
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Donor- and acceptor-functionalized dibenzo[a,e]pentalenes: modulation of the electronic band gap†
J.
Wilbuer
ab,
D. C.
Grenz
a,
G.
Schnakenburg
c and
B.
Esser
 *a
*a
aInstitute for Organic Chemistry, University of Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: besser@oc.uni-freiburg.de
bKekulé-Institute for Organic Chemistry and Biochemistry, University of Bonn, Gerhard-Domagk-Str. 1, 53121 Bonn, Germany
cInstitute for Inorganic Chemistry, University of Bonn, Gerhard-Domagk-Str. 1, 53121 Bonn, Germany
First published on 27th September 2016
Abstract
Dibenzo[a,e]pentalene (DBP) is a low-bandgap organic semiconductor. A versatile synthetic route to DBPs substituted with aryl or alkyl groups in the 5,10-positions and electron-donating or -accepting functionalities in the 2,7-positions is described. Six donor- or acceptor-functionalized DBP derivatives were synthesized that show amphoteric redox behavior and band gaps around 2 eV. Through choice of the 2,7-substituents, the HOMO/LUMO energy levels and band gaps can be adjusted within a range of up to 0.6 eV. In the solid state, the DBP derivatives assume herringbone-type packing structures.
Introduction
Dibenzo[a,e]pentalene (DBP, 1, Chart 1) is a ladder-type conjugated hydrocarbon that has received a significant amount of attention in recent years.1 As organic semiconductors with a low band-gap, derivatives of 1 have been employed in organic field-effect transistors (OFETs), exhibiting p-type behavior.2–6 First synthesized in 1912 by Brand,7 a variety of methods for the synthesis of DBP derivatives have been developed in the past ten years.8 While Brand's original synthesis relies on the addition of Grignard reagents to diphenyl succindandione followed by a two-fold elimination of water,7,9,10 derivatives of DBP (1) can now be accessed from (di)aryl–alkynes by reductive11,12 or Lewis-acid-induced cyclizations13 as well as transition-metal-catalysed or -mediated processes.14–18 These methodologies were also applied to the synthesis of π-extended, ring-fused derivatives of 1.19–22 Related hydrocarbons consisting of fused five- and six-membered rings are indeno[1,2-b]fluorenes,23–26 bispentalenes,27 and other systems that include heteroatoms in the ring structure.28–31 In spite of the recent synthetic developments towards DBP derivatives, systematic studies on the modulation of its electronic properties through substitution have been undertaken only twice. Kawase et al. reported on the synthesis and properties of 2,(3,)7(,8)-aryl-substituted DBPs,16 while Orita and co-workers attached aryl–ethinylenes to the 5,10-positions of the DBP core.32 We herein present the synthesis and optoelectronic properties of six DBP derivatives of type 2 (Chart 1), substituted with aryl–alkynes in the 2,7-positions. Electron-donating or -withdrawing groups R1 were chosen that allow for a modulation of the HOMO and LUMO energy levels. The aryl–alkyne groups were attached through posterior modification of the DBP core starting from 2,7-dibromo-DBPs of type 3. Six derivatives of 3 as precursors to 2 were synthesized carrying different aryl or alkyl groups in 5,10-position that allow for adapting the solubility properties. The solid-state structure of six DBP derivatives of type 2 and 3 are presented and discussed. | ||
| Chart 1 Dibenzo[a,e]pentalene (DBP, 1), donor/acceptor-functionalized DBPs 2 and dibromo-functionalized derivatives 3. | ||
In order to most strongly influence the HOMO energy of the DBP core, which is relevant for an application as p-type material in OFETs, the C2 and C7 positions on the six-membered rings were chosen for attachment of the electron-donating or -withdrawing groups, since these possess the largest MO coefficients in the HOMO.33 The aryl-R1 substituents were connected to the DBP core through alkyne bridges, since these allow for co-planarity of the latter with the electronically modulating aryl group and hence maximum orbital overlap.
At C5 and C10 of the five-membered rings aryl or alkyl substituents R2 were attached to modulate the solubility and crystallinity of the resulting compounds. A high crystallinity would be advantageous for the charge carrier mobility in OFETs, while a good solubility in common solvents would allow for facile processing of the materials.
Results and discussion
Syntheses
For the synthesis of derivatives 3, we chose a method based on Brand's procedure,7 since the more recent methods for DBP synthesis do not give access to derivatives suitable for further functionalization, such as 3 with bromo-substituents.32,34 Bis-brominated diphenyl succindandione 4,6 synthesized in an improved synthetic procedure in three steps and 52% yield from ethyl-4-bromophenylacetate (see ESI†), was reacted with different Grignard or lithiated alkyl or aryl compounds R2M(X) followed by acid-catalysed elimination of water to yield 3a–f (Scheme 1). DBP derivatives 3a (R2 = Me), 3b (R2 = Et), and 3d (R2 = Ph) were synthesized through initial addition of the corresponding Grignard reagent to the keto groups in 4 in refluxing toluene, while 3e (R2 = 4-tert-butylphenyl) was accessed through addition of the corresponding lithiated species. For the synthesis of 3c (R2 = 2-ethylhexyl) and 3f (R2 = mesityl) the addition of cerium trichloride was necessary35 for the Grignard addition step to proceed. 3f with mesityl substituents was chosen for further derivatization, since this compound combined the properties of solubility and crystallinity. 3f (R2 = Mes) showed a solubility of 4.6 g L−1 in dichloromethane and 2.9 g L−1 in cyclohexane, while for 3a (R2 = Me) only 1.8 and 0.8 g L−1, respectively, dissolved. | ||
| Scheme 1 Synthesis of bis-brominated DBP-derivatives 3a–f and aryl–alkyne-substituted DBPs 5a–f (Method a: (i) R2MgBr, toluene, 110 °C, 1–2 h; (ii) HCl aq., rt, 0.5–14 h; Method b: (i) R2MgBr, CeCl3, THF, 0 °C, 1 h; (ii) HCl aq., rt, o.n.; Method c: (i) (4-t-Bu)PhBr, n-BuLi, THF, −50 °C to rt, 2 h; (ii) CF3COOH, rt, 2 d; Method d: 1. R2MgBr, CeCl3, THF, 0 °C, 8 h; 2. p-TsOH, toluene, 110 °C, 3.5 h). | ||
The bis-brominated DBPs 3a, 3b and 3d–f were further investigated by X-ray crystallography; their solid-state structures will be discussed below.
Starting from 3f, six different aryl–alkynes with para-substituents R1 were attached to the C2- and C7-positions of the DBP core through Sonogashira cross-coupling reactions to yield 5a–f (Scheme 1). The conditions employed (Pd(PhCN)2Cl2/P(t-Bu)3) allowed for coupling of the bromides at room temperature.28 The aryl–alkynes were chosen based on their electronic properties: two electron-donating (OMe (5b) and NMe2 (5a)), one “neutral” (H (5c)) and three electron-withdrawing (CO2Me (5d), CN (5e), and CHC(CN)(CO2Et) (5f)) substituents. 5c (R1 = H) was further investigated by X-ray crystallography; the structure will be discussed below.
Optoelectronic properties
(TD)DFT calculations were performed in order to gain information on the orbital energies and electronic transitions in the absorption spectra (see ESI† for details).‡ For calculations all mesityl groups in 5a–f and the ethyl groups in 5f were replaced by methyl groups, denoted with an asterix (5a*–f*). Four different density functionals in combination with D3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37,38 and the def2-TZVP39 basis set were tested with varying amounts of HF exchange from 0% to 50%, since this has been shown to have a pronounced effect:40 BP8641–43 (0%), TPSSH44,45 (10%), B3LYP46,47 (20%) and BHLYP48 (50%). The closest match between TDDFT calculations and experimental absorption spectra was found with B3LYP, while TPSSH gave the best orbital energies (see ESI† for details), and hence these results will be used in the further discussion. Selected calculated orbital energies of 5a*–f* as well as the molecular orbitals of 5c* are shown in Fig. 1. With increasing electron-withdrawing character of the substituent R1 the frontier molecular orbital energies of 5a*–f* decrease. The HOMO energies experience a stronger shift (ΔE = 1.0 eV) compared to the LUMO energies (ΔE = 0.88 eV), which we had anticipated before based on the size of the MO coefficients in the HOMO and LUMO of DBP (1, see above). The order of decrease in energy of the orbitals from 5a* to 5f* corresponds well with the Hammett substitution constants σ of R1: the σpara constants of the substituents R1 amount to −0.83 for NMe2 (5a*), −0.27 for OMe (5b*), 0 for H (5c*), 0.45 for CO2Me (5d*), 0.66 for CN (5e*), and ∼0.8§ for CHC(CN)(CO2Me) (5f*).36
37,38 and the def2-TZVP39 basis set were tested with varying amounts of HF exchange from 0% to 50%, since this has been shown to have a pronounced effect:40 BP8641–43 (0%), TPSSH44,45 (10%), B3LYP46,47 (20%) and BHLYP48 (50%). The closest match between TDDFT calculations and experimental absorption spectra was found with B3LYP, while TPSSH gave the best orbital energies (see ESI† for details), and hence these results will be used in the further discussion. Selected calculated orbital energies of 5a*–f* as well as the molecular orbitals of 5c* are shown in Fig. 1. With increasing electron-withdrawing character of the substituent R1 the frontier molecular orbital energies of 5a*–f* decrease. The HOMO energies experience a stronger shift (ΔE = 1.0 eV) compared to the LUMO energies (ΔE = 0.88 eV), which we had anticipated before based on the size of the MO coefficients in the HOMO and LUMO of DBP (1, see above). The order of decrease in energy of the orbitals from 5a* to 5f* corresponds well with the Hammett substitution constants σ of R1: the σpara constants of the substituents R1 amount to −0.83 for NMe2 (5a*), −0.27 for OMe (5b*), 0 for H (5c*), 0.45 for CO2Me (5d*), 0.66 for CN (5e*), and ∼0.8§ for CHC(CN)(CO2Me) (5f*).36
