
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticellular tumor spheroids: a relevant 3D model for the in vitro preclinical investigation of polymer nanomedicines
Gianpiero
Lazzari
 ,
Patrick
Couvreur
and
Simona
Mura
*
,
Patrick
Couvreur
and
Simona
Mura
*
Institut Galien Paris-Sud, UMR 8612, CNRS, Univ Paris-Sud, Université Paris-Saclay, Faculté de Pharmacie, 5 rue Jean-Baptiste Clément, F-92296 Châtenay-Malabry cedex, France. E-mail: simona.mura@u-psud.fr
First published on 15th May 2017
Abstract
The application of nanotechnology to medicine, usually termed nanomedicine, has given a crucial impulse to the design of various drug-loaded nanocarriers driven by the aim to overcome the limits associated with traditional drug delivery modalities, in particular, in the field of cancer treatment. However, an appropriate preclinical evaluation of the real therapeutic potential of nanomedicines suffers from the lack of relevant models that are well representative of the human disease and good predictors of the therapeutic response in patients. In this context, great emphasis has been directed toward 3D tumor models aiming to surmount the insufficient predictive power of traditional 2D monolayer cultures of cancer cells. This review focuses on multicellular tumor spheroids (MCTS), which are currently the most widely employed 3D tumor model in preclinical studies. After a brief discussion on spheroid construction strategies and analytical/imaging techniques employed in experimental settings, the application of 3D MCTS to the evaluation of nanomedicines displaying various physico-chemical properties is reviewed. Finally, relevant examples of scaffold and microfluidic systems in which MCTS have been included are described.
 Gianpiero Lazzari | Gianpiero Lazzari graduated in Nanobiotechnology from the University of Salento (Italy) in 2015 and then he joined the group of Prof. Patrick Couvreur (Institut Galien Paris-Sud) at the University Paris-Sud (Chatenay-Malabry, France) as a PhD researcher with a Marie Skłodowska-Curie fellowship in the framework of the NABBA project (NAnomedicines to overcome Biological BArriers and to treat severe diseases). His research focuses on the development of relevant in vitro 3D and in vivo models of pancreatic cancer for preclinical evaluation of nanomedicines. |
 Patrick Couvreur | Patrick Couvreur is a Full Professor of Pharmacy at the Paris-Sud University (France). He held the Innovation Technologique chair (2009–2010) at the prestigious Collège de France and received an ERC Advanced Grant (2010–2015). Patrick Couvreur's contributions in the field of nanomedicine are highly recognized and respected around the world (H-index 84 and over 28 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 citations). His research is interdisciplinary, being at the interface between physico-chemistry of colloids, polymer chemistry, materials science and pharmacology with applications in oncology and neurosciences. He is a member of several Academies both in France and abroad.
000 citations). His research is interdisciplinary, being at the interface between physico-chemistry of colloids, polymer chemistry, materials science and pharmacology with applications in oncology and neurosciences. He is a member of several Academies both in France and abroad. Simona Mura | Dr Simona Mura gained her degree in Pharmaceutical Chemistry and Technology in 2005. In 2009, she was awarded her PhD in Chemistry and Technology of Drugs at the University of Cagliari, Italy, working on the design and in vitro evaluation of novel vesicular systems for the topical delivery of drugs. In 2008 she joined the group of Prof. Elias Fattal (UMR CNRS 8612) at the University Paris-Sud, Chatenay-Malabry, France, as a post-doctoral research assistant to study the lung toxicity of biodegradable nanoparticles designed for pulmonary administration of drugs. In 2011 she was appointed Associate Professor within the framework of the “CNRS-Higher Education chairs (Chaire d'excellence)” programme, in the group of Prof. Patrick Couvreur (UMR CNRS 8612) at the University Paris-Sud, Chatenay-Malabry, France. Her research focuses on the design of novel lipid and biopolymer-based nanoscale drug delivery systems for the treatment of severe neoplastic diseases and on the development of valuable 3D in vitro models for the assessment of their therapeutic efficacy. |
1. Introduction
The efficacy of conventional therapies is often limited by a non-specific cell/tissue distribution of drugs, and their rapid metabolization and/or excretion from the body. In this context, nanoscale systems for drug delivery (i.e., nanomedicines) have received in the last few decades tremendous attention because they hold the potential to overcome these limits, providing a solution to medical challenges that urgently require novel therapeutic strategies.1–3 Nanomedicines can indeed improve the therapeutic index of the transported drug by (i) offering protection from degradation, (ii) enabling its controlled release and distribution and (iii) increasing its bioavailability. In particular, with cancer being one of the leading causes of death worldwide, much work has been done in this field with the aim of proposing more efficient treatments.Accordingly, a plethora of well-engineered nanomedicines surface-modified with cell targeting ligands4 or endowed with stimuli-responsiveness5 have been designed. However, despite the encouraging results observed in preclinical experimental models as well as in clinical trials,6,7 the introduction of nanoscale drug delivery systems in clinical practice is not straightforward,8,9 and only a limited number of nanomedicines have reached the marketplace. These include, for instance, lipid and natural protein-based nanomedicines such as Doxorubicin (Doxo)-loaded liposomes (i.e., Doxil®, Myocet®) and paclitaxel albumin-bound nanoparticles (NPs) (i.e., Abraxane®). It is noteworthy that so far no nanomedicine based on synthetic polymers has been introduced in the market despite the high versatility offered by the macromolecular synthesis and the possibility of opportunely tuning the polymers’ properties (e.g., composition, structure, functionalization, degradability etc.).10–13
It has to be noted that reaching the biological targets and ensuring sufficient drug delivery and accumulation is extremely challenging as a consequence of the multiple biological barriers that characterize tumors and their microenvironment, which are crossed by nanomedicines either at low efficiency or not at all.14,15 However, such biological transport barriers are generally not taken into account during the in vitro preclinical evaluation of nanomedicines, the majority of which is still routinely carried out in two dimensional (2D) monocultures of isolated cancer cells. Despite their relative ease of handling, these cultures do not show any structural architecture and lack the complex physiology and the microenvironment of real tumor tissues, which consist of different cell types (e.g., fibroblasts, macrophages, endothelial cells, immune cells) embedded in an extracellular matrix (ECM) mainly composed of fibrous proteins and proteoglycans.16–18 Cells cultured in 2D monolayers display altered gene expression and activation of signaling pathways when compared to cells grown in the native tumor tissue.19 Moreover, growing in a single layer, they do not replicate either (i) the cell-to-cell and cell-to-ECM interactions20–22 or (ii) the oxygen, nutrients and pH gradients,23 which play a crucial role in tumor progression, chemoresistance and metastatic spread.24
Accordingly, there is an urgent need for more relevant models capable of closely mimicking the heterogeneity and the microenvironment of the in vivo conditions, thus allowing a more predictive in vitro evaluation of nanomedicines. In this context, three dimensional (3D) culture models such as multicellular spheroids (MCTS), polymer scaffolds and microfluidic systems have been proposed as an alternative approach to overcome the aforementioned limitations.25–32
3D tumor models are indeed capable of recapitulating some key features of real tumors, thus representing a valuable tool for (i) a more accurate preclinical screening of nanomedicines and (ii) the identification of candidates with the highest chances of success, which would be further evaluated in vivo. According to their predictive capacity, 3D models would enable to limit the number of animals required in preclinical studies allowing to abide by the 3Rs guidelines.33,34
Although the application of 3D models for a systematic assessment of the therapeutic efficacy of nanomedicines is still at the beginning, their superiority to 2D models is nowadays clearly acknowledged. This review will provide the reader with an overview of the application of multicellular tumor spheroids, the widest employed 3D tumor model so far, for the evaluation of nanomedicines, highlighting their usefulness as a discriminating tool. In agreement with the scope of the journal, attention has been focused on drug delivery systems either made of polymers or in which polymers have been used for surface modifications.
2. 3D multicellular tumor spheroids
Multicellular tumor spheroids are scaffold-free spherical self-assembled aggregates of cancer cells displaying an intermediate complexity between 2D in vitro cell cultures (i.e., cell monolayers) and in vivo solid tumors with which they share important similarities. Since their first introduction in the early 1970s by Sutherland and coworkers,35,36 the application of multicellular tumor spheroids in drug discovery has grown exponentially, offering the possibility of screening a large variety of different molecules,37–39 and they are currently considered a suitable 3D model for drug evaluation in the oncology field.37,40The physiological communication and the signaling established between cells growing in close contact make possible to reproduce in spheroids key aspects of the tumor and its microenvironment such as: (i) different cell proliferative rates, (ii) specific gene expression, (iii) deposition of ECM components, (iv) cell-to-cell and cell-to-microenvironment interactions and (v) drug resistance.40–42 Still, one limitation of MCTS is the fact that they mimic only the avascular region of in vivo tumor tissues, thus leaving out relevant aspects of real tumorigenesis such as the surrounding vasculature, the immune system components and the fluid dynamics.38,41
Large spheroids (∼400–500 μm diameter) display an internal layered cell distribution analogous to that observed in solid tumors. This is the result of mass transport limitations, which interfere with the diffusion of oxygen, nutrients and metabolic wastes through the spheroid creating specific gradients.43,44 Accordingly, thanks to the easier access to oxygen and nutrients, highly proliferating cells are located in the external layer of spheroids and correspond to tumor cells close to capillaries in vivo. Conversely, quiescent cells characterize the middle layer, because cell metabolism decreases progressively with the increasing distance from the spheroid periphery. Finally, a critical situation characterizes the spheroid core in which the oxygen depletion (i.e., hypoxia), the nutrient shortage and the metabolic waste accumulation result in cell necrosis (Fig. 1).40,43–45 The organization in cell layers and the presence of diffusive gradients, acting as microenvironmental stresses, force the inner cells to a specific metabolic adaptation,43 responsible for the observed impaired therapeutic efficacy of various anticancer drugs or drug-loaded nanocarriers.40,46 For instance, cells in the hypoxic region are resistant to drugs which promote cellular death through reactive oxygen species, while the existence of necrotic and quiescent cells reduces the therapeutic efficacy of drugs active against the proliferating ones.40 Another important feature of MCTS is the presence of a network of structural (i.e., collagen and elastin) and adhesive (i.e., fibronectin and laminin) ECM proteins embedded in a gel of glycosaminoglycans and proteoglycans.40,42 This microenvironment acts as a regulating factor influencing cell proliferation, differentiation and tumor growth.41,43 In addition, it closely mimics the physical barriers found in real solid tumors, which obstruct the free penetration of drugs through the whole mass.40
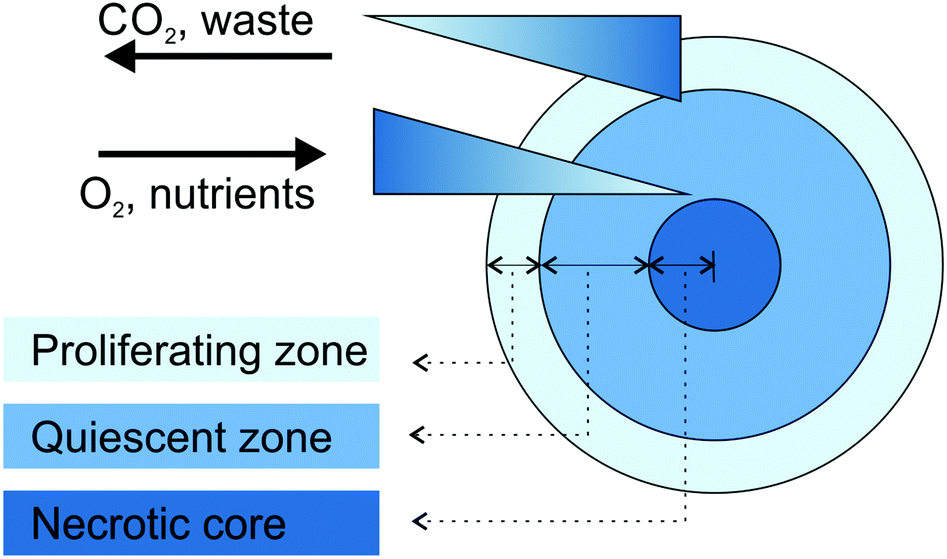 | ||
| Fig. 1 Schematic representation of cell organization and gradients in tumor spheroids. Adapted with permission from ref. 44. Copyright 2008 Wiley-VCH. | ||
Undoubtedly, the presence of this tumor microenvironment represents an advantage compared to the conventional monolayer cultures although it has to be noted that ECM components found in MCTS present a different cellular origin compared to those present in the in vivo tumor tissues. In the former, they are secreted by the same cancer cells forming the spheroids while in the latter they are secreted by tumor-associated fibroblasts (TAF).42 To face this issue, spheroids composed of cancer cells and components of the supportive stroma have been recently proposed.47,48 These hetero-type multicellular spheroids better reproduce the cellular heterogeneity of the tumor tissues and are expected to allow a more reliable evaluation of cancer cell–microenvironment interactions and their impact on the therapeutic outcomes.43,47–49
2.1. 3D MCTS culture methods
Different techniques for spheroid construction are currently available and many of them have been optimized for large-scale production under highly reproducible conditions.40 Principally, they involve the use of cell attachment-resistant surfaces or physical forces to promote cell-to-cell interactions and support the 3D spheroid formation (Fig. 2). These techniques will be briefly discussed in the following paragraphs while for a more detailed and extensive description the interested reader could refer to recently published reviews.38,41,45,50,51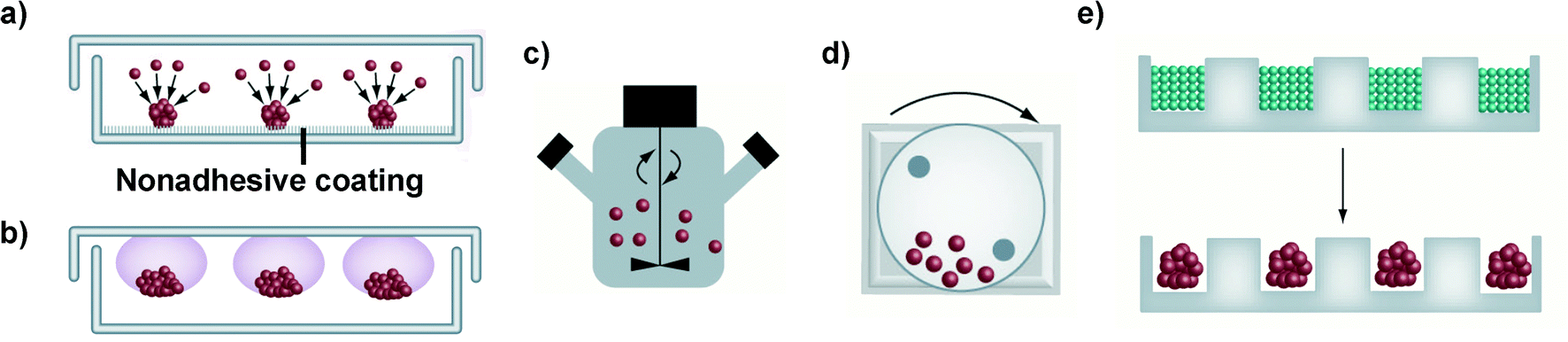 | ||
| Fig. 2 Schematic representation of the main techniques used for spheroid construction: (a) liquid overlay; (b) hanging drop method; (c) spinner flask; (d) NASA bioreactor; (e) micromolding microwells. Adapted with permission from ref. 50. Copyright 2013 American Chemical Society. | ||
It has to be noted that only cell lines that can withstand high shear stress can be cultured in spinner flasks.38,50 For less resistant cells types, the NASA Rotary Cell Culture System characterized by lower shear forces, thanks to the simultaneous rotation of the culture chamber together with its content, should be instead preferred.38,44 However, both approaches require specialized equipment and a large amount of culture media, which therefore limits their wide application.38
Similarly, the formation of cellular aggregates in a controlled environment and with high simplification of the liquid handling procedures has been obtained with microfluidic systems containing various micro-sized chambers and channels.45,50,102 Requiring only limited amounts of cells, media and reagents they are extremely attractive and convenient for drug screening applications; however, the impossibility to retrieve and extensively characterize the formed spheroids is a general drawback of these approaches.38,111 A further level of complexity is achieved with microfluidic devices displaying (i) distinct compartments loaded with different cell types (e.g., epithelial cells, endothelial cells and fibroblast), (ii) collagen gel inserts and (iii) variously shaped channels, which ensure the cell-to-cell chemical communication, thus mimicking the complex in vivo-like organizations.111–113 Nevertheless, to date, the complexity and the costs of the equipment required for their use have hindered their wide application in the preclinical investigation of nanomedicines.
2.2. End-point assessment in 3D MCTS
Thanks to the capacity to recreate key features of real tumors, 3D MCTS have been largely used for the assessment of the efficacy of various therapeutic strategies. In this context, assays and detection methods specific for 3D cultures are highly required to carry out an accurate and predictive evaluation (Table 1). Their detailed description being outside the scope of this review, the most common techniques are only summarized in the next paragraphs, while a comprehensive presentation can be found in recently published articles.37,39–41,45| Analyzed parameter | Assay/detection methods | Description/principle | Ref. |
|---|---|---|---|
| a Preliminary cell dissociation required. b Quantification of metabolically active cells. c Imaging of intact spheroids. | |||
| Cell viability/cytotoxicity | 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT)a,b | Evaluation of intracellular metabolic activity | 71 |
| Reduction of the MTT tetrazolium salt into an insoluble formazan product by the mitochondrial NADPH dehydrogenases | |||
| Absorbance measurement at 570 nm | |||
| 2-(2-Methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2-tetrazolium (WST-1)a,b | Evaluation of intracellular metabolic activity | 122 and 123 | |
| Reduction of the WST-1 into a water soluble formazan product by NADH dehydrogenase and plasma membrane electron transport | |||
| Absorbance measurement at 450 nm | |||
| 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)a,b | Evaluation of intracellular metabolic activity | 65 and 88 | |
| Reduction of the MTS tetrazolium salt into an water soluble formazan product by the mitochondrial NADPH dehydrogenases | |||
| Absorbance measurement at 490 nm | |||
| AlamarBlue®a,b | Evaluation of intracellular metabolic activity | 110 and 146 | |
| Reduction of resazurin to resorufin | |||
| Fluorescence measurement. Ex 540–570 nm/Em 580–610 nm | |||
| Acid phosphatase (APH)b | Quantification of APH activity | 82, 95, 115 and 124 | |
| Hydrolysis of p-nitrophenyl phosphate in p-nitrophenol by the APH enzyme | |||
| Absorbance measurement at 405 nm | |||
| Adenosine triphosphate (ATP)b | Measurement of the intracellular ATP content | 66, 67, 120 and 125 | |
| Oxidation of luciferin by the luciferase enzymes in the presence of intracellular ATP and emission of bioluminescence | |||
| Lactate dehydrogenase (LDH)a | Evaluation of membrane integrity | 53 and 136 | |
| Measurement of the conversion of lactate into pyruvate via NAD+ reduction by LDH. The resulting NADH catalyzes the reduction of a tetrazolium salt to a formazan product | |||
| Absorbance measurement at 450 nm | |||
| DNA quantificationa | Quantification of total cell number | 90 and 91 | |
| Hoechst 33258 staining | |||
| Fluorescence measurement following lyophilization and lysis of spheroids | |||
| Growth inhibition | Optical microscopyc | Measurement of morphometric parameters (e.g., mean diameter, minimum diameter, maximum diameter, volume, area and circularity) | 52, 55–58, 60–65, 70, 73, 74, 77, 84, 89–93, 95, 107, 110 and 114–126 |
| Morphological effects | Scanning electron microscopy (SEM)c | Assessment of spheroid integrity | 46, 55, 57, 63, 64, 70 and 73 |
| Cell death/apoptosis | Trypan Blue exclusiona | Quantification of living cells | 46, 70, 118, 126 and 131 |
| Trypan Blue staining of dead cells | |||
| Live/dead staining | Identification and quantification of live and dead cells | 46, 70, 118 and 133 | |
| Staining with calcein-acetoxymethyl (calcein-AM) and intercalating agents (e.g., propidium iodide (PI) or ethidium homodimer (EthD-1)). Live cells are stained in green following intracellular cleavage of the acetomethoxy group of calcein-AM. Dead cells are stained in red following penetration of the intercalating agents through their permeable membrane | |||
| AnnexinV-FITC staining | Detection of the apoptosis marker phosphatidyl serine on the cell membrane surface with Annexin V-FITC. Used in combination with PI staining to distinguish apoptotic and necrotic cells | 96, 133 and 134 | |
| Caspase-3 activation | Quantification of caspase-3 activity via measurement of fluorescent emission of activate-caspase-3 substrates | 54 and 118 | |
| Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL) | Quantification of DNA fragmentation as a marker of late apoptosis. Detection of double stranded DNA breaks via TdT-mediated incorporation of labeled dUTP to their blunt ends | 56 and 136 | |
| Penetration/uptake of fluorescently labeled nanocarriers and dyes | Confocal Laser Scanning Microscopy (CLSM)c | Imaging of nanocarrier diffusion ability | 48, 53–60, 62–64, 66–69, 72, 74–76, 78–80, 83, 84, 89–91, 94, 95, 105–109, 115, 117–119, 122–126, 131, 133, 134, 136, 137 and 146–157 |
| Pinhole-equipped microscope to remove out-of-focus light and increase optical resolution | |||
| Light penetration depth limited to 100–150 μm from the spheroid surface | |||
| Two-photon microscopy (TPM) and multi-photon microscopy (MPM)c | Imaging of nanocarrier diffusion ability | 70, 81, 85, 120, 137 and 141 | |
| Sample excitation with pulsed long-wavelength photons | |||
| Increase of the image resolution and depth of penetration (up to 500–800 μm) | |||
| Fluorescence-activated cell sorting (FACS)a | Quantification of cell fluorescence | 71, 78, 85, 122, 134 and 136 | |
| Fluorescence microscopy | Imaging of nanocarrier diffusion ability | 46, 71, 88, 96–98, 138 and 139 | |
| Imaging of fixed, optimal cutting temperature (O.C.T.)-embedded spheroids cross-sections | |||
Optical microscopy is the simplest and most widely used technique for the visual assessment of morphological changes in spheroids.52,55–58,60–65,70,73,74,77,84,89–93,95,107,110,114–126 Following exposure to various treatments, the measurement of the variation of morphometric parameters (e.g., mean diameter, minimum diameter, maximum diameter, volume, area and circularity) of spheroids from bright field images is used for a quantitative analysis of the extent of growth inhibition.37,87 After interruption of the treatment, the spheroid growth delay might be calculated in comparison with untreated samples as the time lag required to reach the quintuple spheroid volume of day 4 (5 × Vd4; assuming Vd4 as the starting spheroid volume at the onset of treatment).87
The assays currently used in 2D monolayer cultures as extensively validated methods to assess the drug cytotoxicity mainly rely on the measurement of (i) cell membrane integrity (i.e., LDH assay)127 or (ii) intracellular metabolic activity (e.g., MTT, WTS-1, AlamarBlue® assays).128 Some of them have been adapted to spheroids and are currently commercially available. Among them, the quantifications of the acid phosphatase (APH) activity and the intracellular adenosine triphosphate (ATP) level are well-suited assays that do not require preliminary dissociation of spheroids into single-cell suspensions.37,45 The APH assay is a simple and inexpensive technique based on the quantification of cytosolic acid phosphatase activity in viable cells through the measurement of the absorption of the p-nitrophenol, obtained by APH hydrolyzation of p-nitrophenyl phosphate, at 405 nm using any standard plate detection reader.37,129 The APH assay has been validated to measure spheroid viability and cytotoxic effects of both free drugs87,129 and drug-loaded nanocarriers.82,95,115,124
The ATP assay exploits the oxidation of the luciferin, the substrate of the firefly luciferase enzymes, in the presence of O2, Mg2+ and ATP. This reaction brings the luciferin to an electronically excited state, which then decays with the emission of a photon of light (i.e., bioluminescence). Thus, when substrate and enzyme are added to cell cultures the light output, which is directly proportional to the intracellular ATP content (marker of metabolically active cells), allows to quantify the number of viable cells. Ready-to-use assay kits, which exploit the high sensitivity and the low background of the bioluminescence signal45 (e.g., CellTiter-Glo® 3D Cell Viability Assay (Promega); Molecular Probes® ATP Determination Kit (Invitrogen)), are currently available. The CellTiter-Glo® 3D Cell Viability Assay represents a time-effective and well standardized assay since the addition of a single reagent directly to the 3D cell culture results in cell lysis and simultaneous generation of the bioluminescence.130 This assay is especially formulated with a robust lytic capacity to overcome 3D MCTS obstacles such as tight cell–cell junctions and the presence of the extracellular matrix.130 The relatively simple workflow and data analysis make this ATP assay scalable to high-throughput screenings of drug-loaded nanocarrier efficacy.125
Qualitative assessment of proliferating or dead/necrotic cells can be performed by Confocal Laser Scanning Microscopy (CLSM) imaging following spheroid incubation with various live/dead reagents.46,118,131 Preliminary spheroid dissociation into single cells is instead required for a quantitative measurement by Fluorescence-Activated Cell Sorting (FACS).70 Staining cell dispersions of dissociated spheroids with fluorescently labeled Annexin V (AnnexinV-FITC) and propidium iodide (PI) has also been largely used to detect early apoptotic and dead/necrotic cells, respectively.96,132–134 Apoptosis can be detected also on spheroid cross-sections by using the Terminal deoxynucleotidyl Transferase (TdT) dUTP Nick-End Labeling (TUNEL) method. The TdT recognition of the blunt ends of double stranded DNA breaks, which characterizes the late stages of apoptosis,135 catalyzes the addition of biotinylated dUTPs that are then visualized using streptavidin-conjugated detection agents (i.e., peroxidase or fluorescent markers).56,136
Monitoring of fluorescent drugs and dye-labeled nanocarriers by CLSM has been largely applied for evaluating their uptake and penetration in individual spheroids.63,66,69,76,95,105,133 Unfortunately, compared to conventional 2D cultures, the 3D structure represents a technical challenge for conventional instrumentation. Indeed, the increases of spheroid size and thickness result in a loss of image quality due to the scattering, reflection and absorbance of light.37,41 The layered structure of spheroids limits the inner scanning depth and, as a consequence, the penetration ability of different systems can be compared only in a portion of the spheroid generally corresponding to a depth of 80–100 μm from the spheroid surface.63,76,95,131,137 Although more time consuming, better information on the real depth of penetration can be provided by the acquisition of fluorescent images from serial sections obtained from fixed spheroids.46,71,88,96–98,138,139
Compared to traditional CLSM, significant improvements in the evaluation of the penetration into living tumor spheroids can be obtained with Two-Photon (TPM) or Multi-Photon microscopy (MPM) in which the energies of two (or more) photons are combined to promote the transition of a fluorescent marker to an excited state.140 The use of an excitation wavelength in the NIR region (700–1000 nm) offers the possibility of increasing the laser penetration into 3D MCTS (up to 500–800 μm according to the instrumentation) with low phototoxic effects.50,140 These techniques have already been applied to visualize with high resolution the diffusion through spheroids of various polymer-based nanocarriers.70,81,85,120,137,141 Imaging of the cellular processes and drug delivery in spheroids with an even higher 3D isotropic resolution and with limited photodamage is the promise of Light-Sheet Fluorescent Microscopy (LSFM) methods such as the Selective Plane Illumination Microscopy (SPIM)47,142,143 method already applied for the imaging of large biological samples.144,145 However, so far, LSFM methods have not found any application in the preclinical investigation of polymer nanomedicines.
2.3. Use of 3D MCTS to screen polymer nanocarriers with different physico-chemical properties
The distinguishing characteristics of polymer nanocarriers, such as chemical composition, size, shape and surface properties, might strongly affect their capacity to diffuse into tumors and therefore have a profound impact on their anticancer efficacy.158 Accordingly, these parameters must be taken into account during the design and evaluation of any novel nanomedicine. In this context the 3D MCTS, thanks to their similarity in morphology and biological microenvironment to solid tumors, have already been used as a robust tool for easy polymer nanoparticle screening,70,97 and to accurately predict the in vivo behavior of the nanocarriers as a function of their specific physico-chemical properties.139 The most relevant results will be discussed in the following sections.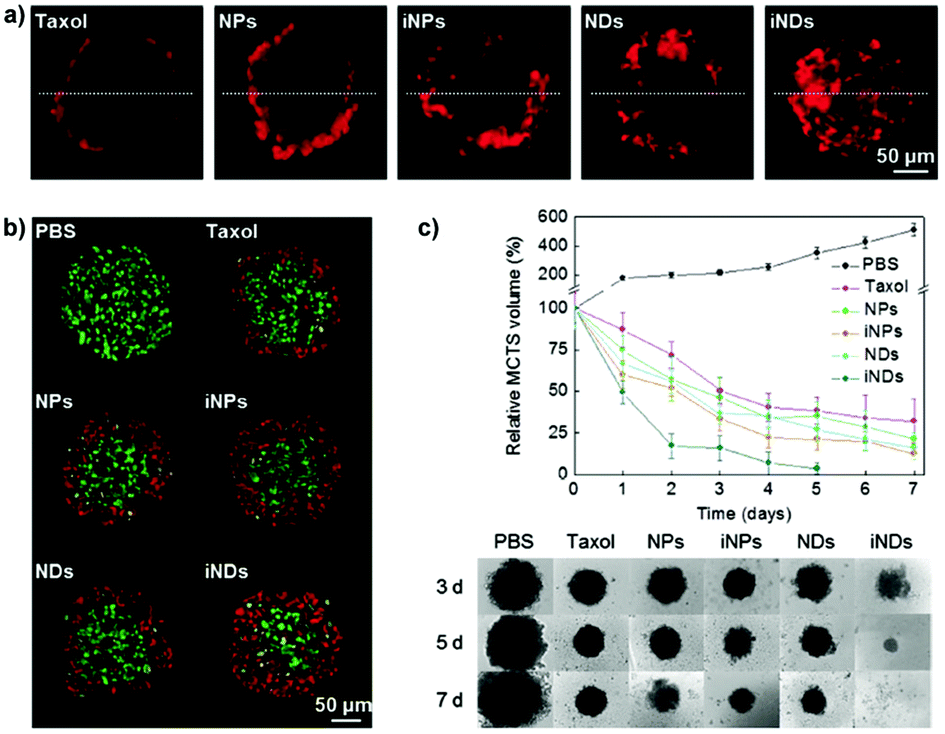 | ||
| Fig. 3 Tissue penetration and antitumor efficacy in 4T1 MCTS of Taxol, ∼70 nm Ptx nanocrystals (NPs), RGD-grafted ∼70 nm Ptx nanocrystals (iNPs), ∼10 nm Ptx nanocrystals (NDs) and RGD-grafted ∼10 nm Ptx nanocrystals (iNDs). (a) CLSM images after 24 h incubation with Cy5-labeled Taxol/NDs/NPs/iNDs/iNPs, (b) CLSM images of TUNEL analysis after 24 h treatment with the different Ptx formulations; dead cells are shown in red and live cells appear in green. (c) Growth inhibitory effect of the different formulations. Reproduced with permission from ref. 56. Copyright 2015 Wiley-VCH. | ||
Despite these results, the identification in vitro of the optimal size of a nanocarrier for achieving the highest drug delivery in vivo is extremely complex because although MCTS closely mimic the tumor tissues, they lack predictive value in terms of pharmacokinetics and biodistribution. Accordingly, the evaluation of nanomedicines in animal tumor models still remains a mandatory step to assess these parameters. It has indeed been shown that the requirements for tumor penetration and tumor retention are often in contradiction with each other. Thus, despite being unfavorable for penetration in the tumor mass, due to their considerable diffusional hindrance,159 nanocarriers with a size of around 100 nm are the most advantageous in improving pharmacokinetics and extravasation.160 On the other hand, smaller nanocarriers show much better penetration in the tumor interstitial space.139,148 However, for extremely small particles (≤5.5 nm), rapid clearance from tumors and short half-life have been observed.161 To face this issue, the ideal drug delivery system should be able to shrink and adapt its size in response to the encountered microenvironment. Interestingly, size-switchable stimuli-responsive nanoparticles able to overcome multiple tumor barriers have been developed by various groups.58,76,119,133,134,138,141 For instance, pH sensitive nanoparticles were formulated by the molecular assembly of platinum (PtIV)-prodrug conjugated polyamidoamine (PAMAM) dendrimers with two amphiphilic polymers containing either ionizable pH-responsive amide bonds (i.e., polycaprolactone-2-propionic-3-methylmaleic anhydride (PCL-CDM), PCL-CDM-PAMAM/Pt nanoparticles)134 or tertiary amine groups (i.e., poly(ethylene glycol)-b-poly(2-azepane ethyl methacrylate) (PEG-b-PAEMA) PEG-b-PAEMA-PAMAM/Pt) (Fig. 4a).133 Size variation as a function of pH has been evaluated on pancreatic cancer multicellular spheroids (BxPC3 cells). At physiological pH the clustered nanoparticles (i.e., pH-sensitive cluster nanobombs (SCNs/Pt)) displayed a size of around 100 nm, while the pH drop (from 7.4 to 6.5–7) in the tumor extracellular space triggered an instantaneous disassembly of these pH sensitive nanoparticles in small Pt-PAMAM prodrugs (≈5 nm) able to penetrate deeply and uniformly into the spheroid mass (Fig. 4b). Then, once internalized, the intracellular redox environment led to the release of the active molecule (i.e., Pt(II) species) resulting in significant cell apoptosis. As expected, a higher cell viability was observed when spheroids were incubated with pH-insensitive nanoparticles (i.e., ICNs), which demonstrated a limited capacity of penetration and drug delivery as a consequence of their stable size (Fig. 4b). Whether such advantage was maintained in vivo was then assessed after an intravenous injection of both nanoparticles in an experimental model of pancreatic cancer (BxPC3 cancer cells) characterized by an important desmoplastic reaction and a limited permeability.162 Tumor accumulation studies confirmed a higher capacity of penetration of pH-sensitive nanoparticles, which diffused in the tumor interstitium, after vessel extravasation, for several hundreds of nanometers. In contrast, the pH-insensitive NPs accumulated in the tumor vessels with little penetration in the tumor mass (Fig. 4c) leading to an inefficient drug delivery. In agreement with the in vitro results, an extensive apoptosis was detected in tumor sections of mice treated with the pH-sensitive nanoparticles, thus confirming the predictive potential of the 3D multicellular spheroids.
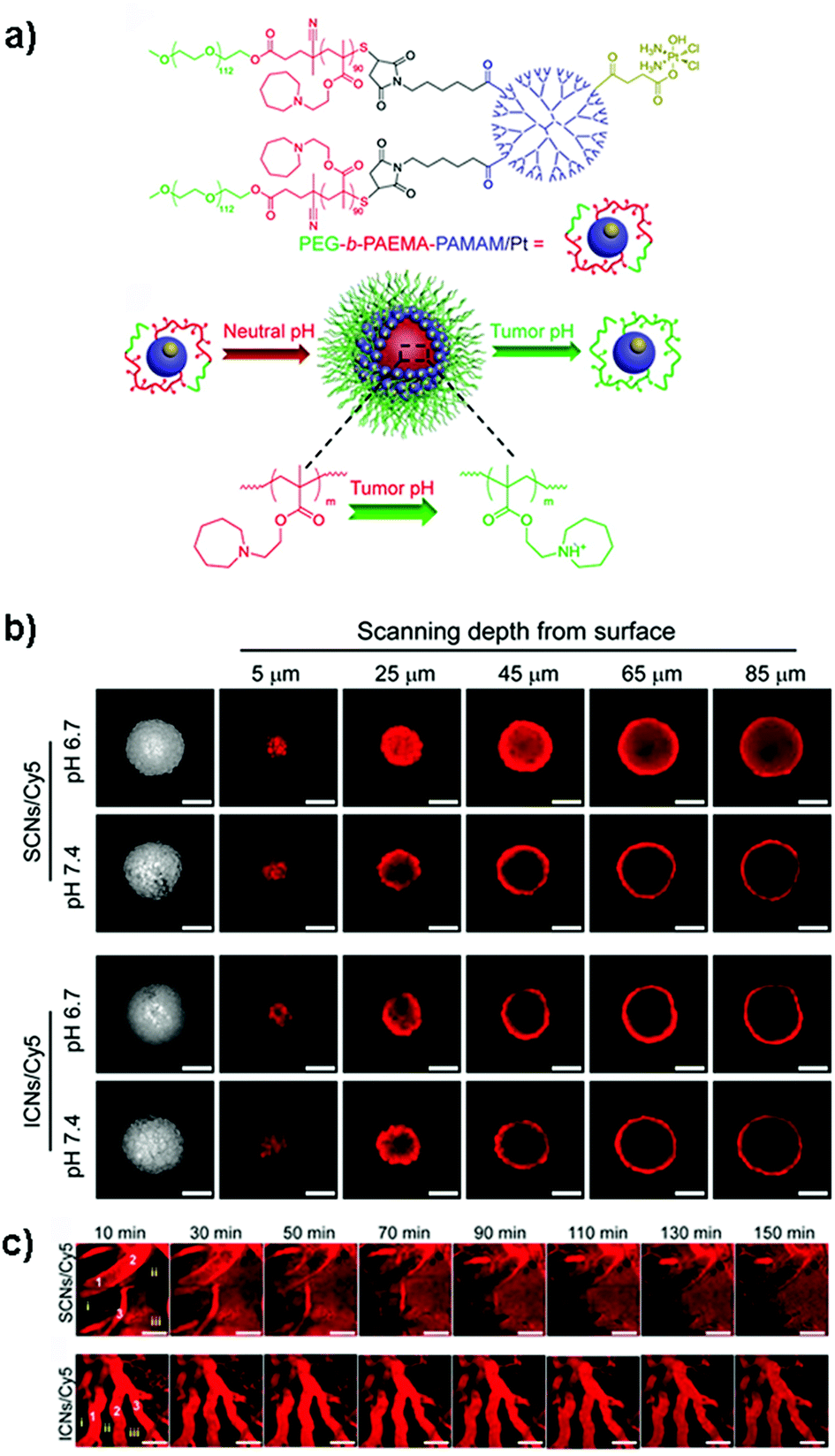 | ||
| Fig. 4 (a) Structure of PEG-b-PAEMA-PAMAM/Pt and schematic illustration showing the self-assembly into the pH-sensitive cluster nanobombs (SCNs/Pt) at neutral pH and the disintegration into small particles at tumor acidic pH. (b) CLSM images showing in vitro penetration of fluorescently-labeled SCNs/Cy5 and ICNs/Cy5 in BxPC-3 multicellular spheroids. Scale bar: 100 μm. (c) In vivo real-time microdistribution of SCNs/Cy5 and ICNs/Cy5 in BxPC-3 xenografts after intravenous administration. Scale bar: 100 μm. Reproduced with permission from ref. 133. Copyright 2016 American Chemical Society. | ||
The influence of length was studied by Stenzel and coworkers, who investigated rod-shaped poly(1-O-methacryloyl-β-D-fructopyranose)-b-poly-(methyl methacrylate) (poly(1-O-MAFru)-b-PMMA)-based micelles, clearly showing that, among the various fructose-coated rod-like micelles, the shortest ones displayed the highest capacity of penetration in a 3D MCTS model of breast cancer cells (MCF-7).147 Similarly, in order to better highlight the role of these parameters, poly(ethylene glycol) diacrylate (PEGDA)-based anionic nanohydrogels were synthesized by jet and flash imprint lithography in the form of disc-shaped nanocylinders and cuboidal nanorods of two different sizes (low and high aspect ratio; aspect ratio (H/D) = height/diameter).85 The resulting nanohydrogels displayed negative charge of ∼−55 mV, which should limit the interactions with cell membranes and serum proteins and promote their penetration into HEK293 spheroids (human embryonic kidney cells). Fluorescence intensity analysis of two-photon microscopy images revealed two fold higher accumulation near the spheroid outer half of the disc-shaped nanocylinders with the lowest aspect ratio (H/D ∼0.3, 325 nm diameter and 100 nm height) (Fig. 5f) compared to both nanocylinders with higher aspect ratio (H/D ∼0.45) (Fig. 5e) and nanorods (Fig. 5g and h). Such a preferential penetration might be attributed to their larger surface contact area, which promoted the interaction with the cells and the diffusion (either passive or active) across the 3D tumor mass.
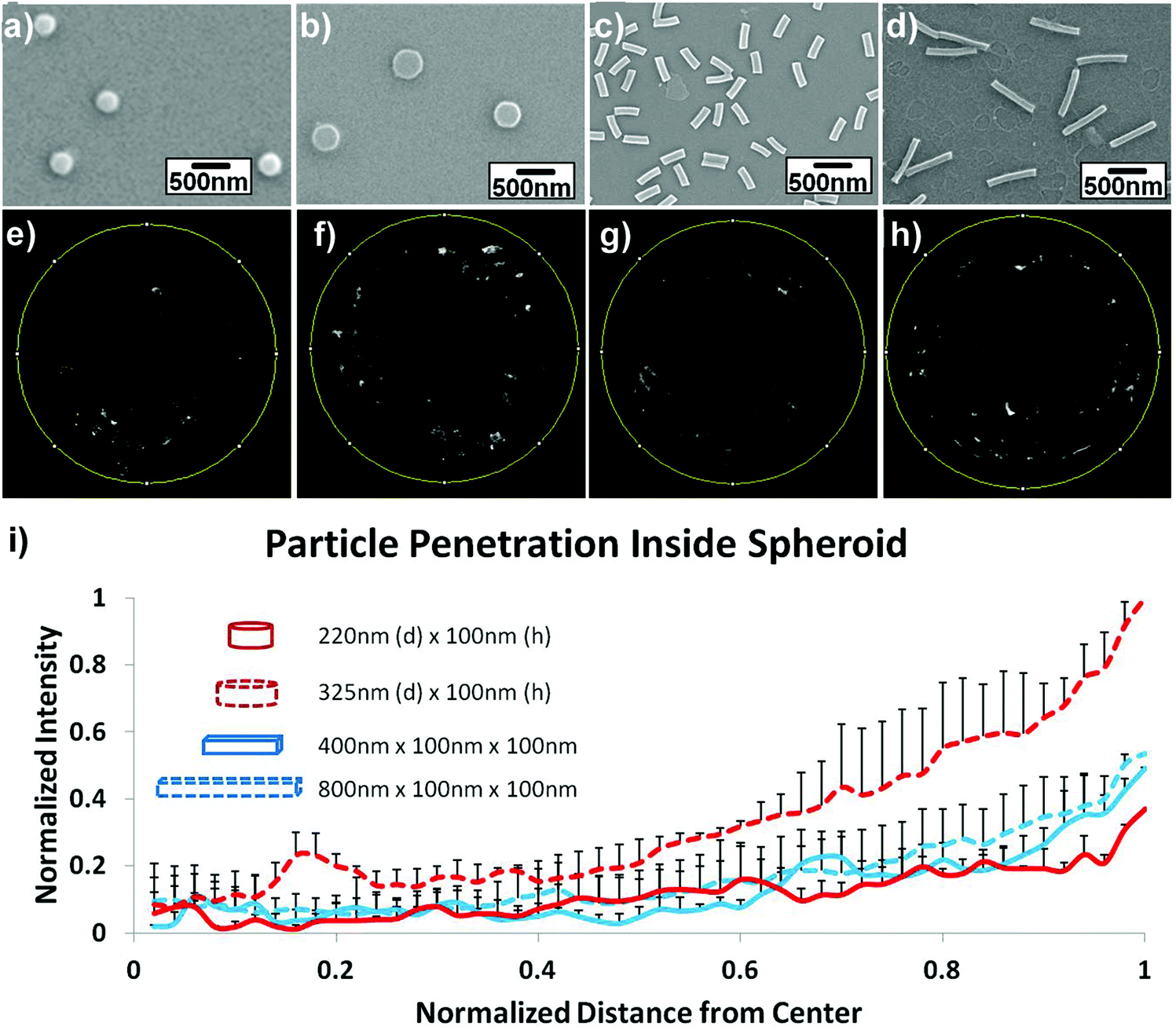 | ||
| Fig. 5 Uptake and penetration of shape specific particles in spheroids. Scanning Electron Microscopy (SEM) images of (a) 220 nm × 100 nm discs (H/D ∼ 0.45), (b) 325 nm × 100 nm discs (H/D ∼ 0.3), (c) 400 nm × 100 nm × 100 nm rods and (d) 800 nm × 100 nm × 100 nm rods. (e–h) Two-photon pictures of spheroids incubated with the discs or rods in the correspondent upper panel. (i) Normalized radial intensity distribution as a function of distance from the center of the spheroid. Reproduced with permission from ref. 85. Copyright 2015 Wiley-VCH. | ||
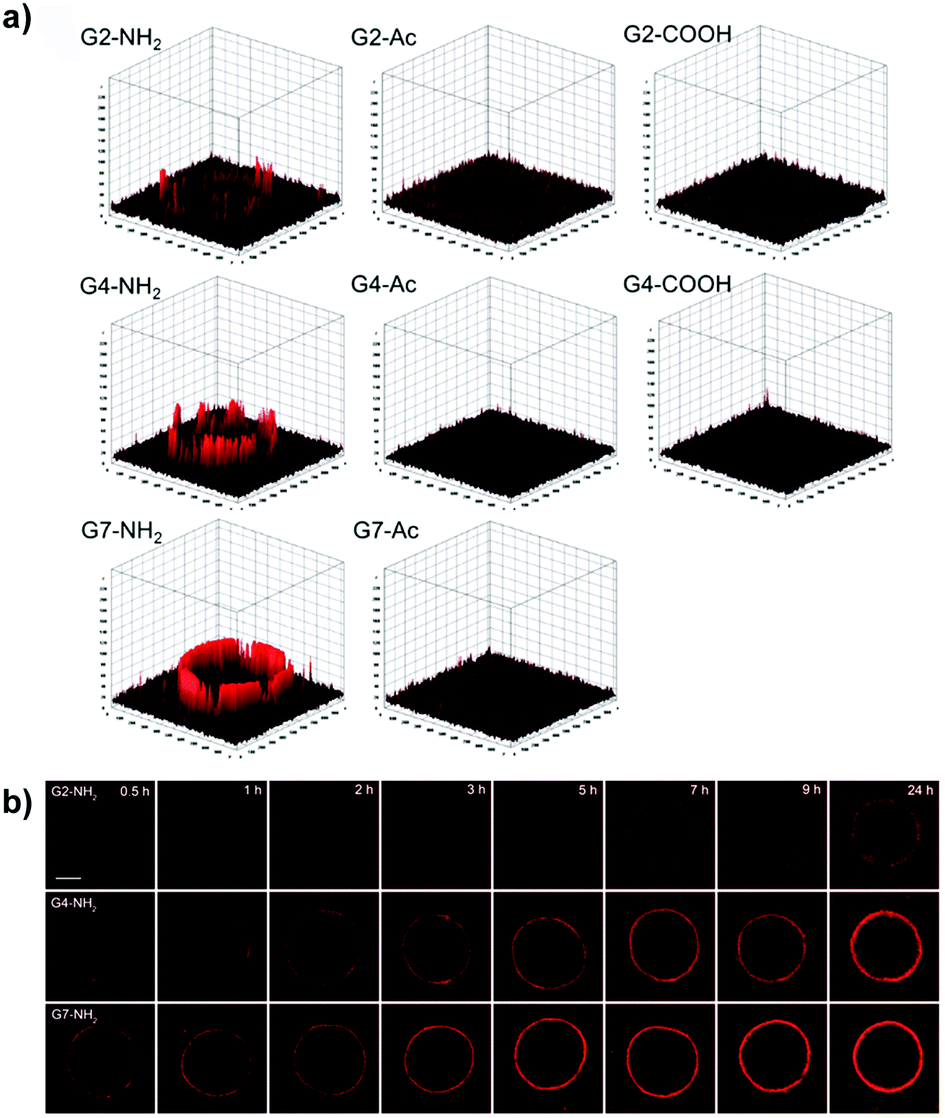 | ||
| Fig. 6 (a) Accumulation and permeation behaviors of G2, G4, and G7 PAMAM dendrimers in MCF-7 MCTS as a function of the surface charge. (b) Tumor penetration of amine-terminated G2, G4, and G7 PAMAM dendrimers. Reproduced with permission from ref. 72. Copyright 2016 American Chemical Society. | ||
The key role of a positive surface charge in the accumulation into multicellular spheroids has been shown also by comparing gold nanorods (AuNRs) (55 nm length × 14 nm diameter) coated with either (i) cetyltrimethylammonium bromide (CTAB) and poly(diallyldimethylammonium chloride) (PDDAC), or (ii) polystyrene sulfonate (PSS) displaying a surface charge of +40–50 mV or −25 mV, respectively.82 When incubated with MCF-7 breast cancer MCTS, the coating with a cationic polymer ensured the highest gold accumulation, which was expected to induce strong photothermal cytotoxicity after NIR irradiation as was observed in 2D monolayer cultures. However, despite their tendency to be largely retained, irradiation of these positively charged AuNRs led to a 40% lower hyperthermia efficacy compared to the negatively charged PSS-coated AuNRs. Such unexpected behavior was related to the penetration capacities of the different AuNRs. Indeed, cationic polymer-coated AuNRs highly accumulated in the spheroids but only in the outer region, and simple surface adsorption was also observed. Moreover, the interaction with negatively-charged serum proteins led to an increase of their size, preventing diffusion. In contrast, the negative charge of PSS-coated AuNRs allowed a more homogeneous distribution in the spheroid core, which resulted in higher viability loss and destruction of the inner compact spheroid structure.
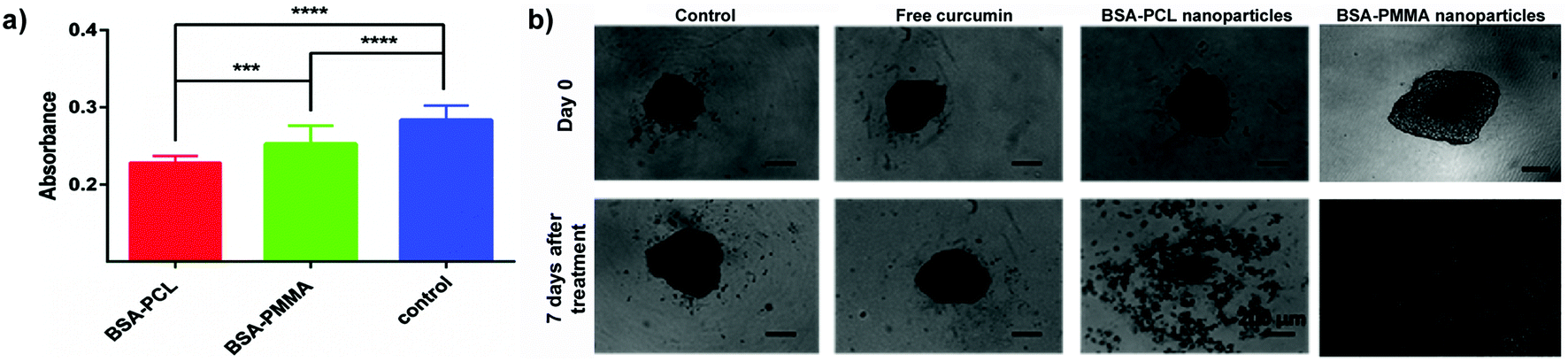 | ||
| Fig. 7 (a) Absorbance values (mean ± standard error) corresponding to the DNA content in LNCaP spheroids treated with curcumin-loaded nanoparticles for 7 days. (Curcumin concentration: 30 μM.) ***p < 0.001. ****p < 0.0001. (b) LNCaP spheroids treated with free curcumin and curcumin-loaded nanoparticles for 7 days. (Curcumin concentration: 150 μM.) Scale bars: 200 μm. Reproduced with permission from ref. 91. Copyright 2016 Royal Society of Chemistry. | ||
Other examples of anticancer drug-loaded biodegradable nanocarriers evaluated on 3D spheroids included milk protein nanoparticles153 and micelles composed of either (i) pseudo block copolymers formed by the assembly of β-cyclodextrin terminated multiarmed poly(N-vinylpyrrolidone) (PVP) and adamantane functionalized polycaprolactone (PCL)155 or (ii) poly (ethylene oxide)-poly [(R)-3-hydroxybutyrate]-poly (ethylene oxide) triblock copolymers.96 The capacity of penetration of these nanosystems in 3D models was consistent with further in vivo experiments, thus supporting the predictive capacity of the tumor spheroids.96,153,155
Whether the crosslink could affect the mechanism and depth of penetration as well as the cytotoxicity of drug-loaded micelles has been recently investigated by the group of Stenzel using pancreatic (AsPc-1 cells) multicellular tumor spheroids.90 Hence, 1,8-diaminooctane-crosslinked poly(N-(2-hydroxypropyl) methacrylamide-co-methacrylic acid)-block-poly(methyl methacrylate) (P(HPMA-co-MAA)-b-PMMA) micelles (CKM) were compared to their uncrosslinked version (UCM) (Fig. 8a). Results revealed that CKM were capable of moving through the cell layers via a transcellular process and delivered higher doxorubicin amounts to the spheroid core which resulted in greater cytotoxicity compared to the UCM (Fig. 8b–d). The latter quickly disassembled after penetration into the cells of the outer layers, releasing the loaded drug and causing cell death. As a result, no further micelle transcytosis could occur. The lower efficacy of the free drug (evaluated in terms of DNA content and inhibition of spheroid growth) correlated with its limited diffusion.90
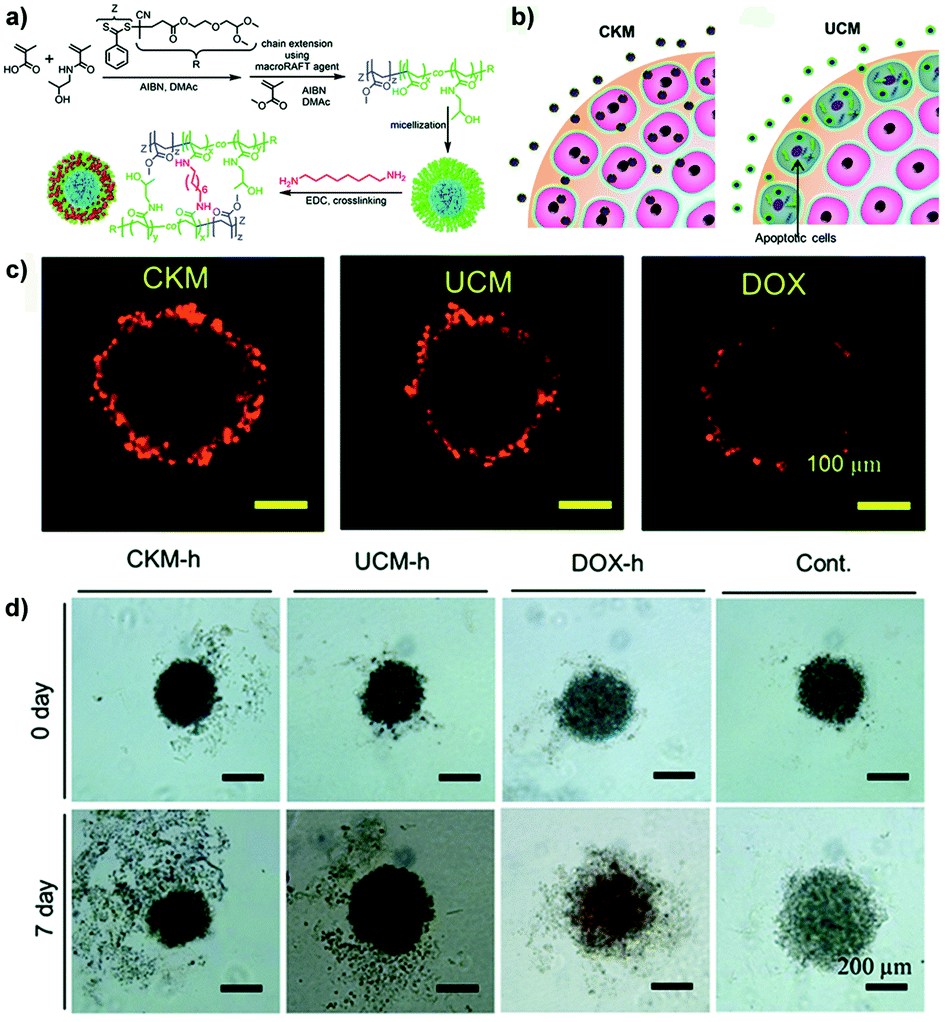 | ||
| Fig. 8 (a) Schematic representation of the synthesis of block copolymer and formation of UCM and CKM. (b) Schematic hypothesis of penetration differences for Doxo-loaded CKM and UCM. (c) Doxo delivery into pancreatic MCTS by CKM and UCM revealed by CLSM. (d) Inhibition of the pancreatic MCTS growth by Doxo-loaded micelles. Microphotographs of pancreatic spheroids before and after treatment with Doxo-loaded micelles, free drug (Doxo-h) or untreated (Cont). Reproduced with permission from ref. 90. Copyright 2015 Royal Society of Chemistry. | ||
Nevertheless, crosslinking is not always the best strategy to improve drug cytotoxicity. Thus, the evaluation of the effect of the reversible disulfide core-crosslinker cystamine in micelles formed by the poly(ethylene glycol methyl ether acrylate)-b-poly(carboxyethyl acrylate) (POEGMEA-b-PCEA) block copolymer95 led to results that were in clear contrast to those previously published by the same group of Stenzel.90,124 Indeed, micelles with the highest level of crosslinking were capable of the deepest penetration in LNCaP prostate multicellular tumor spheroids, but displayed the lowest cytotoxicity. This behavior could be explained by their too compact structure that hindered intracellular reductive agents (e.g., glutathione) from diffusing in the core of the micelles, thus slowing down cross-linked micelle disassembly and release of the loaded drug.95 Compared to the 2D monolayer cultures, the 3D spheroid model would provide a reliable correlation between micelle penetration and cytotoxicity, thus allowing figuring out the real contribution of the crosslinking in terms of drug delivery efficiency.
2.3.6.1. Transferrin-targeted nanocarriers. Overexpression of transferrin receptor (TfR) has been detected in different types of rapidly proliferating tumors.168 Accordingly, opportune surface functionalization with transferrin (Tf) has been applied to efficiently deliver drugs to cancer cells. Such modification clearly enhanced the cellular uptake and penetration depth of polyethylene glycol-phosphatidyl ethanolamine (PEG-PE) micelles67,68 and poly(amidoamine) (PAMAM) dendrimers61 in MCTS models of ovarian carcinoma67,68 and glioma61 compared to the non-functionalized counterparts. More efficient delivery of the loaded drugs (paclitaxel (Ptx)67,68 or doxorubicin (Doxo))61 resulted in a significant inhibition of cell proliferation confirmed by reduction of spheroid volume and metabolic activity (ATP content). A 6-fold reduction of the IC50 value (8.92 μM vs. 1.35 μM) was measured on the 3D spheroids incubated with Tf-functionalized Ptx-loaded PEG-PE micelles (as compared to non-targeted ones) but these values were higher than those observed in 2D monolayers. These results reflect a reduction of efficacy due to the existence in the MCTS of physical barriers to diffusion and a different cell sensitivity due to the 3D spatial organization.67
Reflectance confocal microscopy and synchrotron X-ray fluorescence microscopy (XFM) have been used to monitor, in a quantitative and qualitative manner, the capacity of penetration into 3D breast cancer spheroids (MCF-7 cells) of transferrin-decorated polymer-modified gold nanoparticles (100 nm).105 Images revealed that although functionalization with human Tf increased the amount of internalized NPs compared to controls (i.e., bovine transferrin functionalized particles and naked ones), after 48 h the penetration of functionalized NPs was limited to a depth of 50 μm, thus representing a real issue for further therapeutic applications due to their incapacity to diffuse in the tumor core.105
2.3.6.2. Folic acid-targeted nanocarriers. As previously observed for the transferrin receptor, also the folate receptor is largely expressed in cancer cells, thus making the functionalization with folic acid (FA) a widely applied approach to enhance the ligand-mediated uptake by cancer cells.55,57,94,107 3D tumor spheroids have been employed to assess the influence of the folic acid density at the NP surface on the cell targeting capacity, internalization and tumor penetration of polymer/DNA complexes (polyplexes) made by the assembly of DNA with poly(amidoamine)-poly(ethylenimine) (PME) copolymers conjugated to FA functionalized PEG (PME-(PEG-FA)). FA functionalization should enable overcoming the reduction of cellular uptake caused by the PEG chains (the so-called PEG-dilemma)169 and make these systems valuable tools for efficient gene delivery. Divalent modification (PME-(PEG3.4k-FA2)1.72) resulted in a higher receptor mediated uptake and a better penetration in HEK293 T human embryonic kidney spheroids compared to mono-functionalized (PME-(PEG3.4k-FA1)1.66) and non-functionalized nanocarriers (PME-(PEG3.5k)1.69) (Fig. 9a–c).94 Indeed, although all polyplexes were detected up to 380 μm depth, the divalent FA modification allowed achieving a better cell internalization (Fig. 9d).
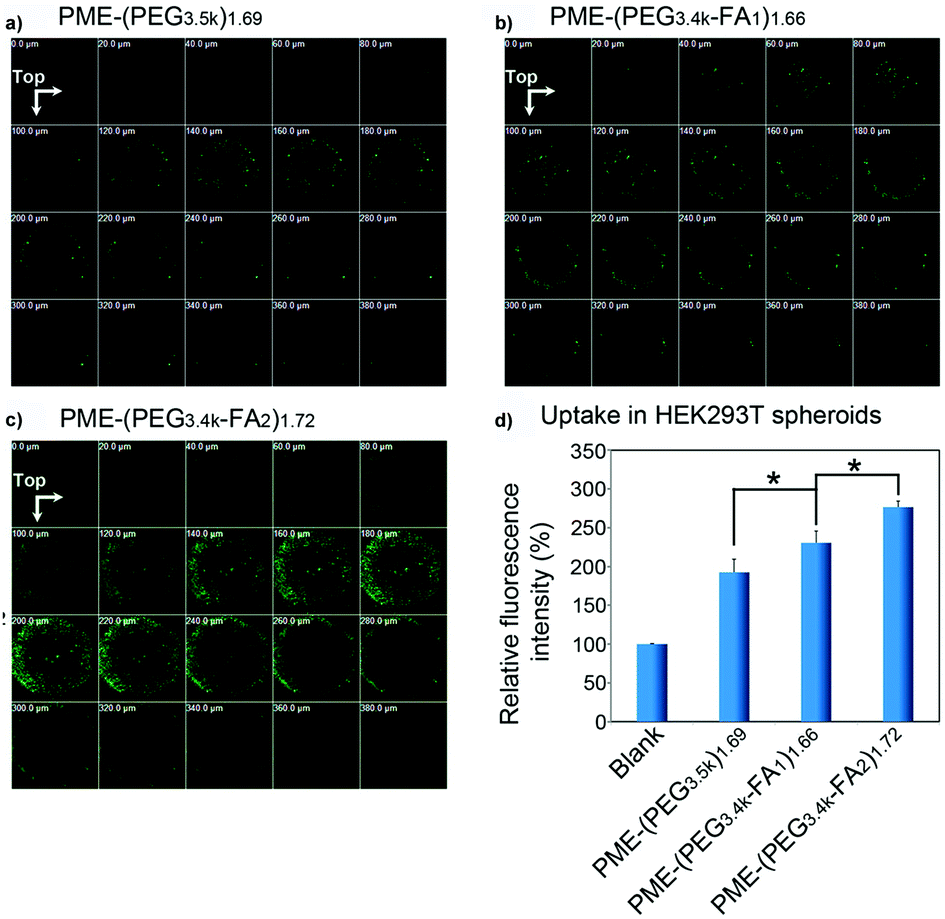 | ||
| Fig. 9 CLSM images of HEK293T multicellular spheroids 22 h after transfection with (a) PME-(PEG3.5k)1.69; (b) PME-(PEG3.4k-FA1)1.66, or (c) PME-(PEG3.4k-FA2)1.72 complexes. (d) Relative fluorescence intensity (treated group/blank) of HEK293T multicellular spheroids treated with PME-(PEG3.5k)1.69, PME-(PEG3.4k-FA1)1.66, or PME-(PEG3.4k-FA2)1.72 complexes for 4 h and for a further 18 h culture. Reproduced with permission from ref. 94. Copyright 2015 American Chemical Society. | ||
The in vitro–in vivo predictive capacity of the 3D models in the evaluation of the FA functionalized nanocarriers was recently reported. Indeed, the higher capacity of penetration in vitro into 3D neuroblastoma spheroids (SH-SY5Y cells) of FA-decorated Doxo-loaded soy protein NPs (SP-NPs)55 or carboxymethyl chitosan-N-3-acrylamidophenylboronic acid (CMCS-PAPBA)57 NPs resulted in the highest inhibition of tumor growth in H22 tumor-bearing mice.
2.3.6.3. Carbohydrate-targeted nanocarriers. The natural affinity of dextran for highly glycosylated surfaces has been exploited for achieving targeted delivery of ald-dex-Doxo nanoparticles (i.e., aldehyde-dextran polymer conjugated to doxorubicin via a pH sensitive bond) to SK-N-BE(2) cells by the interaction with the glycocalyx at their surface.126 Investigation of these NPs in 3D neuroblastoma spheroids (∼400–500 μm) enabled to discern their potential as drug delivery systems, while it was not evident under 2D conditions where the free drug displayed superior cytotoxicity. Such a difference disappeared in the spheroid model in which a drastic reduction of the free Doxo efficacy was observed while that of the ald-dex-Doxo nanoparticles remained unvaried and resulted in a more efficient reduction of tumor outgrowth. Such a difference was related to the different capacity to overcome the encountered biological barriers: after 24 h the free Doxo penetrated up to 50 μm in the spheroids while nanoparticles were detected at that depth after 1 h only and diffused in the whole mass at 4 h. Interestingly, such a capacity was exclusive of ald-dex-Doxo nanoparticles and was not observed when cells were incubated with dextran based NPs in which the drug has been only physically loaded and not covalently linked to the polymer.
Chitosan has been instead used in the formulation of nanoparticles for specific targeting of the CD44 receptor in cancer stem-like cells (CSLCs),53,60,77,89,106,122 a rare tumor cell population whose resistance to therapeutic agents is a major cause of anticancer treatment failure.170 While non-stem cancer cell resistance in 2D cultures is mainly the result of the overexpression of transmembrane P-glycoprotein transporters,171 in CSLCs a key role is played also by the tumor microenvironment.172 Accordingly, relevant preclinical investigations of such functionalized nanocarriers required a model capable of mimicking the complex relationship between cancer cells and the surrounding environment. Thus, 3D mammary tumor spheroids (i.e., mammospheres) enriched with CSLCs have been successfully created by culturing MCF-7 cells with different growth factors and chemicals in order to promote stemness (i.e., self-renewal) and epithelial-to-mesenchymal transition (EMT) as demonstrated by the downregulation of the estrogen receptors (ER) expression whose role appears to be pivotal in maintaining the epithelial differentiation.122,173 These CD44-expressing mammospheres, which are currently one of the most advanced examples of the 3D systems used for polymer nanocarrier evaluation, allowed assessing the capacity of chitosan-decorated Pluronic127 nanoparticles to efficiently deliver the loaded doxorubicin and to bypass the CSLC drug resistance in vitro. Chitosan functionalization resulted in nanoparticles able to selectively target CD44-overexpressing cells in mammospheres (Fig. 10a) while only a minimal targeting was observed in 3D spheroids made of normal human adipose-derived stem cells.122 The better in vitro penetration of such functionalized nanocarriers was also confirmed in vivo revealing their highest capacity of inhibition of tumor growth (Fig. 10b and c).
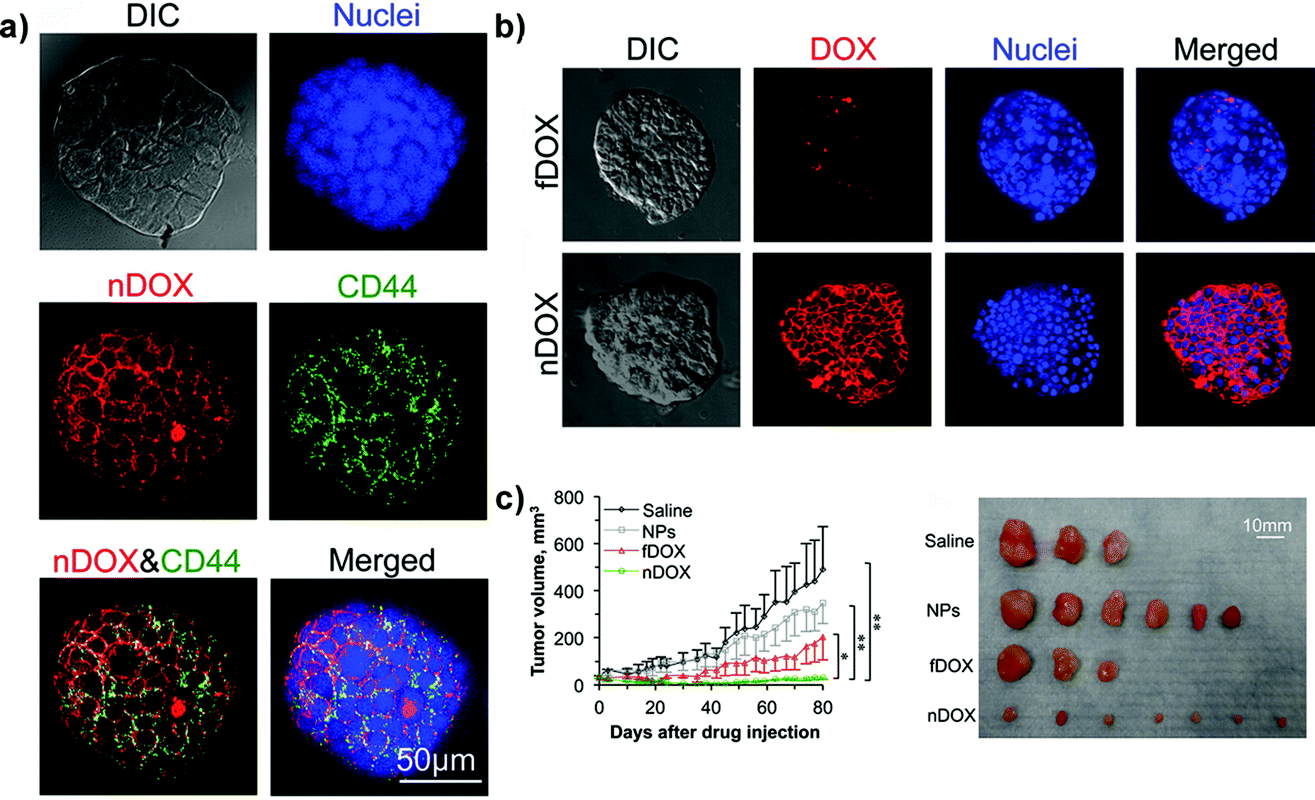 | ||
| Fig. 10 Structured illumination microscopy (confocal-like) images of the specific binding between chitosan-decorated doxorubicin-loaded nanoparticles (nDOXO) and mammosphere cells: (a) co-localization of nDOXO and CD44 receptors; (b) binding between nDOXO and free Doxo (fDOXO) with mammosphere cells. (c) Tumor volume as a function of time for four different treatments and image of tumors collected on day 80 after the initial drug administration. Reproduced with permission from ref. 122. Copyright 2015 American Chemical Society. | ||
CD44 receptors have been targeted also by chondroitin sulfate A-deoxycholic acid-(3-aminomethylphenyl) boronic acid (CSA-DOCA-AMPB) NPs, which in addition exploited the interaction between boronic acid and sialic acid for efficient delivery of Doxo to human lung adenocarcinoma A549 tumors spheroids.60 Penetration observed in vitro by Confocal Laser Scanning Microscopy (CLSM) was confirmed in vivo in A549-tumor bearing mice by three-dimensional near-infrared fluorescence (NIRF) imaging and resulted in significant suppression of tumor development.
2.3.6.4. Monoclonal antibody-targeted nanocarriers. Among the plethora of possible strategies for active targeting, the 2C5 monoclonal antibody (mAb 2C5) has been employed for its capacity to target several types of tumor cells thanks to the interaction with the nucleosomes originating from neighboring apoptotically died tumor cells.174 Nucleosomes are specifically bound to the surface of tumor cells and are always present in the spent media of growing tumor-cell lines as well as in the extracellular fluid of cancer patients.175 Thus, mAb2C5-functionalized Doxo-loaded polyethylene glycol-phosphatidyl ethanolamine (PEG-PE) micelles have been formulated and evaluated in vitro on ovarian cancer MCTS constructed by using NCI-ADR-RES cells.136 The key role played by this Doxo-resistant MCTS model relied on the Bcl-2 gene overexpression associated with the three dimensional organization of the tumor cells, which closely mimicked the real situation found in patients. Consistent with the beneficial effect of mAb2C5 monoclonal antibody conjugation, uniform Doxo distribution throughout the spheroids was achieved only with the targeted micelles. These same PEG-PE micelles have been also functionalized with the single chain fragment variable (scFv) of the monoclonal antibody against glucose transporter-1 (GLUT-1) whose overexpression in cancer cells relies on their continuous requirement for glucose supply. Accordingly, selective targeting of this transmembrane protein might promote the ligand mediated delivery of anticancer drugs.66,73 The in vitro evaluation on a 2D monolayer of U87MG glioblastoma cells revealed that Doxo and curcumin co-encapsulation in GLUT-1-targeted micelles resulted in a significant enhancement of caspase 3 and 7 activity as compared to un-targeted micelles (mono-drug-loaded and two-drug-loaded).66 Moreover, GLUT-1 targeting improved the penetration of PEG-PE micelles into 3D glioblastoma spheroids of U87MG cells in which the Doxo and curcumin synergistic effect was confirmed by the highest cytotoxicity (approx. 70% cell death) after 5 days of treatment.66
An estrogen receptor alpha (ER-α) mAb has been instead used to functionalize the surface of polyacrylic acid (PAA)-coated ion doped NaYF4:Yb,Er upconversion nanoparticles (UCNPs), which have been investigated as a potential early-stage cancer detecting agent.83 Such functionalization enabled targeting of MCF7 breast cancer cell spheroids (∼500 μm) transplanted in a chick embryo chorioallantoic membrane (CAM) aiming at modelling an early stage (i.e., diameter smaller than 2 mm) breast cancer. This model not only mimicked the cell-to-cell and cell-to-microenvironment interactions but also displayed a novel vascularization around the transplanted spheroid. Being more convenient and easy to handle compared to in vivo animal models, this simplified system allowed a direct microscopy study of the UCNPs-mAb's ability to target cancer cells and thus detect tumors in vivo at an early-stage. Indeed, following a systemic administration of UCNPs-mAb via venule injection under a stereomicroscope, a strong upconversion luminescence was observed in the spheroid mass. It should be noted that only cancer cells were targeted and that no accumulation in the other surrounding tissues of the embryo was observed, thus demonstrating the optimal selective capacity of such modified nanoparticles.
2.3.6.5. Aptamer-targeted nanocarriers. Nucleic acid aptamers (DNA and RNA) capable of recognizing with high specificity the epithelial cell adhesion molecule (EpCAM), a type I membrane protein expressed on the surface of a variety of cancer cells,176 have been used for surface functionalization of poly(lactide-co-glycolide) (PLGA)75 and alginate-coated chitosan nanoparticles (CHNPs)74 whose efficiency has been evaluated in vitro on 3D MCTS models of breast75 and colon cancers.74 Decoration with locked nucleic acid (LNA) aptamers allowed CHNPs to reach colon cancer EpCAM-expressing cells deeply in the core of both 3D MCTS and tumor experimental models in mice.74 Proof of their being potential efficient drug carriers was provided using CHNPs loaded with the apoptotic agent SR9, a survivin antagonist. Compared to non-functionalized nanoparticles, their efficient targeting and penetration capacity resulted in a 5-fold reduction of spheroid volume in vitro (after 72 h exposure) and up to 4 times lower tumor volume in colon cancer xenografts in mice (at d = 70 post tumor induction).74
2.3.6.6. Peptide-targeted nanocarriers. Specific recognition of ανβ3 integrins has been demonstrated to endow RDG-functionalized nanocarriers with a targeting capacity toward cancer cells facilitating their internalization.177 Accordingly, the linear or cyclic version of the RGD has been covalently linked to:
(i) micelles made by the assembly of the enzyme-sensitive peptide-linked poly(ethylene glycol) and partially hydrolyzed poly(β-benzyl L-aspartate) (PEG-GPLGVRGDG-P(BLA-co-Asp)) co-polymer;138
(ii) mesoporous silica nanoparticles (MSNs) coated with poly(ethylene glycol) (PEG), polyethyleneimine (PEI) or chitosan;84
(iii) PEG-poly(trimethylene carbonate) (PEG-PTMC) nanoparticles;63,64
(iv) poly(amidoamine) (PAMAM) dendrimers.146,149,150
The influence of the extent of RGD functionalization was also investigated. For instance, the transfection efficiency of PAMAM-RGD dendrimers displaying various levels of RDG ligands was evaluated in a 3D spheroid model of glioma in comparison with the conventional 2D culture of U87MG cells.149,150 In 2D cultures no advantage was observed compared to the naked PAMAM, probably as a consequence of the predominant non-specific interaction mediated by the positively charged dendrimers with the cell membrane. However, the evaluation in 3D spheroids highlighted the capacity of PAMAM-RGD to strongly interfere with the ανβ3 integrin-mediated interaction of cells with the ECM, which was directly correlated to the number of ligands conjugated to PAMAM. As a consequence of the reduced adhesion, RGD functionalization facilitated the penetration and the uptake of PAMAM dendrimers into the spheroid model although it did not result in a significant gene silencing.149
In addition to the capacity of interaction with integrins, conferred by the RGD sequence, the so-called tumor penetrating peptides (TPP)177,178 display a C-terminal sequence R/KXXR/K known as the C-end rule (CendR).62,63,80 Together, these two sequences mediate an active transport through tumor vessels and within the extravascular tumor tissue by interaction first with the αvβ3-integrin and then with the neuropilin-1 receptor (NRP).59,80 Clearly such modification represents a valuable approach for increasing the penetration of nanocarriers in the tumor mass.56,62,117
Among the TPP, the iRGD has been physically adsorbed onto boronic acid-rich chitosan-poly(N-3-acrylamidophenyl boronic acid) (CS-PAPBA) nanoparticles,117 or covalently linked to Pluronic F127 coated paclitaxel nanocrystals.56 CLSM images of spheroids exposed to fluorescently TPP-functionalized nanocarriers revealed an intense signal, which spread from the periphery toward the center of the spheroids, demonstrating their superior capacity of penetration and accumulation leading to an efficient drug delivery confirmed in vitro by the reduction of spheroid volume and in vivo by the inhibition of tumor progression (Fig. 11).56,62,89,117
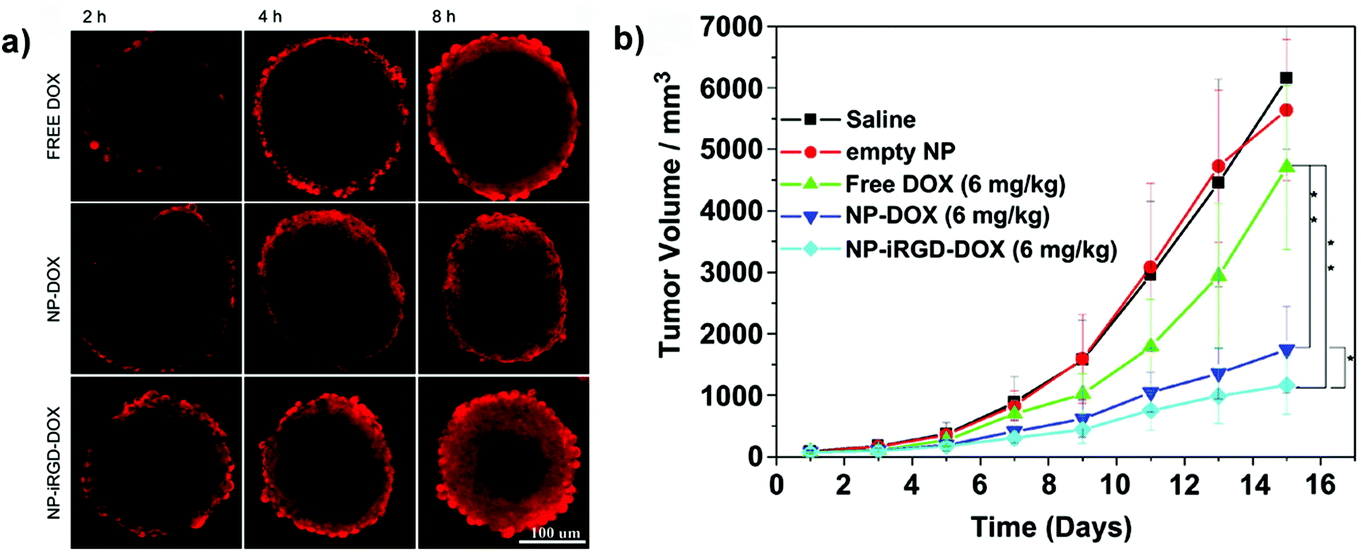 | ||
| Fig. 11 (a) CLSM images of SH-SY5Y MCTS incubated with free Doxo, Doxo-loaded CS-PAPBA NPs and Doxo-loaded iRGD-CS-PAPBA NPs for 2 h, 4 h and 8 h, respectively. (b) In vivo tumor growth curves of H22 tumor-bearing mice that received different treatments. Data are presented as the mean ± SD (n = 10). * represents P < 0.05 since the 7th day and ** represents P < 0.01 since the 11th day. Reproduced with permission from ref. 117. Copyright 2013 American Chemical Society. | ||
The use of the simple CendR motif with the RGERPPR sequence has also been proposed to increase the penetrating capacity of poly(ethylene glycol)-polyethylenimine/plasmid DNA complexes resulting in a higher accumulation into a glioma spheroid model (U87MG cells) compared to the non-functionalized counterpart. Nevertheless whether or not the peptide could also be efficient at improving the transfection efficacy in this 3D model still needs to be verified.80
Cell penetrating properties are also displayed by the interleukin-13 peptide (IL-13p) capable of specific recognition of the IL13Rα2, a tumor-restricted receptor overexpressed in gliomas. Functionalization of poly(ethylene glycol)-poly(ε-caprolactone) (PEG-PCL) nanoparticles with this peptide (ILNPs) resulted in an enhanced cell-uptake and penetration in U87MG spheroids.78 A 3-fold higher tumor accumulation was obtained in vivo compared to un-functionalized nanoparticles, which resulted in a better docetaxel delivery and a significant reduction of tumor weight compared to the other treatments (saline, free drug and drug-loaded naked NPs).78 Endowing these NPs with dual targeting properties by functionalization with both IL-13p and RGD peptides further improved their penetration ability in C6 glioma spheroids as well as in vivo in orthotopic glioma-bearing mice.79
2.4. Use of 3D MCTS to evaluate macromolecules-loaded nanomedicines
Polymer-based nanomedicines have been investigated for the delivery of proteins or DNA/RNA molecules, whose application in clinical settings is strongly limited by (i) inappropriate size and surface charge; (ii) low stability against enzymatic degradation and (iii) low cell membrane permeability.179,180A variety of polymer/siRNA and pDNA polyplexes have been constructed and evaluated in 3D models demonstrating their efficiency as drug carriers. Complexation has been realized for instance with (i) poly(amidoamine) (PAMAM) dendrimers;92,149 (ii) folate-functionalized-poly(ethylene glycol)-polyamidoamine-polyethylenimine (PME-(PEG-FA)) copolymers94 (see also section 2.3.6.2); (iii) poly(L-lysine);118 (iv) PEG-b-poly(N-substituted asparagine) copolymers(PEG-b-P[Asp(DET));131 (v) triblock poly(2-ethyl-2-oxazoline)-poly(L-lactide)-g-poly(ethylenimine) (PEOz-PLA-g-PEI) polymers;108,109 (vi) mPEG-PEI (CendR-penetrating-peptide-modified methoxy poly(ethylene glycol)-polyethylenimine) copolymers80 (see also section 2.3.6.6); (vii) folate-poly(ethylene glycol) (PEG)-Amino Acid Modified Chitosan (CM-PFA)107 or (viii) poly[(N,N-dimethylamino) ethyl methacrylate] (PDMAEMA)-derivatized albumin.93,116
The pioneering studies of Kataoka and coworkers have highlighted the utility of human hepatocarcinoma multicellular spheroids (HuH-7 cells) for the long-term evaluation of the transfection efficacy achieved using polyplex micelles as gene delivery systems.118,131 In particular, core–shell type micelles assembled through electrostatic interactions between poly(L-lysine) and lactosylated poly(ethylene glycol)-siRNA conjugate (lac-PEGylated polyplexes)118 or pDNA and PEG-block-poly(N-asparagine) copolymers (PEG-b-P[Asp(DET)])131 have been investigated. Only the 3D MCTS spheroids, which can be maintained in culture for several weeks, allowed carrying out an extended analysis of gene expression/suppression under conditions close to those observed in vivo in solid tumors.118 In contrast, such time dependent studies could not be performed on 2D monolayer cultures as a consequence of cell–cell contact-induced arrest and viability decrease that prevented the monitoring of a prolonged gene expression.131 Thus, it was possible to observe that the delivery of the RecQL1 siRNA using the lac-PEGylated polyplexes allowed an efficient suppression of gene expression, which resulted in the inhibition of spheroid growth for up to 21 days.118 Using the same 3D model it was possible to demonstrate that pDNA-loaded PEGylated polyplexes penetrated in the spheroids and stably induced the expression of the encoded yellow fluorescent protein Venus for more than 10 days.131
A successful prediction of in vivo transfection efficiency was obtained following an evaluation in MDA-MB-231 breast cancer spheroids of heptafluorobutyric acid modified generation 4 (G4) poly(amidoamine) (PAMAM) dendrimers (G4-F735) loaded with a plasmid encoding for the TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) gene.92 Improved gene delivery and better performance, compared to both naked pTRAIL and conventional transfection reagent poly(ethylene imine) (PEI), were confirmed in vitro by the complete degradation of the MDA-MB-231 spheroids after 7 days of treatment (Fig. 12a) and in vivo by suppression of tumor growth in a subcutaneous model of luciferase expressing MDA-MB-231 cells (Fig. 12b).92
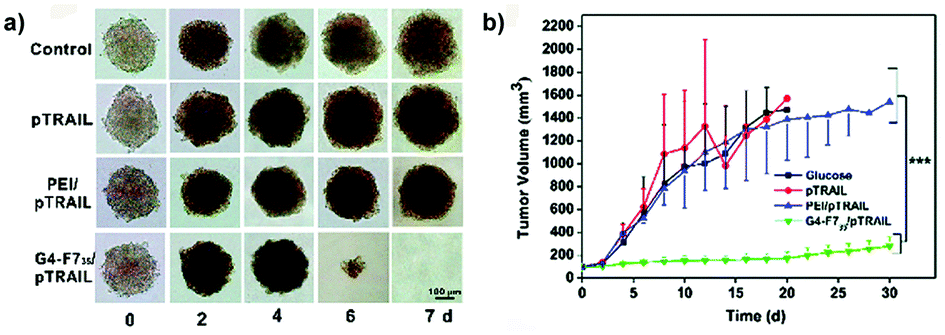 | ||
| Fig. 12 (a) Optical images of MDA-MB-231 MCTS treated with a fresh medium (control), naked pTRAIL (pTRAIL), poly(ethylene imine)/pTRAIL complex (PEI/pTRAIL) and G4-F735/pTRAIL complex (G4-F735/pTRAIL) at different time points. (b) Time-elapsed evolution of tumor sizes in vivo. Reproduced with permission from ref. 92. Copyright 2016 Royal Society of Chemistry. | ||
The usefulness of stimuli-responsive approaches was assessed by Gaspar and coworkers.108,109 Using minicircle DNA (mcDNA), micelleplexes have been constructed by self-assembly of poly(2-ethyl-2-oxazoline)-poly(L-lactide)-g-poly(ethylenimine) (PEOz-PLA-g-PEI) triblock co-polymer or its bioreducible analogue (PEOz-PLA-g-PEI-SS), which allowed the formulation of stimuli-responsive micelles thanks to the introduction of redox sensitive bonds.108 Both systems displayed a good penetration ability and negligible cytotoxicity in 3D MCTS models of melanoma (B16F12 cells), cervix carcinoma (HeLa cells)108 and breast cancer (MCF-7 cells).109 Nevertheless, confocal images of 3D HeLa and B16F10 spheroid sections revealed that bioreducible micelleplexes enhanced GFP gene expression thanks to a higher mcDNA release following rapid intracellular reduction of the disulfide linkages.108
2.5. Miscellaneous nanocarriers evaluated on 3D MCTS
Surface modification of inorganic nanoparticles with various polymers might make possible their use as theranostic systems for efficient delivery of therapeutic molecules and precise monitoring of the response.81 In this view, coating of iron oxide nanoparticles (IONPs, MRI contrast agent) with poly(4-O-acryloyl benzaldehyde)-poly(oligoethylene glycol acrylate) P(HBA)-b-P(OEGA) block copolymers allowed their stabilization as well as the covalent conjugation of doxorubicin via a pH-sensitive bond, which assured drug release in an acidic environment (pH 5.5).81 3D optical sectioning by multi-photon microscopy of multicellular spheroids made of lung (H129) or breast (MCF7) cancer cells clearly showed the fluorescence of Doxo-loaded IONP@P(HBA)-b-P(OEGA) nanoparticles uniformly spread across the spheroid tissue highlighting a complete penetration after 17 h of incubation; in contrast, free Doxo accumulated in the periphery reaching a maximal depth of 40 μm only (Fig. 13).81 | ||
| Fig. 13 3D images of MCF-7 spheroids after incubation with (a) free Doxo and (b) Doxo-loaded IONP@P(HBA)-b-P(OEGA) for 17 h. The representative confocal images (left) were taken every 5 μm section from the top to bottom in the middle of an intact spheroid, whereas the 3D image (right) was reconstructed using Imaris software. Reproduced with permission from ref. 81. Copyright 2013 American Chemical Society. | ||
Real time monitoring of tumor response to treatment is the goal pursued by Oishi and coworkers with the formulation of a PEGylated nanogel containing gold nanoparticles in the cross-linked poly[2-(N,N-diethylamino)ethyl methacrylate] (PEAMA) core in which fluorescein isothiocyanate (FITC) was linked to the PEG chains using the Asp-Glu-Val-Asp (DEVD) peptide sequence as a caspase-3-cleavable linker.156 Based on the Fluorescence Resonance Energy Transfer (FRET) between gold nanoparticles (i.e., fluorescent quencher) and FITC, such a system behaved as a caspase-3-responsive apoptosis sensor for precise in vitro monitoring of the activity of apoptosis-inducing agents. The proof of concept has been provided using human hepatocyte MCTS (HuH-7 cells), which have been incubated with nanogels for 24 h prior to exposure to the apoptotic drug staurosporine for 4 h. Intracellular caspase activation triggered FITC release from the nanogels and the dequenching of the fluorescent signal, which allowed assessing the early stage activation of the induced apoptotic pathway.
The application of nanodiamonds (ND) as drug delivery systems is currently strongly limited by their tendency to agglomerate and precipitate in solution. Thus whether surface modification with polymers might offer a beneficial effect has been explored by the grafting of poly(1-O-methacryloyl-2,3:4,5-di-O-isopropylidene-β-D-fructopyranose) (poly-(1-O-MAipFru)62) onto the surface of amine-functionalized ND. Such an approach should improve their stability and allow a successful loading of doxorubicin.157 When evaluated in 2D monolayer cultures of MCF-7 and MDA-MB-231 breast cancer cells, a clear superiority of the free drug over the NDs was observed. However, in 3D breast cancer MCTS (MCF-7 cells) long term exposure (8 days) to the Doxo-loaded poly(1-O-MAFru)62-ND resulted in higher cytotoxicity compared to the free drug, which can be justified by the deeper penetration of NDs in spheroids and the sustained release of the drug. In contrast, the free Doxo was rapidly internalized by the proliferating cells of the outer layers of the spheroids and the consequent cell death hindered further penetration.157
3. Combining 3D MCTS and polymer scaffolds
3D tumor models made of multicellular spheroids surrounded by a polymer scaffold have also been proposed as an alternative to simple spheroids suspended in cell culture medium. In these systems the scaffold building materials create a matrix around the spheroid capable of mimicking in vitro the microenvironment surrounding the tumors in vivo. Accordingly, this additional barrier may offer the possibility of discerning better the capacity of various nanocarriers to diffuse and to reach the cancer cells.One of the simplest strategies consisted in embedding preformed spheroids into a collagen gel. Such a system enabled for instance highlighting the better efficacy of paclitaxel loaded into pH sensitive NPs (i.e., espansile NPs)181 compared to the free drug, while this difference did not appear in 2D cultures. Indeed, the latter did not display any difference in the cell response independently of the drug administration method (free drug solution vs. drug-loaded nanocarrier).65 In contrast, in 3D cultures, drug-loaded nanoparticles induced a more important slowing of the spheroid growth, which mirrored the inhibition of tumor progression obtained in vivo in tumor-bearing mice.181
In another 3D spheroid design a single cell suspension has been mixed with collagen before gelification and cells aggregated over time inside the matrix in the form of spheres. Again, while no differences were observed in 2D between the free drug (5-fluorouracil) and the drug-loaded micelles, in contrast, thanks to the presence of the collagen matrix which mimicked the tumor ECM, the 3D model enabled revealing the limited diffusive capacity of micelles (152 nm) compared to the small molecules that led to a lower cytotoxicity.182 The same strategy has been applied by mixing prostate cancer cells with hyaluronic acid,183 one of the components of the stroma associated with this tumor in vivo, which not only provided structural support but also strongly influenced tumor cell morphology, gene expression and tumorigenic potential.184 For instance, cells cultured in this 3D scaffold displayed higher expression of multidrug resistance proteins, probably as a result of the limited availability of oxygen and nutrients. Accordingly, while in the 2D cultures the free drug (doxorubicin) easily reached the nucleus and exerted its therapeutic activity, in 3D the sensitivity to the free doxorubicin was reduced (5 μM vs. 15 μM, respectively). Contrariwise, independently of the culture conditions the response to Doxo-loaded PEG-PCL NPs was not modified and analogous IC50 values were measured (11 μM vs. 12.3 μM, respectively) probably thanks to the capacity of these NPs (54 nm diameter) to overcome the MDR while the free drug undergoes a rapid efflux, which reduced its efficacy.183
4. Combining 3D MCTS and microfluidic devices
Undoubtedly, compared to simplistic 2D cultures the abovedescribed 3D systems enabled a more predictive in vitro screening of nanoscale systems for drug delivery. Nevertheless, due to the lack of fluid dynamics these setups mimic only a static condition. To face this issue, microfluidic devices, which combine 3D culture and controlled flow conditions, have been recently developed with the aim: (i) to assess how physico-chemical parameters influence the transport through tumor biological barriers under dynamic conditions and (ii) to provide information on the optimal design required to achieve a successful tumor accumulation.185,186For instance, a tumor-on-a-chip device allowed the passage from a static spheroid culture to a dynamic situation by placing a spheroid in the channel of a two layer poly(diméthylsiloxane) (PDMS) chip (Fig. 14a).185 By tuning the flow rate in the device, fluid velocities and shear stresses similar to the blood flow in capillary vessels (75–675 μm s−1) or the interstitial flow inside a tumor (0.1–3 μm s−1) could be reproduced in a controllable manner. Exposure of spheroids to PEGylated nanoparticles of different sizes under stationary flow revealed interstitial accumulation only of the smaller NPs (40 nm) while the larger ones (110 nm), bigger than the ECM pores, were excluded (Fig. 14b and c). However, accumulation was only transient and nanoparticles flowed out after flushing, confirming that surface modification with PEG chains hindered the establishment of specific interactions with cells and ECM components. In contrast, no efflux was observed following NP functionalization with transferrin and 40 nm nanoparticles showed up to 15-fold increase of tumor accumulation compared to non-functionalized NPs. It was also observed that an increase of the flow rate resulted in accumulation in the external spheroid layer, forming a tissue–fluid interface reservoir but that did not affect the depth of NP penetration. When the NPs were then tested in vivo in a tumor bearing mice, the same size discriminating effect was recorded, with a better accumulation of small NPs (50 nm) compared to the larger ones (160 nm). However, functionalization did not lead to any significant advantage and both targeted and non-targeted 50 nm NPs displayed similar tumor accumulation levels (Fig. 14d and e). Such contradiction with the results obtained in vitro clearly highlighted a limit of the 3D models, which cannot fully reproduce the complexity of living organisms and the behavior of NPs after intravenous administration. Thus, although it is evident that 3D models would allow a more relevant preclinical screening of nanomedicines compared to 2D cultures, at present they cannot completely replace the in vivo experimentation.
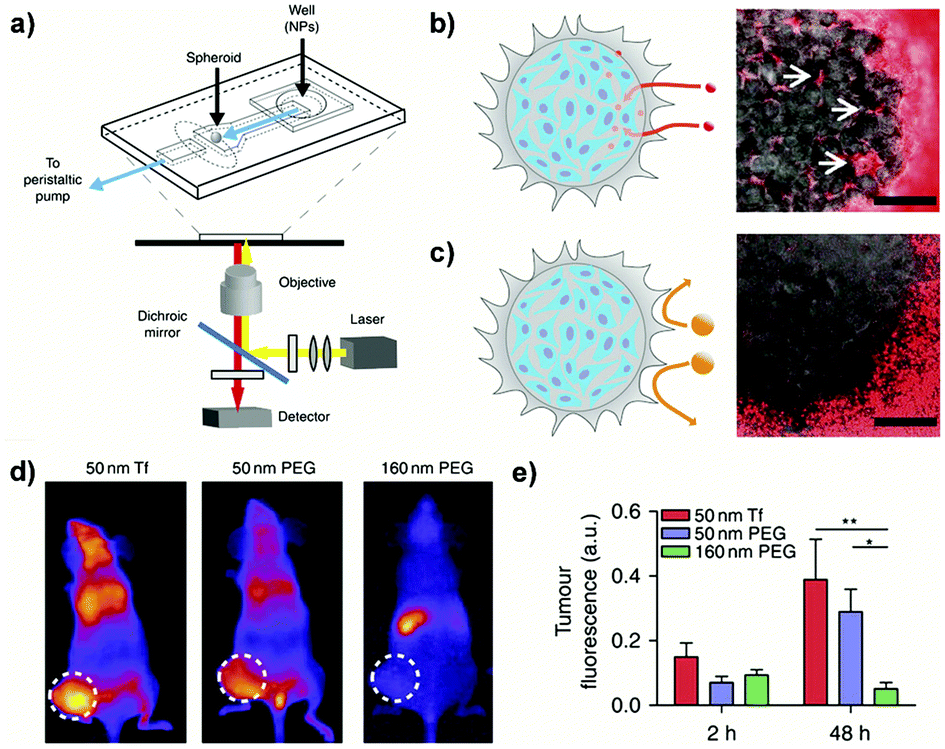 | ||
| Fig. 14 (a) Schematic of the PDMS microfluidic device on a microscope stage. (b) Schematic (left) and image (right) of 40 nm fluorescent PEG-NPs administered for 1 h at 50 μL h−1 entering the spheroid and accumulating in the interstitial spaces (arrows). Scale bar: 100 μm. (c) Schematic (left) and image (right) of 110 nm fluorescent PEG-NPs administered for 1 h at 50 μL h−1 being excluded from the spheroid. Scale bar: 100 μm. (d) Representative images of tumor fluorescence from mice injected with NPs in the tail vein at 48 h post-injection. (e) Quantification of animal fluorescence at 2 and 48 h using whole animal images. Reproduced with permission from ref. 185. Copyright 2013 Nature Publishing Group. | ||
Another proposed approach consisted in the loading of Matrigel-embedded preformed spheroids in the central channel of a microfluidic device, while continuous medium addition in the lateral channels recreated some blood flow conditions. This allowed to assess under dynamic conditions treatment responses to doxorubicin in free form and loaded into micelles (Fig. 15).187
 | ||
| Fig. 15 (a) Bright field image of the microfluidic channel. The black arrow shows the direction of medium flow. Scale bar: 300 μm. (b) Bright-field image of the MCTS in the microfluidic channel. (top) The MCTS in Matrigel prior to Doxo-HCl treatment. (bottom) Real-time, enlarged views of the MCTS during 24 h of treatment with Doxo-HCl. Scale bar: 50 μm. Reproduced with permission from ref. 187. Copyright 2013 American Chemical Society. | ||
5. Conclusion and perspectives
Through many literature examples this review has unambiguously highlighted that simple 2D monolayers cultures do not allow a complete understanding of the therapeutic potential of polymer nanomedicines while the application of 3D tumor models in preclinical evaluation would provide more accurate results, predictive of the in vivo pharmacological efficacy. The key features of the MCTS (e.g., presence of ECM, diffusive gradients, complex cell signaling, drug resistance and metabolic adaptation) undoubtedly enable evaluating the nanomedicines under conditions closer to the clinical reality. But it cannot be ignored that the majority of tumor spheroids used in the literature are made of cancer cells alone and therefore they just represent a rather simplified model of real tumorigenesis. Due to the complexity of the tumor tissues and the cross-talk between cancer cells and their microenvironment, advanced models including several cell types (e.g., endothelial cells, immune cells, fibroblasts etc.) and components of the ECM are urgently required. Thus, 3D co-cultures47,48,188 and microfluidic devices111–113,185,186,189 have been developed but further improvements are still needed in order to allow wider application in preclinical investigation. In this context, the accurate characterization (e.g., cell number, long term viability of each cell component, type of ECM protein, etc.) of any developed system is mandatory for allowing a reliable interpretation of the obtained results. It is indeed well acknowledged that the spheroid size43,46 as well as the presence of stroma components47,48,188 strongly affect the response to treatment according to the possible development of penetration barriers,46–48,188 chemical gradients and/or necrosis.43The availability of microscopy techniques suitable for high resolution imaging of 3D cell cultures is another challenge. Indeed, although widely used, CLSM does not allow the in toto study of large 3D samples and the obtained results refer only to penetration into the spheroids at the depth of a maximum of 100–150 μm. A significant improvement in 3D imaging should result from Two-Photon (TPM) and Multi-Photon Microscopy (MPM) studies but the low spatial resolution along the optical axis and the incompatibility of certain fluorophores with the multi-photon excitation still restrict the applicability of these methods.140,190 In this challenging panorama, LSFM approaches (e.g., Selective Plane Illumination Microscopy (SPIM)) are emerging as techniques of choice in life sciences for the imaging of complex and highly scattering samples.142–145 Offering the possibility of visualizing the spheroids in their entirety with a sub-cellular resolution191,192 the SPIM technique clearly enables achieving a superior degree of information in the screening of nanomedicines’ pharmacological efficiency.47 The penetration ability of nanomedicines can be assessed over the whole 3D MCTS mass and a dynamic study of the anticancer response is also possible with the time lapse imaging of living spheroids. Unfortunately, such advanced techniques require highly specialized technologies whose availability still remains limited.
It is evident that labor-intensive handling, time-consuming procedures, instrumental limitations and costs still hinder the routine use of advanced 3D models in drug discovery programmes. It is noteworthy that, to the best of our knowledge, no promising nanomedicine currently in clinical trials has been tested in 3D MCTS during in vitro preclinical studies. Nevertheless, advances in the near future are expected to rapidly support their widespread use, thus making the in vitro drug screenings more predictive and able to sieve out underperforming compounds in the early preclinical stage.
Acknowledgements
The authors acknowledge financial support from the European Union's Horizon 2020 research and innovation programme under Marie Skłodowska Curie grant agreement no. 642028, the CNRS and the French Ministry of Research.Notes and references
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- M. W. Tibbitt, J. E. Dahlman and R. Langer, J. Am. Chem. Soc., 2016, 138, 704–717 CrossRef CAS PubMed.
- P. Couvreur and C. Vauthier, Pharm. Res., 2006, 23, 1417–1450 CrossRef CAS PubMed.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- C. A. Schütz, L. Juillerat-Jeanneret, H. Mueller, I. Lynch and M. Riediker, Nanomedicine, 2013, 8, 449–467 CrossRef PubMed.
- A. K. Lytton-Jean, K. J. Kauffman, J. C. Kaczmarek and R. Langer, Cancer Treat. Res., 2015, 166, 293–322 CAS.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2016, 1, 10–29 Search PubMed.
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- P. Lu, V. M. Weaver and Z. Werb, J. Cell Biol., 2012, 196, 395–406 CrossRef CAS PubMed.
- K. M. Bussard, L. Mutkus, K. Stumpf, C. Gomez-Manzano and F. C. Marini, Breast Cancer Res., 2016, 18, 84 CrossRef PubMed.
- M. W. Pickup, J. K. Mouw and V. M. Weaver, EMBO Rep., 2014, 15, 1243–1253 CrossRef CAS PubMed.
- A. Birgersdotter, R. Sandberg and I. Ernberg, Semin. Cancer Biol., 2005, 15, 405–412 CrossRef PubMed.
- E. Cukierman, R. Pankov, D. R. Stevens and K. M. Yamada, Science, 2001, 294, 1708–1712 CrossRef CAS PubMed.
- D. R. Albrecht, G. H. Underhill, T. B. Wassermann, R. L. Sah and S. N. Bhatia, Nat. Methods, 2006, 3, 369–375 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- C. H. Heldin, K. Rubin, K. Pietras and A. Ostman, Nat. Rev. Cancer, 2004, 4, 806–813 CrossRef CAS PubMed.
- L. G. Griffith and M. A. Swartz, Nat. Rev. Mol. Cell Biol., 2006, 7, 211–224 CrossRef CAS PubMed.
- M. C. Cox, L. M. Reese, L. R. Bickford and S. S. Verbridge, ACS Biomater. Sci. Eng., 2015, 1, 877–894 CrossRef CAS.
- C. Fischbach, R. Chen, T. Matsumoto, T. Schmelzle, J. S. Brugge, P. J. Polverini and D. J. Mooney, Nat. Methods, 2007, 4, 855–860 CrossRef CAS PubMed.
- C. R. Thoma, M. Zimmermann, I. Agarkova, J. M. Kelm and W. Krek, Adv. Drug Delivery Rev., 2014, 69–70, 29–41 CrossRef CAS PubMed.
- L. B. Weiswald, D. Bellet and V. Dangles-Marie, Neoplasia, 2015, 17, 1–15 CrossRef PubMed.
- X. Xu, M. C. Farach-Carson and X. Jia, Biotechnol. Adv., 2014, 32, 1256–1268 CrossRef CAS PubMed.
- A. Nyga, U. Cheema and M. Loizidou, J. Cell Commun. Signal., 2011, 5, 239–248 CrossRef PubMed.
- W. Asghar, R. El Assal, H. Shafiee, S. Pitteri, R. Paulmurugan and U. Demirci, Mater. Today, 2015, 18, 539–553 CrossRef CAS PubMed.
- K. A. Fitzgerald, M. Malhotra, C. M. Curtin, F. J. O'Brien and C. M. O'Driscoll, J. Controlled Release, 2015, 215, 39–54 CrossRef CAS PubMed.
- J. Tannenbaum and B. T. Bennett, J. Am. Assoc. Lab. Anim. Sci., 2015, 54, 120–132 Search PubMed.
- A. Abbott, Nature, 2003, 424, 870–872 CrossRef CAS PubMed.
- R. M. Sutherland, J. A. Mccredie and W. R. Inch, J. Natl. Cancer Inst., 1971, 46, 113–120 CAS.
- R. Sutherland, J. Carlsson, R. Durand and J. Yuhas, Cancer Res., 1981, 41, 2980–2984 Search PubMed.
- D. V. LaBarbera, B. G. Reid and B. H. Yoo, Expert Opin. Drug Discovery, 2012, 7, 819–830 CrossRef CAS PubMed.
- S. Breslin and L. O'Driscoll, Drug Discovery Today, 2013, 18, 240–249 CrossRef CAS PubMed.
- J. C. Lovitt, B. T. Shelper and M. V. Avery, Biology, 2014, 3, 345–367 CrossRef PubMed.
- E. C. Costa, A. F. Moreira, D. de Melo-Diogo, V. M. Gaspar, M. P. Carvalho and I. J. Correia, Biotechnol. Adv., 2016, 34, 1427–1441 CrossRef PubMed.
- E. Fennema, N. Rivron, J. Rouwkema, C. van Blitterswijk and J. de Boer, Trends Biotechnol., 2013, 31, 108–115 CrossRef CAS PubMed.
- G. Hamilton, Cancer Lett., 1998, 131, 29–34 CrossRef CAS PubMed.
- F. Hirschhaeuser, H. Menne, C. Dittfeld, J. West, W. Mueller-Klieser and L. A. Kunz-Schughart, J. Biotechnol., 2010, 148, 3–15 CrossRef CAS PubMed.
- R.-Z. Lin and H.-Y. Chang, Biotechnol. J., 2008, 3, 1172–1184 CrossRef CAS PubMed.
- G. Mehta, A. Y. Hsiao, M. Ingram, G. D. Luker and S. Takayama, J. Controlled Release, 2012, 164, 192–204 CrossRef CAS PubMed.
- A. Kang, H. I. Seo, B. G. Chung and S.-H. Lee, Nanomedicine, 2015, 11, 1153–1161 CrossRef CAS PubMed.
- P. Sethi, A. Jyoti, E. P. Swindell, R. Chan, U. W. Langner, J. M. Feddock, R. Nagarajan, T. V. O'Halloran and M. Upreti, Nanomedicine, 2015, 11, 2013–2023 CrossRef CAS PubMed.
- D. L. Priwitaningrum, J.-B. G. Blondé, A. Sridhar, J. van Baarlen, W. E. Hennink, G. Storm, S. Le Gac and J. Prakash, J. Controlled Release, 2016, 244(Part B), 257–268 CrossRef CAS PubMed.
- J. Friedrich, R. Ebner and L. A. Kunz-Schughart, Int. J. Radiat. Biol., 2007, 83, 849–871 CrossRef CAS PubMed.
- P. Benien and A. Swami, Future Oncol., 2014, 10, 1311–1327 CrossRef CAS PubMed.
- N. R. Patel, B. Aryasomayajula, A. H. Abouzeid and V. P. Torchilin, Ther. Delivery, 2015, 6, 509–520 CrossRef CAS PubMed.
- Q. Yang, Y. Yang, L. Li, W. Sun, X. Zhu and Y. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 6661–6673 CAS.
- X. Wei, T. H. Senanayake, G. Warren and S. V. Vinogradov, Bioconjugate Chem., 2013, 24, 658–668 CrossRef CAS PubMed.
- H. Lei, S. C. Hofferberth, R. Liu, A. Colby, K. M. Tevis, P. Catalano, M. W. Grinstaff and Y. L. Colson, J. Thorac. Cardiovasc. Surg., 2015, 149, 1417–1424 CrossRef CAS PubMed.
- X. Cheng, X. Wang, Z. Cao, W. Yao, J. Wang and R. Tang, Mater. Sci. Eng., C, 2017, 71, 298–307 CrossRef CAS PubMed.
- D. Ni, H. Ding, S. Liu, H. Yue, Y. Bao, Z. Wang, Z. Su, W. Wei and G. Ma, Small, 2015, 11, 2518–2526 CrossRef CAS PubMed.
- Z. Cao, X. Wang, X. Cheng, J. Wang and R. Tang, Int. J. Polym. Mater. Polym. Biomater., 2017, 66, 495–506 CrossRef CAS.
- G. Yan, Q. Zha, J. Wang, X. Wang, X. Cheng, W. Yao and R. Tang, Polymer, 2017, 111, 192–203 CrossRef CAS.
- S. Ma, J. Zhou, Y. Zhang, Y. He, Q. Jiang, D. Yue, X. Xu and Z. Gu, ACS Appl. Mater. Interfaces, 2016, 8, 28468–28479 CAS.
- J.-Y. Lee, S.-J. Chung, H.-J. Cho and D.-D. Kim, Adv. Funct. Mater., 2015, 25, 3705–3717 CrossRef CAS.
- Y. Li, H. He, X. Jia, W.-L. Lu, J. Lou and Y. Wei, Biomaterials, 2012, 33, 3899–3908 CrossRef CAS PubMed.
- Q. Hu, X. Gao, G. Gu, T. Kang, Y. Tu, Z. Liu, Q. Song, L. Yao, Z. Pang, X. Jiang, H. Chen and J. Chen, Biomaterials, 2013, 34, 5640–5650 CrossRef CAS PubMed.
- X. Jiang, H. Xin, J. Gu, X. Xu, W. Xia, S. Chen, Y. Xie, L. Chen, Y. Chen, X. Sha and X. Fang, Biomaterials, 2013, 34, 1739–1746 CrossRef CAS PubMed.
- X. Jiang, X. Sha, H. Xin, X. Xu, J. Gu, W. Xia, S. Chen, Y. Xie, L. Chen, Y. Chen and X. Fang, Biomaterials, 2013, 34, 2969–2979 CrossRef CAS PubMed.
- K. M. Charoen, B. Fallica, Y. L. Colson, M. H. Zaman and M. W. Grinstaff, Biomaterials, 2014, 35, 2264–2271 CrossRef CAS PubMed.
- C. Sarisozen, S. Dhokai, E. G. Tsikudo, E. Luther, I. M. Rachman and V. P. Torchilin, Eur. J. Pharm. Biopharm., 2016, 108, 54–67 CrossRef CAS PubMed.
- C. Sarisozen, A. H. Abouzeid and V. P. Torchilin, Eur. J. Pharm. Biopharm., 2014, 88, 539–550 CrossRef CAS PubMed.
- W. Zou, C. Sarisozen and V. P. Torchilin, J. Drug Targeting, 2016, 25, 225–234 CrossRef PubMed.
- X. Cao, X. Zhou, Y. Wang, T. Gong, Z.-R. Zhang, R. Liu and Y. Fu, J. Mater. Chem. B, 2016, 4, 3216–3224 RSC.
- H. L. Ma, Q. Jiang, S. Han, Y. Wu, T. J. Cui, D. Wang, Y. Gan, G. Zou and X. J. Liang, Mol. Imaging, 2012, 11, 487–498 CAS.
- H. E. Colley, V. Hearnden, M. Avila-Olias, D. Cecchin, I. Canton, J. Madsen, S. MacNeil, N. Warren, K. Hu, J. A. McKeating, S. P. Armes, C. Murdoch, M. H. Thornhill and G. Battaglia, Mol. Pharmaceutics, 2014, 11, 1176–1188 CrossRef CAS PubMed.
- J. Bugno, H.-J. Hsu, R. M. Pearson, H. Noh and S. Hong, Mol. Pharmaceutics, 2016, 13, 2155–2163 CrossRef CAS PubMed.
- X. Jiang, H. Xin, J. Gu, F. Du, C. Feng, Y. Xie and X. Fang, J. Pharm. Sci., 2014, 103, 1487–1496 CrossRef CAS PubMed.
- K. Roy, R. K. Kanwar, C. H. A. Cheung, C. L. Fleming, R. N. Veedu, S. Krishnakumar and J. R. Kanwar, RSC Adv., 2015, 5, 29008–29016 RSC.
- M. Das, W. Duan and S. K. Sahoo, Nanomedicine, 2015, 11, 379–389 CrossRef CAS PubMed.
- G. L. Hu, Y. Wang, Q. He and H. L. Gao, RSC Adv., 2015, 5, 85933–85937 RSC.
- S. Sharma, J. Singh, A. Verma, B. V. Teja, R. P. Shukla, S. K. Singh, V. Sharma, R. Konwar and P. R. Mishra, RSC Adv., 2016, 6, 73083–73095 RSC.
- H. Gao, Z. Yang, S. Zhang, S. Cao, Z. Pang, X. Yang and X. Jiang, J. Controlled Release, 2013, 172, 921–928 CrossRef CAS PubMed.
- H. Gao, Y. Xiong, S. Zhang, Z. Yang, S. Cao and X. Jiang, Mol. Pharmaceutics, 2014, 11, 1042–1052 CrossRef CAS PubMed.
- J. Wang, Y. Lei, C. Xie, W. Lu, Z. Yan, J. Gao, Z. Xie, X. Zhang and M. Liu, Int. J. Pharm., 2013, 458, 48–56 CrossRef CAS PubMed.
- J. S. Basuki, H. T. T. Duong, A. Macmillan, R. B. Erlich, L. Esser, M. C. Akerfeldt, R. M. Whan, M. Kavallaris, C. Boyer and T. P. Davis, ACS Nano, 2013, 7, 10175–10189 CrossRef CAS PubMed.
- S. Jin, X. Ma, H. Ma, K. Zheng, J. Liu, S. Hou, J. Meng, P. C. Wang, X. Wu and X.-J. Liang, Nanoscale, 2013, 5, 143–146 RSC.
- K. Liu, J. A. Holz, Y. Ding, X. Liu, Y. Zhang, L. Tu, X. Kong, B. Priem, A. Nadort, S. A. Lambrechts, M. C. Aalders, W. J. Buma, Y. Liu and H. Zhang, Nanoscale, 2015, 7, 1596–1600 RSC.
- Y. You, H. Hu, L. He and T. Chen, Chem. – Asian J., 2015, 10, 2744–2754 CrossRef CAS PubMed.
- R. Agarwal, P. Jurney, M. Raythatha, V. Singh, S. V. Sreenivasan, L. Shi and K. Roy, Adv. Healthcare Mater., 2015, 4, 2269–2280 CrossRef CAS PubMed.
- A. Ivascu and M. Kubbies, J. Biomol. Screening, 2006, 11, 922–932 CrossRef CAS PubMed.
- J. Friedrich, C. Seidel, R. Ebner and L. A. Kunz-Schughart, Nat. Protoc., 2009, 4, 309–324 CrossRef CAS PubMed.
- M. Cui, D. J. Naczynski, M. Zevon, C. K. Griffith, L. Sheihet, I. Poventud-Fuentes, S. Chen, C. M. Roth and P. V. Moghe, Adv. Healthcare Mater., 2013, 2, 1236–1245 CrossRef CAS PubMed.
- D.-S. Liang, H.-T. Su, Y.-J. Liu, A.-T. Wang and X.-R. Qi, Biomaterials, 2015, 71, 11–23 CrossRef CAS PubMed.
- H. X. Lu, R. H. Utama, U. Kitiyotsawat, K. Babiuch, Y. Jiang and M. H. Stenzel, Biomater. Sci., 2015, 3, 1085–1095 RSC.
- Y. Y. Jiang, H. X. Lu, A. Dag, G. Hart-Smith and M. H. Stenzel, J. Mater. Chem., 2016, 4, 2017–2027 RSC.
- Y. Wang, M. Wang, H. Chen, H. Liu, Q. Zhang and Y. Cheng, J. Mater. Chem. B, 2016, 4, 1354–1360 RSC.
- Y. Jiang, C. K. Wong and M. H. Stenzel, Macromol. Biosci., 2015, 15, 965–978 CrossRef CAS PubMed.
- D. Cao, S. Tian, H. Huang, J. Chen and S. Pan, Mol. Pharmaceutics, 2015, 12, 240–252 CrossRef CAS PubMed.
- A. W. Du, H. Lu and M. Stenzel, Mol. Pharmaceutics, 2016, 13, 3648–3656 CrossRef CAS PubMed.
- T. H. Kim, C. W. Mount, W. R. Gombotz and S. H. Pun, Biomaterials, 2010, 31, 7386–7397 CrossRef CAS PubMed.
- T. T. Goodman, P. L. Olive and S. H. Pun, Int. J. Nanomed., 2007, 2, 265–274 CrossRef CAS PubMed.
- T. T. Goodman, J. Chen, K. Matveev and S. H. Pun, Biotechnol. Bioeng., 2008, 101, 388–399 CrossRef CAS PubMed.
- S. S. Verbridge, A. Chakrabarti, P. DelNero, B. Kwee, J. D. Varner, A. D. Stroock and C. Fischbach, J. Biomed. Mater. Res., Part A, 2013, 101, 2948–2956 CrossRef PubMed.
- T. Okuyama, H. Yamazoe, N. Mochizuki, A. Khademhosseini, H. Suzuki and J. Fukuda, J. Biosci. Bioeng., 2010, 110, 572–576 CrossRef CAS PubMed.
- H. Hardelauf, J.-P. Frimat, J. D. Stewart, W. Schormann, Y.-Y. Chiang, P. Lampen, J. Franzke, J. G. Hengstler, C. Cadenas, L. A. Kunz-Schughart and J. West, Lab Chip, 2011, 11, 419–428 RSC.
- Y. Morimoto, A. Y. Hsiao and S. Takeuchi, Adv. Drug Delivery Rev., 2015, 95, 29–39 CrossRef CAS PubMed.
- M. Cordey, M. Limacher, S. Kobel, V. Taylor and M. P. Lutolf, Stem Cells, 2008, 26, 2586–2594 CrossRef PubMed.
- J. Fukuda, A. Khademhosseini, Y. Yeo, X. Yang, J. Yeh, G. Eng, J. Blumling, C.-F. Wang, D. S. Kohane and R. Langer, Biomaterials, 2006, 27, 5259–5267 CrossRef CAS PubMed.
- T. Liu, I. Kempson, M. de Jonge, D. L. Howard and B. Thierry, Nanoscale, 2014, 6, 9774–9782 RSC.
- S. Ohta, S. Hiramoto, Y. Amano, M. Sato, Y. Suzuki, M. Shinohara, S. Emoto, H. Yamaguchi, H. Ishigami, Y. Sakai, J. Kitayama and T. Ito, Bioconjugate Chem., 2016, 27, 504–508 CrossRef CAS PubMed.
- V. M. Gaspar, E. C. Costa, J. A. Queiroz, C. Pichon, F. Sousa and I. J. Correia, Pharm. Res., 2015, 32, 562–577 CrossRef CAS PubMed.
- V. M. Gaspar, P. Baril, E. C. Costa, D. de Melo-Diogo, F. Foucher, J. A. Queiroz, F. Sousa, C. Pichon and I. J. Correia, J. Controlled Release, 2015, 213, 175–191 CrossRef CAS PubMed.
- V. M. Gaspar, C. Gonçalves, D. de Melo-Diogo, E. C. Costa, J. A. Queiroz, C. Pichon, F. Sousa and I. J. Correia, J. Controlled Release, 2014, 189, 90–104 CrossRef CAS PubMed.
- T. Anajafi, M. D. Scott, S. You, X. Yang, Y. Choi, S. Y. Qian and S. Mallik, Bioconjugate Chem., 2016, 27, 762–771 CrossRef CAS PubMed.
- S. I. Montanez-Sauri, D. J. Beebe and K. E. Sung, Cell. Mol. Life Sci., 2015, 72, 237–249 CrossRef CAS PubMed.
- R. Sudo, S. Chung, I. K. Zervantonakis, V. Vickerman, Y. Toshimitsu, L. G. Griffith and R. D. Kamm, FASEB J., 2009, 23, 2155–2164 CrossRef CAS PubMed.
- K. E. Sung, N. Yang, C. Pehlke, P. J. Keely, K. W. Eliceiri, A. Friedl and D. J. Beebe, Integr. Biol., 2011, 3, 439–450 RSC.
- S. Ghosh, S. Mohapatra, A. Thomas, D. Bhunia, A. Saha, G. Das, B. Jana and S. Ghosh, ACS Appl. Mater. Interfaces, 2016, 8, 30824–30832 CAS.
- W. L. Lee, W. M. Guo, V. H. B. Ho, A. Saha, H. C. Chong, N. S. Tan, E. Y. Tan and S. C. J. Loo, Acta Biomater., 2015, 27, 53–65 CrossRef CAS PubMed.
- Y. Jiang, H. Lu, F. Chen, M. Callari, M. Pourgholami, D. L. Morris and M. H. Stenzel, Biomacromolecules, 2016, 17, 808–817 CrossRef CAS PubMed.
- X. Wang, X. Zhen, J. Wang, J. Zhang, W. Wu and X. Jiang, Biomaterials, 2013, 34, 4667–4679 CrossRef CAS PubMed.
- M. Oishi, Y. Nagasaki, N. Nishiyama, K. Itaka, M. Takagi, A. Shimamoto, Y. Furuichi and K. Kataoka, ChemMedChem, 2007, 2, 1290–1297 CrossRef CAS PubMed.
- J. Li, Y. Han, Q. Chen, H. Shi, S. ur Rehman, M. Siddiq, Z. Ge and S. Liu, J. Mater. Chem. B, 2014, 2, 1813–1824 RSC.
- U. Till, L. Gibot, P. Vicendo, M.-P. Rols, M. Gaucher, F. Violleau and A.-F. Mingotaud, RSC Adv., 2016, 6, 69984–69998 RSC.
- Y. Jiang, H. Lu, Y. Y. Khine, A. Dag and M. H. Stenzel, Biomacromolecules, 2014, 15, 4195–4205 CrossRef CAS PubMed.
- W. Rao, H. Wang, J. Han, S. Zhao, J. Dumbleton, P. Agarwal, W. Zhang, G. Zhao, J. Yu, D. L. Zynger, X. Lu and X. He, ACS Nano, 2015, 9, 5725–5740 CrossRef CAS PubMed.
- H. Lu, B. M. Blunden, W. Scarano, M. Lu and M. H. Stenzel, Acta Biomater., 2016, 32, 68–76 CrossRef CAS PubMed.
- A. W. Du, H. Lu and M. Stenzel, Biomacromolecules, 2015, 16, 1470–1479 CrossRef CAS PubMed.
- Y. Zhang, P. Lundberg, M. Diether, C. Porsch, C. Janson, N. A. Lynd, C. Ducani, M. Malkoch, E. Malmstrom, C. J. Hawker and A. M. Nystrom, J. Mater. Chem. B, 2015, 3, 2472–2486 RSC.
- S. M. Sagnella, H. Duong, A. MacMillan, C. Boyer, R. Whan, J. A. McCarroll, T. P. Davis and M. Kavallaris, Biomacromolecules, 2014, 15, 262–275 CrossRef CAS PubMed.
- X. Han, R. Gelein, N. Corson, P. Wade-Mercer, J. Jiang, P. Biswas, J. N. Finkelstein, A. Elder and G. Oberdörster, Toxicology, 2011, 287, 99–104 CrossRef CAS PubMed.
- R. Scherließ, Int. J. Pharm., 2011, 411, 98–105 CrossRef PubMed.
- J. Friedrich, W. Eder, J. Castaneda, M. Doss, E. Huber, R. Ebner and L. A. Kunz-Schughart, J. Biomol. Screening, 2007, 12, 925–937 CrossRef CAS PubMed.
- M. Zanoni, F. Piccinini, C. Arienti, A. Zamagni, S. Santi, R. Polico, A. Bevilacqua and A. Tesei, Sci. Rep., 2016, 6, 19103 CrossRef CAS PubMed.
- M. Han, Y. Bae, N. Nishiyama, K. Miyata, M. Oba and K. Kataoka, J. Controlled Release, 2007, 121, 38–48 CrossRef CAS PubMed.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutellingsperger, J. Immunol. Methods, 1995, 184, 39–51 CrossRef CAS PubMed.
- H.-J. Li, J.-Z. Du, J. Liu, X.-J. Du, S. Shen, Y.-H. Zhu, X. Wang, X. Ye, S. Nie and J. Wang, ACS Nano, 2016, 10, 6753–6761 CrossRef CAS PubMed.
- H.-J. Li, J.-Z. Du, X.-J. Du, C.-F. Xu, C.-Y. Sun, H.-X. Wang, Z.-T. Cao, X.-Z. Yang, Y.-H. Zhu, S. Nie and J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4164–4169 CrossRef CAS PubMed.
- K. Kyrylkova, S. Kyryachenko, M. Leid and C. Kioussi, Methods Mol. Biol., 2012, 887, 41–47 CAS.
- F. Perche and V. P. Torchilin, J. Controlled Release, 2012, 164, 95–102 CrossRef CAS PubMed.
- R. I. Dmitriev, S. M. Borisov, H. Düssmann, S. Sun, B. J. Müller, J. Prehn, V. P. Baklaushev, I. Klimant and D. B. Papkovsky, ACS Nano, 2015, 9, 5275–5288 CrossRef CAS PubMed.
- W. Ke, J. Li, K. Zhao, Z. Zha, Y. Han, Y. Wang, W. Yin, P. Zhang and Z. Ge, Biomacromolecules, 2016, 17, 3268–3276 CrossRef CAS PubMed.
- A. S. Mikhail, S. Eetezadi, S. N. Ekdawi, J. Stewart and C. Allen, Int. J. Pharm., 2014, 464, 168–177 CrossRef CAS PubMed.
- F. Helmchen and W. Denk, Nat. Methods, 2005, 2, 932–940 CrossRef CAS PubMed.
- M. Cantisani, D. Guarnieri, M. Biondi, V. Belli, M. Profeta, L. Raiola and P. A. Netti, Colloids Surf., B, 2015, 135, 707–716 CrossRef CAS PubMed.
- P. J. Verveer, J. Swoger, F. Pampaloni, K. Greger, M. Marcello and E. H. K. Stelzer, Nat. Methods, 2007, 4, 311–313 CAS.
- E. H. K. Stelzer, Nat. Methods, 2015, 12, 23–26 CrossRef CAS PubMed.
- M. Mickoleit, B. Schmid, M. Weber, F. O. Fahrbach, S. Hombach, S. Reischauer and J. Huisken, Nat. Methods, 2014, 11, 919–922 CrossRef CAS PubMed.
- Q. Fu, B. L. Martin, D. Q. Matus and L. Gao, Nat. Commun., 2016, 7, 11088 CrossRef CAS PubMed.
- A. Yuan, B. Yang, J. Wu, Y. Hu and X. Ming, Acta Biomater., 2015, 21, 63–73 CrossRef CAS PubMed.
- J. Zhao, H. Lu, P. Xiao and M. H. Stenzel, ACS Appl. Mater. Interfaces, 2016, 8, 16622–16630 CAS.
- R. Akasov, T. Borodina, E. Zaytseva, A. Sumina, T. Bukreeva, S. Burov and E. Markvicheva, ACS Appl. Mater. Interfaces, 2015, 7, 16581–16589 CAS.
- C. L. Waite and C. M. Roth, Bioconjugate Chem., 2009, 20, 1908–1916 CrossRef CAS PubMed.
- C. L. Waite and C. M. Roth, Biotechnol. Bioeng., 2011, 108, 2999–3008 CrossRef CAS PubMed.
- X. Liu, M. Wu, Q. Hu, H. Bai, S. Zhang, Y. Shen, G. Tang and Y. Ping, ACS Nano, 2016, 10, 11385–11396 CrossRef CAS PubMed.
- V. H. B. Ho, N. K. H. Slater and R. Chen, Biomaterials, 2011, 32, 2953–2958 CrossRef CAS PubMed.
- X. Zhen, X. Wang, C. Xie, W. Wu and X. Jiang, Biomaterials, 2013, 34, 1372–1382 CrossRef CAS PubMed.
- X. Zhen, X. Wang, C. Yang, Q. Liu, W. Wu, B. Liu and X. Jiang, Macromol. Biosci., 2014, 14, 1149–1159 CrossRef CAS PubMed.
- C. Xie, P. Zhang, Z. Zhang, C. Yang, J. Zhang, W. Wu and X. Jiang, Nanoscale, 2015, 7, 12572–12580 RSC.
- M. Oishi, A. Tamura, T. Nakamura and Y. Nagasaki, Adv. Funct. Mater., 2009, 19, 827–834 CrossRef CAS.
- J. Zhao, H. Lai, H. Lu, C. Barner-Kowollik, M. H. Stenzel and P. Xiao, Biomacromolecules, 2016, 17, 2946–2955 CrossRef CAS PubMed.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- L. Tang, X. Yang, Q. Yin, K. Cai, H. Wang, I. Chaudhury, C. Yao, Q. Zhou, M. Kwon, J. A. Hartman, I. T. Dobrucki, L. W. Dobrucki, L. B. Borst, S. Lezmi, W. G. Helferich, A. L. Ferguson, T. M. Fan and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15344–15349 CrossRef CAS PubMed.
- S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer and W. C. W. Chan, Nano Lett., 2009, 9, 1909–1915 CrossRef CAS PubMed.
- H. S. Choi, W. Liu, F. Liu, K. Nasr, P. Misra, M. G. Bawendi and J. V. Frangioni, Nat. Nanotechnol., 2010, 5, 42–47 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- A. Theumer, C. Gräfe, F. Bähring, C. Bergemann, A. Hochhaus and J. H. Clement, J. Magn. Magn. Mater., 2015, 380, 27–33 CrossRef CAS.
- B. Kim, G. Han, B. J. Toley, C.-k. Kim, V. M. Rotello and N. S. Forbes, Nat. Nanotechnol., 2010, 5, 465–472 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813–827 CrossRef CAS PubMed.
- R. Savic, L. Luo, A. Eisenberg and D. Maysinger, Science, 2003, 300, 615–618 CrossRef CAS PubMed.
- T. R. Daniels, E. Bernabeu, J. A. Rodríguez, S. Patel, M. Kozman, D. A. Chiappetta, E. Holler, J. Y. Ljubimova, G. Helguera and M. L. Penichet, Biochim. Biophys. Acta, 2012, 1820, 291–317 CrossRef CAS PubMed.
- S. Mishra, P. Webster and M. E. Davis, Eur. J. Cell Biol., 2004, 83, 97–111 CrossRef CAS PubMed.
- J. E. Visvader and G. J. Lindeman, Nat. Rev. Cancer, 2008, 8, 755–768 CrossRef CAS PubMed.
- D. Sobot, S. Mura and P. Couvreur, J. Mater. Chem. B, 2016, 4, 5078–5100 RSC.
- S. Chuthapisith, J. Eremin, M. El-Sheemey and O. Eremin, Surg. Oncol., 2010, 19, 27–32 CrossRef PubMed.
- I. K. Guttilla, K. N. Phoenix, X. Hong, J. S. Tirnauer, K. P. Claffey and B. A. White, Breast Cancer Res. Treat., 2012, 132, 75–85 CrossRef CAS PubMed.
- T. A. Elbayoumi and V. P. Torchilin, Eur. J. Nucl. Med. Mol. Imaging, 2006, 33, 1196–1205 CrossRef CAS PubMed.
- V. P. Torchilin, L. Z. Iakoubov and Z. Estrov, Trends Immunol., 2001, 22, 424–427 CrossRef CAS PubMed.
- M. Munz, P. A. Baeuerle and O. Gires, Cancer Res., 2009, 69, 5627–5629 CrossRef CAS PubMed.
- F. Danhier, A. Le Breton and V. Préat, Mol. Pharmaceutics, 2012, 9, 2961–2973 CrossRef CAS PubMed.
- T. Teesalu, K. N. Sugahara and E. Ruoslahti, Front. Oncol., 2013, 3, 216–224 Search PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- D. A. Braasch, Z. Paroo, A. Constantinescu, G. Ren, O. K. Oz, R. P. Mason and D. R. Corey, Bioorg. Med. Chem. Lett., 2004, 14, 1139–1143 CrossRef CAS PubMed.
- R. Liu, D. M. Gilmore, K. A. Zubris, X. Xu, P. J. Catalano, R. F. Padera, M. W. Grinstaff and Y. L. Colson, Biomaterials, 2013, 34, 1810–1819 CrossRef CAS PubMed.
- V. M. Le, M. D. Lang, W. B. Shi and J. W. Liu, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 540–544 CrossRef CAS PubMed.
- X. Xu, C. R. Sabanayagam, D. A. Harrington, M. C. Farach-Carson and X. Jia, Biomaterials, 2014, 35, 3319–3330 CrossRef CAS PubMed.
- V. B. Lokeshwar, D. Rubinowicz, G. L. Schroeder, E. Forgacs, J. D. Minna, N. L. Block, M. Nadji and B. L. Lokeshwar, J. Biol. Chem., 2001, 276, 11922–11932 CrossRef CAS PubMed.
- A. Albanese, A. K. Lam, E. A. Sykes, J. V. Rocheleau and W. C. Chan, Nat. Commun., 2013, 4, 2718 Search PubMed.
- K. Shin, B. S. Klosterhoff and B. Han, Mol. Pharmaceutics, 2016, 13, 2214–2223 CrossRef CAS PubMed.
- C. S. Shin, B. Kwak, B. Han and K. Park, Mol. Pharmaceutics, 2013, 10, 2167–2175 CrossRef CAS PubMed.
- L. Miao, J. M. Newby, C. M. Lin, L. Zhang, F. Xu, W. Y. Kim, M. G. Forest, S. K. Lai, M. I. Milowsky, S. E. Wobker and L. Huang, ACS Nano, 2016, 10, 9243–9258 CrossRef CAS PubMed.
- B. Kwak, A. Ozcelikkale, C. S. Shin, K. Park and B. Han, J. Controlled Release, 2014, 194, 157–167 CrossRef CAS PubMed.
- R. K. P. Benninger and D. W. Piston, Curr. Protoc. Cell Biol., 2013, 59, 4.11.1–4.11.24 Search PubMed.
- F. Pampaloni, N. Ansari and E. H. K. Stelzer, Cell Tissue Res., 2013, 352, 161–177 CrossRef PubMed.
- C. Lorenzo, C. Frongia, R. Jorand, J. Fehrenbach, P. Weiss, A. Maandhui, G. Gay, B. Ducommun and V. Lobjois, Cell Div., 2011, 6, 22 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |