product was purified in flow by passing through a final scavenger column for removing side-products. As the catalysts, reagents used in different steps are separated in space (in different columns) and due to the possibility of using scavenger columns between or following the reactors, there are no undesired interactions and consequently no unfavourable effects on the performance of the reaction components; thus, the products may be obtained in high quality. Despite these advantages, these reactions may not be considered one-pot reactions, and are accordingly out of the scope of the present review. Moreover, the development of such processes is still in its infancy, mostly due to the laborious design of the necessary experimental setups, making the one-pot batch methods more accessible for laboratory chemists.
The examples surveyed in the above subsection, in which heterogeneous chiral organocatalysts are applied in one-pot reactions have demonstrated the potential of using such catalysts for the convenient preparation of chiral fine chemicals. Although many of these solid catalysts were applied in combination with homogeneous catalysts, several materials were developed, which contain all the catalytic species immobilized on a single support, leading to isolation of the active sites needed in different steps. Accordingly, such catalysts could give improved results as compared with the homogeneous catalysts. In a few reactions, the support was also involved in the activation of one or more substrates or intermediates. Thus, the role of the heterogenization of the chiral organocatalysts is often not merely to facilitate the recovery and reuse of these materials, but also to improve the performances of the chiral catalysts. The known examples also show that the latter goals may be reached only by meticulous catalyst design.
2.3. Asymmetric one-pot reactions over chirally modified solid catalysts
A convenient method to obtain heterogeneous chiral catalysts is the modification of catalytic surfaces by optically pure compounds, the so-called chiral modifiers.17–24 The chiral compounds used have no catalytic activity in the reaction and will have “solely” the role of directing the substrate on the catalyst surface. Usually, the simple adsorption of the chiral modifier is sufficient to reach appropriate results and often the addition of the modifier to the reaction slurry, i.e. the in situ modification, gives similar results to those obtained with catalyst premodified before introduction in the systems.Most of the efficient chirally modified heterogeneous catalysts are supported metals, which are used in enantioselective hydrogenations of ketones and olefins.20–24 However, metal oxides are also used as enantioselective catalysts following modification with chiral compounds.193,194 Still, only one example of an asymmetric one-pot sequential reaction applying chirally modified metal oxide catalyst is known.195 Nanocrystalline chirally modified MgO was used in the Claisen–Schmidt condensation and asymmetric epoxidation reaction sequence. The surface of the oxide nanoparticles was modified in situ with (+)-diethyl tartrate (CM1*) following the condensation step as illustrated in Scheme 43, when the oxidation agent was also introduced. The same sequence was also catalysed by preadsorbed poly-L-leucine on hydrotalcite CT21*, as shown previously (Scheme 34), where the oligopeptide was already present in the system in the first condensation step and was the active catalyst in the second asymmetric Juliá–Colonna epoxidation.177 In contrast, CM1* has no catalytic activity, it was anchored on the surface of the MgO by hydrogen-bonding and controlled the direction of delivery of the nucleophilic oxygen of the surface-bonded peroxide (Mg–OOtBu) to the intermediates formed from the chalcone derivatives 121, as shown by the structure in Scheme 43. In this one-pot sequential reaction, lower yields and enantioselectivities were obtained as compared to the use of CT21* or even with the two-pot method using the same MgO-NPs and CM1*. However, due to its simplicity and the catalyst recyclability, the procedure is an attractive way of preparing chiral ketoepoxides 122.195
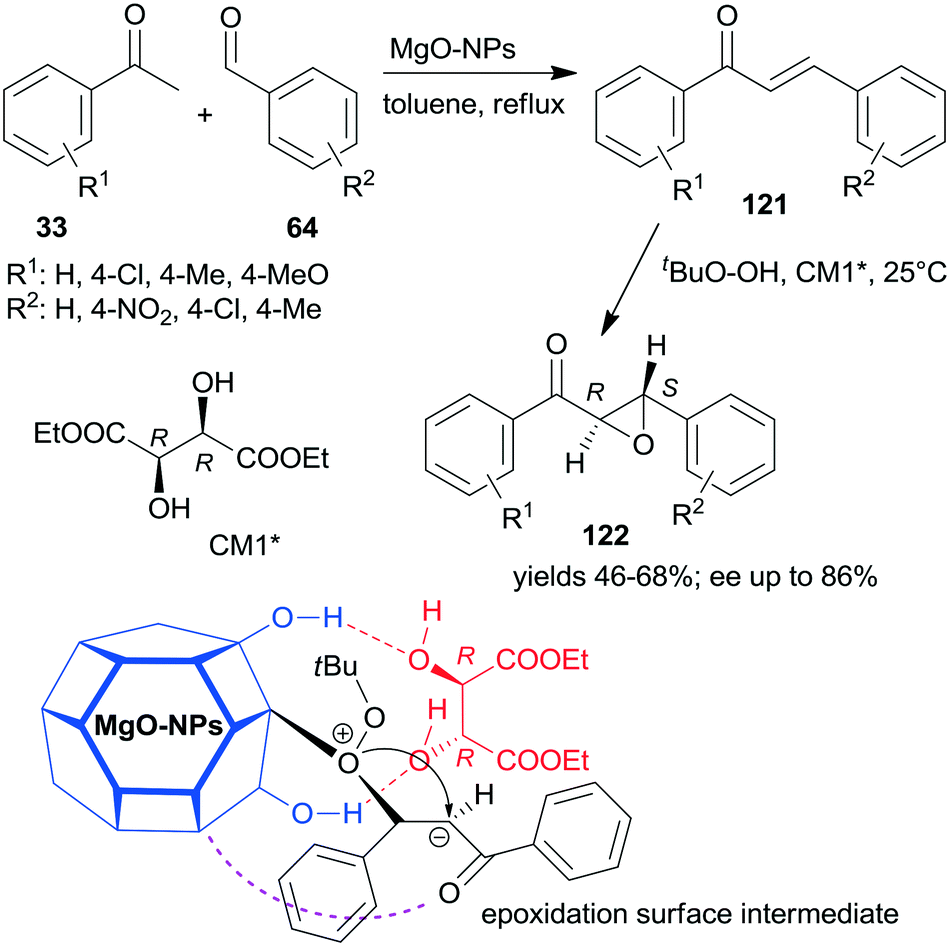 | ||
| Scheme 43 Claisen–Schmidt condensation and asymmetric epoxidation one-pot sequential reaction catalysed by chirally modified nanocrystalline MgO and the suggested structure of the surface intermediate.195 | ||
The use of heterogeneous chirally modified metal catalysts in enantioselective hydrogenations became extensively studied at the turn of this century. Among these, three catalytic systems were found to be highly efficient: the tartaric acid-modified Ni catalysts for the hydrogenation of β-keto esters and the Pt and Pd catalysts modified by cinchona alkaloids used in the enantioselective hydrogenation of α-keto esters and activated olefins, respectively.20–24 However, the application of these catalysts in one-pot sequential or cascade reactions was retarded by the narrow substrate scope of these reactions and by the sensitive nature of the active chiral surfaces.
The first one-pot cascade reaction using a chirally modified metal catalyst exploited the facile cyclization of γ-hydroxycarboxylic acids to the corresponding chiral butyrolactones. Enantioselective hydrogenation of 2-oxoglutaric acid 147 over Pt/Al2O3in situ modified by cinchona alkaloids gave 2-hydroxyglutaric acid 148 as the intermediate product, which by cyclization resulted in the formation of 5-oxotetrahydrofuran-2-carboxylic acid 149 (Scheme 44).196 Hydrogenation in the same catalytic system of the diethyl ester of 147 also resulted in the chiral alcohol in up to 96% ee, however, the cyclization of the product needed a subsequent step using TsOH as catalyst.197 The cascade reaction of the dicarboxylic acid 147 could be carried out in water; the best results, i.e. up to 92% ee at full conversion, were obtained at low temperature, under 25 atm H2 pressure using dihydrocinchonidine methyl ether as modifier (CM2*). The second step of the cascade reaction may occur either on the catalyst surface or in the liquid phase, however, this was not investigated in the report.196
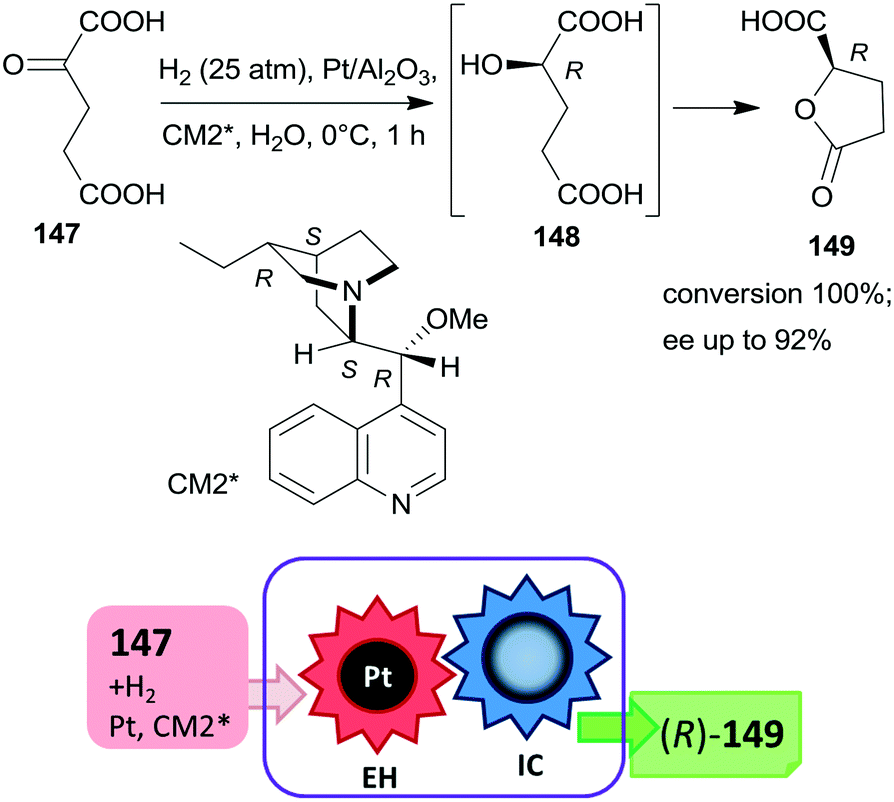 | ||
| Scheme 44 Cascade enantioselective hydrogenation (EH) and intramolecular cyclization (IC) of 2-oxoglutaric acid using Pt catalyst modified by cinchona alkaloid CM2*.196 | ||
Cinchonidine (CM3*) was found to be the most efficient chiral surface modifier for the enantioselective hydrogenation of α,β-unsaturated carboxylic acids over supported Pd catalysts,198 among which substituted (E)-2,3-diphenylpropenoic acids were hydrogenated in up to 96% ee.199–201 During the hydrogenation of (E)-2,3-diphenylpropenoic acids bearing 2-nitro substituents on the 3-phenyl ring (150), the nitro group reduction, enantioselective hydrogenation and intramolecular amidation cascade took place, leading to 3-phenyl-3,4-dihydroquinolin-2(1H)-ones 153 (Scheme 45).202 Although close to complete conversions of the acids to 153 were detected, the enantioselectivities were low due to the unfavourable effect of the substituents in the ortho position of the β-phenyl ring, similar to acids substituted with other groups in the same position.199,201 It must be noted that the first two reductive steps of these reactions are competitive; however, both are necessary for the formation of the final cyclic product. The order of these steps could be deduced from the high initial H2 up-take rate characteristic of the nitro group reduction and confirmed by the ee values, which suggest the presence of electron donating groups (–NH2) during the hydrogenation of the olefinic bond. There was no indication of whether the final step takes place on the catalyst surface or in solution; however lack of detection of the saturated intermediate 152 in the solution at any time may suggest the former pathway.
