
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNa-promoted Ni/ZrO2 dry reforming catalyst with high efficiency: details of Na2O–ZrO2–Ni interaction controlling activity and coke formation†
M.
Németh
 a,
D.
Srankó
a,
J.
Károlyi
a,
F.
Somodi
a,
Z.
Schay
a,
G.
Sáfrán
b,
I.
Sajó
c and
A.
Horváth
*a
a,
D.
Srankó
a,
J.
Károlyi
a,
F.
Somodi
a,
Z.
Schay
a,
G.
Sáfrán
b,
I.
Sajó
c and
A.
Horváth
*a
aInstitute for Energy Security and Environmental Safety, Centre for Energy Research, Konkoly-Thege M. street 29-33, H-1121 Budapest, Hungary. E-mail: horvath.anita@energia.mta.hu
bInstitute for Technical Physics and Materials Science, Centre for Energy Research, Konkoly-Thege M. street 29-33, H-1121 Budapest, Hungary
cSzentágothai Research Centre, University of Pécs, Ifjúság street 20, H-7624 Pécs, Hungary
First published on 7th August 2017
Abstract
Herein, a 0.6 wt% Na-promoted 3% Ni/ZrO2 dry reforming catalyst and its unpromoted counterpart are discussed in detail. Structural investigations were carried out using TEM, TPR, XRD, XPS, and CO pulse chemisorption followed by TPD and DRIFTS methods. In the presence of a dry reforming mixture, bidentate carbonates were detected on Na-promoted Ni/ZrO2, while on Ni/ZrO2 effective hydrogenation by metallic Ni converted bicarbonates to formate species. In continuous flow atmospheric catalytic tests in a high excess of methane, a reactive-type coke was formed on the promoted sample, which did not cause significant deactivation. Temperature ramped 13CO2 isotope labeled dry reforming experiments in a closed loop sub-atmospheric circulation system revealed 13CH4 formation on Ni/ZrO2, while in the case of the promoted catalyst methanation was retarded until the complete consumption of oxidants (from 13CO2). In isothermal experiments in the same circulation system carbon monoxide disproportionation was observed on Ni/ZrO2 leaving carbon on Ni, besides the coke formed from the CH4 source, while on the promoted catalyst carbonaceous deposit under the same conditions did not form from CH4. The superb catalytic properties of Na-promoted Ni/ZrO2 are explained by a proposed catalytic cycle compiling the dynamic participation (formation and decomposition) of the surface Na2CO3 or NaHCO3 species surrounding the NiOxHy active sites on a ZrO2 support that is able to accommodate the labile Na2O promoter capturing and releasing CO2 oxidant.
Introduction
Carbon dioxide reforming or dry reforming of methane (DRM) is a target reaction of high importance in CO2-rich natural gas or biogas conversion. Although a few pilot plants have already been working worldwide under dry reforming or – combined with steam – bi-reforming conditions,1 there is still much to improve or even understand at the level of basic research. The strongly endothermic dry reforming reaction (1)| CH4 + CO2 ⇌ 2CO + 2H2 | (1) |
| CO + H2O ⇌ CO2 + H2. | (2) |
Concerning the reaction mechanism, the general view is that methane dissociates on the metal surface,3,4 and CO2 is activated on the support,5,6 at the metal–support interface7,8 or even on the metal surface,6,9 depending on the reaction conditions and type of support and active metal.10,11 In the next step, the surface CHx fragments (x = 0–3) react with active O or OH species and form a CHxOs like intermediate that after decomposition produces CO and H2 products.12–14 The suggested rate-determining steps vary with the catalyst system and reaction conditions.15
The hardest problem is tackling with the coke formation under the harsh, oxygen lean reaction conditions (low CO2 or water concentration) that usually cause fast deactivation of Ni-based catalysts.16–18 Coke deposition takes place and nanotubes or encapsulating graphitic layers form if the rate of surface carbon formation surpasses its gasification rate. Surprisingly, significant coke deposits can still let the catalyst work.19–22 This is why the amount of deposited coke cannot be simply correlated with the catalytic performance and actually there is no straightforward relationship between the quantity of carbon deposition and the activity. The location of carbon can be quite different: according to the simplest picture, if carbon covers the metal surface, the activity decreases, but if the carbon transfers to the support surface or is produced at the metal–support interface, the dry reforming reaction may proceed further. According to Efstathiou and his co-workers,23 we can distinguish between active and inactive carbon. Inactive carbon deposits are produced via polymerization of surface carbon species to graphite layers and carbon whiskers. The active surface carbon (Cs) can react with oxygen containing surface species (Os or OHs) and form CO desorbing into the gas phase. We should point out that only steady state transient kinetic analysis (SSITKA) is able to measure the usually very low surface coverage of active carbon that truly participates in the formation of CO by using labeled carbon dioxide or methane. As for the inactive surface carbon, Efstathoiu et al.24,25 determined the relative contribution of a CO2 activation route towards its formation via applying a labeled reactant under strictly controlled conditions. The share of the CO2 or CH4 activation route in yielding carbon deposit depended strongly on the reaction temperature and the chemical composition of the support for Ni supported on reducible Pr–Zr oxides or Ce–Zr oxides.23–25
Reducible supports such as CeO2 with high oxygen mobility,19 basic supports enhancing the CO2 adsorption/activation step4,10,26,27 or addition of alkaline (K, CaO) promoters to the support5,6,28,29 may help to diminish coke formation, because carbonates formed by CO2 adsorption at the metal–support perimeter sites are considered as scavengers of carbon.6
Among alkali promoters, the effect of potassium or sodium promotion was studied mostly in the methanation reaction, and dry reforming studies in this respect are less in number. The overall view of these investigations was that the alkali/sodium promoter decreases the amount of deposited coke; however, the concentration of promoter and the synthesis method can greatly influence the catalytic properties. CO hydrogenation was studied on co-precipitated Na–Mn–Ni catalyst and compared to the reference Ni/SiO2, a good methanation catalyst: here, Na was found to decrease the methanation activity and also the CO dissociation.30 Potassium-modified Ru/SiO2 was tested for methanation and Fischer–Tropsch synthesis, and again, selective poisoning of methanation and more strongly bound bridged CO were observed.31 Catalytic oxidation of formaldehyde at room temperature was studied on 1% Pd/TiO2 promoted by 2% Na by co-impregnation, and a negatively charged Pd surface was detected by XPS supposedly due to electron donation by sodium.32
As for dry reforming specifically, Lovell and his co-workers20 pointed out that the Na content of Ni/MCM caused lower DRM activity and higher RWGS contribution. In this case the Na-modified support was first prepared and then impregnated with the Ni precursor in a second step. Higher stability was observed by Ballarini and his co-workers28,29 upon Na addition to Pt/ZrO2 and Pt/Al2O3 due to the creation of basic sites, but a positive effect of the sodium species was observed on Pt/Al2O3 only above 0.5 wt% Pt content. In this case Na or K was added again to the support followed by calcination and impregnation with the Pt salt.
A deeper and more adequate investigation of the effect of alkali promotion of 10% Ni/Al2O3 was published by Mori et al.33,34 They declared that oxides of Na, K, Mg, and Ca markedly suppress carbon deposition during CO2 reforming via the decrease of the CH4 decomposition ability of nickel (note that alkaline promoters were post impregnated onto the calcined nickel catalyst). Based on the determined reaction orders of CH4 and CO2, it was declared that the surface of a 10% CaO-modified Ni/Al2O3 catalyst was supposed to be abundant in adsorbed CO2 instead of methane. The kinetics of the individual steps of reforming were further examined34 on Ni/Al2O3 loaded with 0–10 wt% K. Although the adsorption of CO2 was enhanced by the presence of potassium, the dissociation of CO2 to CO and Os was not significantly influenced. This suggested that the enhancement of the oxidation of CHx by increasing the concentration of Os is not the cause of the carbon-free CO2 reforming but the physical blockade of Ni ensemble by potassium.
Until now, different supported metal catalysts for dry reforming have been studied in our laboratory.19,26,27 Sodium-promoted 1% Ni, 3% Ni and 1% Pt/ZrO2 catalysts proved to be very effective in short range low temperature activity tests,35 and this inspired us to conduct further research on how the Na2O promoter acts under different reaction conditions and longer time on stream compared to an unpromoted sample. In the present manuscript we will pay special attention to the Ni–Na2O–ZrO2 interface that is thought to influence the catalytic properties, taking the highly active and stable 3% Ni/ZrO2 catalyst promoted with 0.6 wt% Na as an example. The differences between the promoted and the unpromoted sample before and after the catalytic runs are studied by X-ray powder diffraction (XRD), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and diffuse reflectance infrared spectroscopy (DRIFTS). The catalytic behavior is evaluated in a fixed bed tubular reactor in high excess (70%) of methane and in a closed loop circulation system at sub-atmospheric pressure, using labeled 13CO2 reactant that allows us to follow the interconversion of carbon atoms of both reactants. Transmission electron microscopy and temperature programmed oxidation of deposited coke after the reactions can shed some light on the intimate interaction of Ni–Na2O–ZrO2 components leading to an active and stable catalyst.
Experimental
Sample preparation
Zr hydroxide from MEL Chemicals was calcined at 600 °C resulting in ZrO2 with about 40 m2 g−1 surface area. The preparation of base 3% Ni/ZrO2 was done by incipient wetness impregnation of ZrO2 with Ni(NO3)2 solution (denoted as Ni/ZrO2 sample). The preparation of the Na2O-promoted 3% Ni/ZrO2 catalyst (denoted as Na–Ni/ZrO2) is described in ref. 35. Briefly, the pH of the aqueous suspension of ZrO2 powder and Ni nitrate precursor was set to pH 6.5 using NaHCO3 (resulting in a final 0.6 wt% Na loading) and heated to 70 °C, then the water was slowly evaporated over 3–4 hours, and the solid was dried. A parent Na-promoted support was prepared in the same way without addition of Ni nitrate (denoted as Na-ZrO2). Dried samples were calcined in air at 600 °C, reduced in H2 at 600 °C for 2 hours and stored in air before further use (these are referred to as as-received samples from here on).Catalyst characterization
The reduction properties of the oxidized catalyst samples were investigated in a Micromeritics AutoChem 2920 catalyst characterization system. TCD response was calibrated in a separate measurement before the temperature programmed reduction (TPR) run. 50 mg as-received catalysts were first oxidized at 700 °C for 1 h in a 10% O2/He stream, cooled under Ar and reduced in a 10% H2/Ar stream with a 10 °C min−1 temperature ramp up to 700 °C followed by a 1 hour isothermal hold. Reducibility was calculated based on the H2 consumption supposing that NiO was reduced during the TPR.CO pulse chemisorption measurements were conducted with 10% CO/He pulses in a Micromeritics AutoChem 2920 flow system after the TPR run. The Ni dispersion in % was calculated by determining the moles of CO molecules adsorbed from the pulses at standard temperature and pressure/moles of Ni in the sample, assuming CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni = 1 chemisorption stoichiometry (this means each metallic surface Ni atom is able to bind a CO molecule that does not dissociate at the temperature of measurement and the chemisorption is strong and irreversible under the flow of the inert He gas). After the CO pulses, temperature programmed desorption (CO-TPD) experiments under He were carried out up to 700 °C and monitored using QMS analysis.
:Ni = 1 chemisorption stoichiometry (this means each metallic surface Ni atom is able to bind a CO molecule that does not dissociate at the temperature of measurement and the chemisorption is strong and irreversible under the flow of the inert He gas). After the CO pulses, temperature programmed desorption (CO-TPD) experiments under He were carried out up to 700 °C and monitored using QMS analysis.
The phase composition of crystalline components was investigated by X-ray powder diffraction (XRPD) analyses after the above listed measurements (TPR and CO TPD). The diffraction patterns were obtained using a Phillips model PW 3710 based PW 1050 Bragg–Brentano parafocusing goniometer using CuKα radiation (λ = 0.15418 nm) with a graphite monochromator and a proportional counter. The digitally recorded XRD scans were evaluated for quantitative phase composition using a full profile fit method with corrections for preferred orientation and microabsorption. This method let us estimate the Ni particle size even if the diffraction peaks of metal and oxide are overlapped.
The morphology and structure of the catalysts and the carbon contamination after dry reforming tests were studied by transmission electron microscopy using a PHILIPS CM 20 conventional 200 kV TEM and a high resolution JEOL 3010 microscope operating at 300 kV with point resolution of 0.17 nm (HRTEM). The samples were prepared by drop drying the aqueous suspensions on carbon-coated micro grids.
For the determination of surface composition, X-ray photoelectron spectroscopy measurements were applied using a KRATOS XSAM 800 XPS machine equipped with an atmospheric reaction chamber. An Al Kα characteristic X-ray line, 40 eV pass energy and FAT mode were applied for recording the XPS lines of Ni 2p, C 1s, O 1s, Zr 3d, and Na 1s. Adventitious carbon C 1s binding energy at 284.8 eV was used as reference for charge compensation. The samples were measured after in situ calcination in synthetic air at 600 °C for 30 min (10 °C min−1) and after the subsequent in situ H2 treatment at 600 °C for 30 min (10 °C min−1) (an atmospheric pretreatment chamber connected to a UHV chamber with a load lock gate allows us to do pretreatments without allowing the sample to come in contact with air).
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was applied to study the catalyst samples under different conditions. Spectra were collected on a Nicolet iS50 infrared spectrometer equipped with a MCT detector and a Specac DRIFT environmental chamber with a ZnSe window. The maximum allowed temperature of the cell was 500 °C. The introduced gas flow directly passed over the upper surface of the catalyst placed in the heatable sample holder of the DRIFTS cell facing the zinc selenide window. Spectra were obtained by collecting 64 scans with a resolution of 4 cm−1 and presented as log(1/R) mode, where R is the reflectance. For in situ reduction, the sample in the DRIFTS cell was heated to 500 °C under a 5% H2/Ar atmosphere at a rate of 10 °C min−1 and kept at this temperature for 30 min; then, it was cooled to the desired temperature of the measurement. CO chemisorption measurements at room temperature were done using 1% CO in He. Temperature programmed DRIFTS measurements were conducted in the presence of a CH4:CO2 = 70:30 DRM reactant mixture (flow rate: 50 cc min−1) after the in situ reduction treatment. Spectra were taken from 300 °C to 500 °C.
Catalytic measurements
:CO2:Ar = 68:31:1 mixture (from here on this is referred to as DRM mixture for the sake of simplicity). An extremely high concentration of methane was set to mimic biogas composition. 10 mg of catalyst along with 100 mg of diluting quartz beads were placed in the tubular quartz reactor where the reactant mixture was introduced at a flow rate of 20 mL min−1 (120 L h−1 gcat−1). At the beginning of the short catalytic tests, the as-received samples were ramped at 10 °C min−1 in a 30 mL min−1 90% H2/Ar stream to 600 °C and kept at this temperature for 30 min. After reduction, the sample was purged with He (30 mL min−1) while it cooled to room temperature. Next, the He flow was changed to the reactant gas mixture and the temperature was increased to 600 °C at 10 °C min−1 followed by a 2 h hold time. The other type of catalytic experiments was the long-term isothermal stability test lasting for 24 hours. For these experiments, a new portion of the as-prepared sample was reduced with 90% H2/Ar by heating the catalyst to 750 °C at a rate of 10 °C min−1 and maintaining this temperature for 30 min. Subsequently, the sample was cooled to 675 °C in 8 min while it was purged with He, then the flowing gas was switched to the DRM mixture. A quadrupole Pfeiffer Prisma mass spectrometer was connected via a differentially pumped quartz capillary to the reactor outlet. Due to the reaction stoichiometry that causes a volume flow increase at the outlet of the reactor, argon was used as an internal reference gas for calculation of the outlet mass flows to determine the H2/CO ratio and methane and carbon dioxide conversion values after adequate calibration.
The quantitative analysis was based on the following equation:
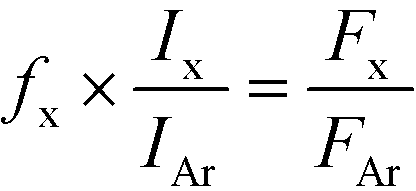
The following mass signals as representatives of the gas components were measured: 2-H2, 15-CH4, 28-CO, and 44-CO2. In the quantification of mass signal m/z = 28 (CO), the actual fragmentation of m/z = 44 (CO2) was taken into account. The error in the carbon balance in the effluent was within ±5% in all measurements. The relative difference between two repeated catalytic runs was about 5%. Note that sampling was very fast and the linked symbols showing QMS signals in any of the figures are the results of skipping several measured points for the sake of clarity.
Temperature programmed oxidation (TPO) measurements were conducted in the same flow system to detect and measure carbon deposits formed in the DRM reaction. At the end of the short catalytic run, the gas flow was switched to He and the system was cooled to room temperature. Then, the samples were oxidized in a 30 mL min−1 O2:He:Ar = 10:89:1 mixture by heating from ambient temperature to 600 °C at a rate of 10 °C min−1 followed by a 30 min isothermal hold. After the stability test, the temperature during the TPO runs was increased to 750 °C. The CO2 signal was used for quantification of coke after a calibration procedure.
:1 ratio of the reactants, precisely 25 ± 0.5 mbar CH4 (containing 2% Ar) and 25 ± 0.5 mbar 13CO2 (99% purity, Consigour Ltd.), was applied. The system could be separated into the gas blending space and the reactor space, and these two could be merged when needed. Reactants were introduced by measuring the sequential pressure using a capacitive pressure gauge in the gas blending space. Circulation was carried out using a double-acting piston pump. During the experiments, the overall system containing a certain amount of gases (p = 50 mbar for DRM and p = 200 mbar for TPO) was monitored through a fine glass capillary connected to a Pfeiffer Prisma quadrupole mass spectrometer. DRM experiments in this circulation system are able to provide only qualitative results due to the nature of the capillary inlet system and the significant pressure increase during the reaction (MS calibration of all components would be elusive). The following mass signals were taken as representatives of the gas components: 2-H2, 15-CH4, 17-13CH4, 28-CO, 29-13CO, 40-Ar, 44-CO2, and 45-13CO2. It is also well known that H2O has a small contribution at m/z = 17 and CO2 molecules have a low intensity fragment signal at m/z = 28 (or in the case of labeled 13CO2 at m/z = 29). However, it was ascertained that the m/z = 17 signal during dry reforming corresponded to the change of 13CH4 concentration and not to that of H2O. At the beginning of the experiments, 10 mg of the sample was placed into a U-shaped quartz reactor tube. After an in situ reduction treatment in H2 flow at 600 °C for 30 min, the sample was cooled to 150 °C and evacuated to about 1.5 × 10−2 mbar pressure. There were 2 types of catalytic experiments ending with TPO measurements. In one case, the premixed reactants were introduced into the reduced, evacuated sample at about 150 °C; afterwards the temperature was ramped up to 600 °C at a 10 °C min−1 rate and held there for 30 min, and then evacuated and cooled close to room temperature before the TPO measurements (ramp-hold type experiment). The other type of measurements was the isothermal runs at 600 °C, which means that the reduced sample in the reactor space was heated in vacuum to 600 °C; then the reactant mixture was introduced into the catalyst, and after 30 min it was evacuated and then cooled for the start of TPO. Terminal TPO measurements were carried out by adding 200 mbar oxygen to the evacuated catalyst and ramping the temperature again to 600 °C. The CO2 signal (CO alone was not formed) detected at m/z = 44 and m/z = 45 was differentiated to get a peak-shaped curve instead of the original integral signal. In this case the CO2 signal could be calibrated and the surface coke evolving as gas phase labeled or unlabeled carbon dioxide was quantified.
Results and discussion
Catalyst characterization
First, the temperature programmed reduction measurements will be discussed. A considerable difference was seen in the reduction behavior of the oxidized samples as depicted in Fig. 1. TPR experiments revealed 96.2% reducibility in the case of the Na–Ni/ZrO2 sample and 96.9% for the Ni/ZrO2 catalyst, supposing NiO to be the reducible compound. This means that a nearly complete reduction of the Ni oxide phase has already taken place below 600 °C in both samples. The NiO in weak interaction with the support was present in a considerable amount on Ni/ZrO2 and was reduced at around 315 °C. The NiOx species in stronger interaction with the support37 were reducible at 428 °C on this sample but only at 490 °C in the case of the sodium-containing counterpart. It seems that the Na incorporation resulted in a NiOx phase that is relatively hard to reduce.
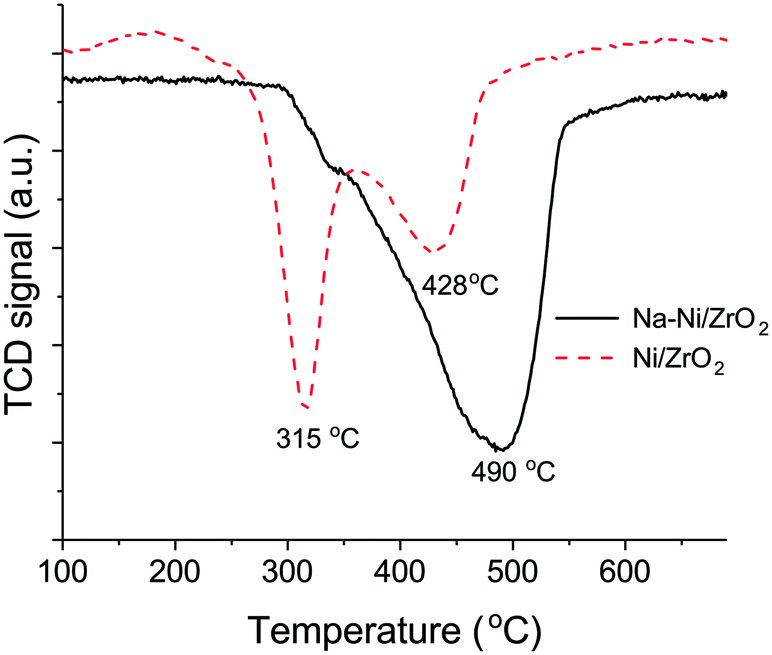 | ||
| Fig. 1 Results of temperature programmed reduction on preoxidized Ni/ZrO2 and Na–Ni/ZrO2 samples. | ||
Determination of Ni size in the ZrO2 supported samples turned to be difficult using TEM due to the lack of sufficient contrast between the crystalline oxide support and the Ni particles. Metal particle size estimated by XRD could not provide firm results either, because the Ni (111) and the monoclinic baddeleyite ZrO2 peaks overlapped after calcination/reduction treatment at 600 °C.35 XRD measurements were done on the samples after the TPR run as well, with the aim of detecting considerable sintering if any happened. The size of the Ni particles was estimated to be around 20 nm in both Ni samples (Table 1). No separate Ni peaks could be detected as seen in the XRD patterns in Fig. S2.† The only small difference is that a minute tetragonal ZrO2 phase originating from the support preparation process is still seen for the Ni/ZrO2 besides the prevailing monoclinic structure, while it is absent for Na–Ni/ZrO2.
| Sample name | Ni particle size by XRDa | Ni dispersion by CO pulse chemisorptiona | XPS surface concentrationb | |
|---|---|---|---|---|
| Ni/Zr | Na/Zr | |||
|
a After TPR measurements. Particle size was calculated with CO:Ni = 1 stoichiometry supposing hemispherical shape.
b After in situ calcination/reduction treatment at 600 °C.
|
||||
| Na–Ni/ZrO2 | 20 nm | 0.9%/114 nm | 0.07 | 0.83 |
| Ni/ZrO2 | 20 nm | 2.2%/46 nm | 0.04 | 0.08 |
CO pulse chemisorption (see Table 1) after TPR revealed low Ni dispersion values for both samples. We must emphasize that these data were obtained by assuming a CO:Ni = 1 chemisorption stoichiometry. However, the dispersion values are underestimated, because real CO bonding is not linear, but CO binds to more than 1 surface Ni atom in a bridged form (see FTIR data later). The other reason for the extremely low dispersion of Na–Ni/ZrO2 may be that sodium (oxide) suppresses the CO chemisorption if the Ni surface is partially covered after the high temperature reduction as was reported for Li, Na or K-doped Ni catalysts.34,38,39 A similar conclusion was drawn in the case of Ni–CaO–ZrO2 catalysts prepared by co-precipitation, when decoration of Ni by the support itself was thought to be the reason for the low dispersion measured by H2 chemisorption.37
Balakos and Chuang30 also pointed out that changes during surface composition under reduction and reaction conditions give rise to uncertainty in H2 chemisorption based activity data. According to our results and the adequate literature, we might assume that a part of the Ni surface is electronically influenced or physically covered by Na2O or Na2CO3 patches during CO chemisorption on the promoted catalyst.
Table 1 summarizes the Ni and Na surface concentrations obtained by XPS investigation after in situ calcination/reduction treatment at 600 °C. Note that the ZrO2 support itself contains an insignificant amount of Na impurity, but the NaHCO3 addition during sample preparation causes a 10-fold increase in the Na/Zr ratio. If we consider that 0.6 wt% Na was added during preparation, the Na/Zrbulk would be 0.03, but it is straightforward that Na is segregated and well dispersed on the surface because Na/Zrsurf = 0.83 in the reduced state. A simple rough estimation supposing monolayer coverage of our ZrO2 with Na2O reveals that a maximum of 20% of the ZrO2 surface might be covered (37 m2 g−1 BET surface area of pure ZrO2 was used in the calculation). Thus, it should be accepted that sodium either exists as small Na2O islands on the support or is integrated into the ZrO2 network forming diluted structures of Na2ZrO3. This possibility is really important since bulk Na2ZrO3 was found to be a highly efficient high temperature (T > 500 °C) CO2 absorbent that can bind and release CO2 depending on the temperature and partial pressure of the gas (CO2 + Na2ZrO3 ⇌ Na2CO3 + ZrO2), according to ref. 40 and 41.
Investigating the C 1s region, the following statements could be drawn. The C 1s peak beside the main adventitious carbon component at 284.8 eV had a shoulder at 288.8 eV in the calcined state of the promoted sample, corresponding to a carbonate type carbon (see Fig. S3†), while in the case of the unpromoted sample that shoulder was absent. It means that some carbonates are strongly held by the sodium dispersed on the ZrO2 support even after air treatment at 600 °C and evacuation. After in situ reduction, however, they are gone and only a little adventitious carbon remains on the surface (Fig. S3b†). The Na 1s peak after calcination and reduction was detected at 1071.9 eV (see Fig. S3c and d†). This Na2O must be spread over the ZrO2 and connected to ZrO2via Na–O–Zr entities. The small shoulder at an unusually low binding energy of 1068 eV (Fig. S3d†) in the promoted reduced sample supposedly belongs to sodium species in intimate contact with nickel.
Comparing the surface distribution of Ni obtained by XPS measurements (Ni/Zr in Table 1), the Na2O-promoted sample seems to be more disperse based on the higher Ni/Zr ratio (in contrast to the CO pulse chemisorption data). Let us consider now the Ni states in both samples after in situ calcination treatment as shown in Fig. 2a and b (the fitted curves together with the measured ones are shown in Fig. S3†). The peak detected at 853.9 eV in the Ni 2p region can be assigned to Ni2+ in the NiO phase in the case of the bare catalyst, while over the sodium-promoted sample the nickel signal could be fitted with 2 components, the one at 854.1 eV is Ni2+ present as NiO and the other component at 855.9 eV is assignable to Ni hydroxide. This Ni(OH)2 is supposedly responsible for the stronger oxide–support interaction as was observed during TPR measurements. The reduced Ni state seen in Fig. 2c and d significantly differs for the two samples. In the unpromoted catalyst, Ni is metallic (B.E. at 852.8 eV), but the sodium containing sample – besides metallic Ni – still has a significant contribution at 855.8 eV, corresponding to Ni2+ most probably in a NiOxHy species. This surface hydroxide-like nickel compound seems to be very stable even under a reducing atmosphere, although TPR suggested almost complete reduction of Ni oxide at 600 °C. This might be explained if we assume a well dispersed, thin layered form of Ni hydroxide. The other issue to note is the extremely low intensity satellite peak (Fig. 2d) compared to what one would expect based on the intensity of the apparent Ni(OH)2 (∼NiOxHy) component. This feature suggests that some unique electronic interaction prevails between nickel and other element(s) or even could be the sign of a mixed phase or alloy formation. We suppose that sodium ion is embedded in the NiOxHy–Ni matrix, resulting in decreased satellite intensity. This assumption is based on literature analogues. According to Roberts et al.,42 the interaction of potassium with an oxidized Ni(110) single crystal during annealing resulted in the suppression of the NiO satellite structure due to the incorporation of alkali promoter into the oxide lattice. At 300 °C they observed Ni3+ species present probably as potassium nickelate (K2O2 + 2NiO = 2KNiO2). We cannot exclude or prove that some of the Ni is in the 3+ oxidation state after calcination due to the poor resolution and low signal intensity in Fig. 2b. However, under reducing atmosphere, the inclusion of some sodium oxide into the surface metallic/oxidized Ni compounds or just as a surface decoration is certainly reasonable based on these XPS results. We may even suggest that under a reducing atmosphere an interaction similar to the classic strong metal support interaction (SMSI) develops in the promoted catalyst and part of the metallic Ni surface is surrounded/buried by a Na–O–Zr network.
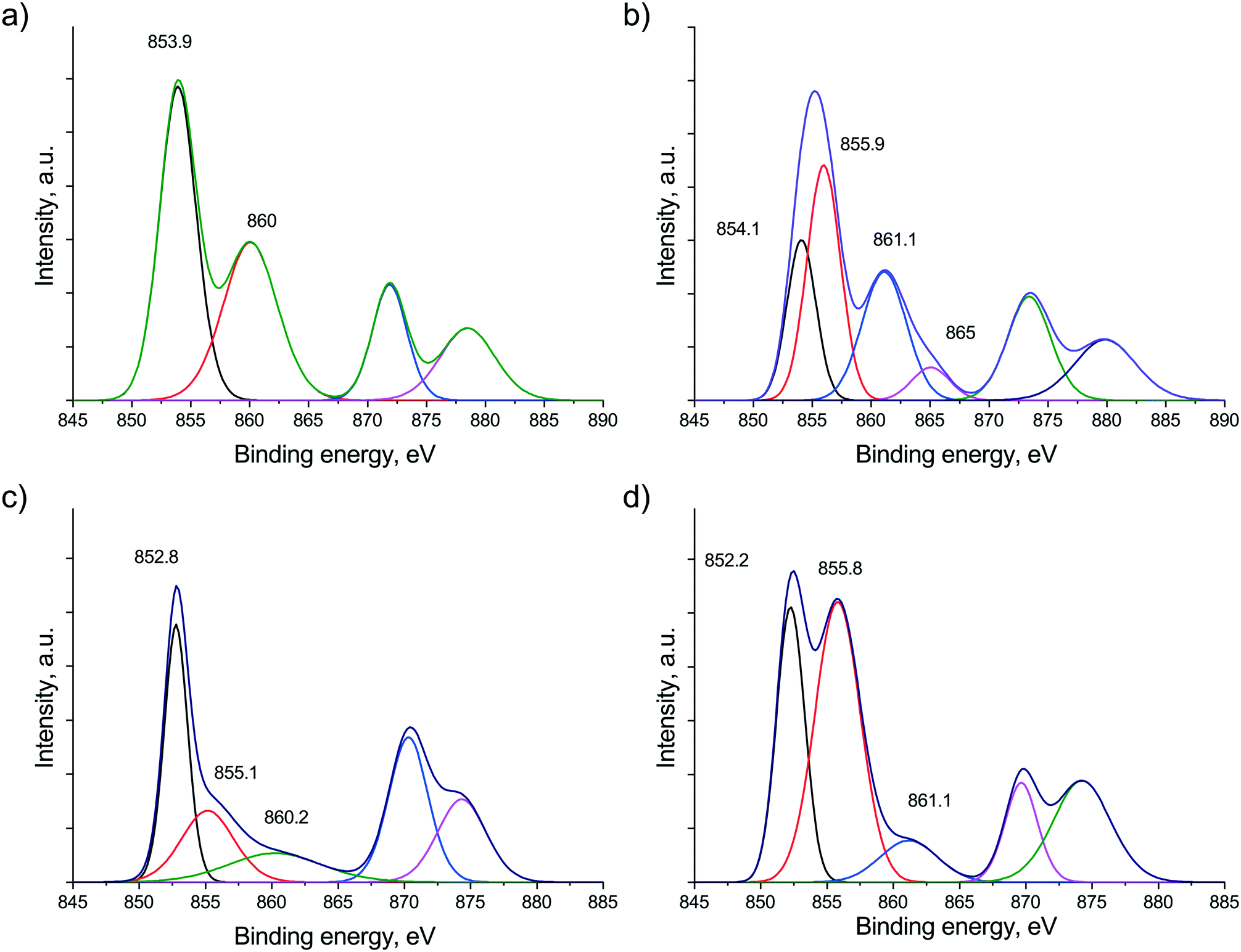 | ||
| Fig. 2 Ni 2p region measured by XPS after calcination at 600 °C on (a) Ni/ZrO2 and (b) Na–Ni/ZrO2 and after reduction at 600 °C on (c) Ni/ZrO2 and (d) Na–Ni/ZrO2. Fitted curves are shown. | ||
The different surface properties of the catalysts were studied by means of CO-TPD experiments carried out in He after CO pulse chemisorption and depicted in Fig. 3a–d. The desorbing species were CO, CO2 and H2O from both catalysts, while H2 desorbed exclusively from the sodium promoted sample. The water desorption curve has a maximum at 150 °C followed by a broad tail and a distinct shoulder at 250 °C in the case of the sodium promoted sample (Fig. 3b), which means that molecular water or hydroxyl groups with a certain and quite uniform environment leave the surface at low temperature. Considering the presence of chemisorbed CO on Ni/NiOxHy and the hydroxyls of the support, a surface water-gas shift reaction may proceed, producing H2 and CO2 through a formate intermediate. The latter is decomposed to H2 and CO2 at low temperature43 but the CO2 re-adsorbs and desorbs together with the stable carbonates of the support at higher temperature only. The high temperature appearance of CO2 with a peak maximum at 450 °C shows the much higher basicity of the sodium promoted sample as expected. The featureless H2O desorption curve with a maximum at around 300 °C for the unpromoted sample points to a wide distribution of strongly bound surface hydroxyls in this case. The absence of desorbing H2 points to the inability of this catalyst to undergo the WGSR under the same conditions.
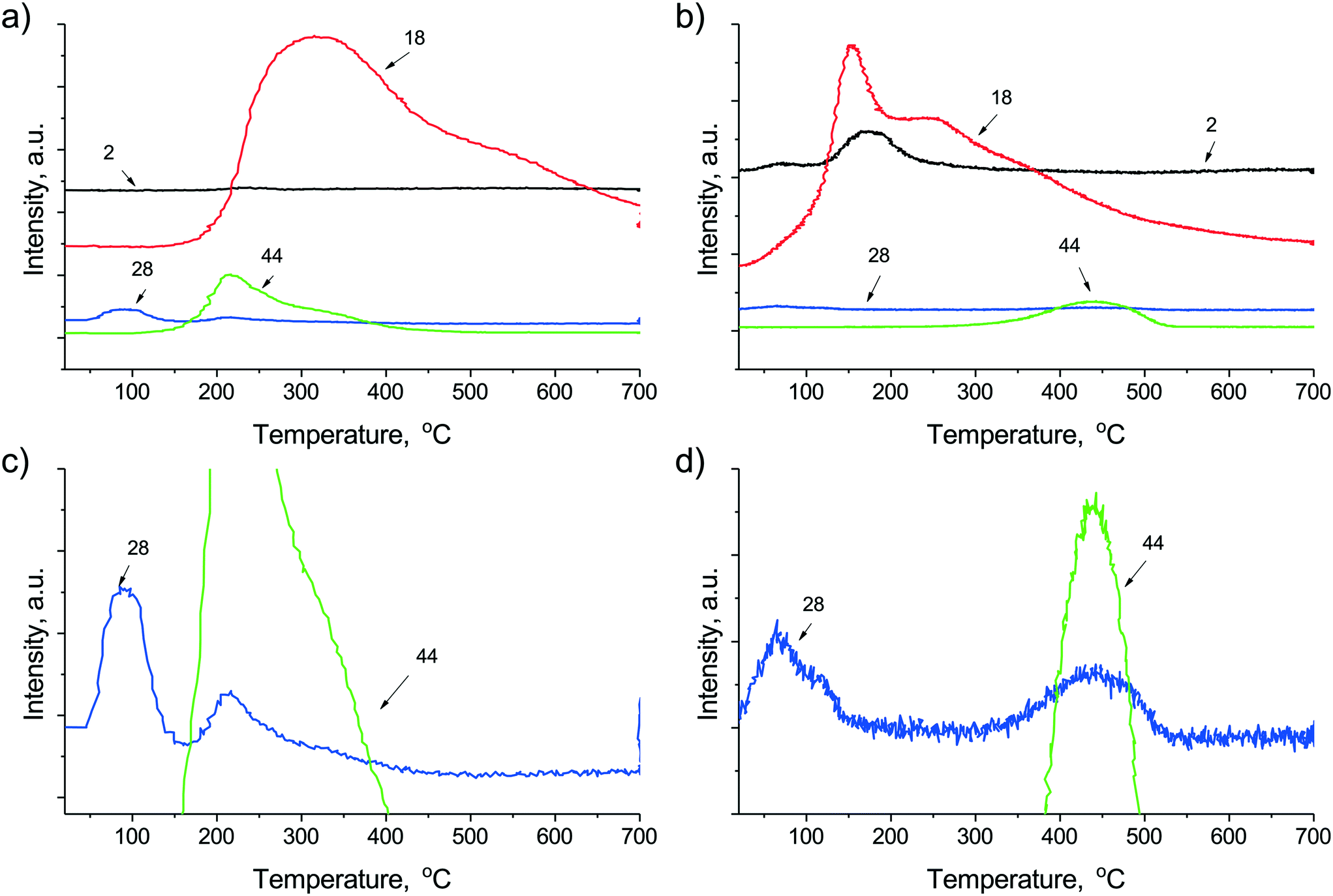 | ||
| Fig. 3 Temperature programmed desorption spectrum in He flow after CO pulse chemisorption carried out on (a) Ni/ZrO2 and (b) Na–Ni/ZrO2 and the corresponding enlarged 28 (=CO) and 44 (=CO2) signals of (c) Ni/ZrO2 and (d) Na–Ni/ZrO2. Note that the second peak in the m/z = 28 signals corresponds only to the fragmentation of CO2 in both cases. | ||
In our previous contribution35 we could not prove or exclude the role of carbonates in the coke oxidizing steps50 because a suitable catalyst without sodium was not available. Our aim was now to follow the conversion of IR visible surface species during temperature increase up to 500 °C in the presence of the DRM reaction mixture under very similar conditions to those of the forthcoming short catalytic tests (atmospheric pressure, CH4:CO2 = 70:30 mixture, 50 cc min−1 flow). Fig. 4 depicts DRIFTS spectra obtained on both samples and the corresponding supports in the presence of a DRM mixture during temperature ramp. The lowest (300 °C) and the highest (500 °C) spectra are compared. The gas phase CH4 and CO2 peaks can be easily discerned at wavenumbers 3015 and 1304 cm−1 and 2360 and 2340 cm−1, respectively. Comparing the unpromoted catalyst with its ZrO2 support, we can conclude that bidentate formate species (2878, 1570, 1365 cm−1) dominate over bicarbonates51 detected at 1627, 1419 and 1224 cm−1 on its surface at 300 °C (Fig. 4a, curves A and B). This means that via finite dissociation of CH4 on Ni particles Hs is produced that accelerates the bicarbonate → formate transition. Chemisorbed CO on Ni is already detected at 2054 cm−1. Inspection of the spectrum at 500 °C taken on Ni/ZrO2 (Fig. 4b, curve B) revealed that formate species may still be present at 1568 cm−1 together with mono- and polydentate carbonates (1537 cm−1).
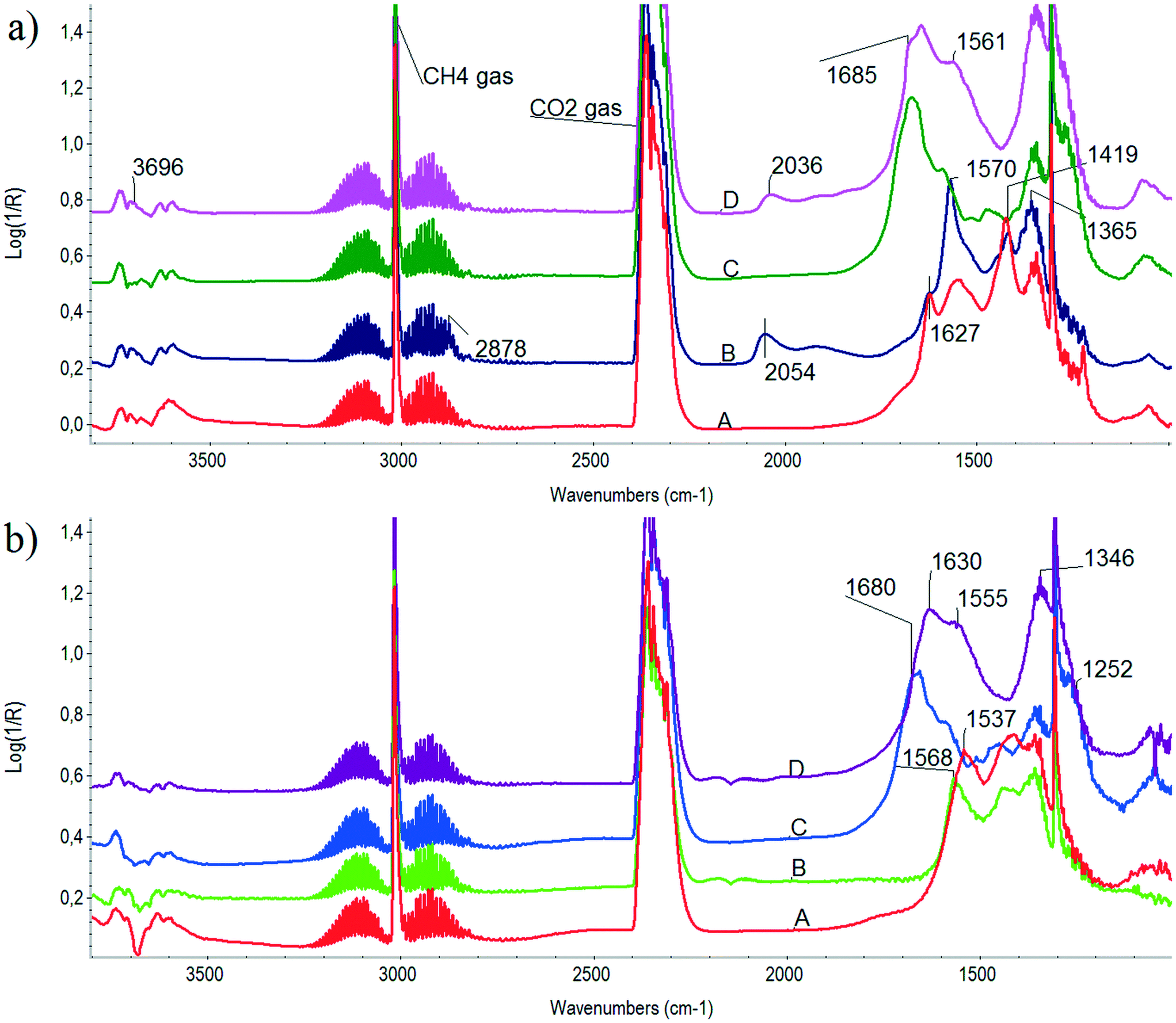 | ||
| Fig. 4 DRIFTS spectra obtained in the presence of CH4:CO2 = 70:30 DRM flow at (a) 300 °C and (b) 500 °C. Curve A: ZrO2 support, B: Ni/ZrO2, C: Na–ZrO2 support, D: Na–Ni/ZrO2. | ||
As for the sodium-promoted case, the difference under a DRM mixture between the support and the catalyst is not so clearly seen. On the Na–Ni/ZrO2 catalyst, a few OH at 3696 cm−1, bidentate carbonates52 at 1645 cm−1, bridged bidentate carbonate53 at 1685 cm−1, monodentate carbonate at 1561 and 1350 cm−1 plus CO chemisorbed on Ni (at 2036 cm−1) are observed at 300 °C. At 500 °C (see Fig. 4b), the situation is very similar except for the missing carbonyls on Ni and the absence of bands at 1680 and 1252 cm−1 on Na–Ni/ZrO2 but still having bands at 1630, 1555 and 1346 cm−1 (bidentate and monodentate carbonate). Although gas phase CH4 obscures the region, we claim that there is no sign of formates at any of the temperatures on Na–Ni/ZrO2 under dry reforming mixture. Moreover, bridged bidentate and bidentate carbonates form only on Na–Ni/ZrO2 due to the presence of sodium on the surface; the former seems to be destabilized (converted) on the Ni-containing catalyst.
Catalytic properties and investigation of the spent samples by TEM and TPO
:CO2:Ar = 68:31:1 mixture at atmospheric pressure in two ways: i) short tests after reduction at 600 °C in H2 consisting of a temperature ramp in DRM mixture to 600 °C followed by an isothermal hold for 2 hours, and ii) stability tests after a 750 °C reduction treatment at constant 675 °C reaction temperature.
Fig. 5 shows the results of the short test. Slightly higher CH4 and lower CO2 conversion could be achieved by the Ni/ZrO2 catalyst at the end of the temperature ramp, and it was deactivated more at 600 °C than the promoted sample. The ratio of CO2/CH4 conversion was about the same for both samples at the end of the experiment, but the H2/CO ratio was lower for Ni/ZrO2 and it changed from 0.71 to 0.55, reflecting some deactivating tendency during the 2 hours of the isothermal part.54 If the H2/CO ratio is lower than the theoretical, it is generally considered that besides DRM, reverse water gas shift happens55 (note that water was not quantified during the measurements, but H2O m/z = 18 signal was fairly low and constant). However, if reverse WGSR would account for the lower H2/CO ratio, the CO2 conversion should be higher on Ni/ZrO2. Clearly, this is not the case. We believe that this small difference in the H2/CO ratio is caused by the different reaction routes producing CO and H2 on the two samples. Most probably the CO2 dissociation into CO is favored on Ni/ZrO2, resulting in more CO.
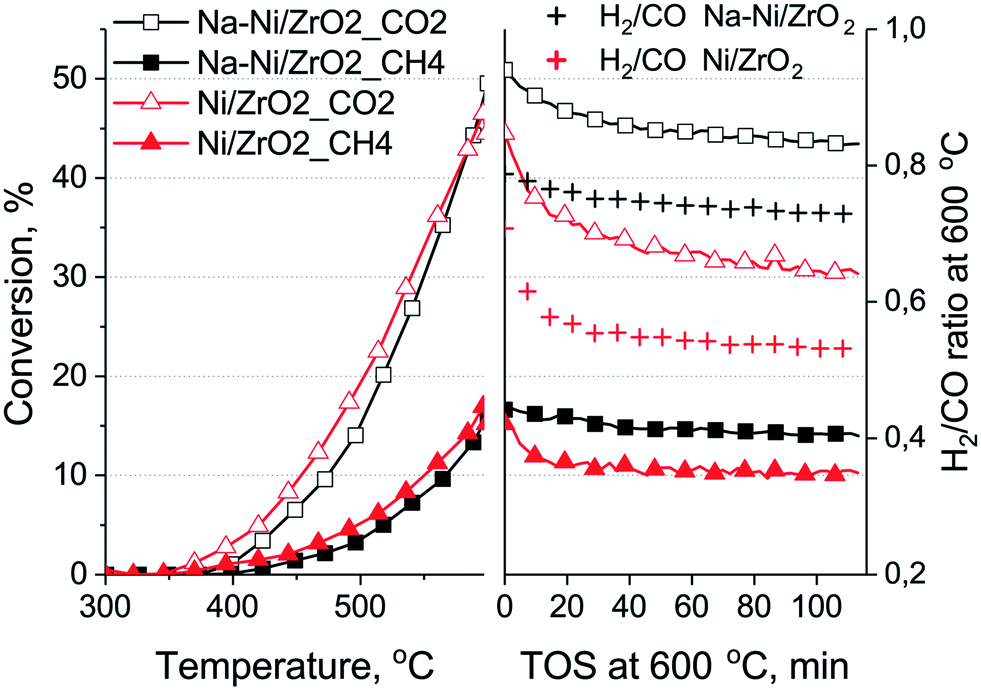 | ||
| Fig. 5 Methane and CO2 conversion curves and the H2/CO ratios during a short term catalytic run on both samples. CH4: full symbols, CO2: empty symbols. Conditions: temperature ramp in CH4:CO2:Ar = 68:31:1 mixture, 120 L h−1 gcat−1, at a 10 °C min−1 rate to 600 °C and then isothermal 120 min hold time. | ||
TEM measurements on the spent catalysts are shown in Fig. 6a–c. Fig. 6a shows a 5 nm size Ni particle (indicated by an arrow) covered by graphitic layers and whirling carbon nanotubes or filaments located also on the support particles of Ni/ZrO2. The carbonaceous deposit on the Na–Ni/ZrO2 sample is composed of disordered carbon nanotubes sometimes with Ni particles at their ends. We observed an amorphous layer or shell on the support particles (indicated by arrows in Fig. 6b) that could also be carbonaceous deposits. The HRTEM image in Fig. 6c shows a 22 nm Ni particle at the end of a carbon nanotube. In the light of the nanotube formation mechanism, the lattice period of 2.00–2.01 Å measured at the outer line or the border of a Ni particle may represent (113) Ni3C, the 2.05 Å might be a strained Ni (111) and the one with 2.08 Å spacing is assigned to NiO (200) lattice. The largest measured period of 2.36 Å fits neither Ni nor Ni carbide; however, it can be indexed as (011) of Ni(OH)2 or (011) of NiOOH spacing. We can thus declare that NiO, probably Ni(OH)2 and Ni/Ni3C, can be observed beside each other in small patches or domains of the metal surface. Indeed, we cannot prove if the oxidized forms of nickel were formed during the reaction or after the handling of the spent sample in air, but we suppose that some of them were already present during the reaction. If so, we tentatively suggest that the neighboring Ni species with different oxidation states keep the particle active, since these kinds of nanodomains – making a “ruffled” Ni look – were also seen on other metal particles of this sample, while none was observed on Ni/ZrO2.
 | ||
| Fig. 6 TEM images taken after short term catalytic tests on (a) Ni/ZrO2 and (b) Na–Ni/ZrO2. (c) HRTEM image of a single Ni particle at the end of a nanotube in the spent Na–Ni/ZrO2. | ||
Fig. 7 shows the results of TPO measurements after the short catalytic tests (together with those after long tests discussed later on). On Na–Ni/ZrO2 4.2 mg carbon/10 mgcat (42 wt% C) was found, while only 2.1 mg C/10 mgcat (21 wt% C) was found on the unpromoted sample, although the latter sample was less active at the end of the test. The different shape of the TPO curves and the descending part of the CO2 signal after the 600 °C temperature plateau reflects a difference in the kinetics of the coke oxidation process, being faster on Na–Ni/ZrO2. This means that although the amount of deposited coke is larger, it is not so strongly held and can be removed more easily. In contrast, the lesser amount of coke (half amount) on Ni/ZrO2 depresses the catalytic activity of Ni sites to a larger extent (lower activity) and it was more difficult to remove. It is widely reported in the literature that Ni particle size or metal loading may increase the amount of coke sometimes without losing catalytic activity.22 In our case, metal dispersion determined by CO pulse chemisorption was higher for the Ni/ZrO2 sample; however, the number of Ni particles observed in the TEM images of the spent samples was not sufficient to determine the average particle size, and we know that reaction conditions might cause extra sintering of the metal. Still, keeping all these in mind, it seems that the similar size Ni particles co-operating with the Na oxide/carbonate promoter transfer a part of the surface coke far from the active Ni sites.
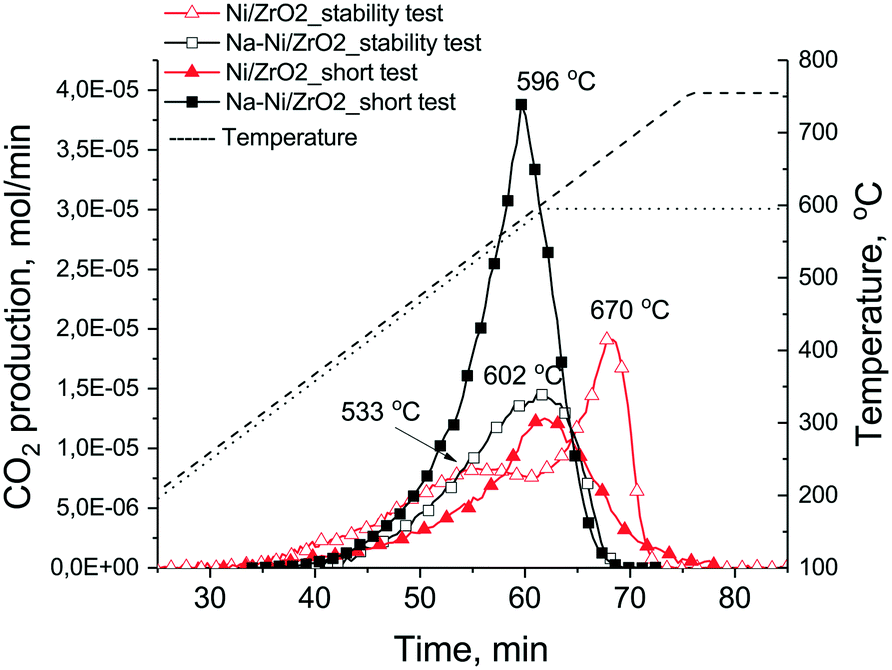 | ||
| Fig. 7 TPO curves of the spent samples obtained after the short and the long term (stability) tests. Short tests: full symbol, long test: empty symbol. CO2 production was measured by QMS and temperature was ramped during TPO to 600 °C or 750 °C after the short or stability tests, respectively. | ||
The results of the long term (24 h) catalytic runs are depicted in Fig. 8. In this case, significant differences were seen: Na–Ni/ZrO2 was active and stable (concerning methane conversion) while the unpromoted catalyst practically died after the daylong reaction at 675 °C in excess methane. On Na–Ni/ZrO2 both the CH4 and the CO2 conversion values at the end of the stability test were 70% of the corresponding starting values, meaning that the rates of Na–Ni/ZrO2 deactivation in terms of CH4 and CO2 should be very similar. This suggests that the CH4 and CO2 activation routes are equally working and well balanced.
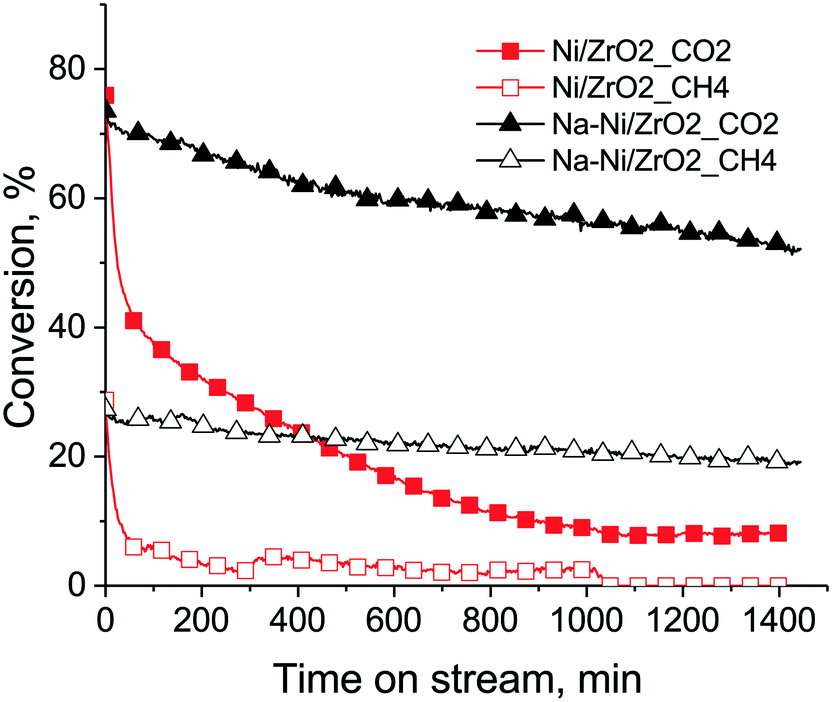 | ||
| Fig. 8 Methane and CO2 conversion curves during long term stability run on both samples. CH4: empty symbols, CO2: full symbols. Conditions: after reduction at 750 °C/0.5 h cooling to T = 675 °C in He, then DRM with CH4:CO2:Ar = 68:31:1 mixture, 120 L h−1 gcat−1. | ||
TEM measurements were used to explore the structure of the samples after the stability test as well. Fig. S5† a shows that numerous long nanotubes were formed on Ni/ZrO2 and some of them contained encapsulated Ni particles at the tips, while fewer carbon nanotube were found on Na–Ni/ZrO2. The deposited coke was oxidized again by TPO measurements (see Fig. 7). The unpromoted sample had a first TPO peak at around 530 °C and a second more definite one at 670 °C, summing all together, 3 mg C/10 mgcat (30 wt% C). The complete deactivation should be explained by the buried Ni particles and not by significant sintering, because d > 50 nm particles (either ZrO2 or Ni) were not observed by TEM. Again, it was more difficult to remove the surface coke from the unpromoted sample (high temperature TPO peak), suspecting a tough, graphitic structure and a less effective coke oxidation ability of the catalyst. The Na-promoted sample produced less (2.3 mg C/10 mgcat – 23 wt% C) carbon deposit now and even less than in the short term catalytic runs with a low 602 °C TPO peak maximum. We might estimate an average rate of coking for these isothermal experiments based on the 24 hour time: it is 2.08 × 10−4 and 1.59 × 10−4 gcarbon gcat−1 min−1 for Ni/ZrO2 and Na–Ni/ZrO2, respectively. This shows again the positive effect of sodium promotion: the coke on the promoted catalyst is a very mobile and reactive type and does not cause significant deactivation. In the case of 5% Ni/Ce–Pr oxide catalysts, the introduction of 20 atom% Pr in the ceria lattice caused a significant reduction in the rate of inactive carbon formation with only a marginal decrease in the catalyst's activity and stability, but further increase of Pr loading caused a decrease in activity with drastic reduction of deposited carbon, which was explained partly by the inhibiting effect of inactive adsorbed carbonates as determined by SSITKA.24 The sodium content in our case is significantly lower (Na/Zr = 0.03); however, a decrease in coking rate is observed already. (When the same sodium promotion was done on our 1% Ni/ZrO2 catalyst,35 coke formation was further decreased with the concomitant decrease of activity as in the case of the above literature reference. Based on this apparent parallel, we might suspect that on that 1% Ni/ZrO2 sample some inactive carbonate species play a role. However, this is not the topic of the present work.)
First, the results of the ramp-hold experiments are discussed (a temperature ramp from about 100 °C up to 600 °C followed by a 30 min isothermal hold, evacuation of the gas phase, then cooling). In Fig. 9, on the left Y axis the intensity of the chosen mass numbers corresponding to the gas phase components versus time are shown (on the X axis 1 MS cycle equals 26 s), and on the right Y axis the reaction temperature versus time (=MS cycles) can be followed. Although quantitative analysis (exact concentrations) cannot be obtained here due to the significant pressure increase during reaction, comparison of the experimental curves normalized to the Ar signal (m/z = 40) can still provide qualitative information about the catalysts. As the reaction starts at about 300 °C, the products such as 13CO (m/z = 29), then 12CO (m/z = 28) and H2 (m/z = 2), can be observed with the concomitant decrease of the mass signal of reactants 12CH4 (m/z = 15) and 13CO2 (m/z = 45). It is easy to see (compare Fig. 9a and b) that Ni/ZrO2 is more active since it converts most of the reactants as the temperature reaches 600 °C, while on Na–Ni/ZrO2 to get the same conversion, longer reaction time is needed; in particular, the CH4 conversion is retarded. During the isothermal part of the experiment (600 °C for 30 min) the transformation of reactants, particularly that of 12CH4, continues on Na–Ni/ZrO2, showing that the system is not at equilibrium; accordingly, 12CO and H2 formation proceeds (designated by arrows), but oxygen providing CO2 molecules are no more present in the gas phase. This suggests that a pool of active oxidants is still available on the surface.
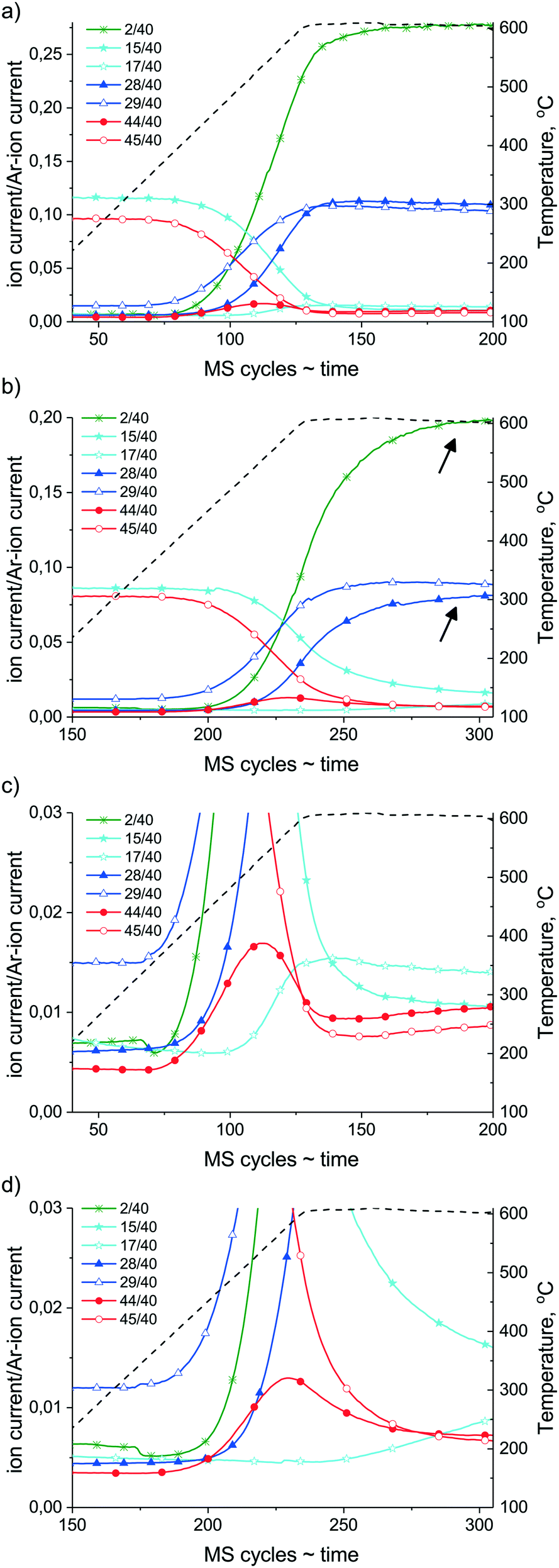 | ||
| Fig. 9 Ramp-hold type dry reforming reaction with a 13CO2:12CH4 = 1 mixture in the closed loop circulation system on (a) Ni/ZrO2 and (b) Na–Ni/ZrO2. (c) Enlargement of low intensity signals of spectrum “a” and (d) enlargement of low intensity signals of spectrum “b”. Conditions: ∼25 mbar each reactant (12CH4 contains 2% Ar), temperature ramp with 10 °C min−1 up to 600 °C then isothermal 30 min at 600 °C. 1 MS cycle = 26 s. | ||
If we take a closer look at the low concentration components formed at the ascending part of the temperature ramp shown in Fig. 9c and d, first 13CO originating from carbon dioxide reactant can be observed on both samples. Then, 12CO coming from methane can be detected, but on Ni/ZrO212CO appears at the same time/temperature like 12CO2 and H2, while in the case of Na–Ni/ZrO2 the 12CO2 and H2 formation precedes by 30 °C the 12CO appearance. This suggests that at the onset of methane dissociation (indicated by the sharp increase of H2 signal intensity), the available oxygen species are in excess and can make possible total oxidation of 12C on the promoted sample. The 12CO2 formation has a local maximum at about 530 °C and 560 °C for Ni/ZrO2 and Na–Ni/ZrO2, respectively. Afterwards, the unlabeled 12CO2 together with the labeled 13CO2 is consumed in dry reforming as the temperature increases. Johnson and Shamsi56 observed during 13CH4 labeled flow dry reforming experiments the formation of 13CO2 at 800 °C.
Eye-catching is the evolution of labeled 13CH4 (m/z = 17) which already happens on Ni/ZrO2 at 490 °C. This 13CH4 is formed by surface hydrogenation of the 13CO2/13CO source. On Ni/ZrO2 the FTIR measurements proved the effective hydrogenation of carbonates leading to formates under dry reforming, that takes place probably at the Ni–ZrO2 interface or by H spillover to further support sites. This hydrogenation route produces methane in the appropriate temperature and concentration window from H13COOs/13CHOs/13Cs. We should keep in mind that the surface of Ni was found fully metallic by XPS, and Ni metal is a requirement of methanation activity. Furthermore, methanation is a structure sensitive reaction and is sensitive to the presence of H2 that induces CO dissociation.57 However, dissociation of CO is inhibited by O traces.58
In the case of the promoted catalyst (Fig. 9d), the 13CH4 formation is retarded until the shortage of oxygen/oxidants (from CO2), although abundant H2 is available. Arena et al.39 reported that surface diffusion of hydrogen is hindered by alkali patches on Ni. This might also be valid for the present case, further suppressing the methanation route. Although we could not go higher than 500 °C during DRIFT measurements, we suppose that some short-lived carbonates are present even at 600 °C due to the Na2O promoter around Ni/Ni(OH)2, and by continuous formation and decomposition they provide oxygen to 12CxHy or Cs removal. Indeed, the HRTEM image in Fig. S6† shows a Ni particle with a shell of Ni(OH)2 and NiOOH patches of the Na–Ni/ZrO2 sample detected after this ramp-hold experiment. TPO measurement after these ramp-hold reactions revealed different types and amounts of deposited coke as depicted in Fig. 10a. For a clearer view, the integral signal of CO2 formation obtained in the closed loop circulation system was differentiated. On Ni/ZrO2, surface carbon originating from the 12CH4 source was oxidized at 330 °C with a significant tail at 520 °C. At the higher temperature range 13CO2 was also detected, meaning that the less reactive carbon deposit was a mixed type (from both 12C and 13C). This means that methane leaves an easily oxidizable (low temperature TPO peak) carbon and a more resistant type of carbon (high temperature tail) on Ni/ZrO2; moreover, a “mixed type” coke product, partly from gas phase 12CO and 13CO, is also formed. In contrast, on Na–Ni/ZrO2 only 12C remained with a wide reactivity/distribution. The detailed quantification of the 12C and 13C ratio of the total deposited carbon on both catalysts and under both types of reactions is collected in Table 2. This analysis helps to distinguish the contribution of the CO2 activation route to the inactive carbon formation for each catalyst and condition as was done in ref. 23–25. It is seen in Table 2 that the amount of deposited carbon was drastically decreased upon sodium promotion, while the contribution of CO2 to the total carbon is similarly low (only 10% on Ni/ZrO2, and zero on the promoted catalyst) in the ramp-hold experiments.
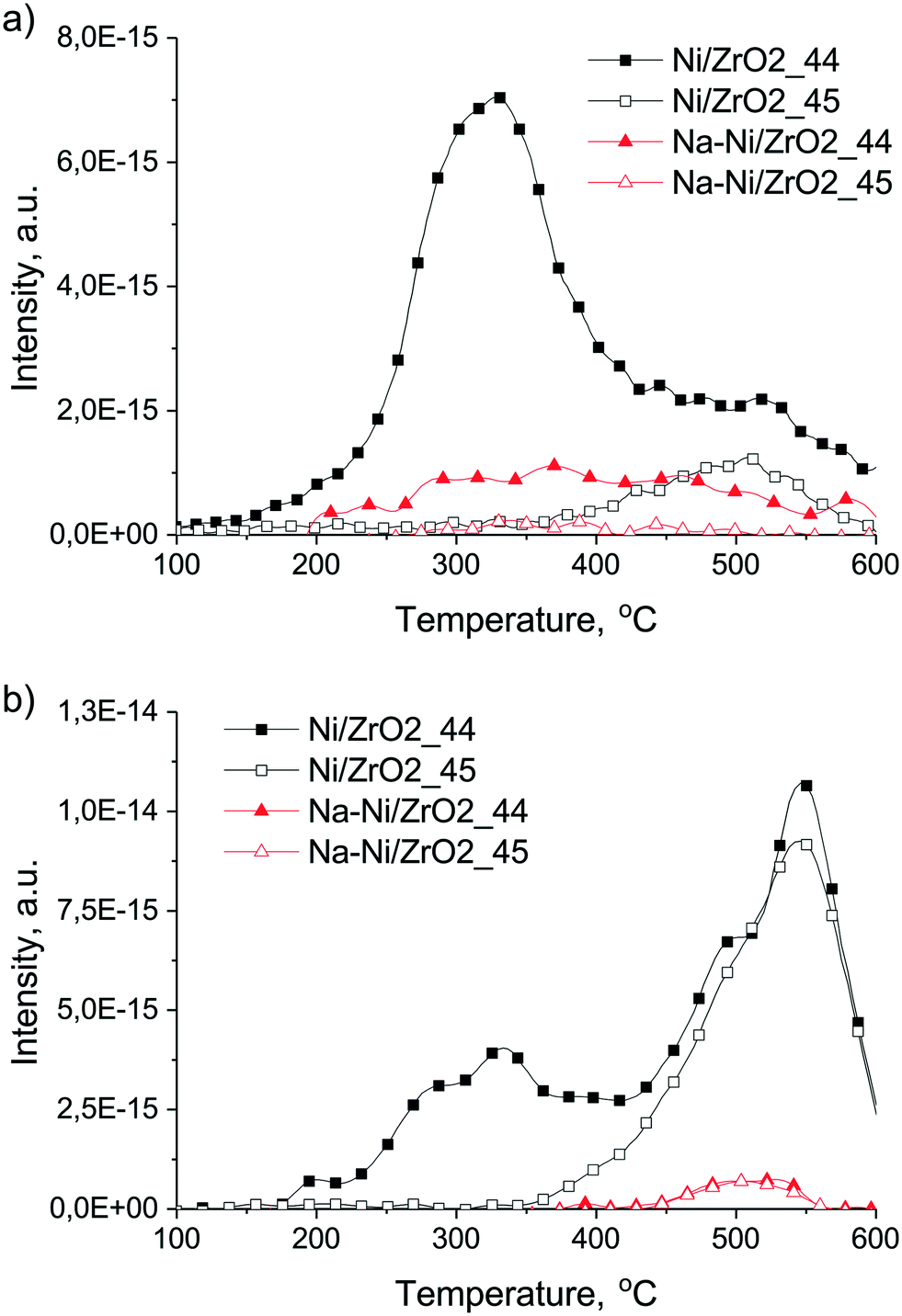 | ||
| Fig. 10 TPO experiments in the circulation system after labeled DRM reaction carried out via the (a) ramp-hold manner and (b) isothermal manner at 600 °C. 12CO2: full symbols, 13CO2: empty symbols. The original measured integral signals are differentiated to get peak-shaped curves. | ||
| Sample | T/°C and type of exp. | 12CO2/μmol g−1 | 13CO2/μmol g−1 | 12C/13C | Total carbon/μmol g−1 |
|---|---|---|---|---|---|
| a % contribution of the CO2 activation route to the total surface carbon derived from both CH4 and CO2 activation routes. b Wt% carbon. | |||||
| Ni/ZrO2 | 600, ramp and hold | 1425.9 | 159.2 | 8.95 (10)a | 1585.1 (1.92)b |
| 600, isothermal | 1030.8 | 647.6 | 1.59 (38.6)a | 1678.3 (2.08)b | |
| Na–Ni/ZrO2 | 600, ramp and hold | 250.0 | 0.0 | n/a (0)a | 250.0 (0.3)b |
| 600, isothermal | 46.3 | 38.0 | 1.22 (45.1)a | 84.4 (0.11)b | |
The other dry reforming experiments carried out in the circulation system were of isothermal type (600 °C reaction for 30 min, then evacuation of the gas phase, and cooling). These results are shown in detail in Fig. S7a–d.† By the end of 30 min, practically all the reactants were converted, but on Ni/ZrO2 at a significantly higher rate. The low concentration components right after the start of the reaction showed exactly the same profile as before: 12CO2 immediately formed and then was consumed (but faster on Ni/ZrO2), while 13CH4 was detected within 1 or after 4.5 min on Ni/ZrO2 and Na–Ni/ZrO2, respectively. The tendentious decrease of both CO products and the increase of CO2 signals on Ni/ZrO2 suggest that CO disproportionation happened at 600 °C leaving some Cs on Ni. In contrast, methane conversion was relatively slow on Na–Ni/ZrO2, and based on the less amount of H2 evolved compared to that on Ni/ZrO2, we assume that Hs/OHs species remained on the catalyst.
The corresponding derived TPO curves of Ni/ZrO2 (Fig. 10b) show that 12CO2 was formed again in 2 ranges, below and above 400 °C, and both peaks contained 2 overlapped components (paired at 280–330 °C and at 490–550 °C). 13CO2 appeared only above 350 °C. The amount of surface carbon removable at low temperature and formed only from methane on Ni/ZrO2 was less now, but the high temperature CO2 peak was larger than that after the ramp-hold run. However, in total, about the same amount of coke formed as in the ramp-hold experiment but the CO2 contribution increased now to about 38.6% (Table 2). Generally, the temperature of the TPO peaks is influenced by the structure, location and oxidation kinetics of coke. The low temperature peaks come from the easily oxidizable and probably CHx or amorphous carbon or carbide species; the high temperature ones must be deeply dehydrogenated, graphitic, and located either on Ni or on ZrO2 support. We propose that the high temperature peaks on Ni/ZrO2 were formed from the CO products via the Boudouard reaction (2CO ⇌ C + CO2) and from 12CH4 at the start of the reaction. The active sites for CO dissociation on this sample seem to be more effective/available during the isothermal experiment than during the ramp-hold type one because they might be less covered by carbon deposits originating solely from CH4 decomposition (see the area of the low temperature peak). This again justifies the general view that at higher temperature less carbon deposit is expected to form.
In the case of the Na–Ni/ZrO2 catalyst we observe now the presence of carbon from the 13CO2 reactant which was not the case during the ramp-hold type experiments: the even more negligible surface carbon was a “mixed type” with 45% contribution of 13C. This deposited carbon is assumed to either originate from CO products or may show that the CH4 and CO2 activation routes produce surface carbon with the same structural characteristics.
In general, we can observe that the contribution of 13CO2 in the deposited coke is increased during the isothermal experiments for both samples, when the reaction starts/proceeds faster. In contrast, on Ni/Ce–Pr oxides at higher reaction temperature (higher rate) the contribution of methane was increased.24 We can hardly compare the two cases because in our isothermal experiments in this circulation system – especially on Ni/ZrO2 – the conversion rate of reactants is too fast and the CO products in the gas phase over the catalyst may also contribute to the deposited coke via the Boudouard reaction. Note the main difference: coke did not form on the promoted sample solely from CH4 reactant as opposed to Ni/ZrO2 under the same type of isothermal reaction. This means that localized Na promotion helps the gasification of a reactive surface carbon – probably CHx type – in a continuous and effective way via the embedded Na2O entities forming carbonates and acting according to the reverse Boudouard reaction: CHx + CO2 ⇌ 2CO + xHs.
The role of sodium promotion in the reaction mechanism and coke removal
Connecting the catalytic properties with all the structural results, we can draw the following statements. The highly hydrated Ni/ZrO2 catalysts active in formate production under low temperature dry reforming conditions will develop deactivating type coke. Formate formation stems from the metal surface providing Hs; thus part of that formate must be in the surrounding of Ni particles occupying/poisoning the adsorption area from the CO2 reactant that could provide new active oxygen species to Cs removal. At high temperature, when the interface of Ni might not play a significant role in CO2 activation, but the metal surface itself, CO disproportionation must be avoided in order to obtain stable catalytic performance.On Na–Ni/ZrO2 formate was not observed; due to its complete absence or its immediate decomposition to carbonates the dry reforming ability was preserved without significant deactivation. In this case the localized Na2O promotion on and around the Ni/Ni(OH)2 entities is of key importance. Even though the existence of a bridged CO species at 1820 cm−1 suggests a decreased C–O bond strength detected on Na–Ni/ZrO2, increased CO dissociation ability was not deducted here. The stable activity under our really demanding dry reforming conditions is explained by the continuously renewing oxygen pool from the balanced interconversion of NaHCO3/Na2CO3/Na2O at the Ni–ZrO2 interface or partially on the surface of Ni particles. Ni – through dissociation of CH4 – provides Hs to the decomposition of Na2CO3 or NaHCO3 to CO2 + O2− + Na+ + H2O. During the process NiOxHy forms that is reduced by Cs or CxHy (from methane) back to Ni (and CO and H2O forms). Then Na2O and the available CO2 result in Na2CO3 again. These tentatively suggested reactions take place simultaneously on small domains of the Ni surface or at the interface. Some metallic Ni is required for CH4 dissociation, Ni(OH)2 or NiOOH (NiOxHy) for carbon oxidation. Our assumption is strongly supported by ref. 40, 41 and 59 reporting about the high CO2 absorption capacity of bulk Na2ZrO3 in the presence of CO2 at temperatures above 500 °C, resulting in a Na2CO3 shell and a ZrO2 layer beneath close to the surface of bulk alkali zirconate as:
| Na2ZrO3 → ZrO2 + O2− + 2Na+ | (3) |
| CO2 + O2− + 2Na+ → Na2CO3. | (4) |
At higher temperature (∼800–850 °C) or at lower CO2 partial pressure the Na2ZrO3 is regenerated by CO2 liberation, forming Na2O that can react with ZrO2 and producing Na2ZrO3 again (the reverse directions of reactions (3) and (4)).40,41,59 We propose that the upwards diffusion of sodium ions in the surface labile oxide lattice to capture CO2 could result in some O2− species being used up by Cs. This must have multiple relevance at high reaction temperatures of our long term catalytic tests. Until there is a small amount of gas phase CO2, the “primary” carbon of methane reactant is removed and not deposited as coke or hydrogenated back to CH4.
Concerning all the results, carbonates on Na–Ni/ZrO2 are suggested to take part in the reaction while on Ni/ZrO2 the formates must be only spectators in the course of dry reforming.
Conclusions
In the present work, 0.6% Na–3% Ni/ZrO2 catalyst was compared to the reference 3% Ni/ZrO2 sample. The localized Na incorporation provided high basicity to the catalyst and resulted in a NiOxHy that is in strong interaction with the support compared to the reference. Na was enriched on the surface and the inclusion of some sodium oxide into the nickel (oxide) phase was suggested to happen and/or the Ni surface became covered by Na2O or Na2CO3 patches based on XPS results and the decrease in CO chemisorption values. CO TPD results suggested WGSR activity on Na–Ni/ZrO2 in contrast to Ni/ZrO2. DRIFTS measurement under flow of the dry reforming mixture revealed the presence of bidentate formate species on Ni/ZrO2, while no sign of formates was discerned on Na–Ni/ZrO2. Apparently, CO2 adsorption and activation processes differ: bidentate carbonate is stabilized by basic oxygen atoms influenced by sodium on Na–Ni/ZrO2 while effective hydrogenation by Ni converts bicarbonates to formate species on Ni/ZrO2.The continuous flow catalytic tests in excess methane revealed coke deposition on both samples, but the activity and stability of Na–Ni/ZrO2 was significantly higher. TPO experiments proved that the surface carbon on the promoted catalyst is a mobile and reactive type one and does not cause significant deactivation.
In 13CO2 labeled stoichiometric DRM experiments in a closed loop circulation system at sub-atmospheric pressure, evolution of labeled 13CH4 was detected on Ni/ZrO2 from the 13CO2/13CO/13C source, possibly due to the effective hydrogenation of carbonates at the Ni–ZrO2 interface and the dissociation of 13CO on Ni. In the case of the promoted catalyst, this methanation (transfer of 13C of 13CO2 to 13CH4) is retarded until the significant shortage of oxygen/oxidants, although abundant H2 is available. During DRM at constant 600 °C, CO disproportionation happened on Ni/ZrO2 leaving some carbon on Ni, besides the coke formed solely from the CH4 source. In contrast, carbonaceous deposit did not form on Na–Ni/ZrO2 solely from the CH4 reactant, only from CO products and moreover in an insignificant amount. We suppose that some short-lived carbonates/hydrocarbonates are present even at high temperatures in contact with the ZrO2-embedded Na2O and Ni/Ni(OH)2 surface, continuously forming and decomposing, thus oxidizing the 12CxHy or Cs surface species to 12CO. This is supported by literature analogies dealing with the high temperature reversible CO2 capture of bulk Na2ZrO3. The tentatively suggested reactions take place simultaneously on small domains of Ni or at the interface. Concerning all the results, carbonates on Na–Ni/ZrO2 are suggested to take part in the reaction while on Ni/ZrO2 the formates are only spectators in the course of low temperature dry reforming. SSITKA experiments are under progress to ascertain this statement.
The ease of sample preparation and the good properties of the resulting Na2O-modified catalyst deserve attention and may propagate further research on carbon resistance of Ni supported on non-reducible ZrO2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to Era-Chemistry and the Hungarian National Research Fund (OTKA NN#107170) for financial support. Professor Johannes Lercher's group at the Technical University of Munich, Department of Chemistry is acknowledged for providing us with the ZrO2 support.References
- P. M. Mortensen and I. Dybkjaer, Appl. Catal., A, 2015, 495, 141 CrossRef CAS.
- M. K. Nikoo and N. A. S. Amin, Fuel Process. Technol., 2011, 92, 678 CrossRef CAS.
- A. Yamaguchi and E. Iglesia, J. Catal., 2010, 274, 52 CrossRef CAS.
- J. Wei and E. Iglesia, J. Catal., 2004, 224, 370 CrossRef CAS.
- N. Sun, X. Wen, F. Wang, W. Peng, N. Zhao, F. Xiao, W. Wei, Y. Sun and J. Kang, Appl. Surf. Sci., 2011, 257, 9169 CrossRef CAS.
- B. B. Baeza, C. M. Pedrero, M. A. Soria, A. G. Ruiz, U. Rodemerck and I. R. Ramos, Appl. Catal., B, 2013, 129, 450 CrossRef.
- S. Sokolov, E. V. Kondratenko, M. M. Pohl and U. Rodemerck, Int. J. Hydrogen Energy, 2013, 38, 16121 CrossRef CAS.
- S. Damyanova, B. Pawelec, K. Arishtirova, M. V. M. Huerta and J. L. G. Fierro, Appl. Catal., B, 2009, 89, 149 CrossRef CAS.
- A. S. Bobin, V. A. Sadykov, V. A. Rogov, N. V. Mezentseva, G. M. Alikina, E. M. Sadovskaya, T. S. Glazneva, N. N. Sazonova, M. Y. Smirnova, S. A. Veniaminov, C. Mirodatos, V. Galvitta and G. B. Martin, Top. Catal., 2013, 56, 958 CrossRef CAS.
- L. M. Aparicio, J. Catal., 1997, 165, 262 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, J. Catal., 1998, 173, 157 CrossRef CAS.
- Y. Z. Chen, B. J. Liaw and W. H. Lai, Appl. Catal., A, 2002, 230, 73 CrossRef CAS.
- M. A. Goula, A. Lemonidou and A. M. Efstathiou, J. Catal., 1996, 161, 626 CrossRef CAS.
- J.-M. Wei, B.-Q. Xu, J.-L. Li, Z.-X. Cheng and Q.-M. Zhu, Appl. Catal., A, 2000, 196, 167 CrossRef.
- Y. Kathiraser, U. Oemar, E. T. Saw, Z. Li and S. Kawi, Chem. Eng. J., 2015, 278, 62 CrossRef CAS.
- C. Papadopoulou, H. Matralis and X. Verykios, in Catalysis for Alternative Energy Generation, ed. L. Guczi and A. Erdőhelyi, Springer Science+Business Media New York, 2012, pp. 57–128 Search PubMed.
- M.-S. Fan, A. Z. Abdullah and S. Bhatia, ChemCatChem, 2009, 1, 192 CrossRef CAS.
- J. W. Han, C. Kim, J. S. Park and H. Lee, ChemSusChem, 2014, 7, 451 CrossRef CAS PubMed.
- A. Horváth, G. Stefler, O. Geszti, A. Kienneman, A. Pietraszek and L. Guczi, Catal. Today, 2011, 169, 102 CrossRef.
- E. Lovell, Y. Jiang, J. Scott, F. Wang, Y. Suhardja, M. Chen, J. Huang and R. Amal, Appl. Catal., A, 2014, 473, 51 CrossRef CAS.
- L. F. Li, C. Xia, C. T. Au and B. S. Liu, Int. J. Hydrogen Energy, 2014, 39, 10927 CrossRef.
- C. Gennequin, M. Safariamin, S. Siffert, A. Aboukaïs and E. Abi-Aad, Catal. Today, 2011, 176, 139 CrossRef CAS.
- M. M. Makri, M. A. Vasiliades, K. C. Petallidou and A. M. Efstathiou, Catal. Today, 2015, 259, 150 CrossRef.
- M. A. Vasiliades, M. M. Makri, P. Djinovic, B. Erjavec, A. Pintar and A. M. Efstathiou, Appl. Catal., B, 2016, 197, 168 CrossRef CAS.
- M. A. Vasiliades, P. Djinović, L. F. Davlyatova, A. Pintar and A. M. Efstathiou, Catal. Today, 2017 DOI:10.1016/j.cattod.2017.03.057.
- A. Horváth, L. Guczi, A. Kocsonya, G. Sáfrán, V. La Parola, L. F. Liotta, G. Pantaleo and A. M. Venezia, Appl. Catal., A, 2013, 468, 250 CrossRef.
- L. Guczi, G. Stefler, O. Geszti, I. Sajó, Z. Pászti, A. Tompos and Z. Schay, Appl. Catal., A, 2010, 375, 236 CrossRef CAS.
- A. Ballarini, F. Basile, P. Benito, I. Bersani, G. Fornasari, S. de Miguel, S. C. P. Maina, J. Vilella, A. Vaccari and O. A. Scelza, Appl. Catal., A, 2012, 433–434, 1 CrossRef CAS.
- A. D. Ballarini, S. R. de Miguel, E. L. Jablonski, O. A. Scelza and A. A. Castro, Catal. Today, 2005, 107–108, 481 CrossRef CAS.
- M. W. Balakos and S. S. C. Chuang, J. Catal., 1992, 138, 733 CrossRef CAS.
- R. D. Gonzalez and H. Miura, J. Catal., 1982, 77, 338 CrossRef CAS.
- C. Zhang, Y. Li, Y. Wang and H. He, Environ. Sci. Technol., 2014, 48, 5816 CrossRef CAS PubMed.
- T. Horiuchi, K. Sakuma, T. Fukui, Y. Kubo, T. Osaki and T. Mori, Appl. Catal., A, 1996, 144, 111 CrossRef CAS.
- T. Osaki and T. Mori, J. Catal., 2001, 204, 89 CrossRef CAS.
- M. Németh, Z. Schay, D. Srankó, J. Károlyi, G. Sáfrán, I. Sajó and A. Horváth, Appl. Catal., A, 2015, 504, 608 CrossRef.
- A. Packter and S. C. Uppaladinni, Krist. Tech., 1975, 10, 985 CrossRef CAS.
- Q. J. Chen, J. Zhang, Q. W. Jin, B. R. Pan, W. B. Kong, T. J. Zhao and Y. H. Sun, Catal. Today, 2013, 215, 251 CrossRef CAS.
- F. Arena, A. L. Chuvilin and A. Parmaliana, J. Phys. Chem., 1995, 99, 990 CrossRef CAS.
- F. Arena, F. Frusteri and A. Parmaliana, Appl. Catal., A, 1999, 187, 127 CrossRef CAS.
- H. G. Jo, H. J. Joon, C. H. Lee and K. B. Lee, Ind. Eng. Chem. Res., 2016, 55, 3833 CrossRef CAS.
- G. G. Santillán-Reyes and H. Pfeiffer, Int. J. Greenhouse Gas Control, 2011, 5, 1624 CrossRef.
- A. F. Carley, S. D. Jackson, J. N. O'Shea and M. W. Roberts, Surf. Sci., 1999, 440, 868 CrossRef.
- T. Shido and Y. Iwasawa, J. Catal., 1993, 141, 71 CrossRef CAS.
- A. Kitla, O. V. Safonova and K. Föttinger, Catal. Lett., 2013, 143, 517 CrossRef CAS PubMed.
- P. Panagiotopoulou and D. I. Kondarides, J. Catal., 2008, 260, 141 CrossRef CAS.
- L. F. Liotta, G. A. Martin and G. Deganello, J. Catal., 1996, 164, 322 CrossRef CAS.
- K. I. Hadjiivanov and G. N. Vayssilov, Adv. Catal., 2002, 47, 307 CAS.
- J. M. Pigos, C. J. Brooks, G. Jacobs and B. H. Davis, Appl. Catal., A, 2007, 319, 47 CrossRef CAS.
- E.-M. Kock, M. Kogler, T. Bielz, B. Klotzer and S. Penner, J. Phys. Chem. C, 2013, 117, 17666 Search PubMed.
- J. H. Bitter, PhD thesis, University of Twente, Enschede, The Nederlands, 1997 Search PubMed.
- S. E. Collins, M. A. Baltanás and A. L. Bonivardi, J. Phys. Chem. B, 2006, 110, 5498 CrossRef CAS PubMed.
- D. Heyl, U. Rodemerck and U. Bentrup, ACS Catal., 2016, 6, 6275 CrossRef CAS.
- R. W. Stevens, R. V. Siriwardane and J. Logan, Energy Fuels, 2008, 22, 3070 CrossRef CAS.
- G. K. Reddy, S. Loridant, A. Takahashi, P. Delichére and B. M. Reddy, Appl. Catal., A, 2010, 389, 92 CrossRef CAS.
- S. Haag, M. Burgard and B. Ernst, J. Catal., 2007, 252, 190 CrossRef CAS.
- A. Shamsi and C. D. Johnson, Catal. Today, 2003, 84, 17 CrossRef CAS.
- M. P. Andersson, F. Abild-Pedersen, I. N. Remediakis, T. Bligaard, G. Jones, J. Engbæk, O. Lytken, S. Horch, J. H. Nielsen, J. Sehested, J. R. Rostrup-Nielsen, J. K. Nørskov and I. Chorkendorff, J. Catal., 2008, 255, 6 CrossRef CAS.
- S. Fujita, M. Nakamura, T. Doi and N. Takezawa, Appl. Catal., A, 1993, 104, 87 CrossRef CAS.
- S. Wang, C. An and Q.-H. Zhang, J. Mater. Chem. A, 2013, 1, 3540 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy01011g |
| This journal is © The Royal Society of Chemistry 2017 |