
Enantioselective acyl-transfer catalysis by fluoride ions†
Ryan
Craig
 ,
Mili
Litvajova
,
Sarah A.
Cronin
and
Stephen J.
Connon
*
,
Mili
Litvajova
,
Sarah A.
Cronin
and
Stephen J.
Connon
*
School of Chemistry Trinity Biomedical Sciences Institute, Trinity College Dublin, 152-160 Pearse Street, Dublin 2, Ireland. E-mail: connons@tcd.ie; Tel: +353 18961306
First published on 20th August 2018
Abstract
The asymmetric nucleophilic catalysis by fluoride ions at a carbon-based electrophile has been demonstrated for the first time. Using a library of ad hoc designed bifunctional phase-transfer catalysts in which both the anion and the cation are directly involved in the reaction, the desymmetrisation of meso-succinic and -glutaric anhydrides is possible. 19F NMR spectroscopic studies support the intermediacy of an acyl fluoride intermediate.
Tetraalkylammonium fluorides have a long history of utility in the field of catalysis, where, due to the hard nature of fluoride, they are almost invariably used (inter alia) as either a base1 (catalytic or stoichiometric) or a desilylating agent.1,2 Chiral ammonium fluorides for applications in asymmetric synthesis are less common but also known.3 It is remarkable that the use of fluoride as a nucleophilic catalyst has received almost no attention. In 1967, Bunton and Fender reported that fluoride ions catalysed the hydrolysis of acetic anhydride (1) in both water and aqueous media (Fig. 1A). Reaction kinetics indicated that the rate of consumption of the starting material outstripped the rate of product formation in a clean reaction, indicating the build-up of a stable intermediate. Aspiration experiments allowed the identification of acetyl fluoride (2) as the intermediate, indicating that acyl transfer proceeded predominantly via nucleophilic catalysis.4,5
 | ||
| Fig. 1 Nucleophilic catalysis by fluoride. | ||
The idea that fluoride can be used as an acyl transfer catalyst has gained little currency since. Superstoichiometric fluoride has been used to catalyse the hydrolysis of nucleotide phosphotriesters,6 and the cleavage of activated amino-acid esters7 and carbamates (e.g. Boc-cleavage, 10 equiv. tetrabutylammonium fluoride (TBAF), THF, reflux, 8 h).8 The mechanism of these transformations was not elucidated but nucleophilic catalysis has been implicated in the latter study. More recently, Chan, Hendrick et al.9 rationalised the fluoride-catalysed of transesterification of aryl ester 5 to furnish 6 in the presence of (for example) benzyl alcohol via nucleophilic catalysis (Fig. 1B), however no mechanistic evidence was provided. In such scenarios, the possibility of the basicity of fluoride dominating catalysis is a potential issue: for instance Edgar et al.10 demonstrated that the TBAF-mediated deacetylation of cellulose-derivative 7 occurs via general base catalysis at C-6 and specific-base catalysis (i.e. E1cB) at C-2/C-3 (Fig. 1B).
We have been interested in the development of the first asymmetric nucleophilic catalysis reactions promoted by fluoride. We were prompted to report our results by a very recent disclosure by Gouverneur et al. detailing the development of an elegant urea-catalysed enantioselective nucleophilic fluoridation reaction involving stoichiometric fluoride, alkyl bromides such as 8 and catalyst 9 (Fig. 1C).11
Herein we report the design of a system that utilises fluoride as an asymmetric nucleophilic catalyst in the desymmetrisation of meso-anhydrides of general type 11 by alcoholysis12–15 (Fig. 1D) to form hemiesters 12via the putative intermediate 11a – with the generation of 4 stereocentres. Ring-opening is mediated by an ionic Phase-Transfer Catalyst (PTC) 13 designed to maximise the potential for delivery of fluoride ion to a catalyst-bound electrophile using cooperation between a hydrogen-bond donating urea moiety (located on the cation) and an alkaloid-derived ammonium bromide unit which forms a nucleophilic chiral ammonium fluoride in situ. To the best of our knowledge it represents the first example of asymmetric nucleophilic catalysis by fluoride ion at a carbon centre.