 | ||
| Scheme 45 Cascade nitro group reduction (NR), EH and IC over cinchonidine modified Pd catalyst.202 | ||
Aromatic nitro group reduction was also one of the steps of a one-pot cascade reaction developed recently over Pt catalyst modified by cinchona alkaloids.203,204 The hydrogenation of 2-nitrophenylpyruvates 154 over chirally modified Pt catalysts resulted in the formation of optically enriched 3-hydroxy-3,4-dihydroquinolin-2(1H)-ones 157 (Scheme 46), instead of the indole-2-carboxylates usually formed via Reissert indole synthesis if other reduction methods are applied. Although in this cascade reaction the first two reductive steps are also competitive, the high yields of the quinolone derivatives 157 showed that the enantioselective hydrogenation accelerated by the presence of the chiral modifier preceded the reduction of the nitro group. The intermediate 156 bearing both hydroxyl and amino groups desorbed from the Pt surface and was further transformed following readsorption after consumption of the pyruvates 154.204 The highest ee values were obtained using CM2* as chiral modifier. Substituents on the phenyl ring of 154 had significant effects on both the yield and the ee by influencing the rate of the –NO2 group reduction, and as a consequence the product distribution and by affecting the substrate-modifier interaction. Substituents next to the nitro group had beneficial influence on the quinolone selectivity.203
 | ||
| Scheme 46 Cascade EH, NR and IC of 2-nitrophenypyruvates over chirally modified supported Pt catalyst.203,204 | ||
It was demonstrated that the chiral modifier has multiple roles; besides inducing differentiation between the molecule enantiofaces by directing the adsorption of 154, it accelerated the hydrogenation of the keto group, a feature characteristic to this catalytic system, and decelerated the reduction of the nitro group by occupying surface active sites. Moreover, it was shown that the presence of the nitro group also contributed to reaching good enantioselectivities by interacting with the adsorbed modifier; the absence of this group diminished the ee value. This heterogeneous cascade reaction was also carried out in a flow system using a Pt catalyst bed, however, the results obtained in batch system could not be equalled, especially as concerning the selectivity of the desired product 157.205 Results obtained in the flow system showed that instantaneous cyclization without desorption of the intermediate from the Pt surface also occurs at higher conversion.
The polymer incarcerated carbon black supported bimetallic Au/Pd NPs, i.e. PI(C-Au/Pd) used as the core of CT30* (Scheme 42) was applied in the oxidation followed by the Horner–Wadsworth–Emmons olefination tandem reaction to obtain α,β-unsaturated esters 159 (Scheme 47).206 The tandem reaction was continued by a sequential asymmetric 1,4-addition of phenylboronic acid derivatives 39 to 159 using polymer incarcerated carbon black supported Rh Nps, i.e. PI(C-Rh), chirally modified with an optically pure diene CM4*. This three-step one-pot reaction afforded chiral β-branched esters 160 in moderate to good yields and outstanding enantioselectivities from various benzylalcohol derivatives and phenylboronic acids.
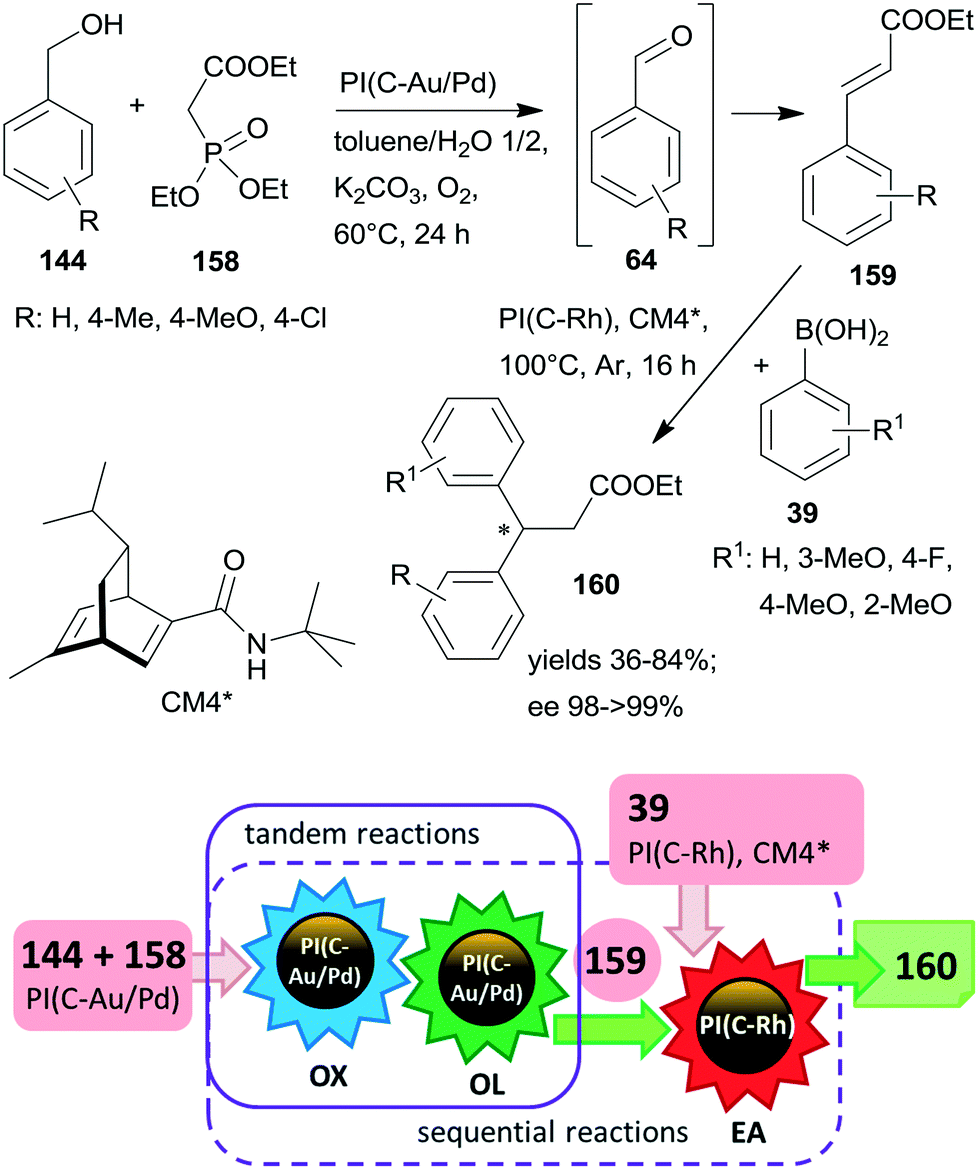 | ||
| Scheme 47 Tandem OX and olefination (OL) followed by sequential one-pot enantioselective 1,4-addition (EA) over chirally modified Rh nanoparticles.206 | ||
Chiral undecagold clusters bearing as ligand (R)- or (S)-(6,6′-dimethoxybiphenyl-2,2′-diyl)bis(diphenylphosphine) in combination with silver salts were used in tandem hydroarylation and cyclization reactions.207 The Ag salt promoted the formation of larger Au NPs, as well as bimetallic gold complexes. Thus, during reaction, chiral surface modified Au was also formed, however, it was suggested that the complexes are the catalytically active species, which afford the target cyclopentane derivative in high yields and up to 44% ee. Utilization of the support chirality was attempted in the one-pot tricomponent aldehyde, alkyne, amine coupling. Deposition of Au NPs into channels of a chiral metal–organic framework prepared from L-lactic acid resulted in catalytically active material, without inducing significant enantioselectivities.208 The latter two examples illustrated alternative approaches to preparing chiral catalytically active metal surfaces; however, these were less successful, as compared to the use of known enantioselective heterogeneous chirally modified catalysts.
In spite of the simplicity of obtaining and applying chirally modified metal catalysts in enantioselective reactions, the few examples of utilizing such materials in one-pot processes showed the difficulties in harmonizing the conditions and the tolerance of the components necessary in different steps of cascade or sequential reactions.
