
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porphyrin-based metal-organic frameworks for solar fuel synthesis photocatalysis: band gap tuning via iron substitutions†
Alex
Aziz
 a,
A. Rabdel
Ruiz-Salvador
b,
Norge C.
Hernández
c,
Sofia
Calero
b,
Said
Hamad
b and
Ricardo
Grau-Crespo
*a
a,
A. Rabdel
Ruiz-Salvador
b,
Norge C.
Hernández
c,
Sofia
Calero
b,
Said
Hamad
b and
Ricardo
Grau-Crespo
*a
aDepartment of Chemistry, University of Reading, Whiteknights, Reading RG6 6AD, UK. E-mail: r.grau-crespo@reading.ac.uk
bDepartamento de Sistemas Físicos, Químicos y Naturales, Universidad Pablo de Olavide, Carretera de Utrera km. 1, 41013 Seville, Spain
cDepartamento de Física Aplicada I, Escuela Técnica Superior de Ingeniería Informática, Universidad de Sevilla, Avenida Reina Mercedes, 41012 Sevilla, Spain
First published on 17th May 2017
Abstract
Photocatalysts based on metal-organic frameworks (MOFs) are very promising due to a combination of high tuneability and convenient porous structure. Introducing porphyrin units within MOFs is a potential route to engineer these natural photosynthesis molecular catalysts into artificial photosynthesis heterogeneous catalysts. Using computer simulations based on density functional theory, we explore how to modify the electronic structure of porphyrin-based MOFs to make them suitable for the photocatalysis of solar fuel synthesis via water splitting or carbon dioxide reduction. In particular, we have investigated the effect that Fe substitutions have on the electronic properties of porphyrin-based metal organic frameworks. By aligning the electron levels with a vacuum reference, we show that Fe at the porphyrin metal centre has the effect of slightly raising the position of the valence band edge, whereas Fe at the octahedral metal node has the ability to significantly lower the position of the conduction band edge on the absolute scale. Iron is therefore a very useful dopant to engineer the band structure and alignment of these MOFs. We find that the porphyrin-based structure with Al in the octahedral sites and Zn in the porphyrin centres has a band gap that is slightly too wide to take advantage of visible-light solar radiation, while the structure with Fe in the octahedral sites has bandgaps that are too narrow for water splitting photocatalysis. We then show that the optimal composition is achieved by partial substitution of Al by Fe at the octahedral sites, while keeping Zn at the porphyrin centres. Our study demonstrates that porphyrin-based MOFs can be engineered to display intrinsic photocatalytic activity in solar fuel synthesis reactions.
Introduction
The solar-driven synthesis of fuels, such as hydrogen1,2 or organic fuels like methanol,3–5 offers great potential for clean and renewable energy, so the design of new efficient and visible-light active photocatalysts for solar fuel synthesis is attracting much research interest. While traditionally inorganic semiconductor materials have received most attention, there is a growing interest in considering alternative photocatalytic materials, such as metal-organic frameworks (MOFs).6–8 MOFs are porous crystalline materials, created by joining organic linkers through coordination nodes, which can be either single metal atoms or metal-containing clusters. They are being widely used in fields like adsorption, separation and catalysis due to their extraordinary properties and versatility.9–15In photocatalysis, MOFs offer the potential capability to perform storage and photochemical conversion of gases in a one-pot approach. The properties of MOFs can be generally tuned by changing the metal atom at the nodes, the organic linkers, and the topology. It is important to understand how these degrees of freedom can be used to tailor the electronic properties of MOFs to target particular photocatalytic reactions. For example, Bordiga, Zecchina and co-workers have found that the Zn-oxocluster in MOF-5 behaves as a ZnO quantum dot semiconductor, where the terephthalic ligand acts as an antenna, sensitizing the inorganic unit.16 Garcia and co-workers showed that such behaviour is responsible for the observed photocatalytic conversion of phenols using MOF-5.17 Fuentes-Cabrera et al. predicted that in MOF-5 the substitution of the metal atoms by metals of groups IIA and d10 metals does not change the band gap.18 If other transition metal atoms, not d10, are introduced, the optical absorption edge (which is related to the band gap) of MOF-5 can be modified, as revealed by experimental UV-Vis data.19,20 Gascon et al.21 showed that by modifying the linker properties the overall bandgap of MOF-5 can be lowered.21 Recently, we have predicted the possibility of finely controlling the electronic structure of zeolitic imidazolate frameworks (ZIFs), by following a linker mix-and-match approach.22 We found that some mixed-linker (e.g. methylimidazole/nitroimidazole) ZIFs exhibit band structures favourable for solar fuel synthesis photocatalysis.
The present work focuses on the electronic properties of porphyrin-based MOFs (PMOFs). Porphyrins are heterocyclic macrocycle organic compounds which are an essential part of the chemistry of life. For example, a Fe-containing porphyrin cycle is responsible for the adsorption of O2 and CO2 in haemoglobin, while light absorption in chlorophyll takes place in a Mg-containing porphyrin cycle. Not surprisingly, porphyrins have been investigated as active molecular centres for artificial photosynthesis,1,23–25 and solar cells.26–28 Rosseinsky and co-workers synthesized PMOFs with Al at the metal nodes (Al-PMOF) which were found to be water-stable.29 In general, MOFs with Al-carboxylate coordination tend to exhibit very high chemical and thermal stability.30,31 In ref. 29, the experimental optical spectra of Al-PMOF resembles that of the isolated porphyrin, which confers the material an almost molecular behaviour. The authors also found interesting photocatalytic properties of the PMOF (aided by auxiliary metal species). It is known that the photocatalytic properties of porphyrin molecules can be modified by the presence of metal cations within the ring,32 and metalation of the porphyrin units by Zn and Cu enhances photocatalytic water splitting29 and CO2 conversion,33 respectively. These observations motivated our previous study on Al-PMOF,34 in which we performed a detailed analysis of the electronic structure of Al-PMOF as a function of the metal M substituted in the porphyrin centre (M = Fe, Co, Ni, Cu, Zn). We found that the energy levels of the frontier orbitals in these materials are in the correct positions for solar fuel synthesis photocatalysis, although the band gaps obtained were still a bit too wide for efficient visible-light photocatalysis of water splitting. Transition metal substitutions at the porphyrin centres did not appreciably change the position of the band edges (except in the case of Fe), therefore such substitutions offered limited opportunities for band structure tuning. Furthermore, the fact that both the valence and conduction band edges of Al-PMOFs are localized in the porphyrin units means that excitations would take place without spatial charge separation, which is not convenient for photocatalysis, as it leads to short electron–hole recombination times.
In this article we report the theoretical investigation of an alternative approach to engineering the electronic band structure of PMOFs, based on the replacement of the Al cations in the PMOFs' octahedral sites (the metal nodes) by Fe cations. This is motivated by the recent experimental success in incorporating iron at both the porphyrin centres and the octahedral sites,35 as well as by the well-known ability of transition metal cations to contribute both occupied and unoccupied d levels around the Fermi energy, thus allowing band gap control. We compare the effects of substituting Fe in the porphyrin and in the octahedral metal centres, as well as in both simultaneously. Finally, we will argue that the optimal electronic properties for photocatalysis, in terms of band gaps and band edge positions, is obtained from partial substitution of the Al cations by Fe in the octahedral sites, while keeping Zn at the porphyrin centres.