Our study began with preliminary experiments to investigate the potential of TBAF as an acyl-transfer catalyst compared to diisopropylethylamine (DIPEA) in the addition of MeOH to meso-anhydride 14 in MTBE solvent16 at ambient temperature (Table 1). In the absence of catalyst no desymmetrisation occurs under the reaction conditions (entry 1). DIPEA (5 mol%) is able to mediate the formation of low levels of 15 after 3 days reaction time (entry 2), however, use of an equivalent loading of TBAF led to quantitative conversion, with smooth generation of 15 (entry 3). Further experiments demonstrated that TBAF could promote the reaction to quantitative conversion in a time frame where use of DIPEA could provide only trace amounts of product (entries 4 and 5). 1 mol% appears to be the lower loading limit under these conditions – providing over 50% conversion in 12 h (entry 6). Consistent with the activity of TBAF emanating from fluoride anion rather than the ammonium cation, a control reaction using TBAB (5 mol%) exhibited no conversion (entry 7).
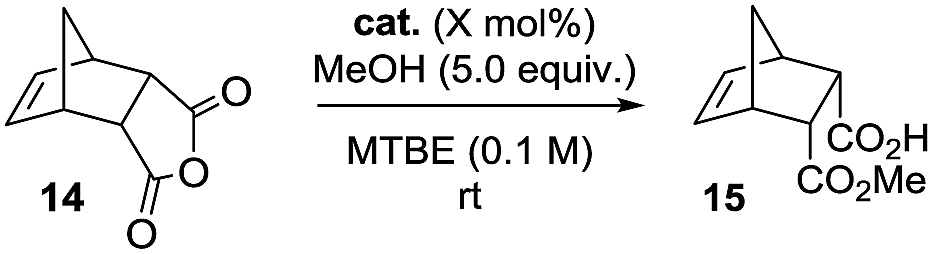
We have shown previously that (thio)urea-modified cinchona alkaloids can catalyse anhydride desymmetrisation by alcoholysis16a – where the hydrogen bond donor presumably stabilises developing negative charge as the alcohol (under the influence of general base catalysis) adds to the anhydride. We therefore reasoned that if such alkaloids were alkylated at the quinuclidine moiety,17 that they could serve as materials capable of both phase-transfer catalysis involving fluoride ion and electrophile activation. Alternatively, they could serve as PTCs capable of binding fluoride ion via the hydrogen bond donating unit and delivering it to the anhydride.
Initial attempts to generate such catalysts produced mixed results. Formation of the ammonium fluorides was possible in solution, but decomposition and/or loss of HF on attempted isolation from solution for characterisation were problematic. It was therefore decided to generate the ammonium fluorides from the corresponding (stable) bromides in situ via anion metathesis. The bromides were expected to be catalytically inert (Table 1, entry 7), so provided that the rates of halide ion exchange were not prohibitively slow, it held promise as an operationally practical methodology. After considerable experimentation, it was found that anhydride 14 underwent methanolysis to 15 in the presence of KF (15 mol%) and ammonium bromides (5 mol%) in THF (0.1 M) at ambient temperature, but no conversion occurred if either the fluoride catalyst or the PTC were absent. These conditions were selected to evaluate a library of alkaloid-based PTCs equipped with hydrogen bond donating moieties and N-alkyl ammonium ions substituents of variable steric/electronic characteristics (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | T (°C) | Conv.a (%) | eeb (%) |
