
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntagonistic effects of selenium against necroptosis injury via adiponectin-necrotic pathway induced by cadmium in heart of chicken†
Jingzeng Cai‡
a,
Yuan Zhang‡a,
Jie Yanga,
Qi Liua,
Rihong Zhaoa,
Sattar Hamida,
Hong Wanga,
Shiwen Xua and
Ziwei Zhang *ab
*ab
aCollege of Veterinary Medicine, Northeast Agricultural University, Harbin 150030, P. R. China
bKey Laboratory of the Provincial Education Department of Heilongjiang for Common Animal Disease Prevention and Treatment, College of Veterinary Medicine, Northeast Agricultural University, Harbin, 150030, P. R. China. E-mail: zhangziwei@neau.edu.cn; Tel: +86 45155190407
First published on 15th September 2017
Abstract
Cadmium (Cd) is one of the most toxic heavy metals having a destructive impact on various organ systems. For example, it induces oxidative stress in heart of chicken. Selenium (Se), in the form of selenoproteins, is known to protect tissues and organs against such heavy metal induced-damage. However, the precise cellular mechanism of the ameliorative role of Se in preventing Cd-induced toxicity in cardiac tissues remains unclear. The aim of this study is to investigate the role of Se in preventing Cd-induced toxicity in the chicken heart and assess the possible cytoprotective mechanism of Se. A total of 128 chickens were divided into four trial groups fed by a standard diet comprising Se, Se+, Cd+, and Se+ + Cd+ for 90 days. qPCR and western blotting were performed to observe the mRNA and protein expression of genes. Correlation analysis (PPI) and heat maps were used for further analysis. The results revealed that the exposure to Cd significantly increased (p < 0.05) the mRNA and protein levels of c-Jun N-terminal kinase (JNK), phosphorylated c-Jun N-terminal kinase (P-JNK), tumor necrosis factor α (TNFα), protein kinase superfamily protein 1 (RIPK1) and mixed lineage kinase domain like pseudokinase (MLKL) in the chicken. On the contrary, the mRNA and protein expression of peroxisome proliferator activated receptor alpha (PPARα), adiponectin (ADIPOQ), adiponectin receptor 1 (AdipoR1), adiponectin receptor 2 (AdipoR2), and adenosine monophosphate-activated protein kinase alpha 1 (AMPKα1) were significantly decreased in the Cd+ group. Furthermore, the mRNA and protein expression levels of PPARα, adiponectin, adipoR1, adipoR2 and AMPK1 were increased (p < 0.05) significantly in the Se+ group. However, the expression of JNK, TNFα and RIPK1 was significantly decreased as compared to that in the group provided with a normal standard diet. Notably, no significant increase in JNK, TNFα, RIPK1 and MLKL expression levels were observed in the chickens provided with a diet comprising Se+ + Cd+, whereas the expression levels of PPARα, adiponectin, adipoR1, adipoR2 and AMPK1 were increased significantly as compared to those in the Cd+ group. These results evidently indicated that Cd could induce severe myocardial damage by activating the necroptosis pathway, whereas Se could play an excellent potential role in preventing Cd-induced myocardial damage through activating adiponectin pathway.
Introduction
Ubiquitously present in natural environments, Cd is known to be a common environmental pollutant that has been associated with the industrial and agricultural production. As a serious toxic heavy metal, Cd is reported as one of the potential threats to human health chiefly from consumption of Cd contaminated food and water.1 Through the production of reactive oxygen species (ROS), Cd could affect various metabolic processes, such as energy metabolism, protein synthesis as well as membrane transport and antioxidant defense mechanisms. Furthermore, it could affect DNA by interference with the genetic control and the repair mechanism.2 Previous studies have indicated that long-term uptake of Cd could cause a systemic damage, at varying degrees, to the cardiovascular tissues, liver, kidney, lungs, testes, placenta bone, as well as the reproductive and immune systems.3–6 In addition, Cd-induced disruption of the blood-testis barrier, testicular damage, germ cell impairment, cardiomyopathy, renal failure, hypertension, arteriosclerotic heart disease6 and immunosuppression in organs including the heart were reported in mice.7,8 Furthermore, a significant decrease of cell viability and Cd cytotoxicity to chicken hepatocytes were observed in the cells treated with Cd.9 The results obtained in our previous study inferred that the extensive damage of hens' ovary tissue following Cd supplementation could be connected to the toxicity of Cd.10 Furthermore, we also proved that the increase of DNA methylation in the hens' liver and kidney was observed following Cd exposure due to the carcinogenic effect of cadmium.11 These effects occur because Cd gets deposited in the heart muscle and produces cardiotoxicity at concentrations as low as 0.1 μM.12 There are numerous risk factors associated with various types of heart diseases, which are the leading cause of deaths worldwide. It has been reported earlier that Cd has pronounced vasculotropic properties, causing the morphological changes of cardiomyocytes on the small blood vessels of the heart and the myocardial interstitial fibrillar collagen network in rats.13 It is proved that on applying a low dose, Cd causes both the inflammation and the cell proliferation but the proliferative changes occur independent of inflammation.14 The result showed that grass carp AIF-1, which is an inflammation responsive protein, could be involved in Cd-induced stress.15 In addition, Cd is known to induce inflammatory cytokines, such as TNF, IL-1, IL-6, interferon and reactive oxygen species (ROS) in the experimental animals.16,17 Notably, the exposure to Cd induces apoptosis in LLC-PK cells and activates signaling pathways that cause cell apoptosis and/or necrosis.18Se, which is an essential micronutrient for the human body, protects the tissues and organs against oxidative damage and improves the immune system in the body. It is an antioxidant that helps in removal of oxygen free-radicals in the cell via various active antioxidant enzymes, including thioredoxin reductase and glutathione peroxidase (GPx).1 Some researchers have reported that the protective role of Se could be associated with the recovery through inhibition activities of glutathione peroxidase (GSH-Px) and thioredoxin reductase (TrxR), scavenging the free radical-mediated lipid peroxidation and regeneration of the glutathione.19 A number of clinical trials have proved that Se is well established as an effective antioxidant with anti-inflammatory, chemo-preventive and antiviral properties.20 In addition, it plays an extensive role in protecting the immune and reproductive system as well as in preventing diabetic, liver and cardiovascular diseases.21 Numerous experiments and clinical studies have confirmed that Se is considered to be the most effective substance for the protection from Cd-induced damage. In the experiments with MMTV-infected female mice, the levels of the Se-antagonistic elements, such as As (arsenic), Pb (lead) and Cd were reduced due to the protecting effects of Se.22 The disruption in the balance of elements such as Cu, Mn, Fe, and Zn, in the body, could be related to the presence of Se in the antioxidant system.23,24 Further, it has been reported that Se reduces the Cd-induced oxidative stress and the histopathological damage in the rat heart.25 We have previously demonstrated that Se could attenuate Cd-induced brain damage in chickens by regulating the iNOS-NO system.26 In addition, the results obtained in our study suggested that Se has an ability to inhibit mitochondrial apoptotic pathway in the oxidative stress mediated kidney dysfunction caused by Cd.1 Liu et al. reported that the overloading of cytosolic calcium could inhibit Cd-induced autophagy that leads to cell death in primary rat proximal tubular cells.27 Furthermore, our recent study has demonstrated that the antagonistic effect of Se could protect endoplasmic reticulum against Cd toxicity via increasing the expressions of selenoprotein K, selenoprotein N, selenoprotein S, and selenoprotein T in the cultured chicken lymphocytes.28 Concomitantly, our previous research results inferred the protective effect of Se against Cd toxicity through changing the level of transcription of HSPs (heat shock proteins) in the chicken splenic lymphocytes.29 However, the information on the exact ameliorative mechanism of Se in preventing Cd-induced cardiotoxicity in the poultry remains unclear.
Secreted from the adipose tissue, adiponectin is a kind of specific collagen that activates multiple signaling pathways. It modulates anti-diabetic, anti-atherogenic and anti-inflammatory functions, and reduces the plasma free fatty acids. Adiponectin carries out its functions through the peroxisome proliferator-activated receptor (PPAR) and adenosine monophosphate-activated protein kinase (AMPK) signaling pathways.30 Comprehensive analysis of the mechanism of adiponectin actions could elucidate its role in a variety of diseases, including diabetes and cardiovascular diseases, where the dysfunction of adiponectin could lead to the pathogenesis of the diseases. Moreover, adiponectin inhibits the expression of TNF-α in adipocytes, whereas both TNF-α and IL-6 inhibit the production of adiponectin.31 Myocardial injury was reduced via adiponectin replacement therapy in mice with viral myocarditis.32 BNP (B-type natriuretic peptide) and adiponectin are two indicators of myocardial damage, which have possible additive effects in cardiovascular homeostasis regulation.33 However, there are few reports on the effect of adiponectin on necrotic pathways. Thus, we intend to conduct studies on this topic, particularly in Cd-induced cardiotoxicity in chickens.
In this study, we try to determine whether the effects of the interaction between Cd+ and Se+ could attenuate the Cd-intoxication in the chicken heart. In the present study, we attempt to provide new insights into the efficiency of Se in enhancing the resistance to Cd-induced cytotoxicity in the chicken heart.
Materials and methods
Poultry and experimental design
All experiments were performed in compliance with relevant laws and guidelines of the Institutional Animal Care, Harbin, China. Animal handling and experimental procedures were approved by the Institutional Animal Care and Use Committee of Northeast Agricultural University. A total of 128 chickens which were kept for 90 days in our experiments, and were randomly divided into four groups (n = 32 per group). Group I (control) was fed with a basic diet containing 0.2 mg per kg Se. Group II (Se+ group) was fed with the basic diet supplemented with 2 mg per kg−1 of Na2SeO3. Group III (Cd+ group) was fed with the basic diet supplemented with 150 mg per kg−1 of CdCl2. Group IV (Cd+ + Se+ group) was fed with the basic diet supplemented with 150 mg per kg−1 of CdCl2 and 2 mg per kg−1 of Na2SeO3. The concentration of Se and Cd in this study was based on our previous experiments.34 Over the entire experimental period, food and water were allowed ad libitum. Feeding, housing and handling of the experimental chickens were conducted according to the established commercial managemental practices. Subsequent to euthanasia with sodium pentobarbital, the heart tissue samples were collected and rinsed with ice-cold sterile deionized water under sterilized conditions, frozen immediately in liquid nitrogen, and stored at −80 °C until further use.Protein–protein interactions (PPI) analysis
The PPI analysis of differentially expressed genes was based on the STRING database (https://string-db.org/) and GeneMANIA obtained from the public databases (http://genemania.org/) of known and predicted interactions. For the species existing in the database, we constructed the networks by extracting the target gene lists, and then the networks were built according to the known interactions of the selected reference species.To obtain their finer functions, we suggest the use of function-specific interaction sub-networks by integrating the functional knowledge of Gene Ontology and the protein–protein interaction network.
RNA extraction
Total RNA was isolated from the heart tissue using Trizol reagent according to the manufacturer's instructions (Invitrogen, Shanghai, China). The dried RNA pellets were re-suspended in 50 μL of diethylpyrocarbonate treated water. The concentration and purity of the total RNA were determined using a spectrophotometer. cDNA was synthesized from 5 μg of the total RNA using oligo dT primers and superscript II reverse transcriptase according to the manufacturer's instructions (Promega, Beijing, China). cDNA was diluted at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 with sterile water and stored at −80 °C.
:5 with sterile water and stored at −80 °C.
Quantitative real-time PCR (qPCR) analysis of genes mRNA levels
Primer Premier software (PREMIER Biosoft International, USA) was used to design specific primers for all of the genes based on the known chicken sequences, including P-JNK, TNFα, RIPK1, MLKL, PPARα, Adiponectin, AdipoR1, AdipoR2 and AMPK1, β-actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Table 1). General PCR was first performed to confirm the specificity of the primers. Quantitative real-time PCR (qPCR) was performed with an ABI PRISM 7500 Detection System (Applied Biosystems, Foster City, CA). The reactions were performed using a 20 μL reaction mixture containing 10 μL of the 2 × SYBR Green I PCR Master Mix (Ta Ka Ra, China), 2 μL cDNA, 0.4 μL of each primer (10 μM), 0.4 μL of 50 × ROX reference Dye II and 6.8 μL of PCR grade water. The PCR procedure for determining JNK, TNFα, RIPK1, MLKL, PPARα, adiponectin, adipoR1, adipoR2, AMPKα1, β-actin and GANPH consisted of the following steps: 95 °C for 30 s followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s and 60 °C for 30 s. For each PCR reaction, Dissociation Curve 1.0 software (Applied Biosystems) was used to analyze dissociation curves in order to detect and eliminate the possible primer-dimers and non-specific amplifications. The relative abundance of each mRNA was calculated according to the Pfaffl method.| Target gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| MAPK8 | TGAACGAATGTCCTATCTCC | CCTCTGGTGCTCTGTAGT |
| JNK | GGCGTTGTCATCTCCATT | ATTCTCCTCTGTCTCATCATC |
| TNFα | GAGGAGACAGTAGGAATCAAT | CGGTCATAGAACAGCACTA |
| RIPK1 | AGATGCTGTAGGAGTGGAT | CGCTGGTTGAAGATGGAT |
| MLKL | CTGCTTCTCCATCGTCAT | CAACTGGCTCCTGGTATC |
| PPARα | TGGAGGAGGATGATATAGGTA | ATGGATGAAGGACTGGATG |
| ADIPOQ | GAGCCAGGTCTACAAGGT | TGCTGTCGTAGTGGTTCT |
| AdipoR1 | AGGAAGAGGAGGAAGTTGT | GAGAGGAAGCCACGAAGTA |
| AdipoR2 | GAGGATAGTAACCACAATGATG | GAGAAGGAGAGGCAGAGTA |
| AMPKα1 | TGGAGGAGGATGATATAGGTA | ATGGATGAAGGACTGGATG |
| β-Actin | CCGCTCTATGAAGGCTACGC | CTCTCGGCTGTGGTGGTGAA |
| GAPDH | AGAACATCATCCCAGCG | AGCCTTCACTACCCTCTTG |
Western blot analysis of protein levels
For total protein extraction, protein lysis solutions were subjected to 15% SDS-polyacrylamide gel electrophoresis under reducing conditions. Then the separated proteins were transferred to nitrocellulose membrane for 2 h at 200 mA in a tank transfer apparatus containing Tris–glycine buffer and 20% methanol. The membrane was blocked with 5% skim milk for 24 h and incubated overnight with diluted primary antibodies against PPARα (1:500, polyclonal antibody produced by our lab), P-JNK (1:500, polyclonal antibody produced by our lab), JNK (1:500, Santa Cruz Biotechnology, USA) and AMPKα1 (1:1000, Santa Cruz Biotechnology, USA) followed by a horseradish peroxidase (HRP)-conjugated secondary antibody against rabbit (PPARα, P-JNK, JNK and AMPKα1) and IgG (1:3000, Santa Cruz Biotechnology, USA). To verify equal loading of the samples, the membrane was incubated with a monoclonal β-actin antibody (1:1500, Santa Cruz Biotechnology, USA), followed by a HRP-conjugated goat anti-mouse IgG (1:3000) secondary antibody. The density of each band was normalized to its respective loading control (β-actin).35 The signal was detected using X-ray films (TransGen Biotech Co., Beijing, China). The optical density (OD) of each band was determined using an Image VCD gel imaging system, and the relative abundance of PPARα, P-JNK, JNK and AMPKα1 proteins was calculated and presented as the ratios of OD of each of these proteins to that of β-actin.
Statistical analysis
Statistical analysis of the entire data was performed using GraphPad Software Prism 5 (version 5.01, GraphPad Software, Inc., La Jolla, USA). When a significant value (p < 0.05) was obtained by one-way analysis of variance, further analysis was performed. All data showed a normal distribution and passed the equal variance testing. Differences between means were assessed by Tukey's honestly significant difference test for post hoc multiple comparisons. In addition, correlation analysis was used to define the relationships between individual variations using the same software.Results
Prediction of protein–protein interaction (PPI) networks
Fig. 1 and 2 display the network of proteins that differ significantly among adiponectin-necrotic pathway-related genes in our present study. The statistical analysis of a total of 9 PPIs with a combined score of >0.4 was obtained using STRING database for automatically detecting enriched functions or processes in the PPI network. The different color lines between proteins show different connections in the PPI networks. The obtained results showed that PPI enrichment p-value is 2.33 × 10−7, and the average of the local clustering coefficient is 0.611. The STRING and GeneMANIA databases automatically highlighted the corresponding nodes between adiponectin-related genes and necroptosis-related genes in the network, where the closer interaction among proteins was observed to occur. It showed that the closer interaction of MAPK8/JNK, TNFα, RIPK1, MLKL, PPARα, adiponectin, adipoR1, adipoR2, and AMPKα1 was specific and meaningful.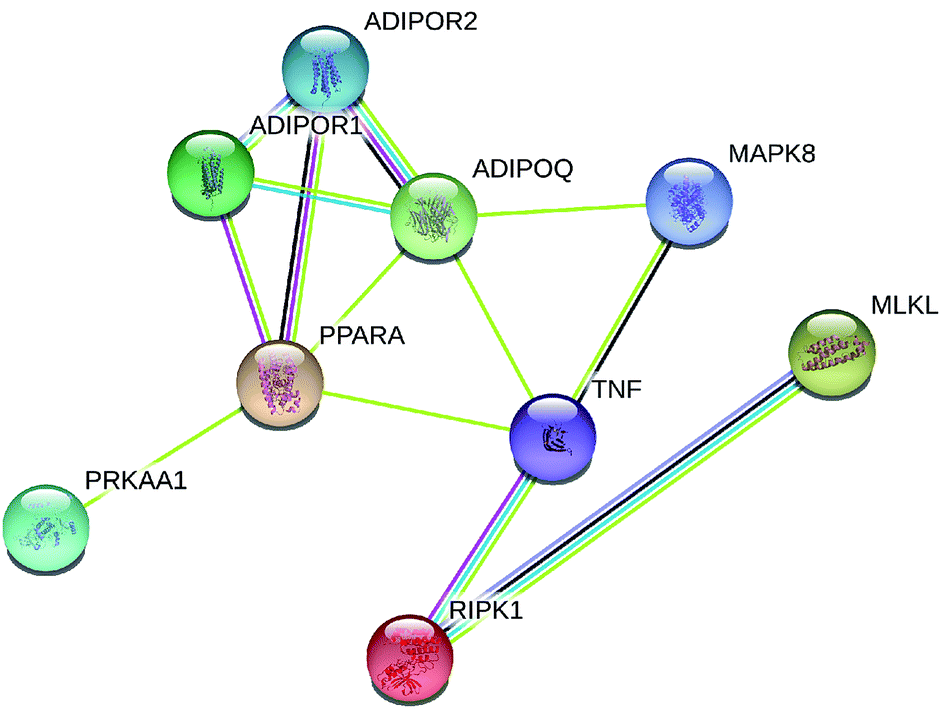 | ||
| Fig. 1 Protein network of proteins regulated among adiponectin-necrotic pathway-related genes in the STRING database. A yellow line means text mining, a grey line means homology, light blue means from the database, a dark blue line means co-occurrence, a pink line means from the experiments and a black line means co-expression. | ||
 | ||
| Fig. 2 Protein network of proteins regulated among adiponectin-necrotic pathway-related genes in the GeneMANIA databases. Small circle of 10 genes and different circle colors represent different pathways. | ||
Effect of treatments on expression of adiponectin-necrotic-associated protein in heart of chicken
Western blot analysis was performed to determine the protein expression of adiponectin and necrotic genes in the chicken cardiomyocytes. The results (Fig. 3 and 4) of the western blot showed that the ratio of P-JNK and JNK was 239% in the chicken heart. However, the expression of AMPK and PPARα was respectively decreased by 12% and 60% in the Cd+ group compared with those in the normal group (p < 0.05). The results (Fig. 3 and 4) also revealed the protein expression of AMPK and PPARα related adiponectin increased by 18% and 710% in the Se+ group, respectively. Moreover, the protein levels of the ratio of P-JNK and JNK increased by 94% in necroptosis pathway when compared to the control group (p < 0.05).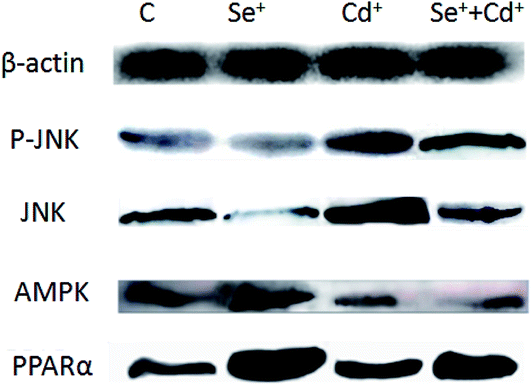 | ||
| Fig. 3 The protein levels of PPARα, P-JNK, JNK and AMPKα1. C indicates the control groups; Se+ indicates the Se groups; Cd+ indicates the Cd groups; Se+ +Cd+ indicates Se + Cd co-treatment. β-actin was selected as the reference. | ||
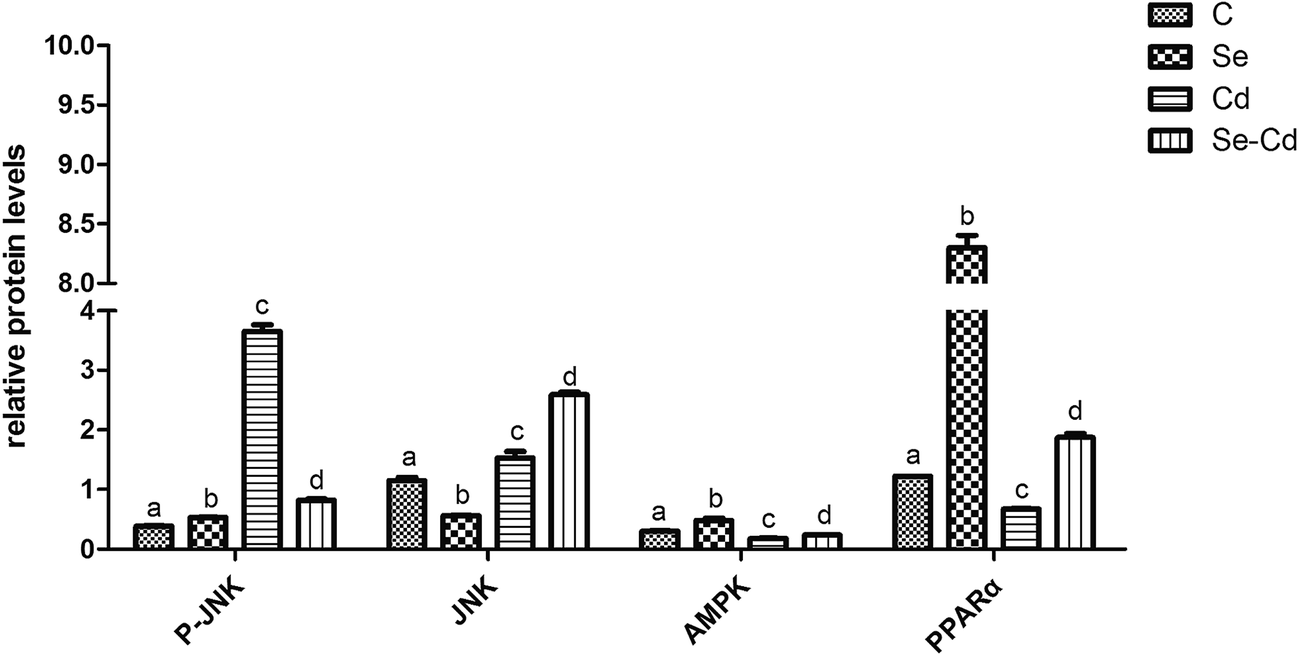 | ||
| Fig. 4 The protein levels verification by western blot. The expression of P-JNK, JNK, AMPK and PPARα affected by Se, Cd, and Se + Cd co-treatment. GADPH and β-actin was selected as the internal reference. The data are expressed as the means ± SD. The mean value between groups without a shared common letter are significantly different (p < 0.05), n = 3, using one-way ANOVA. | ||
The results showed that the ratio of P-JNK and JNK in the heart was 31% in the Se+ + Cd+ group (Fig. 3 and 4). However, there was no significant difference in the protein levels of AMPK between the Se+ group and the Se+ + Cd+ group (p < 0.05). Interestingly, in the Se+ + Cd+ co-treatment group, the content of PPARα increased significantly with the proportion of about 120% in the chicken heart compared with that in the Cd-treated group (Fig. 3 and 4).
Changes of adiponectin-necrotic-associated mRNA in heart of chicken
In order to further investigate the effects of Se on the toxicity after the chicken heart was exposed to Cd, we also examined the RNA expression of JNK, TNFα, RIPK1, MLKL, PPARα, adiponectin, adipoR1, adipoR2 and AMPKα1 for three groups: the Se+ group, the Cd+ group, and the Se+ + Cd+ co-treatment group. Further, we determined whether there were changes in adiponectin- and necrosis-associated mRNA in the chicken heart.The effects of the exposure to Cd on mRNA abundance of necrosis-related genes (JNK, TNFα, RIPK1, MLKL) in the chicken cardiomyocytes are shown in Fig. 5. Fluorescence quantitative PCR assay revealed that mRNA expression of JNK, TNFα, RIPK1 and MLKL increased significantly (p < 0.05) in the Cd+ group. Compared to those in the control group, the mRNA levels of JNK, TNFα, RIPK1 and MLKL in the Cd+ group increased by 292%, 375%, 75% and 234%, respectively. Notably, among them, the expression of TNFα and JNK was the most significant. However, the mRNA expression of PPARα, adiponectin, adipoR1, adipoR2 and AMPKα1 decreased by 14%, 29%, 37%, 58%, and 62%, respectively, in the Cd+ group (p < 0.05).
 | ||
| Fig. 5 Effects of four groups C, Se+, Cd+, Se+ + Cd+ on the mRNA levels of JNK, TNFα, RIPK1, MLKL, PPARα, adiponectin, adipoR1, adipoR2 and AMPKα1 genes in cardiomyocytes. The results were calculated from at least three independent experiments. The data are expressed as the means ± SD. The mean value between groups without a shared common letter are significantly different (p < 0.05), n = 3, using one-way ANOVA. | ||
The results indicating the effects of dietary Se supplementation on mRNA levels of adiponectin-related genes in the chicken heart are also shown in Fig. 5. There were higher mRNA expression levels of PPARα, adiponectin, adipoR1, adipoR2 and AMPKα1 in the Se+ group when compared with those in the control groups (p < 0.05). In addition, the increased expression among them were 249%, 42%, 55%, 63% and 420%, whereas JNK, TNFα, RIPK1 and MLKL mRNA expression levels were decreased by 14%, 38%, 84% and 70%, respectively.
In order to prove the protective role of Se in cardiomyocytes against Cd-induced toxicity and to investigate the changes in adiponectin and necroptosis related genes, we used the Se+ + Cd+ co-treatment group for comparison with the Cd+ group. The results (Fig. 5) showed that the expression of all of the necroptosis-related genes in the Se+ + Cd+ treated chickens was reduced significantly compared with that in the Cd+ group, in which JNK, TNFα, RIPK1 and MLKL were decreased by 236%, 239%, 84% and 262%, respectively (p < 0.05). However, we found that the mRNA expression levels of PPARα, adiponectin, adipoR1, adipoR2 and AMPKα1 were respectively up-regulated by 116%, 30%, 25%, 50% and 50% compared with those in the other two groups (p < 0.05).
Heat map analysis
Heat map (Fig. 6) results revealed that JNK, TNFα, RIPK1 and MLKL genes had a low expression. However, PPARα, adiponectin, adipoR1, adipoR2 and AMPKα1 had a high expression in the Se+ + Cd+ group, which further demonstrated that there was an inseparable relationship between adiponectin and necroptosis in the chicken heart. | ||
| Fig. 6 Heat map of relative mRNA levels of adiponectin and necrotic genes. Rows represent the probe sets. RNA gene expression is shown using the indicated pseudo color scale from blue (0) to red (2.00) relative to values. Red squares represent increased significantly (p < 0.05). Blue squares represent decreased significantly (p < 0.05). Data are presented as the mean ± SD. | ||
Discussion
Over the past few decades, studies on Cd exposure have revealed that it has extremely toxic effects. Numerous reports have indicated that the exposure to Cd led to an augmented risk of cardiovascular disease in mice.6 In the etiology, several experimental studies in vivo have implicated that Cd consumption is the cause for the pathogenesis of hypertension and cardiotoxicity in rats.36,37 Extensive studies have indicated that Cd exposure induced oxidative stress, histological damage, and cardiomyocyte apoptosis in the cardiac tissue of rats.25 Therefore, it could be concluded that Cd induces cardiotoxicity and cell death along with myocardial fiber necrosis in the cardiac tissue. In addition, numerous studies revealed that the exposure to Cd increased the ROS production with oxidative stress resulting in tissue damage in rats.16 Cd-induced [Ca2+] concentration elevation inhibited the degradation of autophagosomes and triggered oxidative stress to aggravate Cd-induced cytotoxicity in primary rat proximal tubular (rPT) cells.27 Furthermore, previous studies have shown that the elevation of [Ca2+] concentration in Pb-exposed rPT cells could evoke apoptosis induced by impaired calcium homeostasis.35 These aforementioned studies proved that the Cd exposure caused hepatic cell death chiefly through the necrotic pathway in addition to the oxidative stress impairments.38 The typical mode of cell death is apoptosis, but necrosis has also been observed.39 Furthermore, previous studies have suggested that Cd induces either apoptosis or necrosis as ERK and JNK promote BNIP3 mediated Cd-induced necrosis in rat neuronal cells.40 Other studies demonstrated that the Cd exposure mediates necrosis of the testis followed by inhibition of the intracellular antioxidant defense system in tissues at early stages in mice.41 Furthermore, Cd exposure induced significant histological changes, which led to myofibrillar loss, oedema, vacuolization of cytoplasm, vitreous degeneration, infiltration of dense focal inflammatory cells and irregularity of myofibrils in the cardiac tissues of chicken.25 In another study, the mode of Cd-induced programmed necrosis in human endothelial cells along with DNA damage has been determined.42 The results of the present study are consistent with those previously reported, which have proved that some genes (JNK, TNFα, RIPK1 and MLKL) related to necroptosis pathway remarkably increased as observed in the Cd+ group in our experiments. mRNA abundance of necrosis-related genes was increased in the Cd+ group, in which necroptosis pathway was triggered by these genes.Extensive studies have indicated that Se was associated with the physiological processes that perform regulatory functions in the cell growth and survival and in the protection from cytotoxicity, transformations of redox regulation and chemical toxicity in living organism.1 Moreover, Se significantly improved the histological appearance of the affected tissues in the Cd+ group. Some reports obtained from the clinic have shown the protective effect of Se against heavy metal toxicity and therapeutic effect.43 Yiin et al.44 reported that Se played an important role in preventing Cd-induced lipid peroxidation with peroxidative damage to liver, heart, and spleen in rats. Moreover, the co-treatment with Se and Cd reduced the alterations of peroxidation and heavy metal levels in the target organ. Previous research has shown that the detoxification effects of Se on Cd-induced toxicity occurred due to the reduction of Cd accumulation in the chicken brain.45 Our previous reports based on the interaction of Cd and Se retention indicated that Cd-induced oxidative stress caused renal injury and Se ameliorated Cd-induced nephrotoxicity, and depressed the rate of apoptosis in the chicken kidneys.46 Se supplementation reduced Cd-induced liver histopathological changes, oxidative stress, and overexpression of NO and apoptosis. This indicated that Cd-induced hepatotoxicity is partly ameliorated by Se in the chicken liver.34 Furthermore, we also proved that the major function of Se was the protective effect against Cd-induced nitric oxide mediated apoptosis caused due to the decreasing Bcl-2 mRNA level and increasing p53 mRNA level in the immune organs of chickens.47 It is reported that Se could ameliorate Cd-induced brain damage with NO production and iNOS mRNA level by regulating the change of iNOS-NO system and oxidative stress in chickens.48 Other research has also showed that Se has an ability to inhibit caspase activation and change the level of VDAC, Bcl-2 and Bax in Cd-induced mitochondrial apoptosis pathway in LLC-PK1 cell in the mice kidney in vivo.1 Liu et al.49 reported that Se had the cytoprotective effects on Cd-induced LLC-PK1 cell apoptosis through activation of P-JNK pathway.
In this paper, JNK, TNF, RIPK1, MLKL, PPAR, adiponectin, adipoR1, adipoR2 and AMPKα1 were screened from a large number of detected genes in Cd-induced cardiotoxicity cells. We observed that Cd-induced necroptosis was suppressed by antagonistic effects of Se, and the JNK, TNF, RIPK1, MLKL expression levels related to necroptosis pathway were increased significantly, which proved that the process of necroptosis occurred in the Cd+ group. The results also provide further evidence that the interaction of MLKL with RIPK1 was enhanced in the chicken cardiomyocytes after stimulation. Our previous studies showed that the regulation of a novel cell death pathway was through necroptosis.50 The important discoveries revealed from these studies are that PPARα, adiponectin, adipoR1, adipoR2 and AMPKα1 expression levels have various degrees of increase in both Se+ group and Se+ + Cd+ group, suggesting that some unknown mechanisms are activated due to the unique role of Se, such as the adiponectin pathway. These findings indicated that the pretreatment with Se in animals has the ability to inhibit Cd-induced necroptosis in the heart tissue. However, little is known about the cytoprotective effects of Se on gallinaceous hearts.
Adiponectin, which is a type of adipocytokines produced from adipocytes, has great potential characteristics, including antidiabetic, anti-atherosclerotic, anti-inflammatory and insulin-sensitizing properties.51 The findings in earlier reports suggest that the antioxidant properties of globular adiponectin could attenuate NADPH oxidase-mediated oxidative damage in the cerebral ische.52 Adiponectin has an intimate connection with oxidative stress for obesity and insulin resistance.53 There was evidence that adiponectin replacement on myocardial damage of leptin-deficient mice could inhibit the development of severe viral myocarditis through augmenting myocyte adiponectin receptor 1 activity.32 Adiponectin inhibits the expression of TNF-α in the adipose tissues of obese mice. However, both TNF-α and IL-6 could reduce the synthesis and secretion of adiponectin,31,54 which further inhibits hypoxia and oxidative stress.55,56 The presence of adiponectin and TNF-α in damaged myocytes could contribute to the processes of myocardial damage occurring in myocardial infarction (MI) or dilated cardiomyopathy (DCM).57 Necroptosis is a new pathway of regulated necrosis which could occur due to the initiation by RIP kinases.58 Recently, more evidence has been obtained showing that necroptosis contributes to the various pathophysiological conditions involving myocardial infarction, ischemia-reperfusion injury, and particularly tissue damage.59–62 An important observation in this study is that the pretreatment of Se in chickens could inhibit the alteration of JNK and RIPK1 proteins and it provided further evidence on the antagonistic effect of Se. In our experiments, the results showed that JNK was significantly activated, causing the increasing ratio of P-JNK and JNK. To explore whether Se played a protective role during this process, we therefore aimed to investigate the effect of the Se treatment on the increasing levels of PPARα, adiponectin, adipoR1, adipoR2 and AMPKα1. The results showed that Se increased adiponectin level and enhanced insulin sensitivity which led to the reduction of the myocardium damage through the activation of RIPK1/MLKL signaling pathway and significantly attenuated the JNK and TNF-α activities.
Furthermore, we applied the protein interaction analysis by using STRING and GeneMANIA databases. The existence of a strong interaction between the increase in PPARα, ADIPOQ, adipoR1, adipoR2 and AMPKα1 proteins and the decrease in JNK, TNFα, RIPK1 and MLKL proteins was observed in the Se+ + Cd+ group, indicating that there was some association between adiponectin pathway and cell necrotic pathway. The GeneMANIA pathway analysis also revealed that the necroptosis-related genes enriched in the RIPK1/MLKL signaling pathways were significantly altered by Se. Meanwhile, the heat map results suggested the same conclusions. We believe that Se has excellent potential roles as an effective nutrient element to protect the cardiomyocytes from being damaged by the effects of Cd exposure. Our study showed that Se activated the adiponectin pathway in the chicken heart as a protective response to Cd-induced toxic insults. Taking all the obtained results into account, we have proved that due to the antagonistic effect on Cd toxicity, Se supplementation not only protected cardiomyocytes from Cd-induced necroptosis but also potentially ameliorated the toxic insults associated with the exposure to Cd. The results of this study chiefly revealed that the protective action mechanism of Se in cardiomyocytes was via stimulation of the adiponectin pathway against Cd-induced necroptosis.
Conclusion
In summary, our study has revealed a novel mechanism to clarify the impact of Cd toxicity on cardiomyocytes and provided new possibilities of using Se in the treatment of cardiovascular diseases related to Cd toxicity. It could be safely concluded that Se ameliorated Cd-induced heart damage in chickens by regulating adiponectin-necrotic pathway changes, and the interactions of Cd and Se could serve as a potential therapeutic for Cd-induced heart failure in the chickens. However, the interactions between selenoproteins and Cd remained enigmatic. Little is known about the role of Se in protection of the gallinaceous hearts from Cd-induced toxicity. It may be related to the mechanistic roles of selenoproteins in conjunction with Cd-induced oxidative stress in the organisms.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This study was supported by the National Natural Science Foundation of China (31402267), the Heilongjiang Province Science Foundation for Youths (QC2014C015), the Heilongjiang Postdoctoral Fund under Grant LBH-Z13028, the Foundation for Young Talents in Higher Education of Heilongjiang, China under Grant UNPYSCT-2015009, the “Young Talents” Project of Northeast Agricultural University under Grant 14QC20, and the degree and graduate education teaching reform project in Heilongjiang province (JGXM_ HLJ_201676).References
- Y. Wang, Y. Wu, K. Luo, Y. Liu, M. Zhou, S. Yan, H. Shi and Y. Cai, Food Chem. Toxicol., 2013, 58, 61–67 CrossRef CAS PubMed.
- A. Szuster-Ciesielska, A. Stachura, M. Slotwinska, T. Kaminska, R. Sniezko, R. Paduch, D. Abramczyk, J. Filar and M. Kandefer-Szerszen, Toxicology, 2000, 145, 159–171 CrossRef CAS PubMed.
- D. M. Templeton and Y. Liu, Chem.-Biol. Interact., 2010, 188, 267–275 CrossRef CAS PubMed.
- N. Yazihan, M. K. Kocak, E. Akcil, O. Erdem and A. Sayal, Hum. Exp. Toxicol., 2011, 30, 391–397 CAS.
- K. M. Kiran Kumar, M. Naveen Kumar, R. H. Patil, R. Nagesh, S. M. Hegde, K. Kavya, R. L. Babu, G. T. Ramesh and S. C. Sharma, Toxicol. Mech. Methods, 2016, 26, 658–666 CrossRef CAS PubMed.
- R. E. Carroll, JAMA, J. Am. Med. Assoc., 1966, 198, 267–269 CrossRef CAS.
- L. Minutoli, A. Micali, A. Pisani, D. Puzzolo, A. Bitto, M. Rinaldi, G. Pizzino, N. Irrera, F. Galfo, S. Arena, G. Pallio, A. Mecchio, A. Germana, D. Bruschetta, R. Laura, C. Magno, H. Marini, F. Squadrito and D. Altavilla, Toxicol. Sci., 2016, 149, 270 CrossRef CAS PubMed.
- S. Kundu, S. Sengupta and S. Chatterjee, et al., J. Inflammation, 2009, 6(1), 19 CrossRef PubMed.
- C. C. Shao, N. Li, Z. W. Zhang, J. Su, S. Li, J. L. Li and S. W. Xu, Ecotoxicol. Environ. Saf., 2014, 106, 109–114 CrossRef CAS PubMed.
- S. Yang, Z. Zhang, J. He, J. Li, J. Zhang, H. Xing and S. Xu, Biol. Trace Elem. Res., 2012, 148, 53–60 CrossRef CAS PubMed.
- J. Zhang, Y. Fu, J. Li, J. Wang, B. He and S. Xu, Environ. Toxicol. Pharmacol., 2009, 27, 345–349 CrossRef CAS PubMed.
- M. L. Ferramola, M. F. Perez Diaz, S. M. Honore, S. S. Sanchez, R. I. Anton, A. C. Anzulovich and M. S. Gimenez, Toxicol. Appl. Pharmacol., 2012, 265, 380–389 CrossRef CAS PubMed.
- A. Veličkov, N. Jančić, N. Điniđić, I. Rančić, N. Bojanić and M. Krstić, Acta Med. Medianae, 2013, 15–22, DOI:10.5633/amm.2013.0203.
- S. Kundu, S. Sengupta, S. Chatterjee, S. Mitra and A. Bhattacharyya, J. Inflammation, 2009, 6, 19 CrossRef PubMed.
- Y. Zhao, R. Li and Y. Lin, Fish Shellfish Immunol., 2015, 47, 444–449 CrossRef CAS PubMed.
- D. Bagchi, P. J. Vuchetich, M. Bagchi, E. A. Hassoun, M. X. Tran, L. Tang and S. J. Stohs, Free Radical Biol. Med., 1997, 22, 471–478 CrossRef CAS PubMed.
- F. Kayama, T. Yoshida, M. R. Elwell and M. I. Luster, Toxicol. Appl. Pharmacol., 1995, 131, 224–234 CrossRef CAS PubMed.
- G. Krumschnabel, H. L. Ebner, M. W. Hess and A. Villunger, Aquat. Toxicol., 2010, 99, 73–85 CrossRef CAS PubMed.
- L. Gan, Q. Liu, H. B. Xu, Y. S. Zhu and X. L. Yang, Biol. Trace Elem. Res., 2002, 89, 165–175 CrossRef CAS PubMed.
- L. V. Papp, J. Lu, A. Holmgren and K. K. Khanna, Antioxid. Redox Signaling, 2007, 9, 775–806 CrossRef CAS PubMed.
- P. Suchý, E. Straková and I. Herzig, Czech J. Anim. Sci., 2014, 59, 495–503 Search PubMed.
- G. N. Schrauzer, J. UOEH, 1987, 9(suppl.), 208–215 CAS.
- A. Al-Waeli, A. C. Pappas, E. Zoidis, C. A. Georgiou, K. Fegeros and G. Zervas, Br. Poult. Sci., 2012, 53, 817–827 CrossRef CAS PubMed.
- H. Wang, S. Li and X. Teng, Biol. Trace Elem. Res., 2016, 171, 437–444 CrossRef CAS PubMed.
- S. Alpsoy, M. Kanter, C. Aktas, M. Erboga, A. Akyuz, D. C. Akkoyun and M. Oran, Biol. Trace Elem. Res., 2014, 159, 297–303 CrossRef CAS PubMed.
- L. L. Liu, C. M. Li, Z. W. Zhang, J. L. Zhang, H. D. Yao and S. W. Xu, Biol. Trace Elem. Res., 2014, 158, 176–185 CrossRef CAS PubMed.
- F. Liu, X. Y. Wang, X. P. Zhou, Z. P. Liu, X. B. Song, Z. Y. Wang and L. Wang, Toxicology, 2017, 383, 13–23 CrossRef CAS PubMed.
- W. Zhao, W. Liu, X. Chen, Y. Zhu, Z. Zhang, H. Yao and S. Xu, Biol. Trace Elem. Res., 2014, 161, 328–333 CrossRef CAS PubMed.
- X. Chen, Y. H. Zhu, X. Y. Cheng, Z. W. Zhang and S. W. Xu, Molecules, 2012, 17, 14565–14572 CrossRef CAS PubMed.
- X. M. Mao, J. Y. Hong and L. Q. Dong, Mini-Rev. Med. Chem., 2006, 6, 1331–1340 CrossRef CAS PubMed.
- N. Maeda, M. Takahashi, T. Funahashi, S. Kihara, H. Nishizawa, K. Kishida, H. Nagaretani, M. Matsuda, R. Komuro, N. Ouchi, H. Kuriyama, K. Hotta, T. Nakamura, I. Shimomura and Y. Matsuzawa, Diabetes, 2001, 50, 2094–2099 CrossRef CAS PubMed.
- T. Takahashi, S. Saegusa, H. Sumino, T. Nakahashi, K. Iwai, S. Morimoto and T. Kanda, J. Int. Med. Res., 2005, 33, 207–214 CrossRef CAS PubMed.
- S. Hasic, A. Hadzovic-Dzuvo, R. Jadric and E. Kiseljakovic, Bosnian J. Basic Med. Sci., 2013, 13, 225–229 CrossRef.
- J. L. Li, C. Y. Jiang, S. Li and S. W. Xu, Ecotoxicol. Environ. Saf., 2013, 96, 103–109 CrossRef CAS PubMed.
- H. Wang, Z. K. Wang, P. Jiao, X. P. Zhou, D. B. Yang, Z. Y. Wang and L. Wang, Toxicology, 2015, 333, 137–146 CrossRef CAS PubMed.
- H. Mollaoglu, A. Gokcimen, F. Ozguner, F. Oktem, A. Koyu, A. Kocak, H. Demirin, O. Gokalp and E. Cicek, Toxicology, 2006, 227, 15–20 CrossRef CAS PubMed.
- S. S. Soares, H. Martins, C. Gutierrez-Merino and M. Aureliano, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2008, 147, 168–178 CrossRef CAS PubMed.
- M. Sinha, P. Manna and P. C. Sil, J. Trace Elem. Med. Biol., 2009, 23, 300–313 CAS.
- W. C. Prozialeck, J. R. Edwards, V. S. Vaidya and J. V. Bonventre, Toxicol. Appl. Pharmacol., 2009, 238, 301–305 CrossRef CAS PubMed.
- B. Wang, J. L. Xiao, Y. H. Ling, X. J. Meng, B. Wu, X. Y. Yang and F. Zou, Toxicol. Sci., 2014, 140, 393–402 CrossRef CAS PubMed.
- A. D. Chiquoine, Anat. Rec., 1964, 149, 23–35 CrossRef CAS PubMed.
- B. Messner, A. Turkcan, C. Ploner, G. Laufer and D. Bernhard, Cell. Mol. Life Sci., 2016, 73, 1699–1713 CrossRef CAS PubMed.
- A. Naganuma and N. Imura, Seikagaku, 1987, 59, 1339–1343 CAS.
- S. J. Yiin, C. L. Chern, J. Y. Sheu and T. H. Lin, Biol. Trace Elem. Res., 2000, 78, 219–230 CrossRef CAS PubMed.
- S. Haider, L. Anis, Z. Batool, I. Sajid, F. Naqvi, S. Khaliq and S. Ahmed, Metab. Brain Dis., 2015, 30, 83–92 CrossRef CAS PubMed.
- L. Liu, B. Yang, Y. Cheng and H. Lin, Biol. Trace Elem. Res., 2015, 167, 308–319 CrossRef CAS PubMed.
- L. L. Liu, J. L. Zhang, Z. W. Zhang, H. D. Yao, G. Sun and S. W. Xu, Biol. Trace Elem. Res., 2014, 159, 199–209 CrossRef CAS PubMed.
- J. L. Li, R. Gao, S. Li, J. T. Wang, Z. X. Tang and S. W. Xu, BioMetals, 2010, 23, 695–705 CrossRef CAS PubMed.
- Y. Liu, S. P. Zhang and Y. Q. Cai, Toxicol. In Vitro, 2007, 21, 677–684 CrossRef CAS PubMed.
- R. S. Al-Lamki, W. Lu, P. Manalo, J. Wang, A. Y. Warren, A. M. Tolkovsky, J. S. Pober and J. R. Bradley, Cell Death Dis., 2016, 7, e2287 CrossRef CAS PubMed.
- M. E. Trujillo and P. E. Scherer, J. Intern. Med., 2005, 257, 167–175 CrossRef CAS PubMed.
- W. Song, T. Huo, F. Guo, H. Wang, H. Wei, Q. Yang, H. Dong, Q. Wang and L. Xiong, Neuroscience, 2013, 248, 136–144 CrossRef CAS PubMed.
- E. Vatansever, E. Surmen-Gur, A. Ursavas and M. Karadag, Sleep Breath., 2011, 15, 275–282 CrossRef PubMed.
- M. Fasshauer, S. Kralisch, M. Klier, U. Lossner, M. Bluher, J. Klein and R. Paschke, Biochem. Biophys. Res. Commun., 2003, 301, 1045–1050 CrossRef CAS PubMed.
- Y. Hattori, K. Akimoto, S. S. Gross, S. Hattori and K. Kasai, Diabetologia, 2005, 48, 1066–1074 CrossRef CAS PubMed.
- N. Hosogai, A. Fukuhara, K. Oshima, Y. Miyata, S. Tanaka, K. Segawa, S. Furukawa, Y. Tochino, R. Komuro, M. Matsuda and I. Shimomura, Diabetes, 2007, 56, 901–911 CrossRef CAS PubMed.
- T. Takahashi, S. Saegusa, H. Sumino, T. Nakahashi, K. Iwai, S. Morimoto, T. Nojima and T. Kanda, J. Int. Med. Res., 2005, 33, 236–244 CrossRef CAS PubMed.
- N. Holler, R. Zaru, O. Micheau, M. Thome, A. Attinger, S. Valitutti, J. L. Bodmer, P. Schneider, B. Seed and J. Tschopp, Nat. Immunol., 2000, 1, 489–495 CrossRef CAS PubMed.
- Z. Dunai, P. I. Bauer and R. Mihalik, Pathol. Oncol. Res., 2011, 17, 791–800 CrossRef CAS PubMed.
- J. D. McCully, H. Wakiyama, Y. J. Hsieh, M. Jones and S. Levitsky, Am. J. Physiol.: Heart Circ. Physiol., 2004, 286, H1923–H1935 CrossRef CAS PubMed.
- T. West, M. Atzeva and D. M. Holtzman, Neurobiol. Dis., 2006, 22, 523–537 CrossRef CAS PubMed.
- Z. Su, Z. Yang, Y. Xu, Y. Chen and Q. Yu, Mol. Cancer, 2015, 14, 48 CrossRef PubMed.
Footnotes |
| † All authors have read the manuscript and agreed to submit it in its current form for consideration for publication in the Journal. |
| ‡ These authors have contributed equally in the paper. |
| This journal is © The Royal Society of Chemistry 2017 |