3. One-pot reactions including biocatalytic asymmetric step combined with heterogeneous chemical catalysis
Biocatalysis is still one of the most important ways to prepare optically pure fine chemicals.12,65 The activities and stereoselectivities of enzymes are enviable. Attempts to reproduce these performances by chemical catalysts necessitate carefully designed catalytic materials. Complementing the substrate specificity of the enzymes with the broad applicability of chemical catalysts was the main driving force for developing one-pot processes combining the two types of catalysts.69–71 In most of these processes applying such catalyst combinations, the stereoselective steps are biocatalysed, leaving the opportunity for choosing convenient heterogeneous chemical catalysts for the other steps. Exceptions to this strategy have already been presented in previous subsections (see Schemes 15, 25 and 33), in which anchored chiral chemical catalysts were applied along with enzymes. Although in DKRs enzymes are often used for the KR and heterogeneous chemical catalysts in the racemization steps,73,75,77,79–82 these processes are beyond the scope of the present review, as indicated in the Introduction section.An early one-pot tandem process was developed by Van Bekkum and co-workers, who combined the enzymatic isomerisation of D-glucose (161) using immobilized glucose isomerase with the heterogeneous catalytic hydrogenation of the 161 and D-fructose (162) mixture (invert sugar) to a mixture of D-glucitol (163) and D-mannitol (164).209,210 Although the process may be considered a DKR, starting from pure 161 it was possible to reach high 164 selectivities using Cu/SiO2 catalyst, suggesting that 161 is transformed in a biocatalytic and heterogeneous chemical catalytic cascade (Scheme 48a). Later, also in carbohydrate chemistry a sequential three-step one-pot process was developed for the transformation of methyl β-D-galactoside (165) to methyl 4-deoxy-6-aldehydo-β-D-glucoside hydrate 168.211 The one-pot process included a sequence of enzymatic oxidation, homogeneous catalytic dehydration and heterogeneous catalytic hydrogenation. Changes in reaction temperature and addition of catalysts was necessary between steps, which could be performed in water, warranting the success of the one-pot procedure (Scheme 48b). During the process, the only step in which a chiral centre was formed was the heterogeneous hydrogenation of the prochiral olefinic bond over Pd/C, and the stereoselectivity of this step was not induced by the catalyst. These sequential heterogeneous catalytic reactions were included in this survey as pioneering examples of using combinations of enzymatic and heterogeneous chemical catalytic steps in one-pot processes.
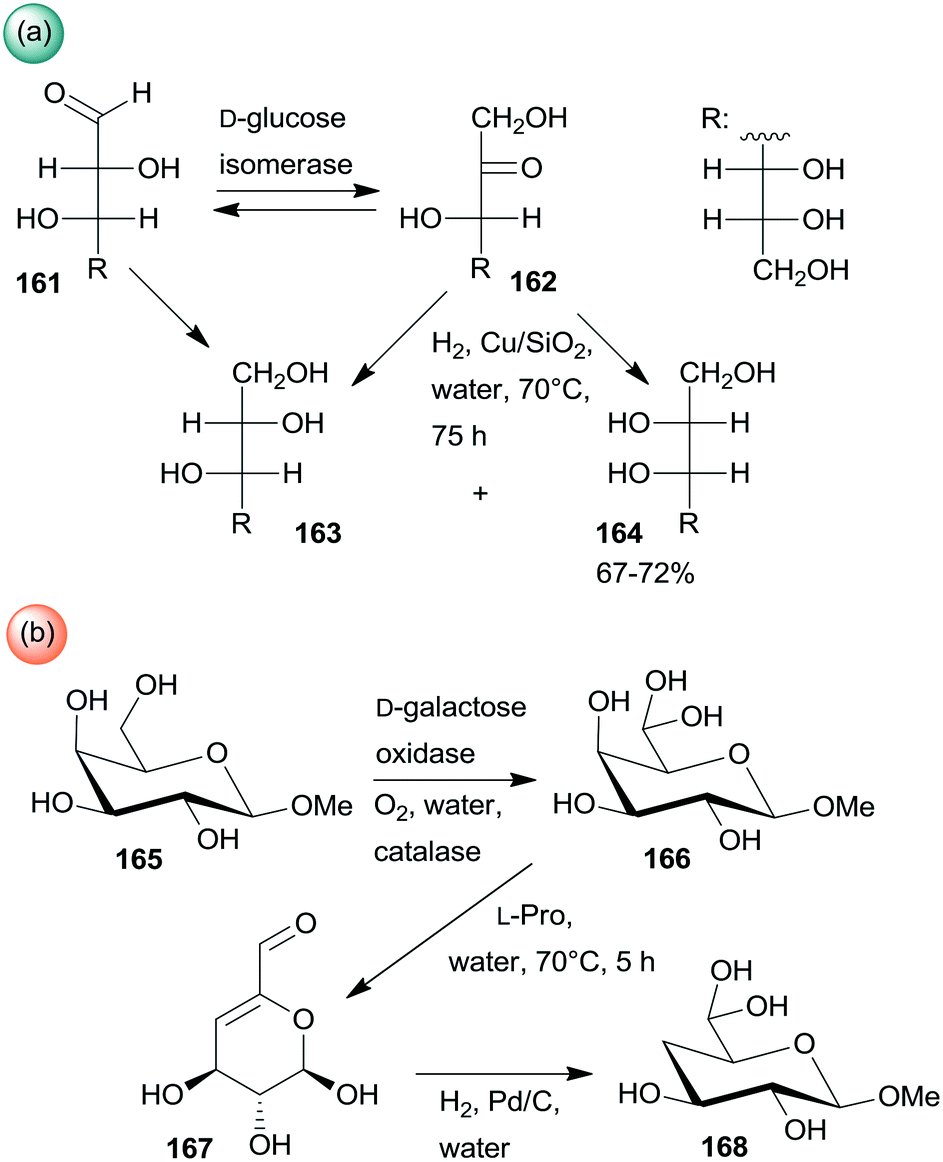 | ||
| Scheme 48 One-pot processes including enzymatic isomerization and heterogeneous catalytic hydrogenation (a); enzymatic oxidation, homogeneous catalytic dehydration and heterogeneous catalytic hydrogenation (b).209–211 | ||
The one-pot combination of heterogeneous catalytic preparation of racemates with their successive DKR was initially described using Pd/C and lipase tandem catalysis.212 Prochiral ketoximes 169 were hydrogenated over Pd/C catalyst, followed by acetylation of the amine intermediate 170 with ethyl acetate using Candida Antarctica lipase B (CALB) immobilized on polyacrylamide polymer (commercially available as Novozym-435) in organic solvent, when the Pd also catalysed the racemization of the unreacted amine enantiomer. By DKR of the amines 170, (R)-N-acetyl amines 171 were prepared in high yields and excellent enantioselectivities using various ketoximes, including carbocyclic and heterocyclic compounds (172–174) (Scheme 49). Notably, the Pd/C catalysed both the hydrogenation and the racemization reactions. Later, home-made Pd nanocatalyst, Pd/AlO(OH), was also applied in this cascade reaction using ethyl methoxyacetate in the acylation step, obtaining similar high yields and enantioselectivities in shorter reaction time (48 h).213
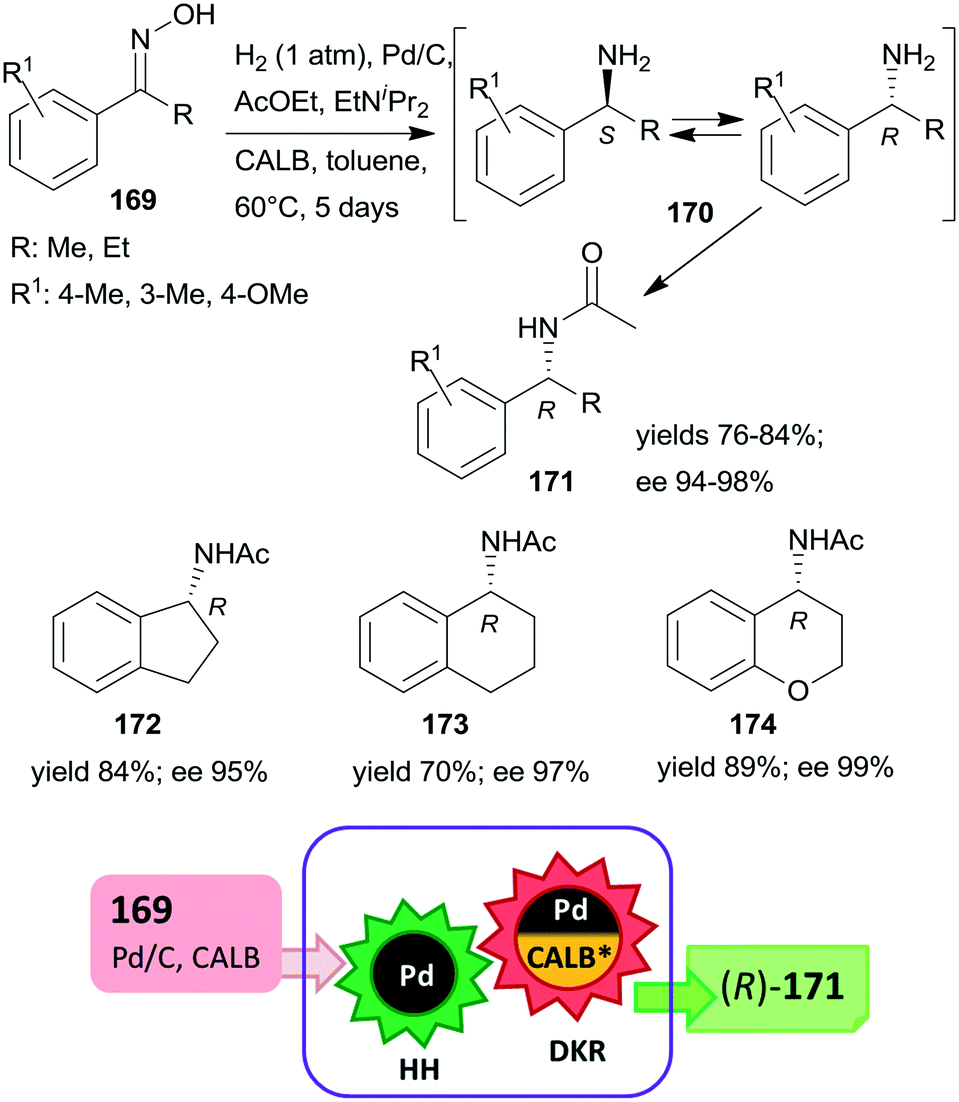 | ||
| Scheme 49 One-pot transformation of prochiral ketoximes by heterogeneous catalytic hydrogenation (HH) and DKR using Pd/C and lipase B (CALB) catalysts.212 | ||
By a similar approach, prochiral ketones were transformed into acylated alcohols through chemo-enzymatic heterogeneous catalytic cascade reactions using supported Pd catalysts in the initial hydrogenation step.214 These reactions, previously carried out using homogeneous hydrogenation catalysts,215,216 were studied in more detail than the above presented one-pot reaction of ketoximes. In most of these studies, acetophenone 33a was transformed into (R)-1-phenylethylacetate (176) by hydrogenation using supported Pd catalysts to produce racemic 1-phenylethanol (175) followed by acylation with ethyl acetate, applying the immobilized CALB (Novozym-435) catalyst.214,217–221 In contrast to the one-pot reaction of ketoximes, in the transformation of 33a it was shown that the Pd catalyst was not efficient in the racemization step. Although optically pure 176 was obtained, the selectivity of the product did not exceeded 50%. It was shown that the chiral product resulted from KR of racemic 175, whereas the S alcohol enantiomer was consumed by side-reactions, such as dehydration to 26a. Moreover, based on results obtained using different supports, it was concluded that by-products decreased the activity of both the metal and the enzyme.214,217,218
The sequential continuous-flow hydrogenation of 33a over Pd/SiO2–Al2O3 and KR of 175 by immobilized CALB or cross-linked enzyme aggregates was carried out in supercritical CO2 using consecutive reactors.219 Later, in order to catalyse the racemization of the unreacted (S)-175, supported Ru catalyst was also introduced in the batch system, which in the absence of Pd did not promote the hydrogenation of 33a.220 Promising results were obtained over Ru/Al2O3 (Scheme 50a).220,221 The scope of the latter cascade reaction was extended on the transformation of 1,2-indanedione 177 using Pd/Al2O3, Ru(OH)3/Al2O3 and lipase AK immobilized on celite as catalysts and trifluoroethyl acetate as acylation agent (Scheme 50b).222 The evolution in time of the composition of the reaction mixture showed the DKR of rac-2-hydroxy-1-indanone 178 formed regioselectively by hydrogenation over Pd/Al2O3. The (R)-2-acetoxy-1-indanone 179 resulted in good enantiomeric purity (86–92% ee), however, only up to 50% yield was achieved.
