
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing heterogeneity of NIR induced secondary fluorescence from DNA-stabilized silver nanoclusters at the single molecule level†
Stefan
Krause
 *,
Miguel R.
Carro-Temboury
,
Cecilia
Cerretani
and
Tom
Vosch
*
*,
Miguel R.
Carro-Temboury
,
Cecilia
Cerretani
and
Tom
Vosch
*
Nanoscience Center and Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark. E-mail: stefan.krause@chem.ku.dk; tom@chem.ku.dk
First published on 11th June 2018
Abstract
In this communication, we investigate optically activated delayed fluorescence (OADF) from DNA-stabilized silver nanoclusters (DNA-AgNCs) at the single molecule level, and we probe the heterogeneity in the primary fluorescence (PF) intensity, NIR induced secondary fluorescence (SF) intensity and SF/PF ratio. Our experiments reveal a heterogeneous distribution in the SF/PF ratio, indicating that engineering of DNA-AgNCs towards a high SF/PF ratio and high OADF signal for background-free imaging might be possible.
In most cases, auto-fluorescence from materials and biological structures will generate an unwanted background signal in fluorescence microscopy imaging.1 The signal-to-background ratio can be particularly low when trying to image single fluorescent molecules in highly auto-fluorescent samples.2 Since increasing the brightness of fluorescent labels is limited, several imaging modalities have been developed to improve the signal-to-background ratio by suppressing the auto-fluorescence background. Such approaches are based on fluorescence upconversion, fluorescence modulation, spectral discrimination and time-gating.3–10 Recently, Fleicher et al. introduced the concept of optically activated delayed fluorescence (OADF, see Fig. 1A), which combines some of the advantages of upconversion and time-gating.11 The imaging modality relies on long-lived (microseconds) dark states in DNA-encapsulated silver nanoclusters (DNA-AgNCs) which are formed upon visible (primary) excitation.9,12–16 Nanoclusters in the long-lived dark states can absorb an additional (secondary) NIR photon, which can bring them back to the emissive state. The following OADF emission will occur on the anti-Stokes side of the secondary NIR excitation pulse.11,17 By choosing an appropriate delay time for the secondary NIR pulse, time-gating can easily eliminate all auto-fluorescence coming from a primary excitation pulse.17 This allows for simple suppression of potential NIR auto-fluorescence by a short-pass filter.6 The intensity of the OADF signal depends essentially on the quantum yield of dark state formation (QD1) and the efficiency to generate the emissive state upon secondary NIR excitation (QD1–S1). Typical QD1 values are of the order of a few percent, which limits the ratio of the OADF signal to primary fluorescence (PF).9,12–16 Therefore, understanding the photo-physics and the factors that influence QD1 and QD1–S1 in DNA-AgNCs is crucial to further improve applications of these emitters for high contrast, background-free fluorescence imaging.
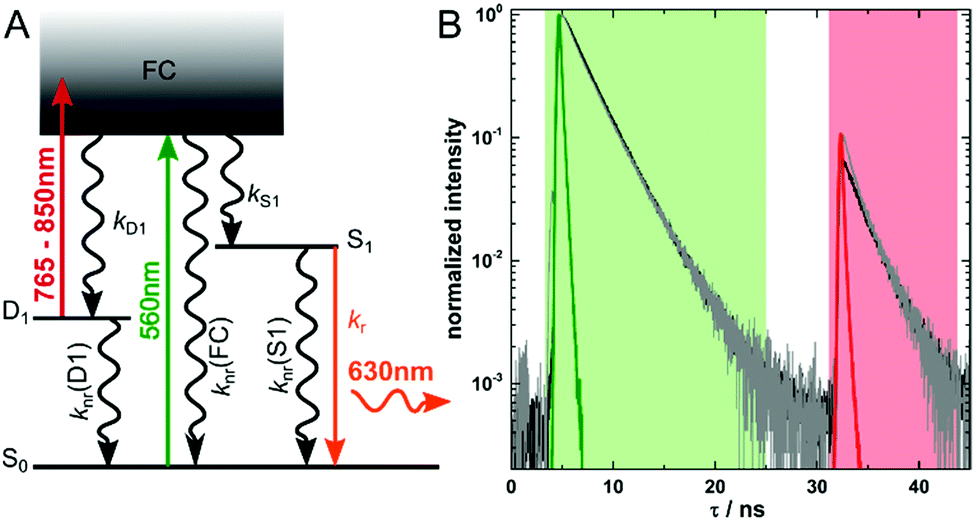 | ||
| Fig. 1 (A) Energy diagram of DNA-AgNCs. Vertical coloured arrows indicate the primary (560 nm) and secondary (765–850 nm) laser and the fluorescence emission at 630 nm, respectively. (B) Fluorescence decay curves illustrating the PF decay at 5 ns and the SF decay at 32 ns. The black curve is from the ensemble sample of DNA-AgNCs in PVA, while the gray curve is the sum of the decay curves of 25 images recorded at single molecule concentration in PVA. The green and red regions indicate the time channels used for determining the intensity of the PF and SF signal, respectively. | ||
The red-emitting DNA-AgNCs used in this publication are synthesized and HPLC-purified in the same way as described previously.18 Steady-state and time-resolved spectroscopic data can be found in the same publication.18 We have already demonstrated that these DNA-AgNCs can undergo OADF and upconversion fluorescence (UCF) when immobilized in a poly(vinyl alcohol) (PVA) matrix.17 The intensity of the NIR pulse used to optically pump the DNA-AgNCs from the D1 to the S1 state can also induce a two consecutive photon absorption process. This UCF process leads from S0via D1 to S1, and results in emission in the same time range as the OADF signal.17 However, at the experimental NIR excitation intensities used in this study, we tried to minimize the UCF contribution. Based on our previous results, we estimate the average contribution of UCF to the SF signal to be maximum 20 percent.17 The SF signal consists of the OADF signal plus the UCF signal and is calculated by summing up all the intensity in the red time channel shown in Fig. 1B. Summing up all values of the green time channel shown in Fig. 1B yields the PF signal. In the present work, we want to focus on the OADF process. We use the SF/PF ratio at the single molecule level to determine whether there are significant differences from molecule to molecule. The phenomenological electronic state diagram of the red-emitting DNA-AgNC can be seen in Fig. 1A. Upon excitation with 560 nm pulsed light, the majority of the DNA-AgNCs will evolve from the Franck–Condon state (FC) to the emissive S1 state resulting in primary fluorescence (PF). A small fraction, however, will evolve from the FC state into a micro-second-lived dark state D1. The initial relaxation processes from the FC state to the S1 and D1 state are ultrafast (sub-picosecond) as previously demonstrated.13,19,20 The black curve in Fig. 1B shows the decay curve measured for an ensemble concentration of DNA-AgNCs embedded in a PVA film. The 560 nm primary excitation pulse arrives at about 5 ns in the measurement window. The secondary and spectrally broad NIR pulse (from 765 nm to 850 nm) arrives at 32 ns and can induce the OADF emission. For these ensemble-like conditions, an SF/PF ratio of 0.07 was obtained. The ensemble only gives an indication of the average SF/PF ratio. Therefore, the solution was further diluted resulting in samples enabling us to investigate the SF/PF ratio at the single molecule level. When summing up the decay data of 25 images recorded at single molecule concentration (see Fig. S1 and S2 for four examples, ESI†) we obtain the gray curve in Fig. 1B. Both decay curves in Fig. 1B are similar. However, an additional fast decay component is present in the SF part of the summed single molecule decay data. This could be due to some minor laser bleed through of the secondary laser which would appear more pronounced for samples of low concentration. Nevertheless, the two curves demonstrate that the average behavior of a limited number of single molecules is similar to the ensemble behavior.
In order to investigate differences in SF/PF ratio among the individual DNA-AgNCs, we acquired macro- and micro-times of every photon during the image scan.21 The photon macro-times and micro-times were then related to each pixel position and allowed us to construct Photon Arrival Time Images (PArTI).8Fig. 2A shows an example emission intensity image (10 × 10 μm2) of individual DNA-AgNCs embedded in PVA. Based on the number of photons arriving in the PF time interval (4–25 ns, green time channel) and in the SF time interval (33–48 ns, red time channel), a false color PArTI image was constructed and is shown in Fig. 2B (see details and additional images in Fig. S1, ESI†). As a result, single molecules with the highest PF and lowest SF intensity will look greenish, while molecules with the lowest PF and highest SF intensity will look reddish in the PArTI image. In this example we use PArTI-imaging to increase the contrast and highlight minor differences in the SF/PF ratio. Fig. S2 (ESI†) shows examples of actual SF/PF ratio images for comparison with the PArTI images of Fig. S1 (ESI†). Two examples of single DNA-AgNCs fluorescence intensity trajectories and decay curves are given in Fig. 2C and D. The discrete intensity jumps to the background level indicate that we are indeed dealing with single emitters, although absolute confirmation of this would require anti-bunching measurements or defocused wide-field imaging.22,23 The average SF/PF ratios are 0.05 (black decay) and 1.86 (purple decay) for the respective molecules. The SF/PF ratio of 0.05 is similar to the PVA ensemble experiment (0.07) shown in Fig. 1B. Surprisingly, the other molecule exceeds the ensemble SF/PF ratio by a factor of 26. This could indicate that this particular molecule has a very high dark state formation quantum yield (QD1). Additionally, the decay times of these two example molecules are rather different. One molecule (black curve) has a PF decay time (τPF) of 3.6 ns and an identical SF decay time (τSF) of 3.6 ns. The other molecule (purple curve) shows shorter decay times of τPF = 0.84 ns and τSF = 0.76 ns. We also collected the emission spectra for a number of DNA-AgNCs to see if there are significant spectral differences between molecules with high and low SF/PF ratio. Fig. S3 (ESI†) shows that the high SF/PF ratio molecules tend to have a slightly broader spectral width than the low SF/PF ratio molecules. In addition to differences in SF/PF ratio and τPF, a number of molecules undergo dynamic changes of the SF/PF ratio during the measurement. An example of such a molecule can be seen in Fig. 2E and F, where the fluorescence intensity trajectory and the fluorescence decay curves are given. Apart from intensity fluctuations, this example shows strong changes in SF/PF ratio during the experiment (ranging from 0.12–2.24, see Fig. 2E). The yellow and blue regions in Fig. 2E were used to create the decay curves in Fig. 2F. From the single molecule examples shown in Fig. 2 we can make two complementary assumptions: τPF and τSF seem to be identical and the SF/PF ratio seems to be inversely related to τPF (and τSF). We will discuss these two assumptions in detail in the next section.
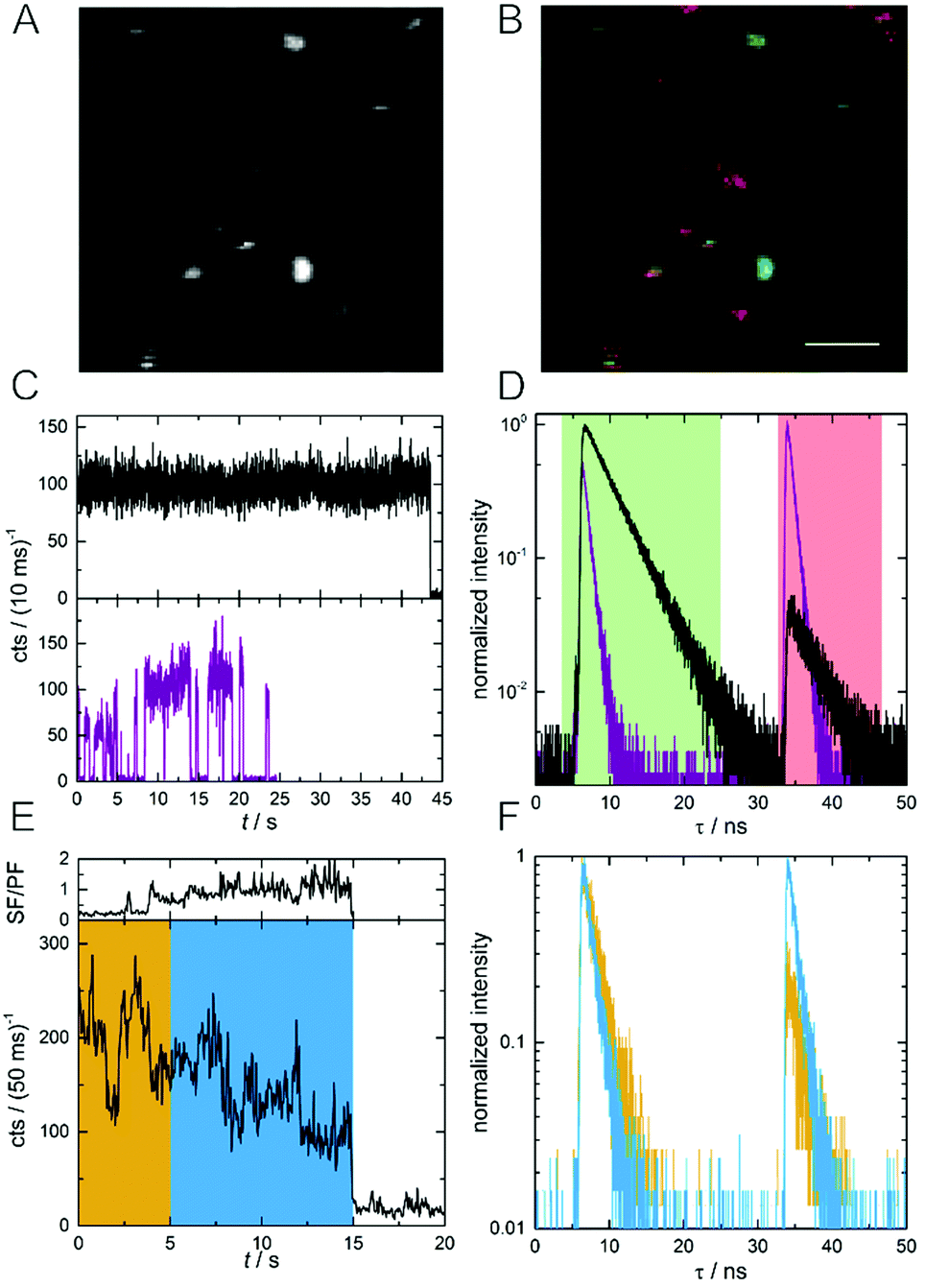 | ||
| Fig. 2 (A) Fluorescence intensity image of single DNA-AgNCs embedded in PVA (10 × 10 μm2). (B) PArTI image of (A). PF and SF have been assigned to the green and red time channel, respectively (see Fig. 1B for time ranges). The scale bar corresponds to 2 μm. (C) Fluorescence intensity trajectories of two DNA-AgNCs, one with low (black curve) and one with high (purple curve) average SF/PF ratio. (D) Normalized decay curves of the two DNA-AgNCs from (C). (E) Fluorescence intensity time trajectory of a single DNA-AgNC with variable SF/PF ratio. The SF/PF ratio is plotted above the intensity trajectory and is based on the number of photons appearing in the green and red time channel in a bin time of 50 ms. (F) Normalized fluorescence decay curves of the single DNA-AgNC shown in (E). The yellow and blue curves represent the photons recorded in the respective time intervals marked with the same colour in (E). | ||
Fig. 3A shows τSF as a function of τPF for 239 individual DNA-AgNCs. The values were obtained by tail fitting a single exponential function to the PF and SF decay curves of all individual DNA-AgNCs. The correlation between τPF and τSF allows us to confirm that indeed both PF and SF result from the same emissive state (here called S1). Clearly, the majority of decay time pairs are situated on a line with slope one. The few molecules that deviate from the straight line are most likely molecules that undergo dynamic changes of the SF/PF ratio during the measurement (see Fig. 2E). Hence, their PF and SF decay times might appear different. Additionally, measuring two close lying molecules with significantly different SF/PF ratios could also cause deviations (see Fig. S4 for a probable example, ESI†). Next, the SF/PF ratio was determined, using the red and green time channels described before, for the same 239 DNA-AgNCs. Fig. 3B shows τPF as a function of the SF/PF ratio. The SF/PF ratios as well as τPF reveal a broad range of values from 0 to 2 and 0.5 ns to 5.7 ns, respectively. Despite a deliberate selection bias towards the more interesting molecules with higher SF/PF ratios, a large number of molecules display average SF/PF ratios from 0 to 0.1. This observation is in agreement with the PVA ensemble data in Fig. 1B. Although from the scheme in Fig. 1A the ratio of SF/PF is not related to τPF (1/(kr + knr(S1))), an interesting trend was observed. It is clear from Fig. 3B that larger τPF values result in lower SF/PF ratios and vice versa. Previously we found that for this DNA-AgNCs in solution, an increase in temperature led to a lower τPF. The corresponding drop in the quantum yield of fluorescence from primary excitation Q (Q = QS1·QF, QS1 is the quantum yield of emissive S1 state formation, QF is the quantum yield of emission from the S1 state) was suggested to be caused by a decrease in QS1 and QF (see also ESI†).18 At this point we do not know the origin of the heterogeneity in the SF/PF ratio, but immobilization in a polymer could trap DNA-AgNCs in different conformations that would yield different rate constants in Fig. 1A. Similar conformational immobilization has been previously observed for single dye molecules in polymers.24,25 Further studies changing parameters like viscosity, dielectric constant, and temperature could provide insight in which parameters affect the SF/PF ratio. Fig. S5 (ESI†) shows that changing the ionic strength by addition of NaNO3 to a final concentration of 0.091 M has no significant effect on the SF/PF ratio or τPF value in solution.
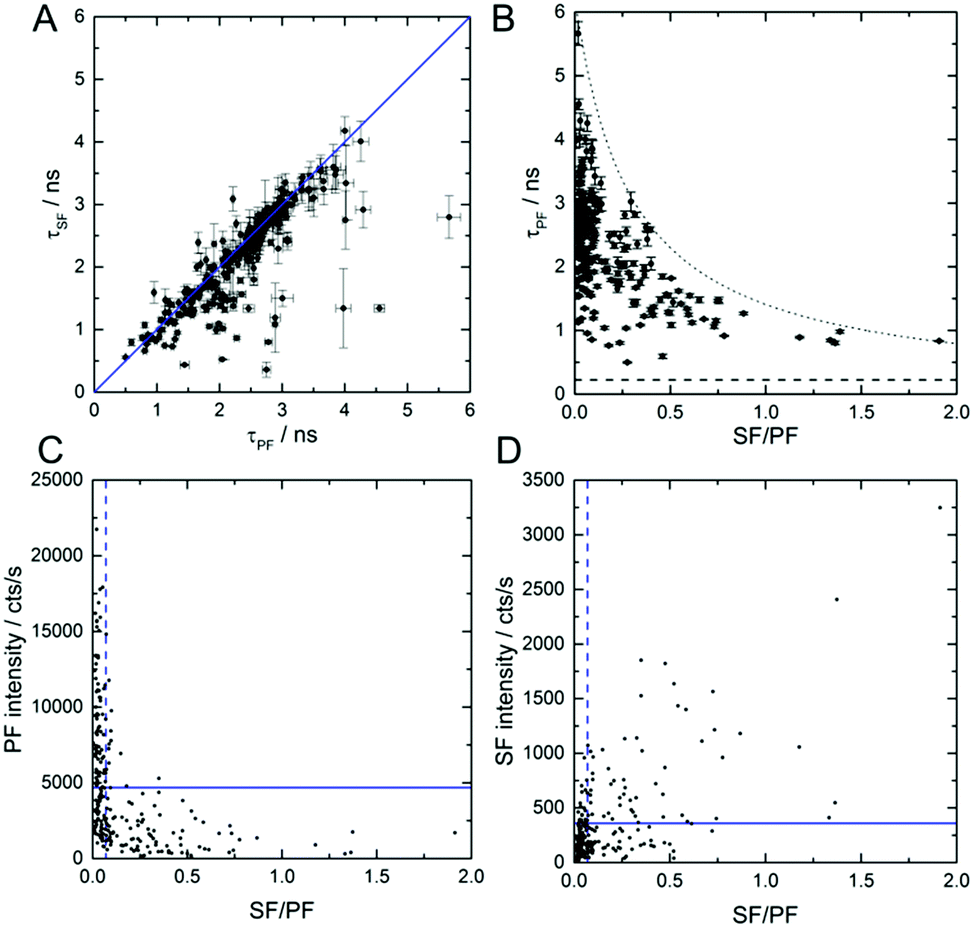 | ||
| Fig. 3 Each τPF, τSF, PF intensity, SF intensity and SF/PF ratio represented here is an average value of an individual molecule. (A) τSF as a function of τPF for 239 DNA-AgNCs. The blue line represents a slope of one. (B) τPF as a function of the SF/PF ratio for 239 DNA-AgNCs. The fluorescence decay times were extracted by mono-exponential tail fitting to each fluorescence decay curve, while the intensities follow from summing up all TCSPC channels belonging to the PF decay or the SF decay, respectively. The horizontal dashed line represents the decay time from tail-fitting the instruments response function while the dotted line serves as a guide for the eye. (C) PF intensity and (D) SF intensity as a function of the SF/PF ratio. The blue dashed vertical lines represent the ensemble SF/PF value of 0.07. The blue horizontal lines indicate the average PF and SF signal from the single molecule measurements. | ||
Fig. 3C and D show the PF and SF intensity as a function of the SF/PF ratio. DNA-AgNCs with high SF/PF ratio have low PF intensities, as can be seen in Fig. 3C. This finding can be explained by an increased QD1. Fig. 3D displays that the SF intensity increases with increasing SF/PF ratio. This can be similarly explained with an increase in the OADF signal upon QD1 increase. The average intensity of the SF signal is 360 cts s−1. This is about one order of magnitude below the average PF signal intensity with 4700 cts s−1. However, for creating contrast in an image, the actual signal intensity is not always the determining factor. Complete suppression of auto-fluorescence in the SF channel can easily surpass the contrast of images constructed from the more intense PF signal.17 Despite the low occurrence of the DNA-AgNCs with high SF/PF ratios we would like to stress the potential of such DNA-AgNCs as interesting labels for high-contrast, background-free imaging.
In conclusion, studying the PF and SF intensity of individual DNA-AgNCs immobilized in PVA revealed heterogeneities in the magnitude of the signals and their ratio. Average SF/PF ratios of up to 1.86 were found – a remarkable increase in comparison to the ensemble value of 0.07.17 An increase of the SF/PF ratio was found to be correlated to a lower QF as can be seen from the shorter τPF and τSF. We speculate that some DNA-AgNCs might be immobilized in different conformations and that this affects the observed photophysical properties. We tentatively associate the increase in SF intensity to an increase in QD1 and hence an increase in the OADF signal. For background-free imaging, a high SF/PF ratio and a high SF signal are beneficial. However, the accompanying shortening of τSF might limit the total detected number of photons in the SF channel. Further investigations might unravel the origin of the flexible nature of the photophysical parameters and relate them to e.g. structural changes/flexibility of the DNA-AgNCs. Additionally, detailed spectroscopic measurements of the S0 to D1 and D1 to S1 excitation spectrum will help to entangle the OADF and the UCF contribution to the SF signal.
The authors gratefully acknowledge financial support from the “Center for Synthetic Biology” at Copenhagen University funded by the UNIK research initiative of the Danish Ministry of Science, Technology and Innovation (Grant 09-065274), bioSYNergy, University of Copenhagen's Excellence Programme for Interdisciplinary Research, the Villum Foundation (Project number VKR023115), HFSP (RGY0081/2014), the Carlsberg Foundation (CF14-0388) and the Danish Council of Independent Research (Project number DFF-7014-00027).
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Neumann and D. Gabel, J. Histochem. Cytochem., 2002, 50, 437–439 CrossRef PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef PubMed.
- S. Lahtinen, S. Krause, R. Arppe, T. Soukka and T. Vosch, Chem. – Eur. J., 2018 DOI:10.1002/chem.201801962.
- L. Xiong, Z. Chen, Q. Tian, T. Cao, C. Xu and F. Li, Anal. Chem., 2009, 81, 8687–8694 CrossRef PubMed.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580 CrossRef PubMed.
- Z. Liao, M. Tropiano, K. Mantulnikovs, S. Faulkner, T. Vosch and T. Just Sorensen, Chem. Commun., 2015, 51, 2372–2375 RSC.
- M. Dahan, T. Laurence, F. Pinaud, D. S. Chemla, A. P. Alivisatos, M. Sauer and S. Weiss, Opt. Lett., 2001, 26, 825–827 CrossRef PubMed.
- Z. Liao, M. Tropiano, S. Faulkner, T. Vosch and T. J. Sorensen, RSC Adv., 2015, 5, 70282–70286 RSC.
- C. I. Richards, J. C. Hsiang, D. Senapati, S. Patel, J. Yu, T. Vosch and R. M. Dickson, J. Am. Chem. Soc., 2009, 131, 4619–4621 CrossRef PubMed.
- G. Marriott, S. Mao, T. Sakata, J. Ran, D. K. Jackson, C. Petchprayoon, T. J. Gomez, E. Warp, O. Tulyathan, H. L. Aaron, E. Y. Isacoff and Y. Yan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17789–17794 CrossRef PubMed.
- B. C. Fleischer, J. T. Petty, J.-C. Hsiang and R. M. Dickson, J. Phys. Chem. Lett., 2017, 8, 3536–3543 CrossRef PubMed.
- T. Vosch, Y. Antoku, J. C. Hsiang, C. I. Richards, J. I. Gonzalez and R. M. Dickson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12616–12621 CrossRef PubMed.
- S. A. Patel, M. Cozzuol, J. M. Hales, C. I. Richards, M. Sartin, J. C. Hsiang, T. Vosch, J. W. Perry and R. M. Dickson, J. Phys. Chem. C, 2009, 113, 20264–20270 Search PubMed.
- I. L. Volkov, P. Y. Serdobintsev and A. I. Kononov, J. Phys. Chem. C, 2013, 117, 24079–24083 Search PubMed.
- J. T. Petty, C. Fan, S. P. Story, B. Sengupta, M. Sartin, J.-C. Hsiang, J. W. Perry and R. M. Dickson, J. Phys. Chem. B, 2011, 115, 7996–8003 CrossRef PubMed.
- J. T. Petty, C. Y. Fan, S. P. Story, B. Sengupta, A. S. Iyer, Z. Prudowsky and R. M. Dickson, J. Phys. Chem. Lett., 2010, 1, 2524–2529 CrossRef PubMed.
- S. Krause, M. R. Carro-Temboury, C. Cerretani and T. Vosch, Chem. Commun., 2018, 54, 4569–4572 RSC.
- C. Cerretani, M. R. Carro-Temboury, S. Krause, S. A. Bogh and T. Vosch, Chem. Commun., 2017, 53, 12556–12559 RSC.
- E. Thyrhaug, S. A. Bogh, M. R. Carro-Temboury, C. S. Madsen, T. Vosch and D. Zigmantas, Nat. Commun., 2017, 8, 15577 CrossRef PubMed.
- S. H. Yau, N. Abeyasinghe, M. Orr, L. Upton, O. Varnavski, J. H. Werner, H. C. Yeh, J. Sharma, A. P. Shreve, J. S. Martinez and T. Goodson, Nanoscale, 2012, 4, 4247–4254 RSC.
- K. D. Weston, M. Dyck, P. Tinnefeld, C. Muller, D. P. Herten and M. Sauer, Anal. Chem., 2002, 74, 5342–5349 CrossRef PubMed.
- E. N. Hooley, V. Paolucci, Z. Liao, M. R. Carro Temboury and T. Vosch, Adv. Opt. Mater., 2015, 3, 1109–1115 CrossRef.
- E. N. Hooley, M. R. Carro-Temboury and T. Vosch, J. Phys. Chem. A, 2017, 121, 963–968 CrossRef PubMed.
- J. Hofkens, T. Vosch, M. Maus, F. Köhn, M. Cotlet, T. Weil, A. Herrmann, K. Müllen and F. C. De Schryver, Chem. Phys. Lett., 2001, 333, 255–263 CrossRef.
- S. Krause, P. F. Aramendia, D. Tauber and C. von Borczyskowski, Phys. Chem. Chem. Phys., 2011, 13, 1754–1761 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, additional PArTI and SF/PF ratio images, emission spectra, single molecule data, quantum yield clarification. See DOI: 10.1039/c8cp02584c |
| This journal is © the Owner Societies 2018 |