Computational methods
Our calculations focus on the orthorhombic crystal structure of porphyrin-based MOF (PMOF) first reported by Fateeva et al.29 The conventional unit cell (space group Cmmm)29 is shown in Fig. 1, with two porphyrins per unit cell. To reduce computational cost, our calculations were performed in the primitive cell, which contains only one porphyrin per cell. Our simulations were based on the density functional theory (DFT), as implemented in the Vienna Ab initio Simulation Package (VASP).36–39 For the geometry optimizations, we used the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional,40 and including van der Waals (vdW) corrections via the DFT-D2 method of Grimme.41 In PBE calculations including Fe atoms, a Hubbard correction was applied to the Fe 3d orbitals, using Dudarev's approach42 based on a single parameter Ueff, which was set to 4 eV. This value has been reported to provide a good description of the electronic structure of iron oxides.43–45 During relaxation, forces on atoms were minimized until they were all less than 0.01 eV Å−1. After geometry optimisation, the screened hybrid functional of Heyd, Scuseria and Ernzerhof (HSE06),46,47 was used to perform single-point electronic structure calculations (now without U or D2 corrections). The HSE06 functional (25% Hartree–Fock exchange and screening parameter 0.2 Å−1) was chosen, as it provides bandgaps in closer agreement with experiment than GGA functionals for a wide range of semiconducting solids.48 This functional has also been shown to provide a good description of the electronic structure of MOFs. For example, the HSE06 bandgap for Zr-UiO-67 is 3.63 eV, which is in agreement with the measured bandgaps (3.5–3.68 eV);49 for HKUST-1 the HSE06 band gap is 3.8 eV, close to the experimental value of 3.6 eV.50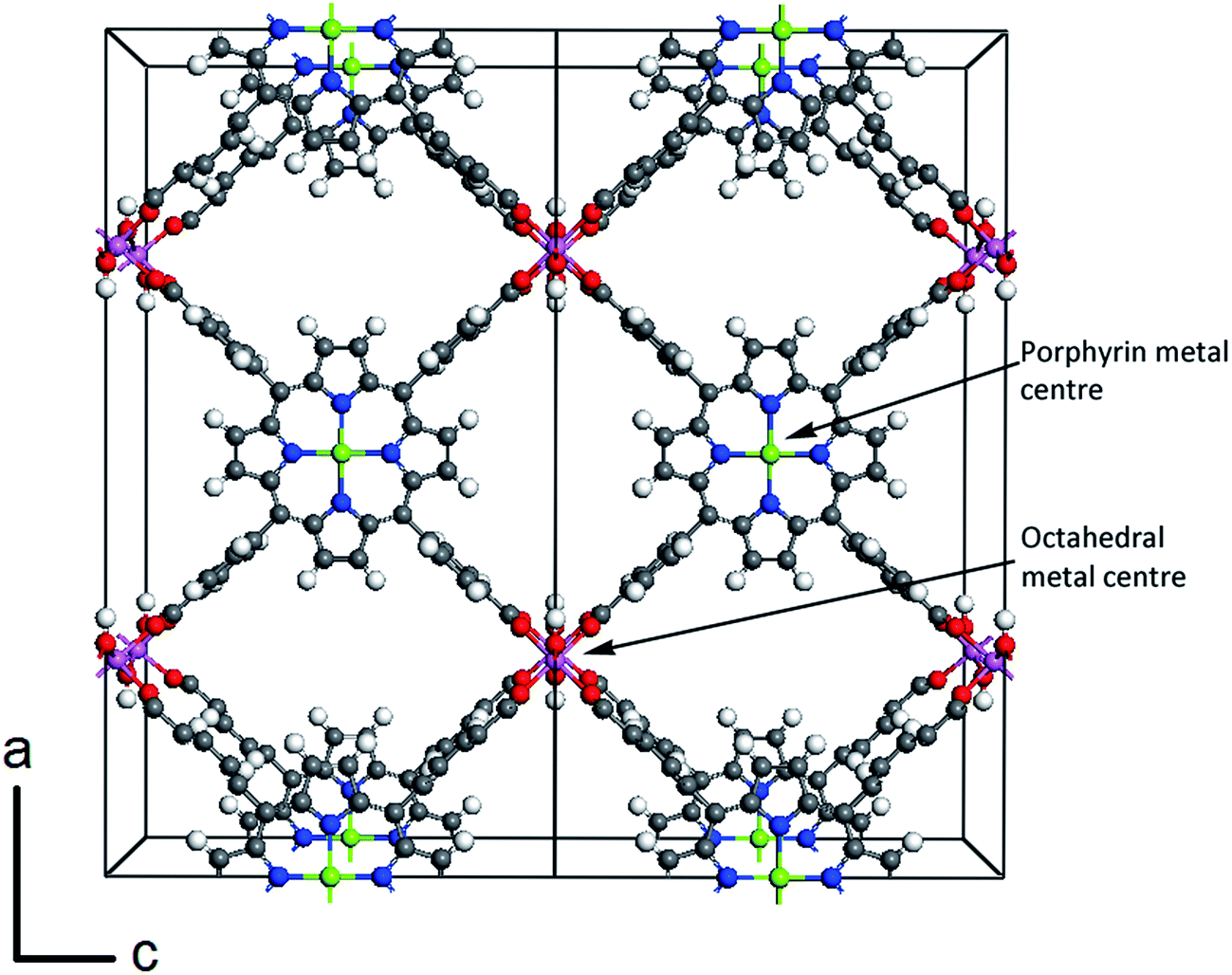 | ||
| Fig. 1 Perspective view of the conventional unit cell (doubled for clarity) for the porphyrin-based MOF investigated in this study, indicating the positions of the octahedral metal centre and the porphyrin metal centre. Colour code: gray = carbon, white = hydrogen, red = oxygen, blue = nitrogen, magenta = octahedral metal centre, green = porphyrin metal centre. A primitive cell, with half the number of atoms of the orthorhombic cell, was used in our calculations. | ||
All calculations including Fe allowed for spin polarisation. We systematically investigated all possible spin states at the Fe centres and report the electronic structure only for the most stable spin state in each case. However, we did not consider different possible relative orientations of the magnetic moments: all calculations correspond to ferromagnetic configurations. Although it is likely that the actual structures are paramagnetically disordered at room temperature, the weak magnetic coupling across distant Fe centres implies that the simulation results would not be affected by the magnetic order.
The projector augmented wave (PAW) method51,52 was used to describe the frozen core electrons (up to 3p for Fe and Zn, up to 2p for Al, and up to 1s for C, N and O) and their interaction with the valence electrons. The kinetic energy cutoff for the plane-wave basis set expansion was set at 520 eV. A Γ-centred grid of k-points was used for integrations in the reciprocal space, where the smallest allowed spacing between k-points was set at 0.5 Å−1. This corresponds to a k-mesh of 2 × 2 × 1 in the reciprocal of the primitive lattice (3 irreducible points sampled). The small number of irreducible k-points is justified by the large size of the unit cell; test calculations showed good convergence of total energies and geometries at that number of k-points. However, since smooth electronic density of state (DOS) plots cannot be obtained with such small number of k-points, we have also calculated DOS and band structures at PBE+U using a much denser grid of k-points for comparison (see Results and discussions and ESI†). Gaussian smearing with width parameter 0.05 eV was used in all calculations.
The calculated band energies given in VASP are relative to the average potential in the crystal. To obtain absolute band energies, which can be used to compare to the potential of the hydrogen and oxygen evolution reactions, it is necessary to align the band energies with the vacuum scale. In this work we take the electrostatic potential in the vacuum region located at the middle of the largest pore to be the vacuum level. This follows the methodology proposed by Butler et al.53 to calculate the vacuum level in MOF structures. In ref. 53, this procedure led to MOF ionization potentials in good agreement with experiment. A Python code (MacroDensity) provided by these authors was employed in our calculations to obtain the potentials.
Results and discussion
Crystal structures and relative stabilities
Our energy minimization calculations provide optimised geometries for the four crystal structures formed by combining two different metal cations in the porphyrin metal centres (M2+ = Zn2+ or Fe2+) with two different metal cations in the octahedral sites (A3+ = Al3+ or Fe3+) of the PMOF. To specify each PMOF composition we will use the notation (A3+, M2+)PMOF.Table 1 shows the main geometric properties of these structures. The calculated cell parameters are close to the experimentally measured values reported for (Al3+, Zn2+)PMOF and (Fe3+, Fe2+)PMOF.29,35 Note that, in contrast with our previous theoretical work,34 where we also considered the (Al3+, Zn2+)PMOF system, our calculations here include vdW corrections, which lead to a small change of cell volume (0.5–0.8%), reducing the difference between computational and experimental values from 3.2 to 2.5%. The cell parameters are noticeably affected by the nature of the cation in the octahedral centre (A3+): the structures with Fe3+ in the octahedral site have ∼5% higher cell volume than the structure with Al3+ in the octahedral site. This is in quantitative agreement with the relative values of the cell volumes found experimentally for (Al3+, Zn2+)PMOF and (Fe3+, Fe2+)PMOF.29,35 (Note that the latter experimental structure is not strictly equivalent to the one modelled by us, because it has pyrazine ligands connecting the porphyrins in the direction perpendicular to the porphyrin planes; the presence of these ancillary ligands can be expected to have little influence on the crystal cell parameters). But the cell parameters are not significantly affected (less than 0.5%) by the nature of the porphyrin metal centre (M2+), which can be expected from the fact that the M–N bond distances at the porphyrin centres are much less variable, due to the surrounding rigid structure of the porphyrin, than the A–O distances at the octahedral centres. The AO6 octahedra exhibit D4h distortions, with two A–O distances (apical) of slightly shorter length than that of the equatorial A–O distances. The magnitude of the D4h distortion, as given by the ratio between the apical and equatorial distances, is not affected by the nature of the metal centre (Al or Fe).
| Porphyrin centre (M2+) | Octahedral centre (A3+) | a (Å) | b (Å) | c (Å) | V (Å3) | d [M–N]a (Å) | Apical d [A–O] (Å) | Equat d [A–O] (Å) |
|---|---|---|---|---|---|---|---|---|
| a Average over 4 bonds in the square-planar coordination. b Experimental cell parameters for the (Zn2+, Al3+)-PMOF are taken from Section 8.5 of the ESI of ref. 29. c Experimental cell parameters for the (Fe2+, Fe3+)-PMOF are taken from those of structure 4B in the ESI of ref. 35 (not identical to the one modelled here due to the presence of ancillary pyrazine ligands in the experimental structure). | ||||||||
| Zn2+ | Al3+ | 32.079 (31.861)b | 6.687 (6.601) | 16.977 (16.895) | 3641.5 (3553.1) | 2.04 | 1.86 | 1.93 |
| Zn2+ | Fe3+ | 32.297 | 6.927 | 17.156 | 3837.9 | 2.04 | 1.96 | 2.04 |
| Fe2+ | Al3+ | 32.020 | 6.687 | 16.958 | 3631.1 | 2.00 | 1.86 | 1.93 |
| Fe2+ | Fe3+ | 32.249 (32.355)c | 6.932 (6.835) | 17.138 (16.896) | 3830.9 (3736.5) | 2.00 | 1.96 | 2.04 |
The relative thermodynamic stabilities of these crystal structures can be discussed in terms of their total energies in comparison with reference phases, for example, the A2O3 oxide phases. Since both Al2O3 and Fe2O3 with corundum structure are stable phases, with metal cations octahedrally coordinated to oxygen as in the PMOF, we consider the energy of the hypothetical cation exchange reaction:
| (Al3+, M2+)PMOF + Fe2O3 → (Fe3+, M2+)PMOF + Al2O3, |
Electronic structure: comparing Fe substitution in porphyrin and octahedral metal centres
We have previously reported the electronic structure of the “parent” compound (Al3+, Zn2+)PMOF and of the compound with Fe substituted in the porphyrin metal centres, (Al3+, Fe2+)PMOF (as well as the effect of substituting other metals in the porphyrin centres),34 but we summarise here their main features in order to establish a comparison with the new materials considered in this study. The electronic density of states of (Al3+, Zn2+)PMOF (Fig. 2a) shows a band gap of ∼2.5 eV. Both the valence and conduction band edges in this structure are contributed by the porphyrin, the occupied 3d orbitals of Zn lie far below the Fermi level. The substitution of Fe at the porphyrin centres, forming (Al3+, Fe2+)PMOF, reduces the band gap by ∼0.2 eV, which is due to the appearance of a β-spin Fe 3d level just above the occupied porphyrin levels (Fig. 2b).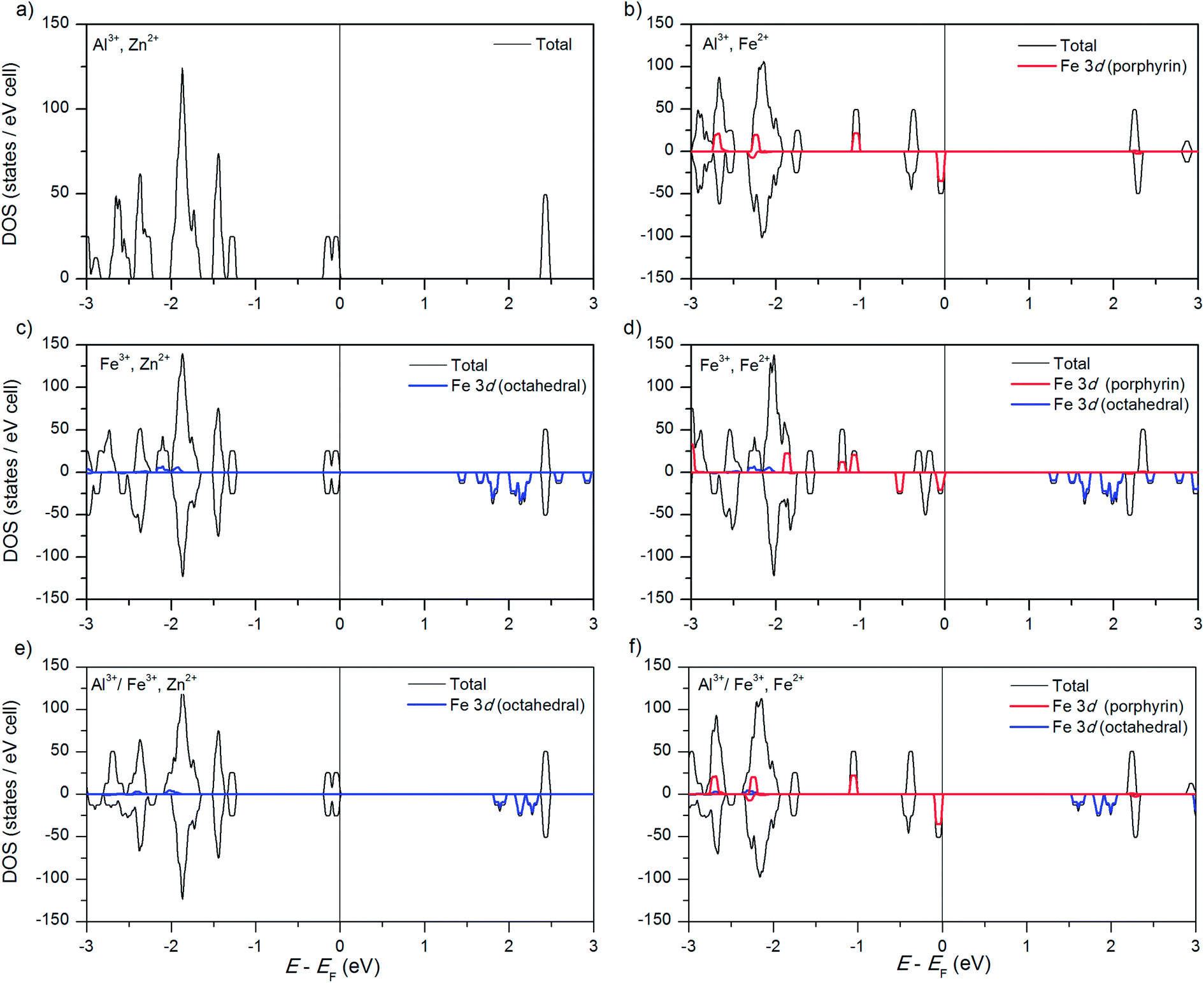 | ||
| Fig. 2 Electronic density of states, as obtained with the screened hybrid functional HSE06, for (a) the “parent” structure (Al3+, Zn2+)PMOF; (b) for the structure with Fe in the porphyrin metal centres, (Al3+, Fe2+)PMOF; (c) for the structure with Fe in the octahedral metal centres, (Fe3+, Zn2+)PMOF; (d) for the structure with Fe both in the porphyrin and the octahedral metal centres, (Fe3+, Fe2+)PMOF; (e) for the structure with a mixture of Al and Fe at the octahedral centres, (Al3+/Fe3+, Zn2+)PMOF, and (f) for the structure with a mixture of Al and Fe at the octahedral centres, and Fe in porphyrin metal centres, (Al3+/Fe3+, Fe2+)PMOF. Positive and negative DOS correspond to α and β spin components, respectively. | ||
The most stable spin state of the Fe2+ cation at the porphyrin centre is one of intermediate spin (IS), with magnetic moment μ(=2S) = 2 μB per Fe2+ cation (4 electrons in α-spin levels, 2 electrons in β-spin levels). The stability of the IS solution compared to the high-spin state with μ = 4 μB (0.3 eV per Fe atom above the groundstate) or low-spin state with μ = 0 (1.4 eV per Fe above the groundstate), was explained in ref. 34 in terms of the levels splitting from the porphyrin ligand field, and is in agreement with previous theoretical results for porphyrin molecules.54 The substitution of other late 3d transition metals (Co, Ni or Cu) at the porphyrin centre does not change the bandgap of the corresponding PMOFs to any significant extent, because their corresponding 3d levels fall below the highest occupied porphyrin levels.34
As shown in Fig. 2c, substituting the Al3+ cation by Fe3+ in the octahedral metal centre (forming (Fe3+, Zn2+)PMOF) does have a much stronger effect on the bandgap, which is now reduced to 1.5 eV. In the (D4h-distorted) octahedral FeO6 coordination, Fe3+ adopts a high-spin state (μ = 5 μB) where all the α-spin levels are occupied and all the β-spin levels are empty (the low-spin state is 0.9 eV per Fe atom higher in energy). The lowest-lying β-spin levels fall below the porphyrin lowest-unoccupied orbitals, thus narrowing the band gap with respect to the PMOFs with Al3+ in the octahedral centre. The observation that the conduction band edge is now contributed by levels from a reducible metal species (Fe3+ → Fe2+) is important from the point of view of potential photocatalytic applications, because it makes easier the creation of an excited state with electron – hole separation, via the promotion of an electron from the porphyrin ligand to the metal node. This ligand-to-metal charge transfer (LMCT) is an essential element for the realisation of photocatalysis in MOFs.55,56
Finally, we consider the simultaneous substitution of Fe into both the porphyrin and the octahedral metal centres, forming (Fe3+, Fe2+)PMOF as in the experimental work by Fateeva et al.35 The PMOF with this composition exhibits a reduced band gap of 1.3 eV, and Fig. 2d shows that this results from a combination of the two effects described above: Fe2+ in the porphyrin centre raises the valence band edge by introducing an occupied 3d level, while Fe3+ in the conduction band lowers the conduction band edge by introducing a low-lying empty 3d level.
We briefly discuss now the precision of our results. The DOS plots reported above are based on calculations with the screened hybrid functional HSE06, which we know to accurately predict bandgaps and d-level energies. We used a k-grid that leads to well-converged geometries and total energies. However, in order to obtain converged DOS plots, a denser k-grid is generally necessary. In standard DFT calculations (e.g. with the PBE function), the grid of k-points can be easily extended by doing non-selfconsistent calculations at extra k-points based on the charge density obtained with a less dense k-grid. In contrast, if hybrid DFT/Hartree–Fock calculations are performed to evaluate the exchange contributions, they should be fully self-consistent. Due to the relative large size of our system, using a much denser k-grid at HSE06 level becomes computationally too expensive. In order to assess the effect of the k-grid we also calculated both DOS and band structure plots using much denser grids at PBE+U level (Fig. S1 in ESI†). The qualitative picture emerging from those plots in terms of the energy position of the Fe 3d levels confirms our conclusions from the HSE06 DOS plots (Fe2+ in the porphyrin centre contributes the valence band edge, and Fe3+ in the conduction band contributes the conduction band edge), although bandgaps are narrower in PBE+U compared with HSE06. The band structure and DOS plots evaluated with a dense k-grid also show that the conduction band formed by Fe 3d levels exhibits some significant dispersion, leading to a continuous and smoother DOS at the conduction band edge, instead of the “peakier” DOS obtained there at the HSE06 level. This indicates a lower effective mass of conduction electrons in the Fe3+-doped system compared with the undoped material (the “intrinsic” conduction band edge is flat). The band structures also show a direct bandgap at the zone centre, indicating that optical transitions can occur without phonon mediation.
The results presented so far suggest that Fe substitution could be a promising approach to engineer the electronic band structure of PMOFs for photocatalytic applications. However, we still need to investigate whether the absolute positions of the band edges are favourable for the photocatalytic reactions of interest. In the following sections we will focus on the band alignments, and will also discuss how to further tune the band gaps.
Estimation of the vacuum potential
For a photocatalytic reaction to occur, the material needs to have the correct band gap, as well as the correct alignment of the bands with respect to the electrode potentials. Investigating this alignment requires knowledge of the absolute values of the electronic energy levels (i.e. with respect to vacuum). However, periodic DFT calculations provide only relative values with respect to an internal reference level, typically the average electron potential in the solid. To solve this problem, Butler et al.53 suggested a method to estimate the vacuum level in porous structures, based on calculating the potential at different positions within the pores. A good approximation to the vacuum level is obtained by calculating the potential along the pores, and choosing a point where the gradient is zero (no electric fields from atoms in the structure). Since the potential along the pore is periodic and continuous, there will always be at least two different points (one maximum and one minimum) of zero gradients. To calculate the vacuum potential level, we chose the zero-gradient point that is farthest apart from the framework atoms.The PMOF structure in our study has large pores perpendicular to the porphyrin planes, which have centres with coordinates (x, x, 0) and intersect other pores parallel to the porphyrin planes, which have centres with coordinates (0.5, 0.5, z) (all coordinates given here refer to fractional coordinates in the primitive cells used in the simulation – optimized coordinates for all structures are given in the ESI†). The calculated electrostatic potential energies along these pore centres are shown in Fig. 3. It is clear that the intersection point (0.5, 0.5, 0) between the two sets of pores is a zero-gradient point which is a good candidate to represent the vacuum region. We have checked that this point is the farthest from all the framework atoms (the nearest atom is at ∼6.2 Å). Other candidate points, like (0.5, 0.5, 0.5) (where the symmetrically equivalent pores along (x, 0.5, 0.5) and (0.5, y, 0.5) intersect), are closer to atoms in the structure (5.2 Å). We therefore take the electrostatic potential at position (0.5, 0.5, 0) of the primitive cell (which corresponds to positions (0, 0.5, 0) or (0.5, 0, 0) in the conventional cell) as our “vacuum” reference level, and subtract this values from the calculated energy levels to put them in an (approximately) absolute scale.
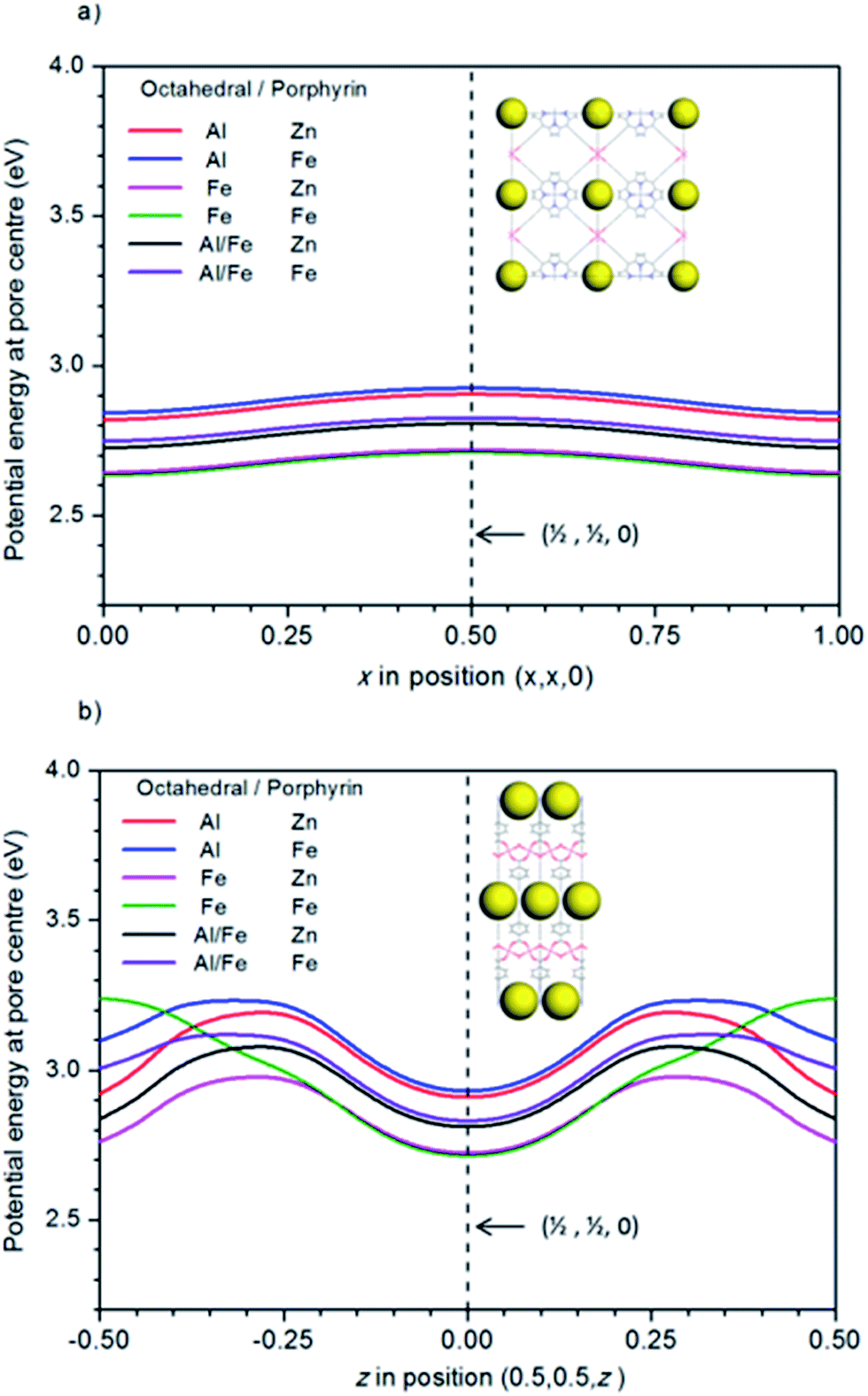 | ||
| Fig. 3 Electrostatic potential energy at the pore centre, as calculated with HSE06 functional along (a) pores perpendicular to the porphyrin planes (coordinates (x, x, 0) in the primitive cell), and (b) pores parallel to the porphyrin plane (coordinates (0.5, 0.5, z) in the primitive cell). The vertical dashed line in both figures corresponds to (0.5, 0.5, 0), which is the point farthest apart from all atoms in the structure and is used to estimate the vacuum potential in this work. It is represented as large yellow spheres within the PMOF structures in the inset figures (shown in the conventional cell for clearer visualisation). | ||
Band alignment with respect to electrode potentials
We will analyse now the suitability of the band positions for the photocatalysis of solar fuel synthesis from H2O or CO2. The splitting of water molecules generates hydrogen, while the reduction of carbon dioxide can give rise to various organic fuels, such as methane, methanol, etc. For water splitting to occur, the band gap must be narrow enough to permit the absorption of large part of the incoming electromagnetic radiation, but at the same time it has to be wide enough for the band edges to straddle the redox potentials for water photolysis.57–59 This means that the valence band edge should be below the energy of the oxygen evolution reaction (OER):| H2O ↔ 2H+(aq) + ½O2(g) + 2e−, | (1) |
| 2H+(aq) + 2e− ↔ H2(g). | (2) |
For CO2 conversion reactions, the condition for the semiconductor valence band is the same, but the relevant reduction reaction to compare the conduction band edge will differ.60,61 In Fig. 4a we show the positions, in the vacuum scale, of the energy levels corresponding to the oxidation and reduction reactions involved in water splitting and in carbon dioxide reduction to produce methane and methanol, at pH = 7 (potentials at finite pH are shifted from the values at pH = 0, by 2.30 × pH × kBT). We also show the positions of the band edges with respect to the vacuum level for the materials discussed so far.
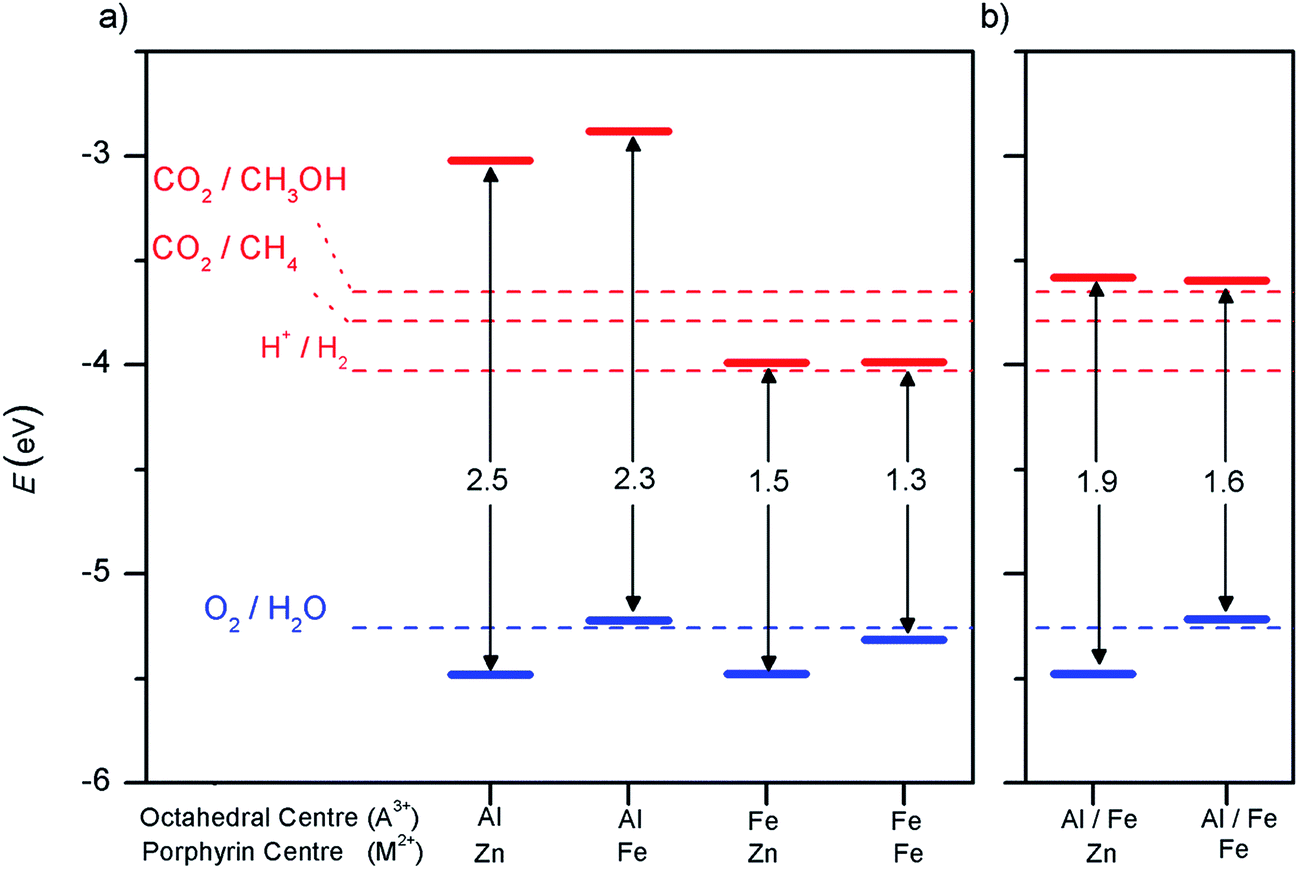 | ||
| Fig. 4 Band gaps and band edge positions with respect to the vacuum level, as calculated with the screened hybrid functional HSE06 for (a) PMOF with only one element (A = Al or Fe) in the octahedral sites, and (b) mixed PMOF with 50% Al and 50% Fe at the octahedral sites. The energy levels corresponding to redox potentials of water splitting and carbon dioxide reduction reactions producing methane and methanol at pH = 7 are also shown with dotted lines. | ||
The parent material (Al3+, Zn2+)PMOF can in principle catalyse both types of reactions (water splitting and carbon dioxide reduction), since the valence band edge is below the OER level and the conduction band edge is above the levels for the different reduction reactions. However, the conduction band edge is a bit too high for the water splitting reaction. The optimum band gap for water splitting photocatalyst is around 2 eV,62 which is the minimum gap that could be used, taking into account the need for additional potential beyond the 1.23 eV per electron required to drive the oxidation and reduction reactions. Although some authors quote a wider range for the ideal band gap, e.g. 1.6–2.4 eV,58 losses in real systems mean that values of at least 2 eV are typically required to drive the reaction.62
Substituting Zn by Fe in the porphyrin centres lowers the band gap, as discussed above, but unfortunately it does so by raising the absolute position of the valence band edge, which is not desirable because it pushes that edge above the OER level. On the other hand, substituting Al by Fe in the octahedral centres lowers the conduction band edge, resulting in band gaps (1.5 eV for (Fe3+, Zn2+)PMOF and 1.3 eV for (Fe3+, Fe2+)PMOF) that are probably too narrow for simultaneously driving the oxidation and reduction reactions in water splitting.
While the absolute band edge positions reported here are only approximate due to inherent uncertainties in our models, it is clear that the band edges are in the desired energy range in the absolute scale. In fact, deviations from the ideal band edge positions in a semiconductor can be corrected via the application of a weak bias voltage, which shifts both band edges with respect to the redox levels, as long as such deviations are small. Therefore, some of the systems discussed above could be already interesting for applications in photocatalysis of water splitting. However, the band gaps reported in the materials discussed so far tend to be either a bit too wide or too narrow for water splitting photocatalysis, therefore a route for further bandgap engineering could be very useful in the design of an optimal photocatalyst.
Properties of mixed PMOFs with both Al and Fe in octahedral centres
With the aim of finding a material with a band gap closer to the optimal value for water splitting, we have finally considered systems with mixed (50% Al3+, 50% Fe3+) occupancy of the octahedral metal centres. In standard semiconductors, the band gap of mixed compounds varies continuously, although generally not linearly, between the pure end-members.63,64 Therefore, any band gap in between that of the pure systems can be obtained in principle by choosing the appropriate composition, as long as the mixed composition is thermodynamically stable. This type of band gap engineering has recently been realised in some organic semiconductors, despite the stronger localization of their electronic states.65 It is therefore interesting to see whether we can obtain an intermediate value of the band gap from mixing the wide-gap and narrow-gap PMOF materials.The high computational cost of screened hybrid functional calculations means that we are restricted to small simulation cells. The simplest ordered mixed structure with composition (Al3+/Fe3+, M2+)PMOF can be created by substituting one of the two Al atoms in the primitive cell of the parent structure (Al3+, M2+)PMOF by an Fe atom. Since the two octahedral sites are equivalent, there is only one symmetrically independent configuration of Al and Fe distribution in the primitive cell. The next larger cell is the conventional orthorhombic cell which contains four octahedral sites. There are six ways of distributing two Fe and two Al atoms over these sites, but only three configurations, which are shown in Fig. 5, are found to be symmetrically independent (using the SOD program).66 In configurations 1 and 2, Fe and Al alternate along the b direction (perpendicular to the porphyrin planes), within the one-dimensional chains of corner-sharing octahedra. In contrast, configuration 3 contains one Al-only chain and one Fe-only chain. We find that configuration 1 has the lowest energy, but the energy of configuration 2 is very close (E2 − E1 = 20 meV), whereas the energy of configuration 3 is somewhat higher (E3 − E1 = 80 meV). These results indicate that the main factor controlling the stability of the configurations is the distribution of Fe/Al along the one-dimensional chains of corner-sharing octahedra. Since the energy differences are small, we cannot assume that the system will exhibit the long-range order of configuration 1. Instead, there will be an equilibrium distribution of cations, with some tendency to Fe/Al alternation in the b direction. Mapping the DFT energies into a simple one-dimensional Ising model, we can estimate that the equilibrium probability of Fe–Al pairs in nearest-neighbour positions at room temperature is p = 73% (see ESI†). Therefore, configurations 1 and 2 (where p = 100%) are better representations of the cation distribution than configuration 3 (where p = 0). We therefore use configuration 1 for the electronic structure and band alignment calculations of the mixed systems, because that configuration has the additional computational advantage that it can be represented within the primitive cell (test calculations show that very similar results are obtained using configuration 2). It is also reassuring that we obtain a very low formation energy (less than 1 kJ per mol of octahedral metal atom) for the mixed system from the corresponding amounts of the pure compounds, which suggests that mixed structures should be stable towards phase separation (they would be stabilised with respect to the pure compounds by the configurational entropy). A more sophisticated and accurate treatment of the cation distribution would require larger supercells, but that becomes too computationally expensive at HSE06 level. Representing the mixed system using alternating Fe/Al ions along the chains of corner-sharing octahedra, even if not a perfect solution, is a good first approximation to explore the potential for compositional band gap engineering in PMOFs.
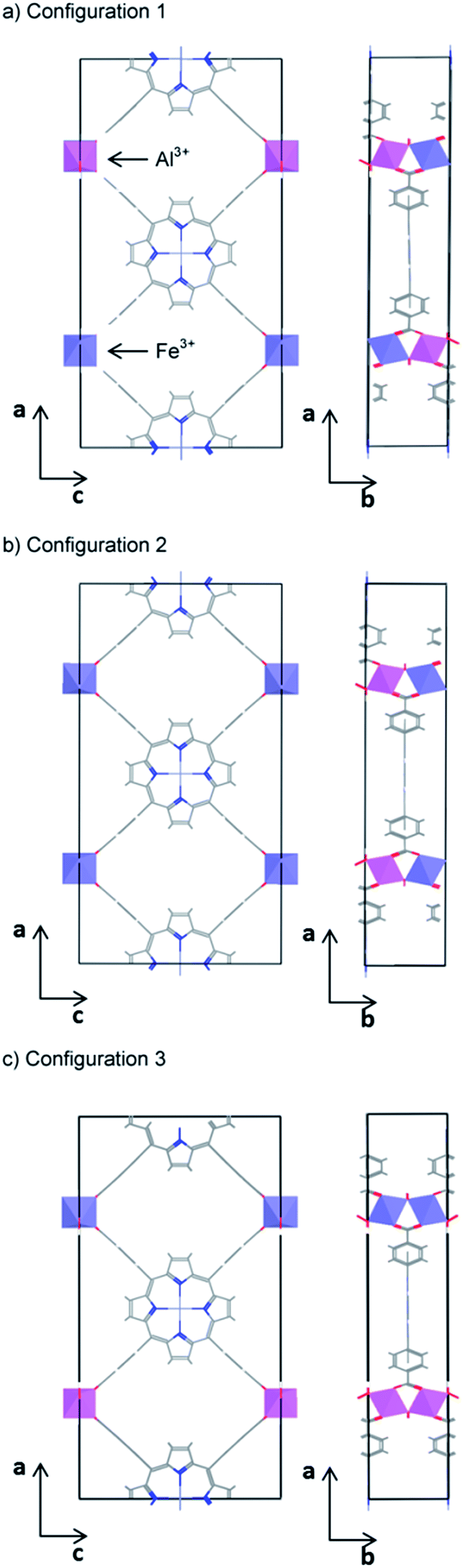 | ||
| Fig. 5 The three symmetrically independent configurations of Fe and Al on the octahedral sites of the PMOF conventional (orthorhombic) cell. | ||
We now discussed the electronic structure of the mixed PMOFs. Fig. 2e shows that, indeed, the obtained band gap (1.9 eV) for (Al3+/Fe3+, Zn2+)PMOF is intermediate between that of (Al3+, Zn2+)PMOF (2.5 eV) and that of (Fe3+, Zn2+)PMOF (1.5 eV). The fact that it is not exactly the average of the values for the two endmembers, but a bit below, indicates a convex “bowing” of the band gap vs. composition curve, which is also common for standard semiconductor alloys.63,64 Comparing Fig. 2e with Fig. 2c we see that in the mixed compound the Fe 3d contributions to the DOS are more localized, and closer to the porphyrin lowest-unoccupied levels, than in the pure Fe compound, leading to a widening of the gap.
Fig. 2f shows the result for the (Al3+/Fe3+, Fe2+)PMOF. In this case, as expected from our previous analysis, the valence band edge is contributed by filled Fe 3d levels from Fe2+ in the porphyrin, while the conduction band has moved to the same intermediate position as in (Al3+/Fe3+, Zn2+)PMOF.
The alignment of the band edges with the redox potentials for photocatalysis is shown in Fig. 4b. In the case of the (Al3+/Fe3+, Zn2+)PMOF, the band edges are in almost ideal position for water splitting, while the bandgap is perfect for absorption of visible light, which is a very encouraging result. In the (Al3+/Fe3+, Fe2+)PMOF system the valence band edge is a bit too high in relation with the O2/H2O redox potential. These results suggest that in order to create an intrinsic PMOF photocatalyst, it might be necessary to limit the Fe doping to the octahedral site, while preventing the Fe doping of the porphyrin site. Although experimentally Fe doping of the PMOF structure has been done at both sites simultaneously,35 we do not anticipate that selective doping of the octahedral site would be too difficult an experimental challenge: several studies have demonstrated the viability of post-synthetic modification of the metal at the porphyrin centre in porphyrin-based MOFs.67–69
We therefore have identified a PMOF composition exhibiting the ideal band structure and alignment required for water splitting photocatalysis. Furthermore, the nature of its valence and conduction band edges are also consistent with the ligand-to-metal charge transfer (LMCT): photons would be absorbed by the porphyrin unit, and an electron would be excited to the reducible Fe3+ centres, allowing for electron–hole separation and potentially long recombination times. At the same time, the porous structure allows diffusion and access of the reactant molecules to the different sites, decreasing the need for high charge mobility towards the external surface, as expected for standard semiconductors. It is clear that this system shows promise as potential photocatalyst for water splitting and merits further investigation.
Conclusions
We have identified the effect that Fe substitutions have in the electronic properties of porphyrin-based metal organic frameworks. While Fe at the porphyrin metal centre has the effect of raising slightly the position of the valence band edge by introducing occupied Fe 3d levels above the highest occupied porphyrin levels, Fe at the octahedral metal node has the ability to lower significantly the position of the conduction band edge. This is due to the introduction of empty Fe 3d levels below the lowest unoccupied porphyrin levels. Fe is therefore an ideal dopant to engineer the band structure of these MOFs.For their potential applications in photocatalysis of water splitting, the original structure with Al in the octahedral sites and Zn in the porphyrin centres has a band gap that is slightly too wide to take advantage of visible-light solar radiation, and not enough spatial separation of the highest-occupied and lowest unoccupied crystal orbitals. On the other hand, the structure with Fe in the octahedral sites has bandgaps that are too narrow for water splitting photocatalysis, and in particular a too low conduction band edge to be able to drive the reduction reactions. We have demonstrated that a promising approach to improve the electronic structure is by partial substitution of Al by Fe at the octahedral sites, while keeping Zn at the porphyrin centres. Such structures seem to be stable with respect to decomposition into the pure phases, have a nearly ideal bandgap (1.9 eV) and correct band edge positions for water splitting photocatalysis, and have well-separated electron and hole localization regions, which could promote longer exciton recombination times. Our study shows that with some careful composition engineering, porphyrin-based MOFs could exhibit intrinsic photocatalytic activity in solar fuel synthesis reactions.
Acknowledgements
We are grateful to the Royal Society for an International Exchange Scheme grant (IE130840). A. A.'s studentship is funded by the EPSRC. Via our membership of the UK's HPC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202), this work made use of the facilities of ARCHER, the UK's national high-performance computing services, which are funded by the Office of Science and Technology through EPSRC's High End Computing Programme. Use of the computer facility ALHAMBRA (Granada, Spain) is acknowledged. This work was also supported by the European Research Council through an ERC Starting Grant (ERC2011-StG-279520-RASPA), by the MINECO (CTQ2013-48396-P) and by the Andalucía Region (FQM-1851).References
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- G. Sneddon, A. Greenaway and H. H. P. Yiu, Adv. Energy Mater., 2014, 4, 1301873 CrossRef.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- J. C. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765–778 RSC.
- C. G. Silva, A. Corma and H. Garcia, J. Mater. Chem., 2010, 20, 3141–3156 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- M. A. Nasalevich, M. van der Veen, F. Kapteijn and J. Gascon, CrystEngComm, 2014, 16, 4919–4926 RSC.
- G. Férey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217–225 CrossRef PubMed.
- A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780–4795 RSC.
- L. M. Rodriguez-Albelo, A. R. Ruiz-Salvador, S. Sampieri, D. W. Lewis, A. Gomez, B. Nohra, P. Mialane, J. Marrot, F. Secheresse, C. Mellot-Draznieks, R. Ngo Biboum, B. Keita, L. Nadjo and A. Dolbecq, J. Am. Chem. Soc., 2009, 131, 16078–16087 CrossRef PubMed.
- H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- S. Bordiga, C. Lamberti, G. Ricchiardi, L. Regli, F. Bonino, A. Damin, K. P. Lillerud, M. Bjorgen and A. Zecchina, Chem. Commun., 2004, 2300–2301 RSC.
- M. Alvaro, E. Carbonell, B. Ferrer, F. X. Llabrés i Xamena and H. Garcia, Chem.–Eur. J., 2007, 13, 5106–5112 CrossRef CAS PubMed.
- M. Fuentes-Cabrera, D. M. Nicholson, B. G. Sumpter and M. Widom, J. Chem. Phys., 2005, 123, 124713 CrossRef PubMed.
- J. A. Botas, G. Calleja, M. Sánchez-Sánchez and M. G. Orcajo, Langmuir, 2010, 26, 5300–5303 CrossRef CAS PubMed.
- C. K. Brozek and M. Dincă, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed.
- J. Gascon, M. D. Hernandez-Alonso, A. R. Almeida, G. P. M. van Klink, F. Kapteijn and G. Mul, ChemSusChem, 2008, 1, 981–983 CrossRef CAS PubMed.
- R. Grau-Crespo, A. Aziz, A. W. Collins, R. Crespo-Otero, N. C. Hernández, L. M. Rodriguez-Albelo, A. R. Ruiz-Salvador, S. Calero and S. Hamad, Angew. Chem., Int. Ed., 2016, 55, 16012–16016 CrossRef CAS PubMed.
- S. L. Gould, G. Kodis, R. E. Palacios, L. de la Garza, A. Brune, D. Gust, T. A. Moore and A. L. Moore, J. Phys. Chem. B, 2004, 108, 10566–10580 CrossRef CAS.
- G. McLendon and D. S. Miller, J. Chem. Soc., Chem. Commun., 1980, 533–534 RSC.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, F. E. CurchodBasile, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, K. NazeeruddinMd and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- M. K. Panda, K. Ladomenou and A. G. Coutsolelos, Coord. Chem. Rev., 2012, 256, 2601–2627 CrossRef CAS.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed.
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834–15842 CrossRef CAS PubMed.
- I. J. Kang, N. A. Khan, E. Haque and S. H. Jhung, Chem.–Eur. J., 2011, 17, 6437–6442 CrossRef CAS PubMed.
- J. B. Allison and R. S. Becker, J. Chem. Phys., 1960, 32, 1410–1417 CrossRef CAS.
- Y. Liu, Y. Yang, Q. Sun, Z. Wang, B. Huang, Y. Dai, X. Qin and X. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 7654–7658 CAS.
- S. Hamad, N. C. Hernandez, A. Aziz, A. R. Ruiz-Salvador, S. Calero and R. Grau-Crespo, J. Mater. Chem. A, 2015, 3, 23458–23465 CAS.
- A. Fateeva, J. Clarisse, G. Pilet, J.-M. Grenèche, F. Nouar, B. K. Abeykoon, F. Guegan, C. Goutaudier, D. Luneau, J. E. Warren, M. J. Rosseinsky and T. Devic, Cryst. Growth Des., 2015, 15, 1819–1826 CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- R. Grau-Crespo, A. Y. Al-Baitai, I. Saadoune and N. H. De Leeuw, J. Phys.: Condens. Matter, 2010, 22, 255401 CrossRef PubMed.
- R. Grau-Crespo, F. Corà, A. A. Sokol, N. H. de Leeuw and C. R. A. Catlow, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 035116 CrossRef.
- G. Rollmann, A. Rohrbach, P. Entel and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 165107 CrossRef.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- T. M. Henderson, J. Paier and G. E. Scuseria, Phys. Status Solidi B, 2011, 248, 767–774 CrossRef CAS.
- C. H. Hendon, J. Bonnefoy, E. A. Quadrelli, J. Canivet, M. B. Chambers, G. Rousse, A. Walsh, M. Fontecave and C. Mellot-Draznieks, Chem.–Eur. J., 2016, 3713–3718 CrossRef CAS PubMed.
- Z.-G. Gu, L. Heinke, C. Wöll, T. Neumann, W. Wenzel, Q. Li, K. Fink, O. D. Gordan and D. R. Zahn, Appl. Phys. Lett., 2015, 107, 183301 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- K. T. Butler, C. H. Hendon and A. Walsh, J. Am. Chem. Soc., 2014, 136, 2703–2706 CrossRef CAS PubMed.
- S. Bhandary, B. Brena, P. M. Panchmatia, I. Brumboiu, M. Bernien, C. Weis, B. Krumme, C. Etz, W. Kuch, H. Wende, O. Eriksson and B. Sanyal, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 024401 CrossRef.
- Y. H. Fu, D. R. Sun, Y. J. Chen, R. K. Huang, Z. X. Ding, X. Z. Fu and Z. H. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen and F. Kapteijn, Sci. Rep., 2016, 6, 23676 CrossRef CAS PubMed.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- A. J. Cowan and J. R. Durrant, Chem. Soc. Rev., 2013, 42, 2281–2293 RSC.
- S. Trasatti, Pure Appl. Chem., 1986, 58, 955–966 CAS.
- J. R. Bolton, S. J. Strickler and J. S. Connolly, Nature, 1985, 316, 495 CrossRef CAS.
- R. Hill, J. Phys. C: Solid State Phys., 1974, 7, 521 CrossRef CAS.
- F. Capasso, Science, 1987, 235, 172–177 CAS.
- M. Schwarze, W. Tress, B. Beyer, F. Gao, R. Scholz, C. Poelking, K. Ortstein, A. A. Günther, D. Kasemann and D. Andrienko, Science, 2016, 352, 1446–1449 CrossRef CAS PubMed.
- R. Grau-Crespo, S. Hamad, C. R. A. Catlow and N. H. de Leeuw, J. Phys.: Condens. Matter, 2007, 19, 256201 CrossRef.
- J. A. Johnson, J. Luo, X. Zhang, Y.-S. Chen, M. D. Morton, E. Echeverría, F. E. Torres and J. Zhang, ACS Catal., 2015, 5, 5283–5291 CrossRef CAS.
- J. A. Johnson, X. Zhang, T. C. Reeson, Y.-S. Chen and J. Zhang, J. Am. Chem. Soc., 2014, 136, 15881–15884 CrossRef CAS PubMed.
- C.-W. Kung, T.-H. Chang, L.-Y. Chou, J. T. Hupp, O. K. Farha and K.-C. Ho, Chem. Commun., 2015, 51, 2414–2417 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ta01278k |
| This journal is © The Royal Society of Chemistry 2017 |