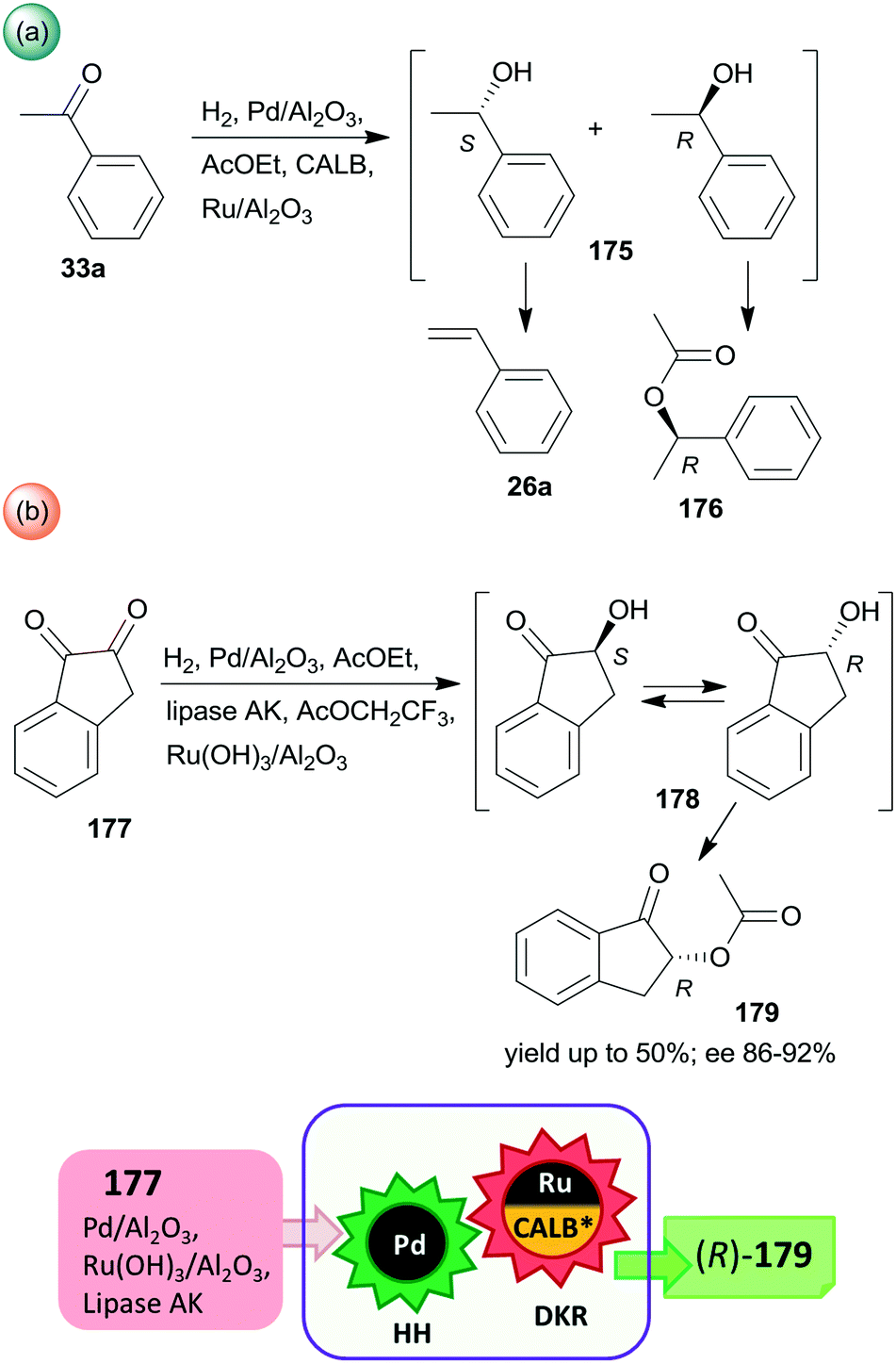 | ||
| Scheme 50 Cascade reaction of prochiral ketones by HH and KR or DKR using supported Pd, Ru and immobilized lipase catalysts.220–222 | ||
Córdova and co-workers disclosed asymmetric one-pot heterogeneous metal – enzymatic relay catalytic processes using Pd NPs anchored on aminopropyl-functionalized mesocellular silica foam catalyst developed previously (Pd NPs/support,97Scheme 3).223 Aromatic ketones 180 were transformed into racemic amines 181 by reductive amination using HCOONH4 for the in situ generation of imines and also the hydrogen source in the reductive step catalysed by Pd. The enzymatic KR of the racemic amines followed this heterogeneous catalytic step by transformation of 181 into amides 182 using lipase (CALB, Novozym-435) catalyst and ethyl methoxyacetate as acylation agent. Changing the solvent between the two catalytic steps and introduction of additional Pd catalyst in the second reaction to decompose the excess HCOONH4 hydrogen donor was necessary. However, it was found that under these conditions, no racemization of the amine occurred, and the corresponding amides were obtained in good optical purities, but low yields. In order to foster the racemization of the unreacted amine enantiomer, the second step was carried out under H2 atmosphere, which promoted this reaction and allowed increased yields of the R amides (Scheme 51a). The reductive amination using the same Pd catalyst was also combined in a one-pot sequential process with enzymatic kinetic resolution of the amines 181. Amine transaminase (ATA-117) and sodium pyruvate together with supported Pd catalyst afforded high yields and optically pure (S)-181 in this catalytic relay process (Scheme 51b).223
 | ||
| Scheme 51 Sequential one-pot catalytic transformations of prochiral ketones by reductive amination and consecutive DKR to amides or kinetic resolution of the amines using supported Pd and CALB or ATA catalysts.223 | ||
Palladium NPs stabilized by thermostable protein (Pd NPs/Te-Dps; Dps: DNA binding protein from starved cells) was used in the one-pot sequential Suzuki–Miyaura cross-coupling and enzymatic enantioselective reduction of phenylboronic acid 39 and phenyl iodide 183 derivatives, with one of the substrates bearing the acetyl group in the para position. The acetyl biaryl intermediates 184 were reduced to chiral biaryl alcohols 40.224 The first step was carried out in water at 100 °C, whereas cooling the mixture to room temperature and addition of iPrOH, alcohol dehydrogenase [(R)-LB-ADH] and NADP+ afforded the optically pure alcohols 40 in high yields after 24 h of stirring, as shown in Scheme 52a. However, attempts to recycle the heterogeneous Pd catalyst were unsuccessful; in the third use, no transformation was observed, which showed that Pd species were removed from the protein.
 | ||
| Scheme 52 Sequential one-pot Suzuki–Miyaura reaction followed by enzymatic enantioselective reduction (a) and stereoselective epoxidation followed by asymmetric enzymatic hydrolysis (b).224,225 | ||
One-pot sequential preparation of a single (1R,2R,4S)-menthene-1,2-diol isomer 187 was possible by epoxidation of (−)-limonene 185 using Ti deposited on ordered or amorphous mesoporous silica (Ti/SBA-15 or Ti/SiO2) catalysts and tBuO-OH as oxidant in PhCF3 solvent followed by selective hydrolysis with recombinantly expressed Rhodococcus erythropolis (Re-LEH) enzyme in potassium phosphate buffer, as illustrated in Scheme 52b.225 The enzyme tolerated the presence of the organic solvent; thus, after cooling the mixture to room temperature, the addition of the aqueous biocatalyst solution to the crude product was sufficient to cause the second hydrolysis step to proceed. Good yields were obtained following the two steps; moreover, starting from (+)-limonene, the opposite diol enantiomer, ent-187, could be obtained.