| a Determined by 1H NMR spectroscopy. b Enantiomeric excess determined by 1H NMR spectroscopic analysis after derivatisation of the appropriate hemiester. c 0.2 M, 50 h. | ||||
| 1 | 16 | rt | 58 | −3 |
| 2 | 17 | rt | 49 | −2 |
| 3 | 18 | rt | 38 | 3 |
| 4 | 19 | rt | 34 | 10 |
| 5 | 20 | rt | 80 | −11 |
| 6 | 21 | rt | 17 | 0 |
| 7 | 22 | rt | 30 | −4 |
| 8 | 23 | rt | 47 | −23 |
| 9 | 24 | rt | 46 | −21 |
| 10 | 25 | rt | 45 | −25 |
| 11 | 26 | rt | 43 | −44 |
| 12 | 27 | rt | 43 | −17 |
| 13 | 28 | rt | 45 | −6 |
| 14 | 29 | rt | 70 | 0 |
| 15 | 30 | rt | 79 | 53 |
| 16 | 31 | rt | 66 | 51 |
| 17 | 32 | rt | 48 | 25 |
| 18 | 33 | rt | 66 | 60 |
| 19 | 34 | rt | 57 | 46 |
| 20 | 35 | rt | 70 | 43 |
| 21 | 36 | rt | 44 | 40 |
| 22 | 37 | rt | 55 | 23 |
| 23 | 38 | rt | 61 | 51 |
| 24 | 39 | rt | 66 | 60 |
| 25 | 39 | −15 | 11 | n.d. |
| 26 | 39 | 50 | 93 | 44 |
| 27c | 39 | rt | 70 | 61.5 |
| 28 | 40 | rt | 72 | 61 |
| 29 | 41 | rt | 77 | 61 |
| 30 | 13 | rt | 79 | 61 |
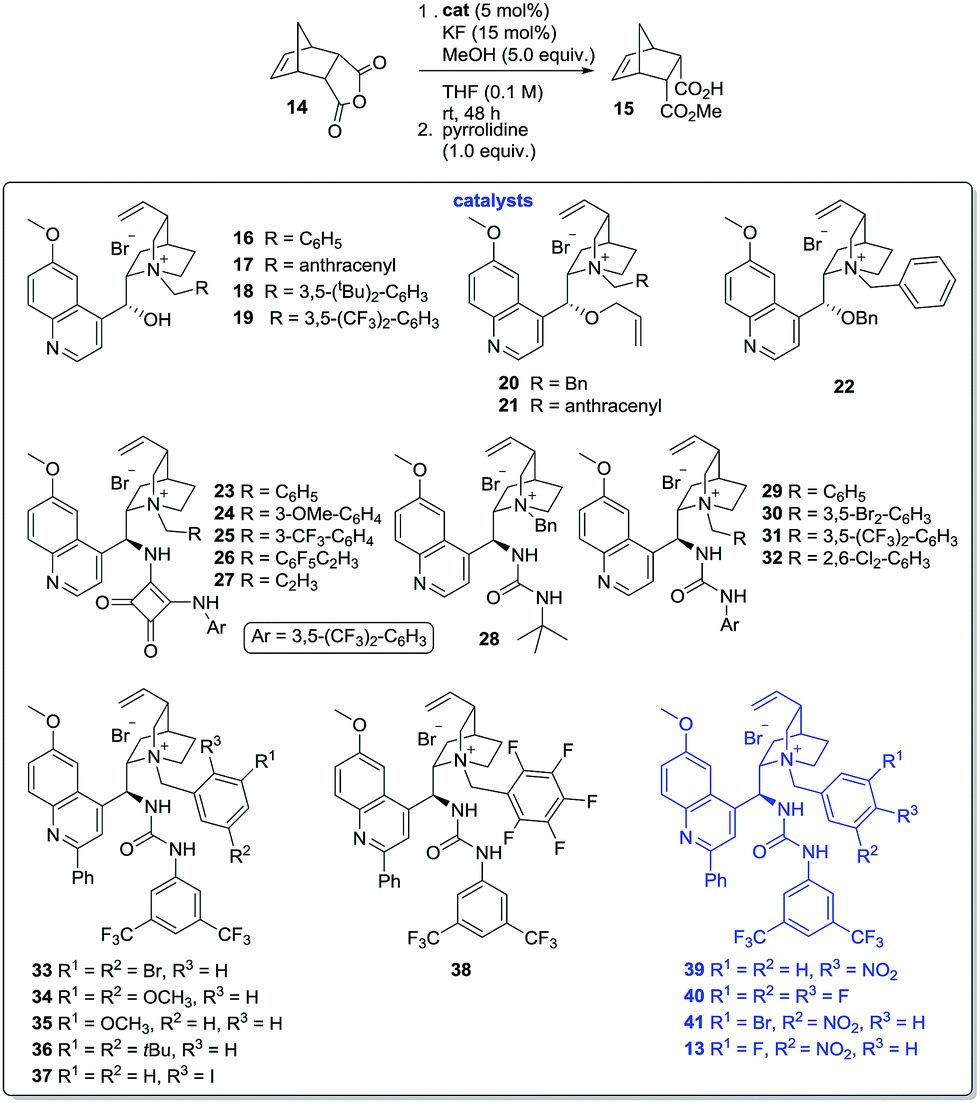
N-Alkyl quinines 16–19 promoted the methanolysis in poor-moderate conversion; with the formation of 15 as a near racemate (entries 1–4).18 Bulky N-alkyl substituents slowed the reaction rate considerably. Deletion of the hydrogen bonding capabilities of these materials via alkylation of the hydroxyl unit (i.e.20–22, entries 5–7) failed to improve selectivity appreciably. Exchange of the C-9 substituent (with concomitant inversion of configuration) with the strongly hydrogen bond donating squaramide moiety17e was advantageous: use of the simple N-benzyl catalyst 23 provided 15 with the highest enantiomeric excess observed up to that point (23%, entry 8), and while the installation of either a single electron donating- or electron withdrawing-group at the N-benzyl moiety led to little change in product ee (i.e.24–25, entries 9 and 10), the pentafluorobenzyl ammonium ion 26 could mediate the desymmetrisation with a greatly improved 44% ee (entry 11). These findings, together with the relative failure of the N-propargyl catalyst 27 (entry 12), underscore the role both the hydrogen bond donating unit and the N-alkyl substituent play in bringing about selective methanolysis of 14.
Accordingly, we next evaluated the use of urea-substituted PTCs – a class of catalyst which recently performed well in the asymmetric alkylation of 3-substituted oxindole derivatives.17f These experiments got off to an inauspicious start: both the bulky tert-butyl urea 28 and the archetypal N-benzyl catalyst 29 accelerated the formation of near optically inactive products (entries 13 and 14). However, variants of 29 characterised by the presence of electron withdrawing groups at the m-positions of the benzyl substituent promoted reactions with >50% ee and improved conversion relative to squaramide-based systems (i.e. catalysts 30–31, entries 15 and 16). o-Substitution (i.e. PTC 32) is less well tolerated (entry 17).
We had previously shown that installation of an aryl substituent at C-2 of the catalyst's quinoline ring can improve the performance of related PTCs,17f and thus prepared a small library of C-2 arylated catalysts 33–38 (entries 18–23). The influence of this modification on efficacy can be divined by comparing catalyst 30 (entry 15) with the superior, substituted analogue 33 (entry 18), which could promote the reaction with 60% ee. The same structural trends were observed as before: N-benzyl moieties containing electron-withdrawing groups lead to better stereocontrol than either electron-rich or bulky analogues, while o-substitution is detrimental. The p-nitro substituted variant 39 proved equal to the hitherto best catalyst in the library (entry 24), although either lowering or raising the reaction temperature failed to improve the process (entries 25 and 26). The difluorobenzyl-PTC 40 (entry 27) promoted the reaction with similar ee and higher conversion, while the incorporation of a bromine atom o- to the nitro functionality of catalyst 39 (i.e.41, entry 29) led to further marginal improvement. Finally, exchange of the bromine atom for the more electronegative fluorine atom (i.e.13, entry 30) provided a catalyst in which the hydrogen-bond donating unit, the N-benzyl moiety and the C-2 quinoline substituent had been modified in near optimal fashion.
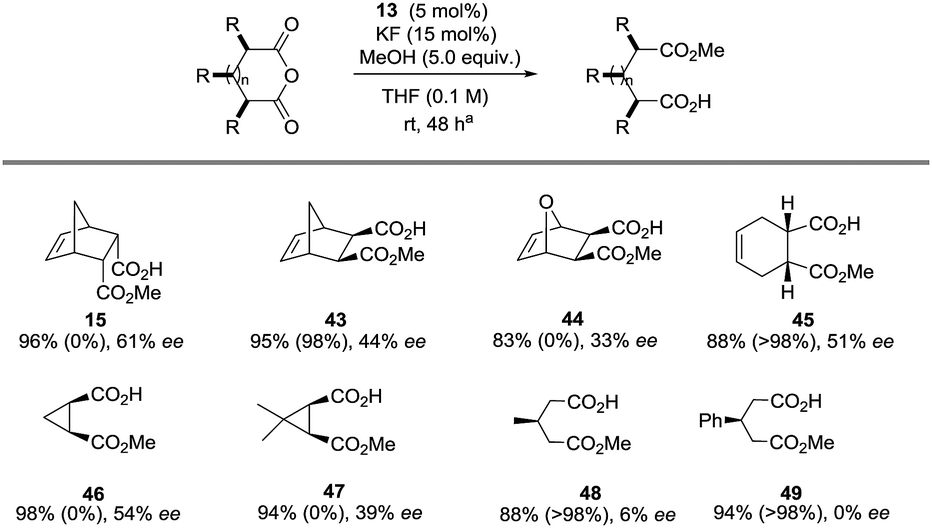
In terms of substrate scope, a range of meso-anhydrides could be converted to the corresponding methyl hemiesters 15 and 43–49 in good-excellent yields at ambient temperature in the presence of methanol, KF and 13 at 5 eq., 15- and 5 mol% levels respectively (Table 3). Enantiomeric excess ranged from trace levels for the glutaric anhydride-derived 47–48 to 33–61% ee for the bicyclic succinic anhydride-derived esters 15 and 43–47. In the case of 43, 45, and 48–49, product optical purity is modulated by competing (occasionally rapid) background methanolysis of the less sterically demanding anhydrides by potassium fluoride alone. No background reaction was observed in the other cases. The fact that (for instance) 45 can be formed in 51% ee despite stiff competition from a racemic pathway is remarkable.
| a Yields are isolated. The value in parenthesis refers to the conversion of a repeat reaction in the absence of 13 under otherwise identical conditions after 48 h. Enantiomeric excess determined by 1H NMR spectroscopic analysis after derivatisation of the appropriate hemiester. |
|---|
|
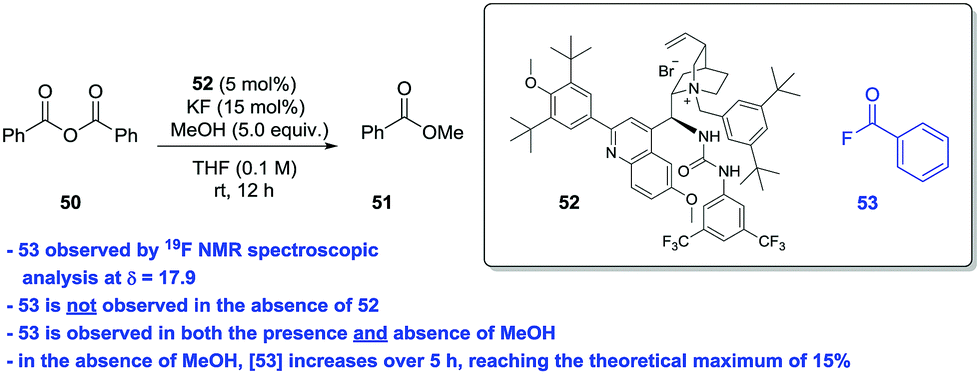
Bunton and Fender (Fig. 1A)4 could not isolate or detect the build-up of acyl fluoride intermediate in the fluoride-catalysed hydrolysis of succinic anhydride – most likely due to either a rapid equilibrium involving ring closure by the carboxylate or general base catalysis of the hydrolysis of the acyl fluoride by the neighbouring carboxylate. Accordingly, we deemed it unlikely that we could isolate or trap an acyl fluoride in the current system. In order to circumvent the problem of interference from a tethered carboxylate ion, we carried out a study on fluoride-catalysed methanolysis of benzoic anhydride (50) in the presence of a PTC (Scheme 1).
 | ||
| Scheme 1 Observation of an acyl fluoride intermediate. | ||
Under conditions identical to those in the desymmetrisation experiments outlined above using PTC 52,17f during the formation of 51 we could detect the presence of 53 by 19F NMR spectroscopy. In the absence of the PTC, 53 is not formed; while the levels of 53 detected were higher in the absence of MeOH than in its presence. In the absence of methanol, we could (using the catalyst's CF3 resonances as an internal standard) track the increase in concentration of 53 over time. After 5 h, all KF is converted to 53. These observations do not exclude a competing general base catalysed pathway, however, it is seems likely that nucleophilic catalysis (Fig. 2) is responsible for much of the observed activity.
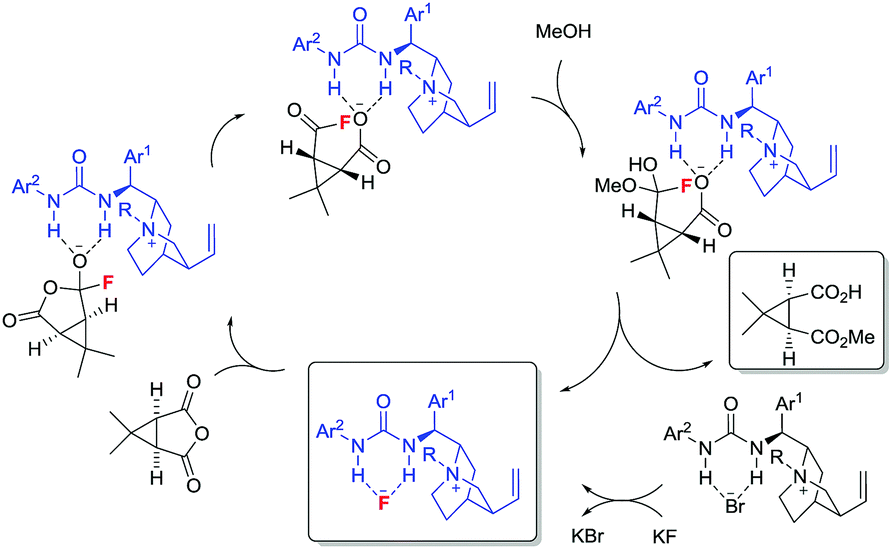 | ||
| Fig. 2 Proposed catalytic cycle. | ||
In summary, the first asymmetric reaction involving nucleophilic catalysis by fluoride ion has been developed. A library of quinine-derived PTCs capable of forming ammonium fluorides in situ were evaluated in the desymmetrisation of a meso-anhydride by methanolysis. The presence of a strong H-bond donating unit at C-9 is essential from an enantioselectivity standpoint. The optimum catalyst designed was capable of promoting the desymmetrisation of 14 with 61% ee. Experiments involving the fluoride-catalysed methanolysis of benzoic anhydride unambiguously demonstrated the formation of the corresponding acyl fluoride, and in the absence of methanol, quantitative formation of 53 was observed.
This publication has emanated from research conducted with the financial support of the Synthesis and Solid State Pharmaceutical Centre (SSPC), funded by Science Foundation Ireland (SFI) under grant numbers 12\RC\2275 and 12-IA-1645 and the European Regional Development Fund (EDRF). Financial support from the Irish Research council is also gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. H. Clark, Chem. Rev., 1980, 80, 429 CrossRef; (b) A. Hameed, R. D. Alharthy, J. Iqbal and P. Langer, Tetrahedron, 2016, 72, 2763 CrossRef.
- I. Kuwajima and E. Nakamura, Acc. Chem. Res., 1985, 18, 181 CrossRef.
- (a) T. Ooi and K. Maruoka, Acc. Chem. Res., 2004, 37, 526 CrossRef PubMed; (b) F. Legros, S. Oudeyer and V. Levacher, Chem. Rec., 2017, 17, 429 CrossRef PubMed.
- C. A. Bunton and J. H. Fendler, J. Org. Chem., 1967, 33, 1547 CrossRef.
- This hypothesis was strengthened by the observation of a much less negative entropy of activation for the fluoride-catalysed hydrolysis than either the corresponding spontaneous hydrolysis or general base-catalysed processes.
- K. K. Oglive and S. L. Beaucage, J. Chem. Soc., Chem. Commun., 1976, 443 RSC.
- (a) M. Namikoshi, B. Kundu and K. L. Rinehart, J. Org. Chem., 1991, 56, 5464 CrossRef; (b) C. J. Salomon, E. G. Mata and C. A. Mascaretti, Tetrahedron, 1993, 49, 3691 CrossRef.
- U. Jacquemard, V. Bénéteau, M. Lefoix, S. Routier, J.-Y. Mérour and G. Coudert, Tetrahedron, 2004, 60, 10039 CrossRef.
- J. M. W. Chan, H. Sardon, A. C. Engler, J. M. García and J. L. Hedrick, ACS Macro Lett., 2013, 2, 860 CrossRef.
- X. Zheng, R. D. Gandour and K. J. Edgar, Biomacromolecules, 2013, 14, 1388 CrossRef PubMed.
- G. Pupo, F. Ibba, D. M. H. Ascough, A. C. Vicini, P. Ricci, K. E. Christensen, L. Pfeifer, J. R. Morphy, J. M. Brown, R. S. Paton and V. Gouverneur, Science, 2018, 360, 638 CrossRef PubMed.
- For seminal reports see: (a) J. Hiratake, Y. Yamamoto and J. Oda, J. Chem. Soc., Chem. Commun., 1985, 1717 RSC; (b) R. A. Aitken, J. Gopal and J. A. Hirst, J. Chem. Soc., Chem. Commun., 1988, 632 RSC.
- For selected reviews detailing anhydride desymmetrisation by alcoholysis see: (a) A. C. Spivey and B. I. Andrews, Angew. Chem., Int. Ed., 2001, 40, 3131 CrossRef; (b) Y. Chen, P. McDaid and L. Deng, Chem. Rev., 2003, 103, 2965 CrossRef PubMed; (c) I. Atodiresei, I. Schiffers and C. Bolm, Chem. Rev., 2007, 107, 5683 CrossRef PubMed; (d) Z. Rodriguez-Docampo and S. J. Connon, ChemCatChem, 2012, 4, 151 CrossRef; (e) A. Borissov, T. Q. Davies, S. R. Ellis, T. A. Fleming, M. S. W. Richardson and D. J. Dixon, Chem. Soc. Rev., 2016, 45, 5474 RSC.
- Selected examples of anhydride desymmetrisation by thiolysis: (a) T. Honjo, S. Sano, M. Shiro and Y. Nagao, Angew. Chem., Int. Ed., 2005, 44, 5838 CrossRef PubMed; (b) A. Peschiulli, B. Procuranti, C. J. O’Connor and S. J. Connon, Nat. Chem., 2010, 2, 380 CrossRef PubMed.
- Selected examples of anhydride alkylative desymmetrisation: (a) E. A. Bercot and T. Rovis, J. Am. Chem. Soc., 2005, 127, 247 CrossRef PubMed; (b) E. E. Stache, T. Rovis and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 3679 CrossRef PubMed.
- We have found ethereal solvents to be superior in reactions involving amine catalysts where the product is a carboxylic acid, for examples see: (a) A. Peschiulli, Y. Gun’ko and S. J. Connon, J. Org. Chem., 2008, 73, 2454 CrossRef PubMed; (b) F. Manoni and S. J. Connon, Angew. Chem., Int. Ed., 2014, 53, 2628 CrossRef PubMed; (c) S. A. Cronin, A. Gutiérrez Collar, S. Gundala, C. Cornaggia, E. Torrente, F. Manoni, A. Botte, B. Twamley and S. J. Connon, Org. Biomol. Chem., 2016, 14, 6955 RSC; (d) F. Manoni, U. Farid, C. Trujillo and S. J. Connon, Org. Biomol. Chem., 2017, 15, 1463 RSC; (e) R. Claveau, B. Twamley and S. J. Connon, Chem. Commun., 2018, 54, 3231 RSC.
- For examples of these types of catalysts see: (a) P. Bernal, R. Fernández and J. M. Lassaletta, Chem. – Eur. J., 2010, 16, 7714 CrossRef PubMed; (b) K. M. Johnson, M. S. Rattley, F. Sladojevich, D. M. Barber, M. G. Nunez, A. M. Goldys and D. J. Dixon, Org. Lett., 2012, 14, 2492 CrossRef PubMed; (c) M. Li, P. A. Woods and M. D. Smith, Chem. Sci., 2013, 4, 2907 RSC; (d) B. Wang, Y. He, X. Fu, Z. Wei, Y. Lin and H. Duan, Synlett, 2015, 2588 Search PubMed; (e) E. Sorrentino and S. J. Connon, Org. Lett., 2016, 18, 5204 CrossRef PubMed; (f) R. Craig, E. Sorrentino and S. J. Connon, Chem. – Eur. J., 2018, 24, 4528 CrossRef PubMed.
- Pyrrolidine is added after 48 h to destroy remaining anhydride.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for the syntheses of starting materials, catalysts and additional data: NMR spectra of the products, etc. See DOI: 10.1039/c8cc05692g |
| This journal is © The Royal Society of Chemistry 2018 |