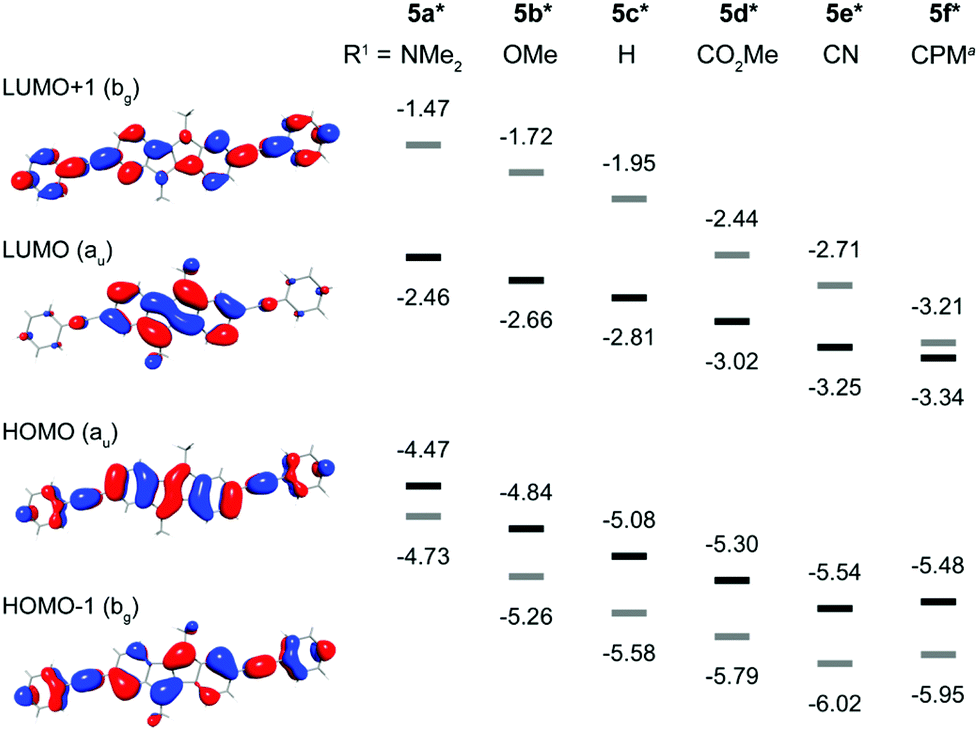 | ||
| Fig. 1 Molecular orbitals of 5c* (R1 = H, left) and energy diagram (eV, right) of calculated MOs of DBP derivatives 5a*–f* (TPSSH-D3/def2-TZVP, aCPM = CHC(CN)(CO2Me)). | ||
The absorption spectra of 5a–f are displayed in Fig. 2. All derivatives show three characteristic absorption bands, which were assigned to electronic transitions using TDDFT calculations (B3LYP-D3/def2-TZVP). The lowest energy bands from 470–700 nm, well visible on the logarithmic scale in the inset in Fig. 2, can be ascribed to the HOMO → LUMO transitions (S0 to S1). These transitions are symmetry forbidden, since both orbitals are of au symmetry (Laporte's rule, see also Fig. 1), which is reflected in the relatively low absorption coefficients of logε = 2.9–3.4. This low energy band appears at very similar wavelengths for all substituents with maxima from 530–534 nm except for 5a and 5f. The NMe2 substituent in 5a affects a bathochromic shift to 567 nm, while the CHC(CN)(CO2Et) substituent in 5f leads to a hypsochromic shift to 511 nm. The second absorption bands from 380–470 nm can be assigned to the HOMO−1 → LUMO, HOMO−2 → LUMO and other transitions, while the most intense (logε = 5.0–5.1) and highest energy absorption bands from 270–380 nm can be attributed to the HOMO → LUMO+1 transitions. The CHC(CN)(CO2Et) substituent presents an exception, since the most intense absorption is the HOMO−1 → LUMO transition at 414 nm. Overall, the NMe2-substituted derivative 5a experiences the strongest bathochromic shift of all absorption bands. This is in accordance with the electrochemical results and calculations that show a strong increase in HOMO (and HOMO−1) energy relative to the other derivatives, while the LUMO energy is less strongly affected (see also Fig. 1 and Table 1). This observation is in agreement with our initial assumption, that substitution at the 2,7-positions of the DBP core would most strongly affect the HOMO energy level (see above). The optical transitions in 5f are different compared to 5a–e, likely due to the extended conjugation into the CHC(CN)(CO2Et) substituent. The band gaps of 5a–f were obtained from the onset of the longest wavelength absorption and are listed in Table 1. None of the DBP derivatives 5a–f showed fluorescence. This has also been observed for the indeno[1,2-b]fluorenes, in which case it was rationalized through transient absorption spectroscopy measurements that showed that the excited state lifetimes were shorter than the timescale at which fluorescence is observed.49
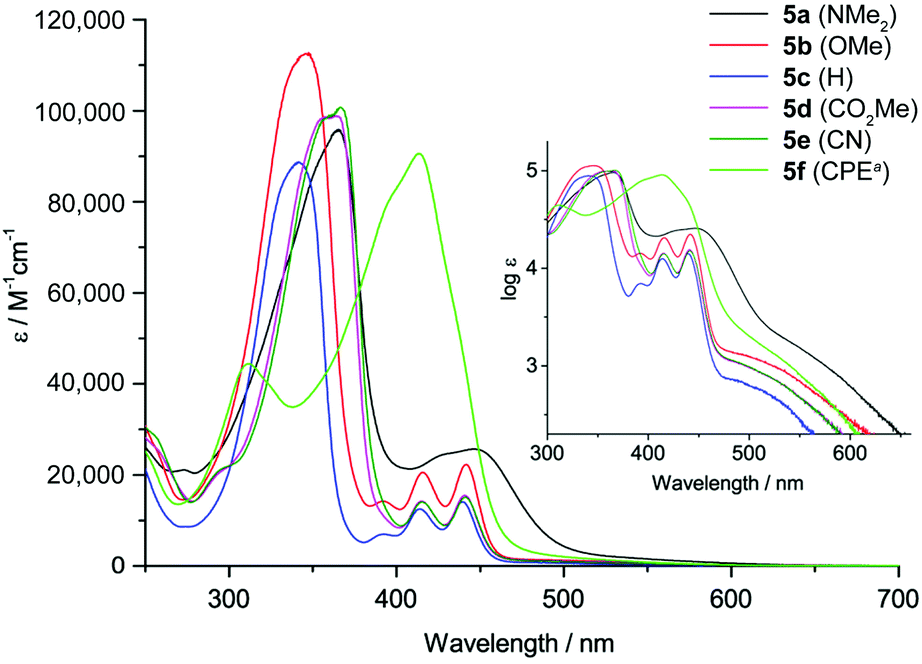 | ||
| Fig. 2 Absorption spectra of 5a–f in CH2Cl2 solution (aCPE = CHC(CN)(CO2Et)). Inset: Spectra plotted on a logarithmic scale. | ||
| E red 1/2 (V) | E ox 1/2 (V) |
E
HOMOa (eV) |
E
LUMOa (eV) |
E g (eV) | E g (opt)b (eV) | |
|---|---|---|---|---|---|---|
| a From the onset of the reduction/oxidation peak vs. Fc/Fc+, assuming an ionization energy of 4.8 eV for ferrocene.50 b From the onset of the longest wavelength absorption band. c Cathodic peak potential. d Anodic peak potential. | ||||||
| 5a | −1.79 | 0.39c | −5.17 | −3.18 | 1.99 | 1.82 |
| 5b | −1.77 | 0.78 | −5.57 | −3.18 | 2.39 | 1.99 |
| 5c | −1.74 | 0.83 | −5.68 | −3.22 | 2.46 | 2.08 |
| 5d | −1.71 | 0.93 | −5.73 | −3.27 | 2.46 | 2.07 |
| 5e | −1.67 | 0.98 | −5.77 | −3.28 | 2.49 | 2.07 |
| 5f | −1.68d | 0.89 | −5.69 | −3.34 | 2.35 | 1.95 |
| −1.79 | ||||||
The electrochemical properties of 5a–f were investigated through cyclic voltammetry measurements in CH2Cl2 solution (Fig. 3 and Table 1),¶ where the first oxidation and reduction can be used to determine the HOMO and LUMO energies, respectively. All DBP derivatives 5a–f show amphoteric redox behaviour. The redox potentials correspond well with the electron-donating or -withdrawing character of the substituents R1: CO2Me (5d), CN (5e) and CHC(CN)(CO2Et) (5f) lead to a positive shift in reduction and oxidation potential relative to 5c with R1 = H, while this trend is reversed for the OMe (5b) and NMe2 (5a) substituents. The NMe2 substituent significantly lowers the oxidation potential and hence the HOMO energy of 5a by ΔEHOMO = 0.51 eV relative to 5c (R1 = H, EHOMO = −5.68 V). For 5f (R1 = CHC(CN)(CO2Et)) two reduction waves were observed, of which the first one was not reversible. This might be related to the close proximity in energy of the LUMO and LUMO+1, as found in DFT calculations (see Fig. 1). For all derivatives the electrochemically determined band gaps are larger than the optical band gaps, which has been observed for other non-alternant hydrocarbons51 before.52
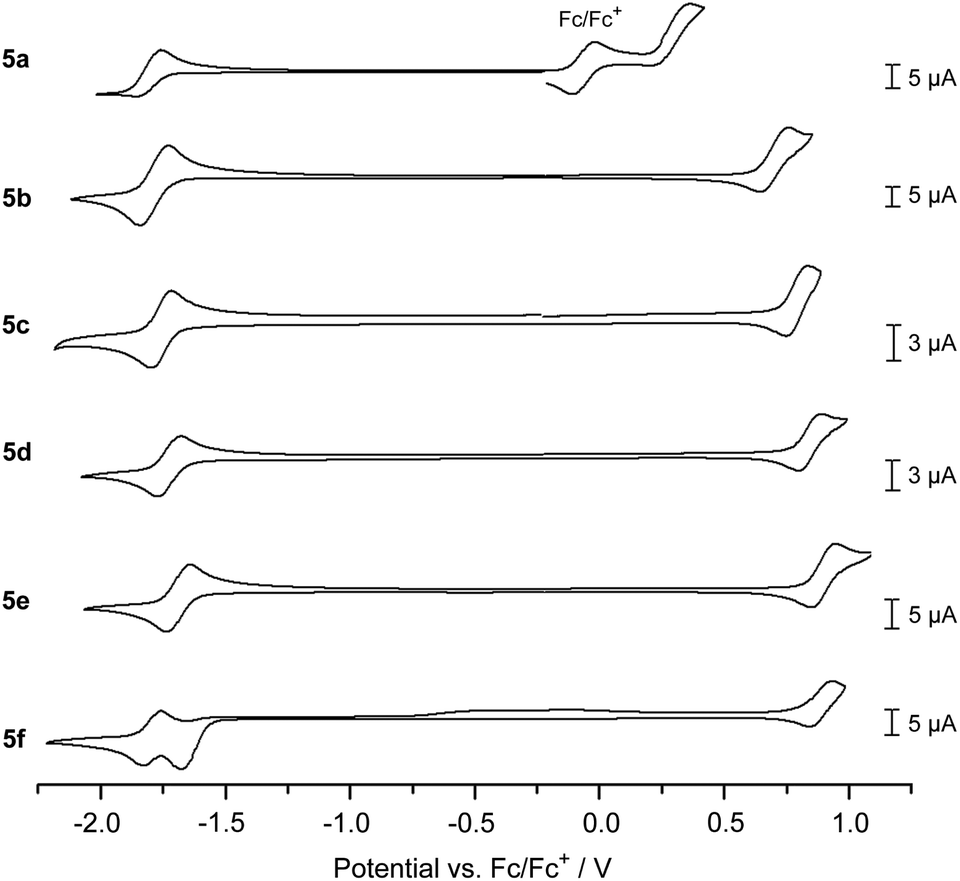 | ||
| Fig. 3 Cyclic voltammograms of 5a–f (1 mm* in CH2Cl2, 0.1 m n-Bu4NPF6, scan rate 0.1 V s−1 (for 5c 0.2 V s−1), 5c, 5d, and 5e with a Pt-electrode, 5b, 5a, and 5f with a GC-electrode).¶ *Due to solubility issues the concentration of 5a was lower. | ||
Our investigations show that by attaching electron-donating or -withdrawing substituents to the 2,7-positions of DBP, its electronic properties can be significantly modulated. The HOMO energies show the largest effect, as anticipated, with a difference of 0.60 eV between 5a and 5e, while the LUMO energies vary within 0.16 eV (5avs.5f).
Structural properties
The molecular packing in the solid state is important for the performance of organic semiconductors in electronics devices. The solid-state structures of six of the synthesized DBP derivatives were investigated by X-ray crystallography.|| Single crystals suitable for X-ray diffraction were obtained by slow evaporation of solutions in CH2Cl2 (3a, d, e), CDCl3 (3b), EtOAc/cyclohexane (3f) or CHCl3 (5c). The molecular structures of 5c, 3a, 3b and 3f are shown in Fig. 4 (see ESI† for 3d and 3e). In all derivatives, the DBP core assumes an almost planar conformation. The phenyl-alkyne substituents in 5c are slightly rotated relative to the DBP core by ∼25°. The dihedral angle between the mesityl groups and the DBP core amounts to ∼70° due to steric hindrance associated with the two ortho-methyl groups. Hence electronic communication through π-orbital overlap between the mesityl groups and the DBP core can be excluded. The bond lengths in the six-membered rings of the DBP core in 5c are relatively homogeneous (1.377–1.431 Å), while in the five-membered rings the C10–C10′ bond is short (1.362 Å) and the C5′–C10′, C4′–C5′ and C1′–C10 bonds are comparably long (1.464–1.477 Å). In the crystal lattice 5c assumes a herringbone pattern, in which the planes of the DBP cores of two non-parallel aligned molecules are oriented at an angle of ∼70° (Fig. 4b). 3a, b, and f also assume herringbone-type packing structures in the solid state (Fig. 4c–e). The distances between to parallel aligned molecules, viewed along the crystallographic a-axis, amount to 3.19 Å for 3a, 3.38 Å for 3b, and 4.26 Å for 3f.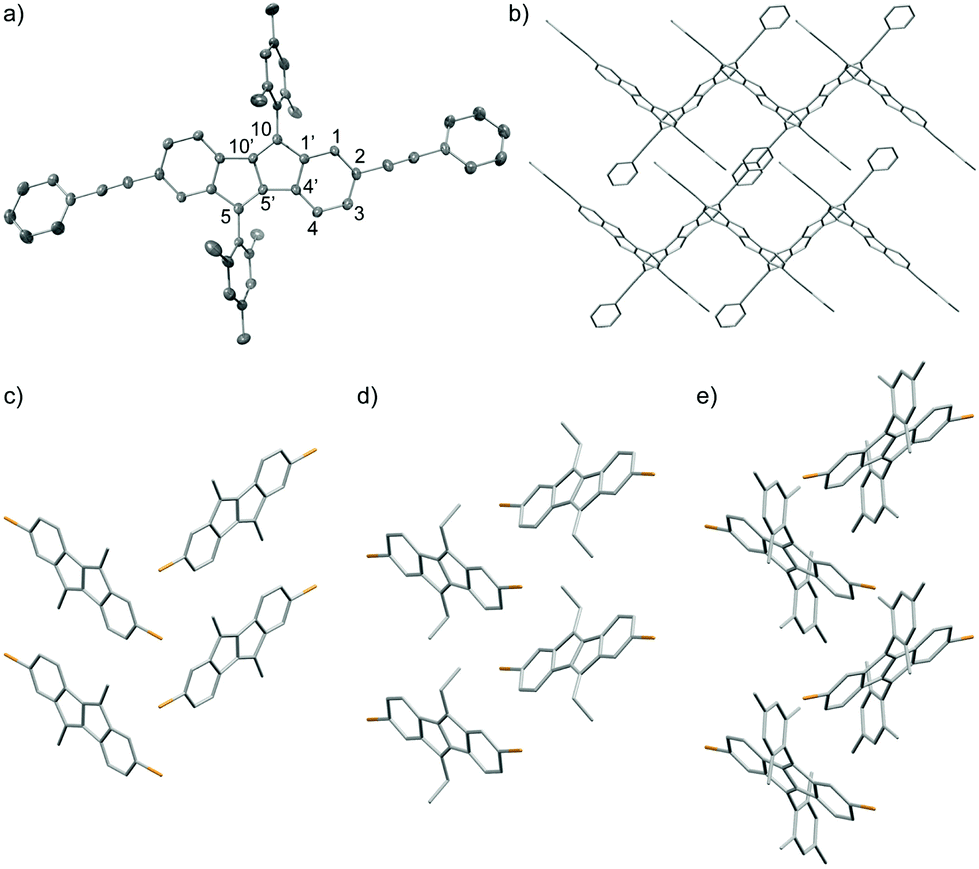 | ||
| Fig. 4 Molecular structure of 5c (a) and packing diagrams of 5c (b), 3a (c), 3b (d), and 3f (e) in the solid state (viewed along the crystallographic a-axis, hydrogen atoms and mesityl groups (in b) are omitted for clarity, ORTEP ellipsoids (in a) are shown at 50% probability). | ||
Conclusions
In summary, we have established a versatile synthetic route to 2,7-dibromo-functionalized dibenzo[a,e]pentalenes 3 with aryl or alkyl substituents in 5,10-position that can be further derivatized through cross-coupling reactions. Six electron-donating or -accepting aryl alkynes were attached through Sonogashira reactions, and the resulting 2,7-aryl–alkyne-substituted DBPs 5a–f were investigated regarding their optoelectronic properties. The choice of the substituent allowed modulating the HOMO or LUMO energy level within a range of 0.60 eV or 0.16 eV, respectively, and the band gap within a range of 0.26 eV. In the solid state, the DBP derivatives 3a, 3b, 3f and 5c assume herringbone-type packing structures. The compounds show amphoteric redox behaviour and are attractive candidates for application in organic electronics devices.Acknowledgements
We thank C. Rödde of the single crystal X-ray diffraction service. Generous support by the German Research Foundation (Emmy Noether program, ES 361/2-1), the Chemical Industry Trust (Liebig Fellowship, Li 189/11) and the University of Bonn (Maria von Linden-program) is gratefully acknowledged.Notes and references
- H. Hopf, Angew. Chem., Int. Ed., 2013, 52, 12224–12226 CrossRef CAS PubMed.
- C. Liu, S. Xu, W. Zhu, X. Zhu, W. Hu, Z. Li and Z. Wang, Chem. – Eur. J., 2015, 21, 17016–17022 CrossRef CAS PubMed.
- G. Dai, J. Chang, W. Zhang, S. Bai, K.-W. Huang, J. Xu and C. Chi, Chem. Commun., 2015, 51, 503–506 RSC.
- T. Kawase, T. Fujiwara, C. Kitamura, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, S. Shinamura, H. Mori, E. Miyazaki and K. Takimiya, Angew. Chem., Int. Ed., 2010, 49, 7728–7732 CrossRef CAS PubMed.
- C. Li, C. Liu, Y. Li, X. Zhu and Z. Wang, Chem. Commun., 2015, 51, 693–696 RSC.
- M. Nakano, I. Osaka, K. Takimiya and T. Koganezawa, J. Mater. Chem. C, 2014, 2, 64–70 RSC.
- K. Brand, Chem. Ber., 1912, 45, 3071–3077 CrossRef.
- M. Saito, Symmetry, 2010, 2, 950–969 CrossRef CAS.
- K. Brand and F. W. Hoffmann, Chem. Ber., 1920, 53, 815–821 CrossRef.
- K. Brand and F. Schläger, Chem. Ber., 1923, 56, 2541–2545 CrossRef.
- M. Saito, M. Nakamura, T. Tajima and M. Yoshioka, Angew. Chem., Int. Ed., 2007, 46, 1504–1507 CrossRef CAS PubMed.
- H. Zhang, T. Karasawa, H. Yamada, A. Wakamiya and S. Yamaguchi, Org. Lett., 2009, 11, 3076–3079 CrossRef CAS PubMed.
- C. Chen, M. Harhausen, R. Liedtke, K. Bussmann, A. Fukazawa, S. Yamaguchi, J. L. Petersen, C. G. Daniliuc, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 5992–5996 CrossRef CAS PubMed.
- A. S. K. Hashmi, M. Wieteck, I. Braun, P. Nösel, L. Jongbloed, M. Rudolph and F. Rominger, Adv. Synth. Catal., 2012, 354, 555–562 CrossRef CAS.
- Z. U. Levi and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 2796–2797 CrossRef CAS PubMed.
- T. Kawase, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata and T. Kubo, Chem. – Eur. J., 2009, 15, 2653–2661 CrossRef CAS PubMed.
- T. Maekawa, Y. Segawa and K. Itami, Chem. Sci., 2013, 4, 2369–2373 RSC.
- J. Zhao, K. Oniwa, N. Asao, Y. Yamamoto and T. Jin, J. Am. Chem. Soc., 2013, 135, 10222–10225 CrossRef CAS PubMed.
- Z. U. Levi and T. D. Tilley, J. Am. Chem. Soc., 2010, 132, 11012–11014 CrossRef CAS PubMed.
- H. Li, X.-Y. Wang, B. Wei, L. Xu, W.-X. Zhang, J. Pei and Z. Xi, Nat. Commun., 2014, 5, 4508 CAS.
- F. Xu, L. Peng, K. Shinohara, T. Nishida, K. Wakamatsu, M. Uejima, T. Sato, K. Tanaka, N. Machida, H. Akashi, A. Orita and J. Otera, Org. Lett., 2015, 17, 3014–3017 CrossRef CAS PubMed.
- J. Shen, D. Yuan, Y. Qiao, X. Shen, Z. Zhang, Y. Zhong, Y. Yi and X. Zhu, Org. Lett., 2014, 16, 4924–4927 CrossRef CAS PubMed.
- D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 1127–1130 CrossRef CAS PubMed.
- D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349–10352 CrossRef CAS PubMed.
- T. Maekawa, H. Ueno, Y. Segawa, M. M. Haley and K. Itami, Chem. Sci., 2016, 7, 650–654 RSC.
- J. L. Marshall, N. J. O'Neal, L. N. Zakharov and M. M. Haley, J. Org. Chem., 2016, 81, 3674–3680 CrossRef CAS PubMed.
- J. Cao, G. London, O. Dumele, M. von Wantoch Rekowski, N. Trapp, L. Ruhlmann, C. Boudon, A. Stanger and F. Diederich, J. Am. Chem. Soc., 2015, 137, 7178–7188 CrossRef CAS PubMed.
- J. D. Wood, J. L. Jellison, A. D. Finke, L. Wang and K. N. Plunkett, J. Am. Chem. Soc., 2012, 134, 15783–15789 CrossRef CAS PubMed.
- X.-Y. Wang, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 7668–7671 CrossRef CAS PubMed.
- A. Konishi, T. Fujiwara, N. Ogawa, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, C. Kitamura and T. Kawase, Chem. Lett., 2010, 39, 300–301 CrossRef CAS.
- G. Dai, J. Chang, J. Luo, S. Dong, N. Aratani, B. Zheng, K. W. Huang, H. Yamada and C. Chi, Angew. Chem., Int. Ed., 2016, 55, 2693–2696 CrossRef CAS PubMed.
- F. Xu, L. Peng, A. Orita and J. Otera, Org. Lett., 2012, 14, 3970–3973 CrossRef CAS PubMed.
- B. Esser, Phys. Chem. Chem. Phys., 2015, 17, 7366–7372 RSC.
- J.-J. Shen, J.-Y. Shao, X. Zhu and Y.-W. Zhong, Org. Lett., 2015, 18, 256–259 CrossRef PubMed.
- T. Imamoto, N. Takiyama, K. Nakamura, T. Hatajima and Y. Kamiya, J. Am. Chem. Soc., 1989, 111, 4392–4398 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- G. Zhang and C. B. Musgrave, J. Phys. Chem. A, 2007, 111, 1554–1561 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 34, 7406 CrossRef.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- B. D. Rose, L. E. Shoer, M. R. Wasielewski and M. M. Haley, Chem. Phys. Lett., 2014, 616–617, 137–141 CrossRef CAS.
- B. W. D'Andrade, S. Datta, S. R. Forrest, P. Djurovich, E. Polikarpov and M. E. Thompson, Org. Electron., 2005, 6, 11–20 CrossRef.
- R. Gleiter and G. Haberhauer, Aromaticity and Other Conjugation Effects, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed.
- X. Yang, X. Shi, N. Aratani, T. P. Goncalves, K.-W. Huang, H. Yamada, C. Chi and Q. Miao, Chem. Sci., 2016, 7, 6176–6181 RSC.
- R. Ahlrichs, M. Bär, H. Marco, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details including syntheses, characterization and X-ray crystallographic data as well as details on DFT calculations. CCDC 1496147–1496152. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00487c |
| ‡ (TD)DFT calculations were performed with the TURBOMOLE 6.6 program package (TURBOMOLE, University of Karlsruhe, Germany, 2012, http://www.turbomole.com).53 |
| § A value of 0.84 was reported for the electronically very similar substituent CHC(CN)(CN).36 |
| ¶ Large peak separations might be due to a temperature effect (CVs were measured in the glove box at 25–30 °C) or the specific cell setup. |
| || CCDC numbers 1496147 (3a), 1496148 (3b), 1496149 (3d), 1496150 (3e), 1496151 (3f), and 1496152 (5c) contain the supplementary crystallographic data for this paper. |
| This journal is © the Partner Organisations 2017 |