Finally, the few examples reported on the application of heterogeneous chemical catalyst and biocatalyst combinations in one-pot preparation of optically pure compounds indicated the difficulties met in employing the two catalyst types concomitantly, due to often met incompatibilities with the components needed in different steps. Such incompatibilities are mostly due to differences in the solvent nature and reaction temperature required in steps catalysed by these very dissimilar catalysts. Due to these dissimilarities, the research of one-pot processes applying these catalyst combinations is directed mainly toward sequential procedures. However, owing to the outstanding stereoselectivities reached in the biocatalytic reactions and the practical advantages of the heterogeneous catalysts, the development of novel one-pot cascade reactions unifying these catalyst types is expected.
Conclusions and outlook
Asymmetric one-pot processes, which include heterogeneous catalytic steps, are presently the most suitable for improving efficiency and sustainability, and decreasing the environmental impact of the methods designed to prepare chiral fine chemicals. These processes may compete or complement the frequently applied biocatalytic methods by uniting the advantages provided by handling heterogeneous catalysts with the benefits of avoiding the intermediate product isolation. In spite of these attractive features, asymmetric one-pot processes using heterogeneous chemical catalysts have rarely been reported until now, although a vast number of highly efficient chiral catalysts used in asymmetric homogeneous one-pot processes are known. This is explained by the often laborious immobilization of these catalysts, the sometimes maleficent presence of the support and by the frequently observed unfavourable effect of reaction components and conditions needed in different steps of one-pot processes.Detailed surveys of the reported results obtained using heterogeneous chemical catalytic systems showed the dominance of the application of catalysts obtained by immobilizing chiral organocatalysts, mostly by covalent bonding on organic polymers or inorganic supports, as shown in Table 1 (the numbers refer to the developed catalysts and the applicability in different reactions is not included). More than twenty catalytic systems have been disclosed with chiral catalysts prepared from amino acid derivatives such as proline and its derivatives, amino alcohol derivatives such as the highly efficient diphenyl prolinol ethers, peptides, cinchona alkaloid derivatives and some other chiral organocatalysts. Obviously, the most efficient, easily available and least sensitive catalytic compounds were anchored for these purposes in these studies, which in many instances ensured cascade reactions, without intervention between steps. However, some processes had to be carried out in a sequential manner, due to incompatibilities of the reactants, catalysts, or owing to the necessity for changing the reaction conditions between steps. The most sophisticated materials designed were solid catalysts bearing more than one kind of catalytic species that are incompatible; however, by site-isolation or compartmentalization, they led to highly efficient, multifunctional catalysts.
| Chiral catalyst type | Type of the one-pot reaction | |
|---|---|---|
| Cascade | Sequential | |
| 1. Soluble chemical catalyst | 3 systems | 2 systems |
| 2. Heterogeneous chemical catalyst | ||
| 2.1. Immobilized metal complexes | 6 catalysts | 9 catalysts |
| 2.2. Anchored organocatalysts | 15 catalysts | 8 catalysts |
| 2.3. Modified solid catalysts | 3 catalysts | 2 catalysts |
| 3. Biocatalyst | 2 systems | 4 systems |
Heterogenized chiral metal complexes were less frequently applied in asymmetric one-pot processes as the anchored organocatalysts (Table 1). In the earliest examples, materials containing the metals that were transformed into the chiral complexes in situ by the addition of the ligand were applied, covalent bonding of the ligands to support prevailed later on. Accordingly, the same or similar immobilization methods could be used for preparing anchored organocatalysts and metal complexes. These common procedures accelerated the research aimed at applying both anchored organocatalyst and metal complexes. Among the one-pot reactions developed using heterogenized chiral metal complexes, those carried out sequentially are more numerous, as compared with the cascade procedures, which is explained by the higher sensitivity of the metal complexes. However, examples of site-isolated materials using chiral metal complexes were also reported, similar to the anchored organocatalysts.
Although simple to prepare, chirally modified catalytically active surfaces were less frequently employed in one-pot reactions, due to the narrow substrate specificity of the few efficient chirally modified catalytic systems. Thus, only few examples of one-pot reactions are known to date, which use such chiral catalysts. Moreover, catalytic systems applying heterogeneous achiral catalysts in combination with soluble chiral catalysts or biocatalysts are also scarcely met in the literature, which is mostly due to the focus on chiral catalyst recycling in these processes.
This survey of the asymmetric heterogeneous catalytic one-pot reactions showed that during the last twenty years significant efforts were devoted to developing such catalytic systems, which culminated in the last few years. The several successful examples disclosed to date have demonstrated the preparative potential residing in such procedures. Undoubtedly, the multiple advantages of these methods will further accelerate the development of asymmetric one-pot reactions relying on the use of heterogeneous chemical catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of the Hungarian National Science Foundation through OTKA Grant K 109278 is appreciated.References
- Methods and Reagents for Green Chemistry, An Introduction, ed. P. Tundo, A. Pedrosa and F. Zecchini, John Wiley & Sons, Inc., Hoboken, New Jersey, 2007 Search PubMed.
- Innovations in Green Chemistry and Green Engineering, ed. P. T. Anastas and J. B. Zimmerman, Springer Sci.+Business Media, New York, 2013 Search PubMed.
- Innovative Catalysis in Organic Synthesis, Oxidation, Hydrogenation and C–X Bond Forming Reactions, ed. P. G. Andersson, WILEY-VCH Verlag & Co., Weinheim, 2012 Search PubMed.
- Fine Chemicals through Heterogeneous Catalysis, ed. R. A. Sheldon and H. van Bekkum, WILEY-VCH Verlag GmbH, Weinheim, 2001 Search PubMed.
- I. R. Baxendale, J. Chem. Technol. Biotechnol., 2013, 88, 519–552 CrossRef CAS.
- I. M. Mándity, S. B. Ötvös, G. Szőllősi and F. Fülöp, Chem. Rec., 2016, 16, 1018–1033 CrossRef PubMed.
- Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, ed. H. U. Blaser and E. Schmidt, WILEY-VCH Verlag GmbH, Weinheim, 2004 Search PubMed.
- Asymmetric Synthesis with Chemical and Biological Methods, ed. D. Enders and K.-E. Jaeger, WILEY-VCH Verlag GmbH, Weinheim, 2004 Search PubMed.
- Asymmetric Organocatalysis – From Biomimetic Concepts to Applications in Asymmetric Synthesis, ed. A. Berkessel and H. Gröger, WILEY-VCH Verlag GmbH, Weinheim, 2004 Search PubMed.
- Enantioselective Organocatalysis: Reactions and Experimental Procedures, ed. P. I. Dalko, WILEY-VCH Verlag GmbH, Weinheim, 3d edn, 2007 Search PubMed.
- Catalytic Asymmetric Synthesis, ed. I. Ojima, John Wiley & Sons, Inc., Hoboken, New Jersey, 2010 Search PubMed.
- Biocatalysis for Green Chemistry and Chemical Process Development, ed. J. Tao and R. Kazlauskas, John Wiley & Sons, Inc., Hoboken, New Jersey, 2011 Search PubMed.
- Enantioselective Organocatalyzed Reactions I: Enantioselective Oxidation, Reduction, Functionalization and Desymmetrization, ed. R. Mahrwald, Springer Sci.+Business Media, Dordrecht, 2011 Search PubMed.
- Enantioselective Organocatalyzed Reactions II: Asymmetric C-C Bond Formation Processes, ed. R. Mahrwald, Springer Sci.+Business Media, Dordrecht, 2011 Search PubMed.
- M. Waser, Asymmetric Organocatalysis in Natural Product Syntheses, Progress in the Chemistry of Organic Natural Products 96, Springer-Verlag, Wien, 2012 Search PubMed.
- Catalytic Methods in Asymmetric Synthesis, Advanced Materials, Techniques and Applications, ed. M. Gruttadauria and F. Giacalone, John Wiley & Sons, Inc., Hoboken, New Jersey, 2012 Search PubMed.
- Chiral Catalyst Immobilization and Recycling, ed. D. E. De Vos, I. F. J. Vankelecom and P. A. Jacobs, WILEY-VCH Verlag GmbH, Weinheim, 2000 Search PubMed.
- M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed.
- Handbook of Asymmetric Heterogeneous Catalysis, ed. K. Ding and Y. Uozumi, WILEY-VCH Verlag GmbH, Weinheim, 2008 Search PubMed.
- M. Studer, H.-U. Blaser and C. Exner, Adv. Synth. Catal., 2003, 345, 45–65 CrossRef CAS.
- M. Bartók, Curr. Org. Chem., 2006, 10, 1533–1567 CrossRef.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- T. Yasukawa, H. Miyamura and S. Kobayashi, Chem. Soc. Rev., 2014, 43, 1450–1461 RSC.
- F. Meemken and A. Baiker, Chem. Rev., 2017, 117, 11522–11569 CrossRef CAS PubMed.
- M. Benaglia, New J. Chem., 2006, 30, 1525–1533 RSC.
- J. M. Thomas and R. Raja, Acc. Chem. Res., 2008, 41, 708–720 CrossRef CAS PubMed.
- J. M. Fraile, J. I. García and J. A. Mayoral, Coord. Chem. Rev., 2008, 252, 624–646 CrossRef CAS.
- B. Pugin and H.-U. Blaser, Top. Catal., 2010, 53, 953–962 CrossRef CAS.
- Enantioselective Homogeneous Supported Catalysis, RSC Green Chemistry No. 15, ed. R. Šebesta, Royal Society of Chemistry, Cambridge, 2012 Search PubMed.
- M. Bartók, Catal. Rev.: Sci. Eng., 2015, 57, 192–255 Search PubMed.
- T. Cheng, Q. Zhao, D. Zhang and G. Liu, Green Chem., 2015, 17, 2100–2122 RSC.
- A. Puglisi, M. Benaglia and V. Chiroli, Green Chem., 2013, 15, 1790–1813 RSC.
- T. Tsubogo, T. Ishiwata and S. Kobayashi, Angew. Chem., Int. Ed., 2013, 52, 6590–6604 CrossRef CAS PubMed.
- D. Zhao and K. Ding, ACS Catal., 2013, 3, 928–944 CrossRef CAS.
- Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492–8509 CrossRef PubMed.
- W. Zhao and F.-E. Chen, Curr. Org. Synth., 2012, 9, 873–897 CrossRef CAS.
- M. O. Sydnes, Curr. Green Chem., 2014, 1, 216–226 CrossRef CAS.
- C. Vaxelaire, P. Winter and M. Christmann, Angew. Chem., Int. Ed., 2011, 50, 3605–3607 CrossRef CAS PubMed.
- C. A. Denard, J. F. Hartwig and H. Zhao, ACS Catal., 2013, 3, 2856–2864 CrossRef CAS.
- L. F. Tietze, G. Brasche and K. M. Gericke, Domino Reactions in Organic Synthesis, WILEY-VCH Verlag GmbH, Weinheim, 2006 Search PubMed.
- D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS.
- J. M. Lee, Y. Na, H. Han and S. Chang, Chem. Soc. Rev., 2004, 33, 302–312 RSC.
- T. Vlaar, E. Ruijter and R. V. A. Orru, Adv. Synth. Catal., 2011, 353, 809–841 CrossRef CAS.
- V. Jeena and R. S. Robinson, RSC Adv., 2014, 4, 40720–40739 RSC.
- Catalytic Cascade Reactions, ed. P.-F. Xu and W. Wang, John Wiley & Sons, Inc., Hoboken, New Jersey, 2014 Search PubMed.
- C. J. Chapman and C. G. Frost, Synthesis, 2007, 1–21 CAS.
- A.-N. Alba, X. Companyo, M. Viciano and R. Rios, Curr. Org. Chem., 2009, 13, 1432–1474 CrossRef CAS.
- H. Pellissier, Adv. Synth. Catal., 2012, 354, 237–294 CrossRef CAS.
- H. Clavier and H. Pellissier, Adv. Synth. Catal., 2012, 354, 3347–3403 CrossRef CAS.
- H. Pellissier, Chem. Rev., 2013, 113, 442–524 CrossRef CAS PubMed.
- H. Pellissier, Tetrahedron, 2013, 69, 7171–7210 CrossRef CAS.
- H. Pellissier, Asymmetric Domino Reactions, RSC Catalysis No. 10, Royal Society of Chemistry, Cambridge, 2013 Search PubMed.
- M. Rueping, J. Dufour and L. Bui, ACS Catal., 2014, 4, 1021–1025 CrossRef CAS.
- D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365–2377 CrossRef CAS PubMed.
- C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef CAS PubMed.
- S. Afewerki and A. Córdova, Chem. Rev., 2016, 116, 13512–13570 CrossRef CAS PubMed.
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed.
- J. Poulin, C. M. Grisé-Bard and L. Barriault, Chem. Soc. Rev., 2009, 38, 3092–3101 RSC.
- C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS PubMed.
- L. Veum and U. Hanefeld, Chem. Commun., 2006, 825–831 RSC.
- F.-X. Felpin and E. Fouquet, ChemSusChem, 2008, 1, 718–724 CrossRef CAS PubMed.
- M. J. Climent, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2014, 4, 870–891 CrossRef CAS.
- P. N. R. Vennestrøm, C. H. Christensen, S. Pedersen, J.-D. Grunwaldt and J. M. Woodley, ChemCatChem, 2010, 2, 249–258 CrossRef.
- M. Filice and J. M. Palomo, ACS Catal., 2014, 4, 1588–1598 CrossRef CAS.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- A. Bruggink, R. Schoevaart and T. Kieboom, Org. Process Res. Dev., 2003, 7, 622–640 CrossRef CAS.
- R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2014, 4, 129–143 CrossRef CAS.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- A. C. Marr and S. Liu, Trends Biotechnol., 2011, 29, 199–204 CrossRef CAS PubMed.
- C. A. Denard, J. F. Hartwig and H. Zhao, ACS Catal., 2013, 3, 2856–2864 CrossRef CAS.
- H. Gröger and W. Hummel, Curr. Opin. Chem. Biol., 2014, 19, 171–179 CrossRef PubMed.
- A. Tai, H. Watanabe and T. Harada, Bull. Chem. Soc. Jpn., 1979, 52, 1468–1472 CrossRef CAS.
- M. Inagaki, J. Hiratake, T. Nishioka and J. Oda, J. Org. Chem., 1992, 57, 5643–5649 CrossRef CAS.
- R. Noyori, M. Tokunaga and M. Kitamura, Bull. Chem. Soc. Jpn., 1995, 68, 36–56 CrossRef CAS.
- M. Studer, H.-U. Blaser and S. Burkhardt, Adv. Synth. Catal., 2002, 344, 511–515 CrossRef CAS.
- K. Szőri, G. Szőllősi and M. Bartók, Adv. Synth. Catal., 2006, 348, 515–522 CrossRef.
- R. Karvembu, R. Prabhakaran, M. M. Tamizh and K. Natarajan, C. R. Chim., 2009, 12, 951–962 CrossRef CAS.
- N. J. Turner, Curr. Opin. Chem. Biol., 2010, 14, 115–121 CrossRef CAS PubMed.
- Y. Kim, J. Park and M.-J. Kim, ChemCatChem, 2011, 3, 271–277 CrossRef CAS.
- K. P. J. Gustafson, R. Lihammar, O. Verho, K. Engström and J.-E. Bäckvall, J. Org. Chem., 2014, 79, 3747–3751 CrossRef CAS PubMed.
- O. Långvik, T. Saloranta, D. Yu. Murzin and R. Leino, ChemCatChem, 2015, 7, 4004–4015 CrossRef.
- O. Verho and J.-E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 3996–4009 CrossRef CAS PubMed.
- D. Y. Murzin and R. Leino, Chem. Eng. Res. Des., 2008, 86, 1002–1010 CrossRef CAS.
- N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211–224 CAS.
- F. Hénin and J. Muzart, Tetrahedron: Asymmetry, 1992, 3, 1161–1164 CrossRef.
- S. J. Aboulhoda, F. Hénin, J. Muzart, C. Thorey, W. Behnen, J. Martens and T. Mehler, Tetrahedron: Asymmetry, 1994, 5, 1321–1326 CrossRef CAS.
- S. J. Aboulhoda, S. Létinois, J. Wilken, I. Reiners, F. Hénin, J. Martens and J. Muzart, Tetrahedron: Asymmetry, 1995, 6, 1865–1868 CrossRef CAS.
- J. Muzart, F. Hénin and S. J. Aboulhoda, Tetrahedron: Asymmetry, 1997, 8, 381–389 CrossRef CAS.
- S. J. Aboulhoda, I. Reiners, J. Wilken, F. Hénin, J. Martens and J. Muzart, Tetrahedron: Asymmetry, 1998, 9, 1847–1850 CrossRef CAS.
- O. Roy, M. Diekmann, A. Riahi, F. Hénin and J. Muzart, Chem. Commun., 2001, 533–534 RSC.
- O. Roy, A. Riahi, F. Hénin and J. Muzart, Eur. J. Org. Chem., 2002, 3986–3994 CrossRef CAS.
- M. A. Baur, A. Riahi, F. Hénin and J. Muzart, Tetrahedron: Asymmetry, 2003, 14, 2755–2761 CrossRef CAS.
- O. Roy, F. Loiseau, A. Riahi, F. Hénin and J. Muzart, Tetrahedron, 2003, 59, 9641–9648 CrossRef CAS.
- J.-F. Detalle, A. Riahi, V. Steinmetz, F. Hénin and J. Muzart, J. Org. Chem., 2004, 69, 6528–6532 CrossRef CAS PubMed.
- P. Kukula, V. Matoušek, T. Mallat and A. Baiker, Tetrahedron: Asymmetry, 2007, 18, 2859–2868 CrossRef CAS.
- P. Kukula, V. Matoušek, T. Mallat and A. Baiker, Chem. – Eur. J., 2008, 14, 2699–2708 CrossRef CAS PubMed.
- L. Deiana, S. Afewerki, C. Palo-Nieto, O. Verho, E. V. Johnston and A. Córdova, Sci. Rep., 2012, 2, 851 CrossRef PubMed.
- M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794–797 CrossRef CAS PubMed.
- Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS PubMed.
- I. Ibrahem and A. Córdova, Chem. Commun., 2006, 1760–1762 RSC.
- D. Enders, M. R. M. Hüttl, C. Grondal and G. Raabe, Nature, 2006, 441, 861–863 CrossRef CAS PubMed.
- D. Enders, M. R. M. Hüttl, J. Runsink, G. Raabe and B. Wendt, Angew. Chem., Int. Ed., 2007, 44, 467–469 CrossRef PubMed.
- H. Li, J. Wang, T. E-Nunu, L. Zu, W. Jiang, S. Wei and W. Wang, Chem. Commun., 2007, 507–509 RSC.
- G.-L. Zhao, F. Ullah, L. Deiana, S. Lin, Q. Zhang, J. Sun, I. Ibrahem, P. Dziedzic and A. Córdova, Chem. – Eur. J., 2010, 16, 1585–1591 CrossRef CAS PubMed.
- S. Lin, G.-L. Zhao, L. Deiana, J. Sun, Q. Zhang, H. Leijonmarck and A. Córdova, Chem. – Eur. J., 2010, 16, 13930–13934 CrossRef CAS PubMed.
- G. Ma, S. Afewerki, L. Deiana, C. Palo-Nieto, L. Liu, J. Sun, I. Ibrahem and A. Córdova, Angew. Chem., Int. Ed., 2013, 52, 6050–6054 CrossRef CAS PubMed.
- L. Deiana, L. Ghisu, O. Córdova, S. Afewerki, R. Zhang and A. Córdova, Synthesis, 2014, 46, 1303–1310 CrossRef.
- L. Deiana, Y. Jiang, C. Palo-Nieto, S. Afewerki, C. A. Incerti-Pradillos, O. Verho, C.-W. Tai, E. V. Johnston and A. Córdova, Angew. Chem., Int. Ed., 2014, 53, 3447–3451 CrossRef CAS PubMed.
- A. Córdova, Pure Appl. Chem., 2015, 87, 1011–1019 CrossRef.
- C. Xu, L. Deiana, S. Afewerki, C. Incerti-Pradillos, O. Córdova, P. Guo, A. Córdova and N. Hedin, Adv. Synth. Catal., 2015, 357, 2150–2156 CrossRef CAS.
- C. Xu, S. Afewerki, C.-W. Tai, A. Córdova and N. Hedin, ChemistrySelect, 2016, 1, 5801–5804 CrossRef CAS.
- H.-S. Yoon, X.-H. Ho, J. Jang, H.-J. Lee, S.-J. Kim and H.-Y. Jang, Org. Lett., 2012, 14, 3272–3275 CrossRef CAS PubMed.
- S. U. Son, K. H. Park, H. Seo, Y. K. Chung and S.-G. Lee, Chem. Commun., 2001, 2440–2441 RSC.
- T. Nemoto, H. Kakei, V. Gnanadesikan, S. Tosaki, T. Oshima and M. Shibasaki, J. Am. Chem. Soc., 2002, 124, 14544–14545 CrossRef CAS PubMed.
- B. M. Choudary, N. S. Chowdari, S. Madhi and M. L. Kantam, Angew. Chem., Int. Ed., 2001, 40, 4619–4623 CrossRef CAS PubMed.
- K. B. Sharpless, W. Amberg, Y. L. Bennani, G. A. Crispino, J. Hartung, K.-S. Jeong, H.-L. Kwong, K. Morikawa, Z.-M. Wang, D. Xu and X.-L. Zhang, J. Org. Chem., 1992, 57, 2768–2771 CrossRef CAS.
- W. Amberg, Y. L. Bennani, R. K. Chadha, G. A. Crispino, W. D. Davis, J. Hartung, K.-S. Jeong, Y. Ogino, T. Shibata and K. B. Sharpless, J. Org. Chem., 1993, 58, 844–849 CrossRef CAS.
- B. M. Choudary, N. S. Chowdari, S. Madhi and M. L. Kantam, J. Org. Chem., 2003, 68, 1736–1746 CrossRef CAS PubMed.
- B. M. Choudary, K. Jyothi, M. Roy, M. L. Kantam and B. Sreedhar, Adv. Synth. Catal., 2004, 346, 1471–1480 CrossRef CAS.
- B. M. Choudary, S. Madhi, N. S. Chowdari and M. L. Kantam, Top. Catal., 2004, 29, 183–187 CrossRef CAS.
- B. M. Choudary, N. S. Chowdari, K. Jyothi, S. Madhi and M. L. Kantam, Adv. Synth. Catal., 2002, 344, 503–506 CrossRef CAS.
- B. M. Choudary, N. S. Chowdari, K. Jyothi, N. S. Kumar and M. L. Kantam, Chem. Commun., 2002, 586–587 RSC.
- K. Takeda, T. Oohara, N. Shimada, H. Nambu and S. Hashimoto, Chem. – Eur. J., 2011, 17, 13992–13998 CrossRef CAS PubMed.
- J. Huang, F. Zhang and H. Li, Appl. Catal., A, 2012, 431–432, 95–103 CrossRef CAS.
- R. Liu, R. Jin, J. An, Q. Zhao, T. Cheng and G. Liu, Chem. – Asian J., 2014, 9, 1388–1394 CrossRef CAS PubMed.
- L. Kong, J. Zhao, T. Cheng, J. Lin and G. Liu, ACS Catal., 2016, 6, 2244–2249 CrossRef CAS.
- D. Zhang, J. Xu, Q. Zhao, T. Cheng and G. Liu, ChemCatChem, 2014, 6, 2998–3003 CrossRef CAS.
- J. Xu, T. Cheng, K. Zhang, Z. Wang and G. Liu, Chem. Commun., 2016, 52, 6005–6008 RSC.
- X. Xia, J. Meng, H. Wu, T. Cheng and G. Liu, Chem. Commun., 2017, 53, 1638–1641 RSC.
- H.-B. Yu, Q.-S. Hu and L. Pu, J. Am. Chem. Soc., 2000, 122, 6500–6501 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2005, 7, 267–278 RSC.
- C. Simons, U. Hanefeld, I. W. C. E. Arends, A. J. Minnaard, T. Maschmeyer and R. A. Sheldon, Chem. Commun., 2004, 2830–2831 RSC.
- C. Simons, U. Hanefeld, I. W. C. E. Arends, R. A. Sheldon and T. Maschmeyer, Chem. – Eur. J., 2004, 10, 5829–5835 CrossRef CAS PubMed.
- D. Zhang, X. Gao, T. Cheng and G. Liu, Sci. Rep., 2014, 4, 5091 CrossRef PubMed.
- X. Xia, M. Wu, R. Jin, T. Cheng and G. Liu, Green Chem., 2015, 17, 3916–3922 RSC.
- F. Song, C. Wang and W. Lin, Chem. Commun., 2011, 47, 8256–8258 RSC.
- Z. Sun, J. Chen, Y. Liu and T. Tu, Adv. Synth. Catal., 2017, 359, 494–505 CrossRef CAS.
- J. Yadav, G. R. Stanton, X. Fan, J. R. Robinson, E. J. Schelter, P. J. Walsh and M. A. Pericas, Chem. – Eur. J., 2014, 20, 7122–7127 CrossRef CAS PubMed.
- A. M. Seayad, B. Ramalingam, C. L. L. Chai, C. Li, M. V. Garland and K. Yoshinaga, Chem. – Eur. J., 2012, 18, 5693–5700 CrossRef CAS PubMed.
- U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496–497 CrossRef CAS.
- Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621 CrossRef CAS.
- K. Hermann and H. Wynberg, J. Org. Chem., 1979, 44, 2238–2244 CrossRef CAS.
- H. Hiemstra and H. Wynberg, J. Am. Chem. Soc., 1981, 103, 417–430 CrossRef CAS.
- Y. Iwabuchi, M. Nakatani, N. Yokoyama and S. Hatakeyama, J. Am. Chem. Soc., 1999, 121, 10219–10220 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- B. List, J. Am. Chem. Soc., 2000, 122, 9336–9337 CrossRef CAS.
- N. Kobayashi and K. Iwai, J. Am. Chem. Soc., 1978, 100, 7071–7072 CrossRef CAS.
- M. Inagaki, J. Hiratake, Y. Yamamoto and J. Oda, Bull. Chem. Soc. Jpn., 1987, 60, 4121–4126 CrossRef CAS.
- M. Benaglia, G. Celentano and F. Cozzi, Adv. Synth. Catal., 2001, 343, 171–173 CrossRef CAS.
- G. Szőllősi, G. London, L. Baláspiri, C. Somlai and M. Bartók, Chirality, 2003, 15, S90–S96 CrossRef PubMed.
- I. Atodiresei, C. Vila and M. Rueping, ACS Catal., 2015, 5, 1972–1985 CrossRef CAS.
- M. Benaglia, M. Cinquini, F. Cozzi and G. Celentano, Adv. Synth. Catal., 2002, 344, 533–542 CrossRef CAS.
- J. Safaei-Ghomi and S. Zahedi, Appl. Organomet. Chem., 2015, 29, 566–571 CrossRef CAS.
- J. Safaei-Ghomi and S. Zahedi, Tetrahedron Lett., 2016, 57, 1071–1073 CrossRef CAS.
- S. Yang and J. He, Chem. Commun., 2012, 48, 10349–10351 RSC.
- Z. An, Y. Guo, L. Zhao, Z. Li and J. He, ACS Catal., 2014, 4, 2566–2576 CrossRef CAS.
- M. Heidlindemann, G. Rulli, A. Berkessel, W. Hummel and H. Gröger, ACS Catal., 2014, 4, 1099–1103 CrossRef CAS.
- G. Rulli, K. A. Fredriksen, N. Duangdee, T. Bonge-Hansen, A. Berkessel and H. Gröger, Synthesis, 2013, 45, 2512–2519 CrossRef CAS.
- E. Alza, S. Sayalero, X. C. Cambeiro, R. Martín-Rapún, P. O. Miranda and M. A. Pericàs, Synlett, 2011, 464–468 CAS.
- H. Miyamura, G. C. Y. Choo, T. Yasukawa, W.-J. Yoo and S. Kobayashi, Chem. Commun., 2013, 49, 9917–9919 RSC.
- L. Deiana, L. Ghisu, S. Afewerki, O. Verho, E. V. Johnston, N. Hedin, Z. Bacsik and A. Córdova, Adv. Synth. Catal., 2014, 356, 2485–2492 CrossRef CAS.
- F. Dai, Z. Zhao, G. Xie, D. Feng and X. Ma, ChemCatChem, 2017, 9, 89–93 CrossRef CAS.
- D. Enders, M. R. M. Hüttl, G. Raabe and J. W. Bats, Adv. Synth. Catal., 2008, 350, 267–279 CrossRef CAS.
- Y. Jia, Z. Mao and R. Wang, Tetrahedron: Asymmetry, 2011, 22, 2018–2023 CrossRef CAS.
- Y. Chi, S. T. Scroggins and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 6322–6323 CrossRef CAS PubMed.
- J. F. Austin and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 1172–1173 CrossRef CAS PubMed.
- S. Wang, J. He and Z. An, Chem. Commun., 2017, 53, 8882–8885 RSC.
- K. Akagawa, T. Fujiwara, S. Sakamoto and K. Kudo, Chem. Commun., 2010, 46, 8040–8042 RSC.
- M. P. Sibi and M. Hasegawa, J. Am. Chem. Soc., 2007, 129, 4124–4125 CrossRef CAS PubMed.
- J. F. Van Humbeck, S. P. Simonovich, R. R. Knowles and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 10012–10014 CrossRef CAS PubMed.
- K. Akagawa, T. Fujiwara, S. Sakamoto and K. Kudo, Org. Lett., 2010, 12, 1804–1807 CrossRef CAS PubMed.
- K. Akagawa, R. Suzuki and K. Kudo, Adv. Synth. Catal., 2012, 354, 1280–1286 CrossRef CAS.
- K. Akagawa, R. Umezawa and K. Kudo, Beilstein J. Org. Chem., 2012, 8, 1333–1337 CrossRef CAS PubMed.
- S. Colonna, H. Molinari, S. Banfi, S. Juliá, J. Masana and A. Alvarez, Tetrahedron, 1983, 63, 1635–1641 CrossRef.
- W. Luo, Z. Yu, W. Qiu, F. Yang, X. Liu and J. Tang, Tetrahedron, 2011, 67, 5289–5292 CrossRef CAS.
- D.-G. Crivoi, A. M. Segarra and F. Medina, J. Catal., 2016, 334, 120–128 CrossRef CAS.
- Cinchona Alkaloids in Synthesis and Catalysis, Ligands, Immobilization and Organocatalysis, ed. C. E. Song, WILEY-VCH Verlag GmbH, Weinheim, 2009 Search PubMed.
- X. Jiang, H. Zhu, X. Shi, Y. Zhong, Y. Li and R. Wang, Adv. Synth. Catal., 2013, 355, 308–314 CAS.
- P. García-García, A. Zagdoun, C. Copéret, A. Lesage, U. Díaz and A. Corma, Chem. Sci., 2013, 4, 2006–2012 RSC.
- A. Leyva-Pérez, P. García-García and A. Corma, Angew. Chem., Int. Ed., 2014, 53, 8687–8690 CrossRef PubMed.
- J. An, T. Cheng, X. Xiong, L. Wu, B. Han and G. Liu, Catal. Sci. Technol., 2016, 6, 5714–5720 CAS.
- C. Li, X. Shu, L. Li, G. Zhang, R. Jin, T. Cheng and G. Liu, Chem. – Asian J., 2016, 11, 2072–2077 CrossRef CAS PubMed.
- G. Xie, D. Feng and X. Ma, Mol. Catal., 2017, 434, 86–95 CrossRef CAS.
- R. P. Jumde, A. Mandoli, F. De Lorenzi, D. Pini and P. Salvadori, Adv. Synth. Catal., 2010, 352, 1434–1440 CrossRef CAS.
- D. Cancogni, A. Mandoli, R. P. Jumde and D. Pini, Eur. J. Org. Chem., 2012, 1336–1345 CrossRef CAS.
- H. Zhu, X. Jiang, X. Li, C. Hou, Y. Jiang, K. Hou, R. Wang and Y. Li, ChemCatChem, 2013, 5, 2187–2190 CrossRef CAS.
- H.-G. Cheng, J. Miguélez, H. Miyamura, W.-J. Yoo and S. Kobayashi, Chem. Sci., 2017, 8, 1356–1359 RSC.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- A. Puglisi, M. Benaglia, R. Porta and F. Coccia, Curr. Organocatal., 2015, 2, 79–101 CrossRef CAS.
- I. Atodiresei, C. Vila and M. Rueping, ACS Catal., 2015, 5, 1972–1985 CrossRef CAS.
- A. M. Hafez, A. E. Taggi and T. Lectka, Chem. – Eur. J., 2002, 8, 4114–4119 CrossRef CAS.
- B. M. Choudary, K. V. S. Ranganath, U. Pal, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2005, 127, 13167–13171 CrossRef CAS PubMed.
- M. L. Kantam, K. V. S. Ranganath, K. Mahendar, L. Chakrapani and B. M. Choudary, Tetrahedron Lett., 2007, 48, 7646–7649 CrossRef CAS.
- B. M. Choudary, M. L. Kantam, K. V. S. Ranganath, K. Mahendar and B. Sreedhar, J. Am. Chem. Soc., 2004, 126, 3396–3397 CrossRef CAS PubMed.
- K. Felföldi, K. Szöri and M. Bartók, Appl. Catal., A, 2003, 251, 457–460 CrossRef.
- K. Balázsik, K. Szöri, K. Felföldi, B. Török and M. Bartók, Chem. Commun., 2000, 555–556 RSC.
- G. Szőllősi, B. Hermán, F. Fülöp and M. Bartók, J. Catal., 2010, 276, 259–267 CrossRef.
- G. Szőllősi, B. Hermán, K. Felföldi, F. Fülöp and M. Bartók, Adv. Synth. Catal., 2008, 350, 2804–2814 CrossRef.
- T. Sugimura, T. Uchida, J. Watanabe, T. Kubota, Y. Okamoto, T. Misaki and T. Okuyama, J. Catal., 2009, 262, 57–64 CrossRef CAS.
- G. Szőllősi, B. Hermán, E. Szabados, F. Fülöp and M. Bartók, J. Mol. Catal. A: Chem., 2010, 333, 28–36 CrossRef.
- G. Szőllősi and M. Bartók, ARKIVOC, 2012,(v), 16–27 Search PubMed.
- G. Szőllősi, Z. Makra, L. Kovács, F. Fülöp and M. Bartók, Adv. Synth. Catal., 2013, 355, 1623–1629 CrossRef.
- G. Szőllősi, L. Kovács and Z. Makra, Catal. Sci. Technol., 2015, 5, 697–704 Search PubMed.
- L. Kovács, G. Szőllősi and F. Fülöp, J. Flow Chem., 2015, 5, 210–215 CrossRef.
- H. Miyamura, A. Suzuki, T. Yasukawa and S. Kobayashi, Adv. Synth. Catal., 2015, 357, 3815–3819 CrossRef CAS.
- E. S. Andreiadis, M. R. Vitale, N. Mézailles, X. Le Goff, P. Le Floch, P. Y. Toullec and V. Michelet, Dalton Trans., 2010, 39, 10608–10616 RSC.
- L. Liu, X. Zhang, S. Rang, Y. Yang, X. Dai, J. Gao, C. Xu and J. He, RSC Adv., 2014, 4, 13093–13107 RSC.
- M. Makkee, A. P. G. Kieboom and H. Van Bekkum, J. Chem. Soc., Chem. Commun., 1980, 930–931 RSC.
- M. Makkee, A. P. G. Kieboom and H. Van Bekkum, Carbohydr. Res., 1985, 138, 237–245 CrossRef CAS.
- R. Schoevaart and A. P. G. Kieboom, Tetrahedron Lett., 2002, 43, 3399–3400 CrossRef CAS.
- Y. K. Choi, M. J. Kim, Y. Ahn and M.-J. Kim, Org. Lett., 2001, 3, 4099–4101 CrossRef CAS PubMed.
- K. Han, J. Park and M.-J. Kim, J. Org. Chem., 2008, 73, 4302–4304 CrossRef CAS PubMed.
- P. Mäki-Arvela, S. Sahin, N. Kumar, T. Heikkilä, V.-P. Lehto, T. Salmi and D. Yu. Murzin, J. Mol. Catal. A: Chem., 2008, 285, 132–141 CrossRef.
- H. M. Jung, J. H. Koh, M.-J. Kim and J. Park, Org. Lett., 2000, 2, 409–411 CrossRef CAS PubMed.
- H. M. Jung, J. H. Koh, M.-J. Kim and J. Park, Org. Lett., 2000, 2, 2487–2490 CrossRef CAS PubMed.
- P. Mäki-Arvela, S. Sahin, N. Kumar, J.-P. Mikkola, K. Eränen, T. Salmi and D. Yu. Murzin, React. Kinet. Catal. Lett., 2008, 94, 281–288 CrossRef.
- P. Mäki-Arvela, S. Sahin, N. Kumar, J.-P. Mikkola, K. Eränen, T. Salmi and D. Yu. Murzin, Catal. Today, 2009, 140, 70–73 CrossRef.
- H. R. Hobbs, B. Kondor, P. Stephenson, R. A. Sheldon, N. R. Thomas and M. Poliakoff, Green Chem., 2006, 8, 816–821 RSC.
- A. Kirilin, P. Mäki-Arvela, M. Rupp, E. Toukoniitty, N. Kumar, K. Kordas, L. M. Kustov, T. Salmi and D. Yu. Murzin, Res. Chem. Intermed., 2010, 36, 193–210 CrossRef CAS.
- A. Kirilin, P. Mäki-Arvela, K. Kordas, A.-R. Leino, A. Shchukarev, D. Boström, J.-P. Mikkola, L. M. Kustov, T. O. Salmi and D. Yu. Murzin, Kinet. Catal., 2011, 52, 77–81 CrossRef CAS.
- O. Långvik, T. Sandberg, J. Wärnå, D. Yu. Murzin and R. Leino, Catal. Sci. Technol., 2015, 5, 150–160 Search PubMed.
- C. Palo-Nieto, S. Afewerki, M. Anderson, C.-W. Tai, P. Berglund and A. Córdova, ACS Catal., 2016, 6, 3932–3940 CrossRef CAS.
- A. Prastaro, P. Ceci, E. Chiancone, A. Boffi, R. Cirilli, M. Colone, G. Fabrizi, A. Stringaro and S. Cacchi, Green Chem., 2009, 11, 1929–1932 RSC.
- C. Palumbo, E. E. Ferrandi, C. Marchesi, D. Monti, S. Riva, R. Psaro and M. Guidotti, ChemistrySelect, 2016, 1, 1795–1798 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |