
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Active sites on graphene-based materials as metal-free catalysts
Sergio
Navalon†
a,
Amarajothi
Dhakshinamoorthy†
 b,
Mercedes
Alvaro
a,
Markus
Antonietti
*c and
Hermenegildo
García
*a
b,
Mercedes
Alvaro
a,
Markus
Antonietti
*c and
Hermenegildo
García
*a
aDepartamento de Química and Instituto de Tecnologia Química (CSIC-UPV), Universitat Politecnica de Valencia, Av. De los Naranjos s/n, 46022 Valencia, Spain. E-mail: hgarcia@qim.upv.es
bSchool of Chemistry, Madurai Kamaraj University, Madurai-625 021, Tamil Nadu, India
cMax Planck Institute of Colloids and Interfaces, Potsdam-Golm, Germany. E-mail: markus.antonietti@mpikg.mpg.de
First published on 1st June 2017
Abstract
Graphenes and related materials have attracted growing interest as metal-free catalysts. The present review is focused on describing the active sites that have been proposed to be responsible for the catalytic activity observed for such systems. It will be shown that diverse defects and chemical functionalities on the graphene layers can catalyze reactions, including oxygenated functional groups, carbon vacancies and holes, edge effects, and the presence of dopant elements. Besides discrete active sites, the catalytic activity arising from the collective properties of graphenes as materials by adsorbing substrates and reagents and activating them by charge transfer is also commented. The review has an introductory general section summarizing the general methodologies that have been used to support the proposed structure of the active sites, including theoretical calculations, comparison of the catalytic activity of graphene samples with different compositions, the use of organic molecules as models of the active centers, and selective masking of functional groups. The review is concluded with our view on future developments in the field.
 Sergio Navalon | Sergio Navalon was born in Valencia (Spain) in 1979. He graduated in Chemical Engineering in 2003 and obtained his PhD in 2010 at the Technical University of Valencia (UPV). Currently, he is working as an Assistant Professor at the Chemistry Department of UPV. His research focuses on the development of heterogeneous (photo)catalysts based on graphene, porous materials and nanoparticles, as well as green chemistry processes. He is the author of about 50 publications. |
 Amarajothi Dhakshinamoorthy | Amarajothi Dhakshinamoorthy received his PhD, degree in 2009 from Madurai Kamaraj University, Madurai-21, India. Later, he worked as a postdoctoral researcher with Prof. Hermenegildo Garcia at the Technical University of Valencia for four years. Then, he returned to India and joined as a UGC-Assistant Professor in June 2013 at the School of Chemistry, Madurai Kamaraj University. His research interests include the catalytic applications of metal–organic frameworks and graphene-related materials. He is the recipient of the Young Scientist Award 2014 for Chemical Sciences by The Academy of Sciences, India. He was also awarded an INSA-DFG international bilateral exchange award to visit Germany for three months in 2015. He has co-authored over ninety publications, two book chapters and one international patent. |
 Mercedes Alvaro | Mercedes Alvaro is a full Professor at the Chemistry Department of the Technical University of Valencia. She has co-authored over 150 papers and has supervised 9 theses. Her actual interests include advanced oxidation techniques for water treatment, applied photochemistry for environmental remediation and organic synthesis. |
 Markus Antonietti | Markus Antonietti has studied Chemistry in Mainz and did his doctorate with Hans Sillescu. His habilitation on nanogels in 1990 fueled his enthusiasm for complex nanostructures based on polymers and carbon. After a professorship at the University of Marburg, he was appointed director for the department of Colloid Chemistry at the MPI of Colloids and Interfaces in 1993. Antonietti's contributions to the chemical community comprise many aspects, but first of all he is devoted to creativity in research. He enjoys cooking and playing in a Rock'n'Roll band. |
 Hermenegildo García | Hermenegildo García was appointed a full Professor at the Technical University of Valencia in 1996 and is a staff member of the Instituto de Tecnología Química, a joint center of the Technical University of Valencia and the Spanish National Research Council. He made postdoctoral stays at the University of Reading with Professor Andrew Gilbert and several sabbatical leaves in the group of Professor J. C. Scaiano at the University of Ottawa. Prof. Garcia has been active in the field of heterogeneous catalysis working with graphene, porous catalysts and nanoparticles, has published over 600 papers and has filed over 25 patents, two of them in industrial exploitation. Prof. Garcia is Doctor Honoris Causa from the University of Bucharest and the recipient of the 2011 Janssen-Cilag award given by the Spanish Royal Society of Chemistry and the 2008 Alpha Gold of the Spanish Society of Glass and Ceramics. |
1. Introduction and scope of the review
Besides acids and bases, homogeneous and heterogeneous catalysis is currently dominated by the use of transition metals, either as free ions, coordination metal complexes, clusters or nanoparticles, acting as active sites.1–13 Some of these transition metals are rare, there being issues of sustainability coming with such choices. There is a large incentive in finding alternative metal-free catalysts, based on abundant elements.14–21 This is one of the reasons why there is growing interest in exploring the potential of carbon-based materials as heterogeneous catalysts.1,15,22–27 Although the use of carbon-based materials in catalysis is not new, and there have been several reactions including deNOx, ozonizations and other oxidations that are known to be catalyzed by active carbons (ACs),28–30 the field has gained a renewed momentum when graphenes (Gs) and more sophisticated carbon nanostructures have become largely available.26,31–33One advantage of Gs and the build-upon products over other allotropic forms of carbon is that they can be obtained in sufficient quantities from commercially available graphite through reliable, well-established preparation procedures.26,34–49 Gs have large surface area and a 2D morphology making accessible most of the atoms that make this material suitable as a catalyst and their preparation allows a certain degree of control on their composition, lateral dimensions and others.
In general terms, transition metals can promote reactions by acting as Lewis acids of appropriate hardness and strength, by redox centers transferring electrons and through the formation of organometallic intermediates.26,50–52 The key issue is that Gs can incorporate into their structure centers that can exhibit similar catalytic properties, by introducing acid and electrophilic centers, redox pairs and by charge transfer interactions.14 In fact, there are by now many classes of reactions, including oxidations with various types of oxidizing reagents, reductions either using molecular hydrogen and other hydrogen sources, couplings and acid catalyzed processes that have been reported to be promoted by Gs.14,15 To favor the development of this area, it would be desirable to gain a complete picture of the catalytic activity of Gs and how this activity compares to that of conventional catalysts. This knowledge on the structure of the active sites combined with methodologies to further enhance this activity by increasing the density of the sites should result in more advanced G catalysts.
In this context, to develop the potential of Gs in catalysis, one of the major issues in the current state of the art is the limited understanding of the nature of the active sites responsible for the observed catalytic activity and how this activity and selectivity can be further increased.53
The aim of the present review is to summarize the types of centers that have been currently proposed as responsible for the catalytic activity of Gs in different reactions, presenting the evidence that supports this claim, describing the reaction mechanism proposed for different reaction types. Thus, after a general section presenting methodologies to gain information about the nature of the active sites promoting different reactions, the main body of the review is focused on describing the catalytic activity of Gs grouped according to the structure of the active centers, including oxygenated functional groups, carbon vacancies and holes, and dopant elements. Two sections emphasizing the importance of the collective properties of Gs in catalysis by adsorbing substrates and activating them through charge transfer have also been included. The final section summarizes the current state of the art and provides our view on future developments in the field.
2. Characterization of active sites on graphenes
One of the major issues that limits the progress of Gs as carbocatalysts is how to determine the nature of the active sites.14 This knowledge on the nature of the active centers, combined with quantitative determination of their population, can serve to accurately estimate turnover numbers (TON) that are the parameters to be used to rank the activity of Gs compared to other alternative materials.14 In addition, knowledge of the structure of the active sites can allow devising the optimal preparation conditions and suitable post-synthetic modification treatments to increase their population. The long term goal would be manipulation of G with atomic precision in such a way that it could be possible to create high populations of isolated single sites on Gs.Several methodologies have been reported in the literature to address the issue of the nature of the active sites responsible for the catalytic activity in Gs and related materials.14 However, all of them have limitations and drawbacks that determine that a certain degree of uncertainty is always associated with the method used to unveil the catalytic centers.16,54,55 As is going to be commented below, part of the problem arises from the limitations inherent to materials sciences as compared to the chemistry of discrete molecules and the difficulty of a complete material characterization.
One of the most powerful tools to propose active sites in G catalysis is to establish valid models of the proposed sites and perform quantum chemical calculations of how these models interact with substrates establishing plausible reaction mechanisms with estimation of the energy barriers.52,56,57 However, there are several issues that limit the validity of this approach such as how close are the models selected in the studies to the G structure, their size and the accuracy level of the calculations. Models should include bidimensional periodicity to better simulate the size of G sheets, reducing the influence of the periphery. In addition, this methodology would require to make models for every possible active site and perform the corresponding calculations for each of them, something that is prohibitive due to the machine time required to carry out these studies. In addition, theoretical studies have shown their highest utility when they are accompanied by experimental data to validate conclusions from calculations.58
A conventional methodology to propose the nature of the active sites is based on the comparison of the activity of a series of different well-characterized samples in which some parameter is gradually changed.58,59 In the present case, this would require the preparation of well-defined G samples, something that is frequently not possible. As an example, one simple case would be to determine the activity of a dopant element and one approach would be to prepare a series of Gs with different dopant contents. The problem is that most frequently oxygen is also present in the composition of the Gs and it can be difficult to alter the dopant content without simultaneously altering also the oxygen percentage and the resulting variations of the catalytic activity can derive from the concurrent alteration of functional groups containing the dopant and oxygen.
One of the most convincing strategies to provide evidence in support of the nature of the active sites is post-functionalization of the parent, active G sample by performing a selective reaction that masks one type of functional group and, then, determining what is the influence on the catalytic activity of this derivatization process.14 If the group masked is involved in the catalysis, a significant decrease in the catalytic activity should be observed, while no influence of derivatization should be expected when the group has no relevant catalytic activity. The problems associated with this protocol are the incomplete masking of all the groups present on G, the possible incomplete selectivity of the derivatization reaction towards a single functional group, combined with the need for characterization of the functionalized G. This strategy has been used by Su and coworkers to gain evidence on the nature of active sites on carbon nanotubes (CNTs) as catalysts for oxidative dehydrogenation of ethylbenzene, observing that only derivatization of the carbonyl groups leads to deactivation of CNTs.60
Another methodology consists in studying the performance of discrete organic molecules that may contain analogous functional groups as those presumed as the active sites for G and observing a similar catalytic activity for the organic molecule and G.61 This approach is really useful since it establishes a parallelism between organocatalysis and carbocatalysis. It is well-known that many organic molecules having acidic or basic properties as well as redox activity can catalyze reactions and, therefore, this information is a useful background when proposing the catalytic centers of G. Generally it has been found that condensed polycyclic aromatic molecules having some functional groups can behave similarly to Gs, promoting different reactions.14,62 One case in which this analogy between organocatalysis and carbocatalysis has been useful is, for example, the proposal of quinone/hydroquinone-like moieties of Gs as redox centers responsible for the activity of Gs as metal-free (photo)Fenton catalysts58,63 that will be commented in the corresponding section below. Among the various possible limitations of this approach, one of them is, however, how to mimic dangling bonds, carbon vacancies, edges and holes with organic molecules. As will be discussed later, carbon vacancies and the periphery of G sheets having dangling bonds are believed to be general active sites in many reactions, such as aerobic oxidations and hydrogenations.64
Obviously all these approaches are complementary and can be used simultaneously to gain insight into a given catalytic reaction and should be combined with kinetic studies about the influence of quenchers or promoters on the catalytic activity of G. For instance, addition of bases or acids can influence the catalytic activity of G by neutralization of the complementary acid or basic sites on Gs acting as active sites.65–67 Also the influence of radical quenchers can be useful to assess the reaction intermediates.58,63
Another general statement at the beginning of such a quantitative discussion is that it is notoriously difficult to compare different heterogeneous catalysts, as accessibility of active sites depends on the specific surface area as well as the dispersion state of the catalysts. Pores which are accessible in one medium cannot be accessed in another, and systematic variation of functional groups also changes hydrophobicity, which may result in activity loss due to aggregation, and other reasons associated with hydrophobicity. Whenever appropriate, we will try to point to those problems, but it is clear that a focus on functional groups must be necessarily a simplification.
3. Overview of the active sites on graphenes
Table 1 compiles the references that will be commented on the review to describe which are the defects on the G sheet that have been proposed as active sites and the corresponding reaction. Emphasis will be made in the discussion on the experimental evidence or calculations that have been obtained to support the active site proposal. The active sites have been ordered by presenting first oxygen-containing active sites, followed by the catalytic activity of periphery, carbon vacancies and even the basal plane and finalizing with doping as catalytic centers as responsible for the observed catalytic activity. Note that beside discrete centers with a local structure, Table 1 also includes references in which the collective 2D structure of graphene has to be considered by itself or in combination with other materials as responsible for the observed catalytic activity.| Active sites | Reaction | Techniques/experiments | Ref. |
|---|---|---|---|
| a Active sites can be a localized group of atoms or they can derive from the collective property. b GO: graphene oxide; PMS: peroxymonosulfate; PS: persulfate; rGO: reduced GO; DFT: Density functional theory; TEMPO: 2,2,6,6-tetramethylpiperidine-N-oxide; XPS: X-ray photoelectron spectroscopy; EPR: electron paramagnetic resonance; STM: scanning tunneling microscope; DMPO: 5,5-dimethyl-1-pyrroline-N-oxide. | |||
| Oxygen functional groups | |||
| Epoxy groups | |||
| GO | Aerobic oxidation of benzyl alcohol to benzaldehyde | DFT calculations | 68 |
| GO | SO2 and NOx oxidation | DFT calculations | 69 |
| Quinone groups | |||
| rGO | Nitrobenzene reduction with hydrazine hydrate | Model compounds | 61 |
| rGO | (Photo)Fenton reaction with H2O2 |
Model compounds
XPS, EPR and DFT calculations |
58 and 63 |
| rGO | Catalytic ozonisation of p-hydroxybenzoic acid | Raman | 70 |
| rGO | PMS activation in water for pollutant degradation |
XPS, DFT calculations
Model compounds Different carbonaceous materials |
59 and 71 |
| rGO | PS activation in water |
Model compounds
Different carbonaceous materials |
72 |
| Benzyl alcohol-type groups | |||
| GO | C–H arylation of benzene | Model compounds, DFT calculations | 73 |
| Acid sites | |||
| Hydrothermally treated GO | Reduction of 4-nitrophenol to 4-aminophenol using NaBH4 | XPS, theoretical calculations and model compounds | 65 |
| GO | Alkylation of arenes with styrenes and alcohols | XPS and characterization of fresh with spent catalyst | 74 |
| GO | Formation of 2,3-dihydroquinazolinones | Model compounds | 75 |
| Sulfur functional groups | |||
| Sulfate groups | |||
| GO | Ring opening of epoxides | Thermal treatment, quenching experiment with pyridine, IR | 66 |
| GO | Acetalization of benzaldehyde | Quenching experiment with pyridine | 67 |
| Sulfonic group | |||
| GO | Dehydration of fructose into 5-hydroxymethylfurfural | Control experiments with catalysts containing SO3H groups, adsorption experiments | 76 |
| GO | Synthesis of polyoxymethylene dimethyl ethers from methanol and trioxymethylene | Control experiments with different GOs by masking experiments, model compounds | 77 |
| GO | Ring opening of chiral epoxides by indoles | Believed to be due to acidic groups | 78 |
| Carbon vacancies | |||
| ba-GO | Oxidation of benzyl amine to imine | STM, 13C-NMR, FT-IR, EPR and model compounds | 64 |
| GO/TEMPO | Oxidation of 5-hydroxymethylfufural to 2,5-diformylfuran | EPR, model compounds | 79 |
| Edges | |||
| G nanofibers | H2 release from NaAlH4 | DFT calculations | 80 |
| Frustrated Lewis pairs | |||
| rGO | Hydrogenation of acetylene | Frustrated Lewis acid–base pairs, control experiments with CO2 or NH3 | 81 |
| rGO and G | Reduction of nitro compounds | Frustrated Lewis acid–base pairs, quenching experiments with acetic acid | 82 |
| (B,N)G | Hydrogenation of acetylene | Theoretical calculations | 83 |
| Collective properties of Gs as materialsa | |||
| g-C3N4 | Alkylation of benzene by methanol | Charge transfer interaction between g-C3N4 as acceptor and benzene as donor | 84 and 85 |
| g-C3N4 | Cyanation of benzene by urea | Charge transfer interaction between g-C3N4 as acceptor and benzene as donor | 86 |
| Graphite oxide/GO | Mukaiyama–Michael addition of 2-(trimethylsiloxy)furan and β-nitrostyrene | DFT calculations | 87 |
| The heterojunction and dyad concepts in catalysisa | |||
|
C@C3N4
N–C@C3N4 HAT–C3N4 |
Selective oxidation of saturated hydrocarbons by dioxygen | Heterojunctions and dyad | 88 |
| Dopant elements | |||
| N-Doping | |||
| N–G (graphitic sp2 N) | Aerobic oxidation of benzyl alcohol | EPR, spin trap experiments with DMPO | 89 |
| N-Doped rGO | PMS activation |
DFT calculations
XPS |
90 and 91 |
| N-Doped rGO | Catalytic wet air oxidation (∼160 °C at 7 bar O2) and catalytic ozonation (room temperature) | XPS | 92 |
| N-Doped G | Reduction of 4-nitrophenol to 4-aminophenol by NaBH4 |
FT-IR
XPS DFT calculations |
93 |
| (S,N)-Doped G | |||
| (S,N)-rGO | PMS activation in water | DFT calculations | 94 |
| (B,N)-Doping | |||
| (B,N–G) | Acetylene hydrochlorination | XPS, Raman, TPD analysis and DFT calculations | 95 |
| P-Doped | |||
| P-Doped graphitic porous carbon | Aerobic oxidation of benzyl alcohol | XPS, theoretical calculations, control experiments | 96 |
| Dopant elements in electrochemical reactions | |||
| N-Doped G | Oxygen reduction reaction, oxygen evolution reaction, hydrogen evolution reaction | Theoretical calculations | 97–99 |
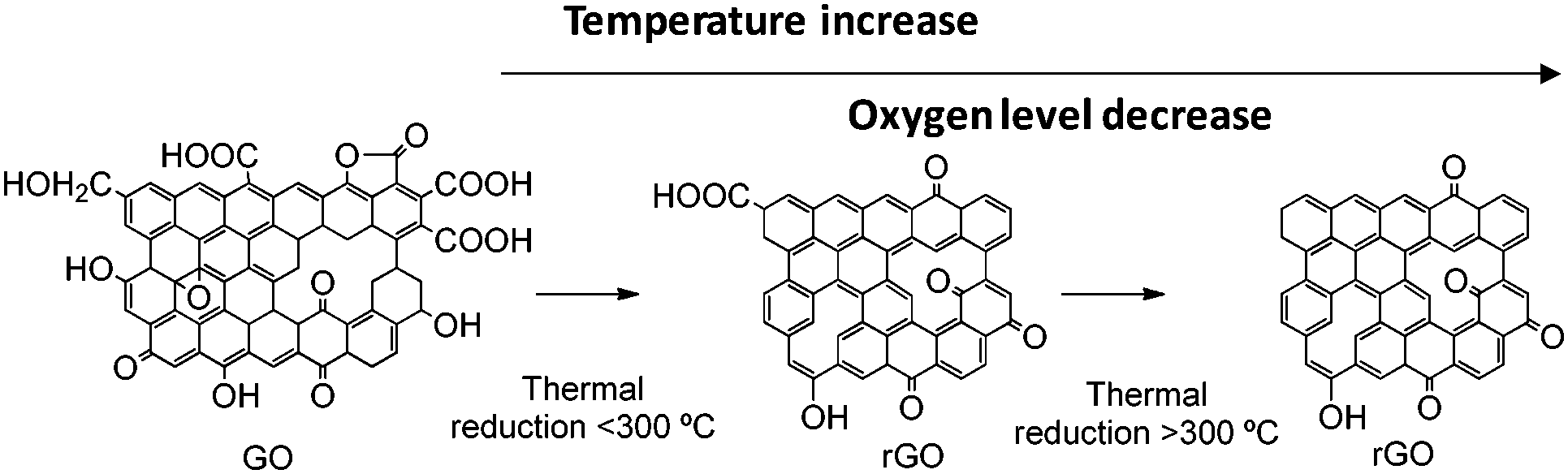
4. Oxygen functional groups
Several oxygen functional groups have been reported as active sites in various carbocatalysts, particularly graphene oxide (GO) and reduced GO (rGO). In this context, density functional theory (DFT) calculations have revealed that the carbonyl oxygen of quinone-like groups is the most nucleophilic site compared to oxygen atoms of carboxyl, 1,2- and 1,3-diketones, isolated ketone or lactone (Fig. 1).100 Ag+-Binding energy calculations were employed to establish theoretically the relative oxygen nucleophilicity order based on electron density parameters, showing again that the quinone group is the most reactive site among the various possible oxygens for the electrophilic attack of Ag+, there being a remarkable difference depending on the armchair or zig-zag configuration of the edge, the best coordinating center being diketone in the zig-zag configuration.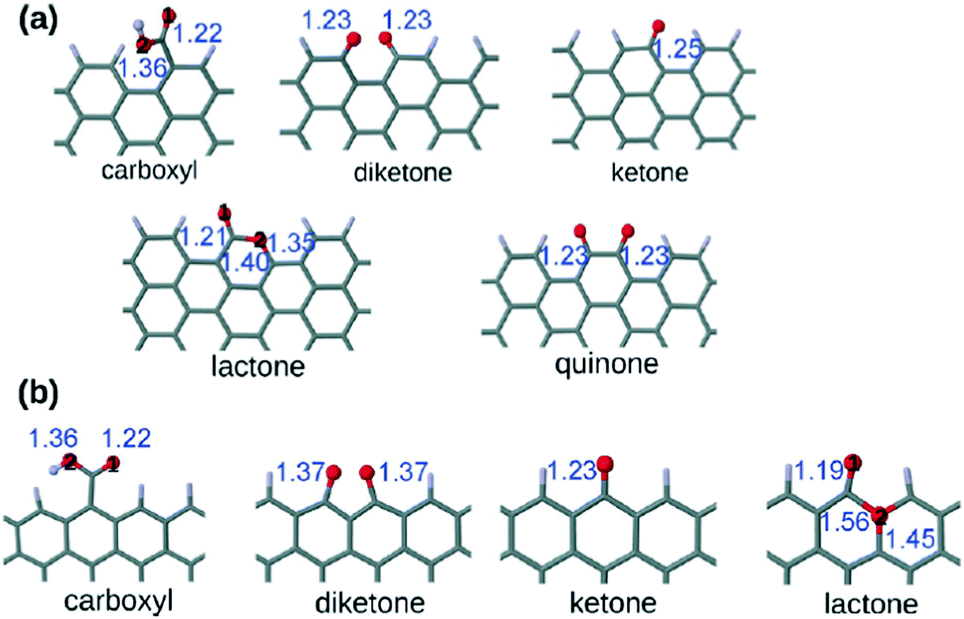 | ||
| Fig. 1 Optimized structures of the carboxyl, diketone, ketone, lactone, and quinone groups at (a) the armchair termination and (b) the zigzag termination. The blue numbers indicate the bond distance in Å. The black numbers appearing inside the atoms indicate the different oxygens in the carboxyl and lactone groups. Red is oxygen and gray is carbon. Reproduced from ref. 100 with permission from Wiley, copyright 2017. | ||
These calculation data are relevant in the context of proposing the active sites when electrophilic species interact with oxygen-containing defective G.
4.1. Epoxy groups
In a pioneering work, Bielawski and co-workers reported the use of GO as a carbocatalyst for the aerobic oxidation of alcohols and olefins.68 Selective oxidation of benzyl alcohol to benzaldehyde was achieved using high GO loadings (20–200 wt%) (Scheme 1). Experiments under oxygen or nitrogen revealed that GO acts as a true carbocatalyst for the aerobic oxidation. However, the carbocatalyst becomes gradually deactivated upon reuse. FT-IR, elemental analysis and powder conductivity indicate that GO suffers a gradual reduction to rGO that is accompanied by catalyst deactivation. Theoretical calculations suggest that epoxide functional groups of GO are the active sites responsible for the catalytic activity.57 Thus, the reaction occurs via transfer of hydrogen atoms from benzyl alcohol to the epoxide oxygens on the GO surface with subsequent epoxide ring opening and final dehydratation of the hydroxyl group. Then, molecular oxygen should promote reoxidation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the partially reduced GO in the presence of water. Note that the fact that the catalytic activity decrease indicates that reoxidation of the CC groups in partially reduced GO by molecular oxygen under the reaction conditions is not complete. In this regard it should be commented that in the first part of the reaction GO acts as a stoichiometric reagent and it is the subsequent reoxidation of CC double bonds that closes the catalytic cycle allowing regeneration of the initial state that becomes ready for a subsequent turn over. Only if turn over occurs can the process be considered as catalytic and not stoichiometric. Interestingly, similar DFT calculations indicate that pristine G or graphite should exhibit substantially higher energy barriers for this reoxidation mechanism with respect to the oxygen-rich GO and, for this reason, they should be much worse catalysts.
C double bond of the partially reduced GO in the presence of water. Note that the fact that the catalytic activity decrease indicates that reoxidation of the CC groups in partially reduced GO by molecular oxygen under the reaction conditions is not complete. In this regard it should be commented that in the first part of the reaction GO acts as a stoichiometric reagent and it is the subsequent reoxidation of CC double bonds that closes the catalytic cycle allowing regeneration of the initial state that becomes ready for a subsequent turn over. Only if turn over occurs can the process be considered as catalytic and not stoichiometric. Interestingly, similar DFT calculations indicate that pristine G or graphite should exhibit substantially higher energy barriers for this reoxidation mechanism with respect to the oxygen-rich GO and, for this reason, they should be much worse catalysts.
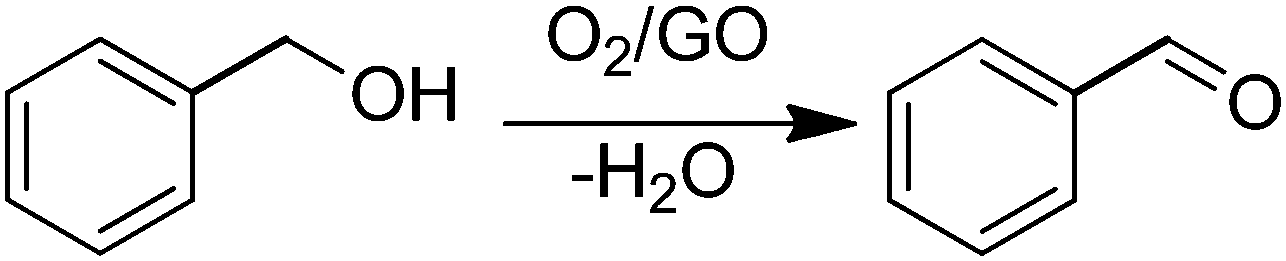 | ||
| Scheme 1 GO as a catalyst for aerobic oxidation of benzyl alcohol to benzaldehyde. | ||
Simultaneous removal of SO2 and NOx at low temperature, <150 °C, is a long term challenge in the treatment of coal-burned flue gases. In this context, calculations at the DFT level predict that both SO2 and NOx should be oxidized by epoxy groups of GO nearly at room temperature.69 The hydroxyl groups on the GO surface should be responsible for enhancing the adsorption and oxidation of SO2 and of NO. In the case of SO2, oxidation would occur through charge transfer taking place between SO2 and epoxide and hydroxyl groups. The oxidation is enhanced by the introduction of more hydroxyl groups near the active site as there are more adsorption sites. In the case of NO a somewhat different mechanism occurs and interaction with the hydroxyl group leads to the formation of covalent N–C bonds between the adsorbed NO molecules and the GO surface. In this way the NO molecule becomes reactive by interaction with the surface hydroxyl groups.
4.2. Quinone groups
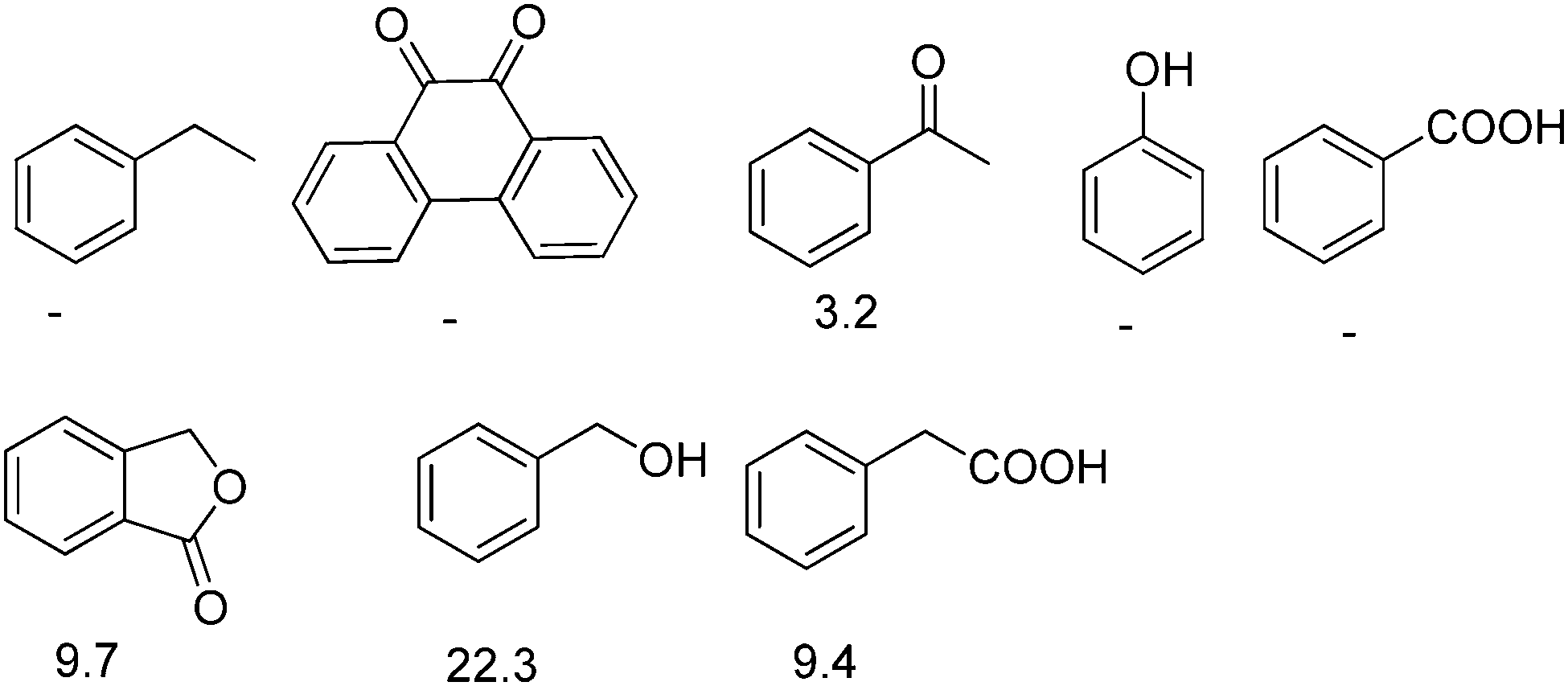
Selective reduction of nitroarenes to the corresponding anilines is an important process from an industrial and a scientific point of view.101 This reaction was known to be catalyzed by natural graphite,102 fullerene,103 and rGO.104 However, one of the problems associated with these catalysts is the identification of the nature of active sites that promote the reduction of nitrobenzene, making it difficult to proceed with the preparation of more efficient carbocatalysts that could compete with metals. In general, mechanistic research over carbonaceous catalysts remains a difficult challenge because of their complex surface structure and the co-existence of various kinds of functional groups. Recently, the role of different oxygen functional groups on a carbon catalyst in the reduction of nitrobenzene by hydrazine has been studied using a series of model molecules (Fig. 2).61 It was observed that carbonyl and hydroxyl groups are the most likely centers in the activation of hydrazine as a reducing molecule. In contrast, the ester, ether and lactone groups seemed to be inactive in promoting this reduction, whereas the carboxylic group has a negative effect, probably due to acid–base interaction with hydrazine.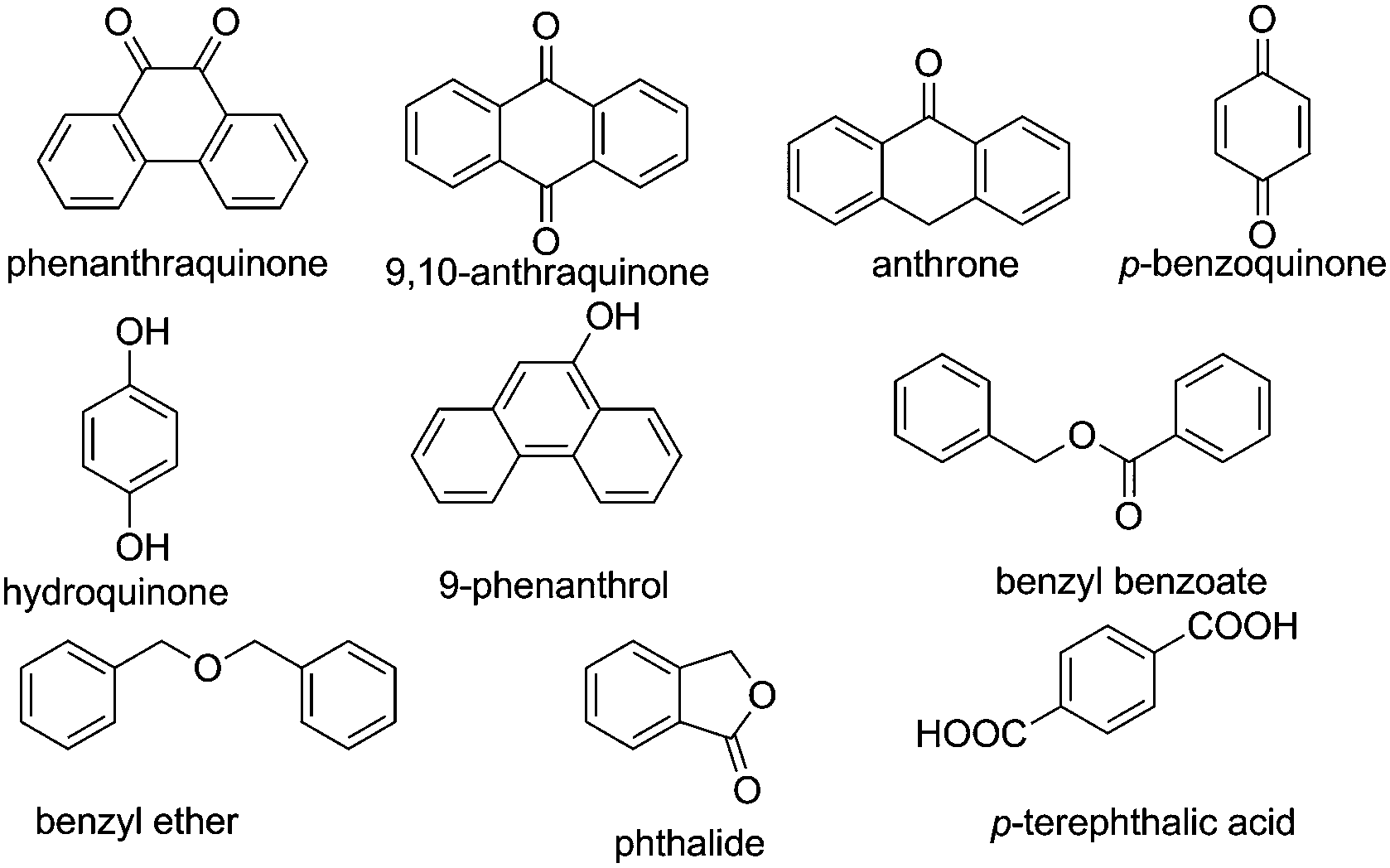 | ||
| Fig. 2 Structures and names of model compounds used to determine the nature of active sites that could be present in carbocatalysts promoting hydrogenation of nitrobenzene by hydrazine. | ||
Aiming at gaining deeper insight into the catalytic behavior of these molecules mimicking the active sites of rGO, it was found that the use of 9,10-anthraquinone as a model catalyst affords 97.7% conversion with 98.4% selectivity to aniline. In general, reduction of CC multiple bonds with hydrazine is believed to proceed through the formation of diimide which is the actual reducing agent generated by the oxidation of hydrazine.105–107 However in the present case, the reduction of nitrobenzene by 9,10-anthraquinone as a model catalyst under a helium atmosphere as well as in air resulted in 96.1% conversion with 99.2% selectivity. The result under the helium atmosphere suggests that the active hydrogen resulting from hydrazine decomposition is the real reducing species and the mechanism does not involve the formation of diimide, since its formation would require the presence of oxygen. It is believed that the reduction of nitrobenzene takes place either through direct or through condensation pathways.108 In the direct pathway, nitrobenzene is reduced to nitrosobenzene, hydroxylamine, and aniline, successively, whereas for the condensation pathway, nitrosobenzene reacts with hydroxylamine to form azoxybenzene, which is further reduced to azobenzene, hydrazobenzene, and aniline. A series of control experiments revealed that the most likely mechanism when using 9,10-anthraquinone as a catalyst proceeds via the direct route rather than through the condensation pathway. Scheme 2 shows a possible mechanism for the reduction of nitrobenzene to nitrosobenzene using 9,10-anthraquinone as the catalyst. The key step in the mechanism is the interaction of unpaired electrons of oxygen atoms in 9,10-anthraquinone with the hydrogen atoms of a hydrazine molecule. As a result, a hydrogen bond is formed, weakening the N–H bond. As nitrobenzene approaches the benzene ring of the catalyst through π–π interactions, the two oxygen atoms of the nitro group abstract two activated hydrogen atoms. In this way, the nitro group is reduced to a nitroso group and a H2O molecule is released. After nitrosobenzene is formed, it is converted further into aniline directly through a noncatalytic process. Moreover, the mechanism when using 1,4-benzoquinone and 1,4-benzenediol as catalysts is also likely to occur through a similar pathway based on N–H⋯O hydrogen bonds.
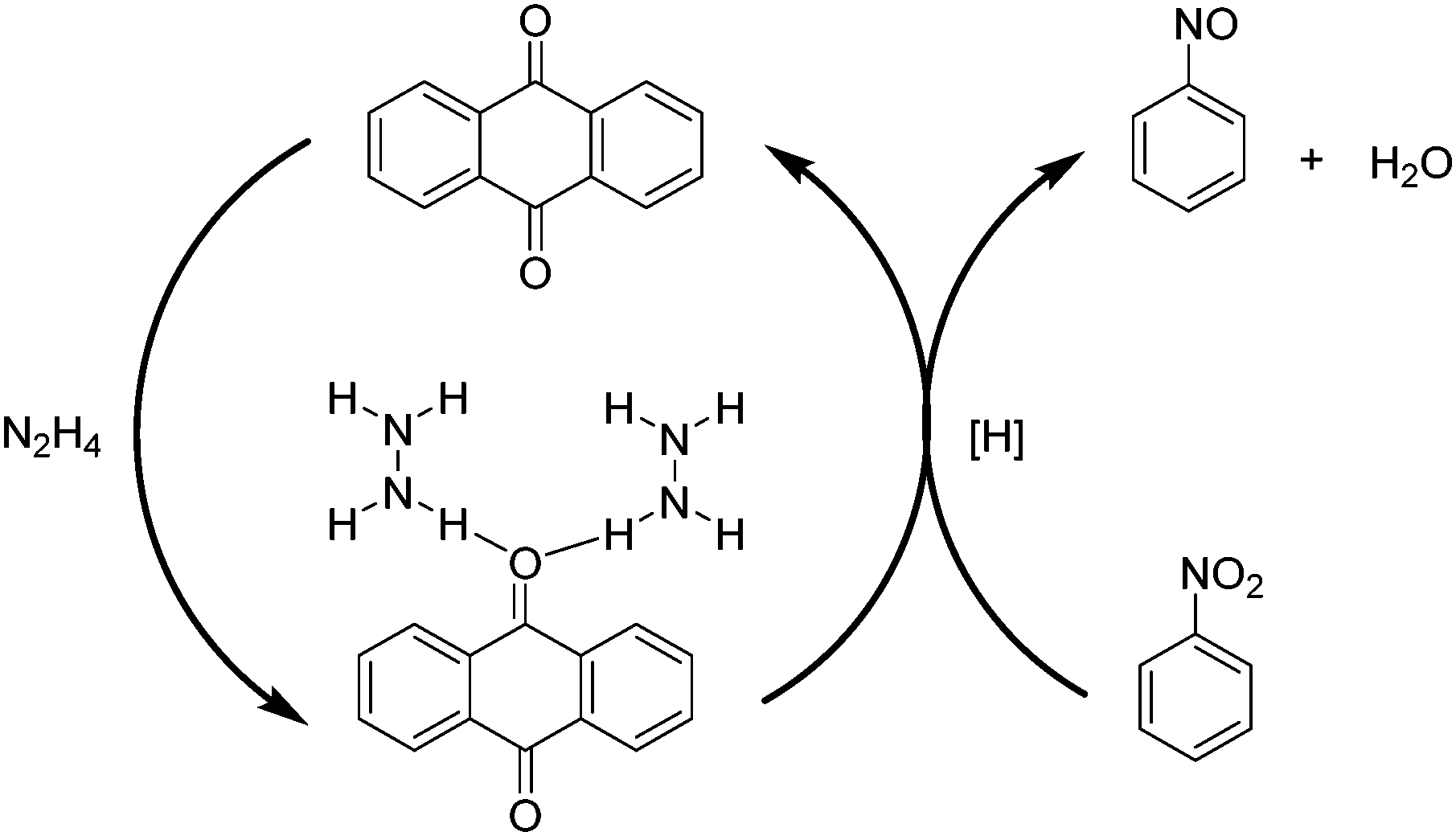 | ||
| Scheme 2 Activation of hydrazine by 9,10-anthraquinone acting as a catalyst resulting in the formation of nitrosobenzene. | ||
Advanced oxidation processes (AOPs) are among the most promising technologies for waste water remediation.109,110 In general, the aim of these processes is the generation of highly aggressive oxidant species such as hydroxyl radicals (HO˙), superoxide (O2˙−) or sulfate radicals (SO4˙−). Among the various AOPs and due to easy implementation in real treatments the (photo)Fenton reaction has attracted considerable attention from academic and industrial points of view.111,112 The Fenton reaction consists in the generation of hydroxyl radicals by reduction of H2O2 using transition metals such as Fe2+ (eqn (1) and (2)).113,114 In order to overcome some of the limitations of this process (requirement of acidic pH, need for stoichiometric amounts of Fe2+, etc.), heterogeneous catalytic (photo)Fenton reaction based on the use of sub-stoichiometric amounts of metals,115 metal oxides,115,116 aluminosilicates,117 or carbonaceous materials has been considered as an alternative.14,118
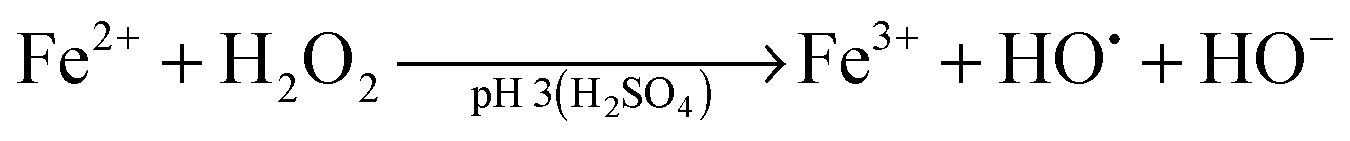 | (1) |
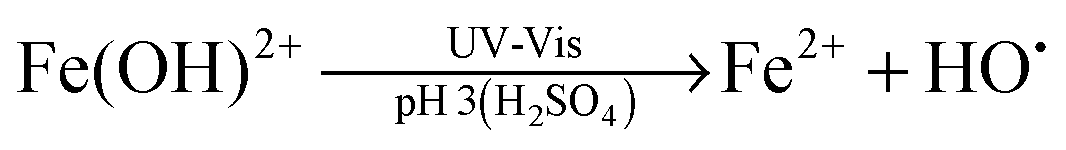 | (2) |
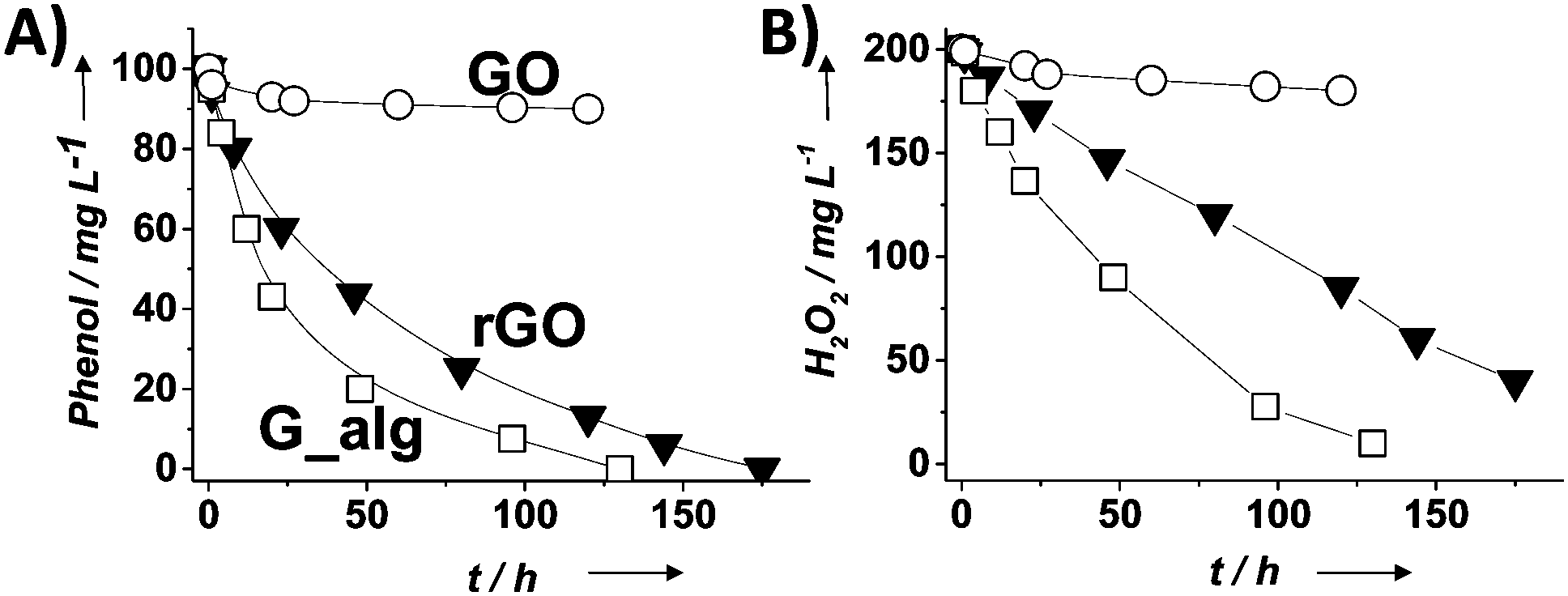 | ||
| Fig. 3 Temporal profiles of phenol degradation (A) and consumed H2O2 (B) using G-based catalysts. Legend: G (□), rGO (▼) and GO (○). Reaction conditions: catalyst (200 mg L−1), phenol (100 mg L−1, 1.06 mM), H2O2 (200 mg L−1, 5.88 mM), pH 3, room temperature. Reproduced from ref. 58 with permission from Wiley, copyright 2017. | ||
For the most active G catalyst having about 8 wt% of oxygen content (derived from pyrolysis of alginate) and rGO, similar kinetic profiles for phenol degradation and H2O2 decomposition were found. In contrast, the activity of GO is negligible for both phenol degradation and H2O2 decomposition. This observation indicates that the total oxygen content does not correlate in this case with the observed catalytic activity and that the oxygenated functional groups present in GO containing mostly sp3 carbons are not active sites for this reaction. In the case of using (N)G as the catalyst, H2O2 decomposes at much higher reaction rates than the disappearance of phenol. This observation agrees with previous work including mechanistic rationalization based on theoretical calculations establishing the H2O2 decomposition towards molecular O2via N–OH intermediates.119 In the case of (B)G an induction period was observed for phenol degradation, but not for H2O2 decomposition.58 This induction period characterized by H2O2 consumption was correlated with the observation of boron leaching from the solid material to the solution as revealed by ICP measurements. Theoretical calculations provided a model for this B leaching from (B)G via B–OH intermediates. Interestingly, using the most active materials G and rGO, the apparent activation energies for phenol degradation and H2O2 decomposition were almost coincident having estimated values of 31 and 30 kJ mol−1, respectively. This observation is compatible with the fact that the generation of HO˙ radicals from H2O2 is the rate determining step of the apparent activation energy for phenol decomposition. Once formed, the HO˙ radicals would react with phenol in a barrierless process. In contrast, for the less active carbocatalysts tested, Ea for phenol degradation is higher than that for H2O2 decomposition indicating that other processes besides the Fenton-like reaction take place. rGO stability was addressed by performing eight consecutive reuse experiments as well as performing an additional run under productivity test conditions with a large excess of phenol (50 mg L−1 rGO, 1 g L−1 phenol, 2 g L−1 H2O2, pH 3). Surprisingly, the observed catalytic activity even slightly increases upon reuse, a fact that was attributed to the better dispersibility in the aqueous medium of the used rGO as a consequence of higher hydrophilicity produced by attack of HO˙ radicals to the basal rGO sheet. The higher hydrophilicity of the five- and eight-times used rGO catalyst with respect to the fresh material agrees with characterization by FT-IR spectroscopy and combustion elemental analyses that reveal the increase of oxygen-functional groups, generated presumably by HO˙ attack. Importantly, the possible contribution of metal traces present on rGO as active sites for the observed catalytic activity was ruled out by performing additional catalytic experiments with the purposeful addition of Mn2+.
Experimental evidence for the generation of HO˙ radicals from H2O2 by rGO was obtained by electron paramagnetic resonance (EPR) spectroscopy using N-tert-butyl-α-phenylnitrone (PBN) as a spin trap as well as by selective hydroxyl radical scavenging using H2O2 decomposition to HO˙ radicals, estimated by quantification of the phenol reaction products (hydroquinone, catechol and p-benzoquinone) and the decomposed H2O2 under conditions of a large phenol excess. Importantly, DFT calculations and the use of simple molecules as organocatalysts suggest that hydroquinone/quinone-like moieties present in rGO could be the active sites for the decomposition of H2O2 to HO˙ radicals in a similar way to the homogeneous metal-promoted Fenton reaction. These hydroquinone/quinone-like substructures should be located in holes or at the periphery of the graphene sheet and, in this regard, terminal OH groups can also correspond to these active sites. In addition, enhanced catalytic activity was achieved by using hydroquinone substituted with electron donating groups such as –OCH3 or –CH3. Quantification by X-ray photoelectron spectroscopy (XPS) of quinone-like centers on rGO based on deconvolution of the C1s peak was used to determine the population of active sites, while the maximum amount of phenol decomposed was determined after the reuse experiments resulting in a TON for phenol degradation and H2O2 decomposition as high as 4540 and 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 023, respectively. Comparison with other carbonaceous materials including multi-walled CNTs (MWCNTs), activated carbon (AC) or graphite reveals that rGO exhibits the highest efficiency for H2O2 decomposition towards HO˙ radicals and subsequent phenol degradation. Interestingly, the generation of hydroquinone/quinone groups in carbonaceous materials such as MWCNTs was found to enhance also their catalytic activity for the Fenton reaction, giving a clue that it should be possible to further increase the catalytic activity of rGO by adequate tuning of its composition.
023, respectively. Comparison with other carbonaceous materials including multi-walled CNTs (MWCNTs), activated carbon (AC) or graphite reveals that rGO exhibits the highest efficiency for H2O2 decomposition towards HO˙ radicals and subsequent phenol degradation. Interestingly, the generation of hydroquinone/quinone groups in carbonaceous materials such as MWCNTs was found to enhance also their catalytic activity for the Fenton reaction, giving a clue that it should be possible to further increase the catalytic activity of rGO by adequate tuning of its composition.
In a similar fashion, the same authors showed that rGO can act as an efficient and reusable carbocatalyst for the photo-Fenton reaction under natural sunlight irradiation.63 Importantly, the reaction rate and the pH of the solution can be increased with respect to dark conditions by light irradiation. Since the Fenton reaction typically requires strong acid solutions, operation of this AOP at quasi-neutral pH values is still a target in this area. In addition, a minimum H2O2 to phenol molar ratio of 5.5 is needed to degrade phenol and its more toxic reaction intermediates formed in the decomposition (hydroquinone, catechol and p-benzoquinone). Again, EPR measurements using the PBN trap and detecting the PBN–OH adduct as well as selective HO˙ radical quenching experiments using DMSO evidenced the generation of HO˙ radicals from H2O2 decomposition when using rGO under natural solar light irradiation. In this study, a relationship was established using three G samples between their oxygen content and the catalytic activity. The lower oxygen content of rGO (∼18%) < rGO (∼33%) < GO (∼44%) resulted in the higher catalytic activity for the photo-Fenton reaction. This observation was interpreted considering that the higher sp2 reconstitution degree of the G layer and the formation of subunits such as p-benzoquinone/hydroquinone should enhance the catalytic activity. Interestingly, the use of methoxyhydroquinone as an organocatalyst resulted in higher catalytic activity than the parent hydroquinone for the photo-Fenton reaction under natural solar light irradiation. In both cases, hydroquinone and methoxyhydroquinone, the influence of the presence of DMSO as a selective hydroxyl radical scavenger decreasing the reaction rate supports the photo-Fenton mechanism by generation of HO˙ radicals. Scheme 3 shows the proposed reaction mechanism for H2O2 activation using rGO under solar light irradiation.63
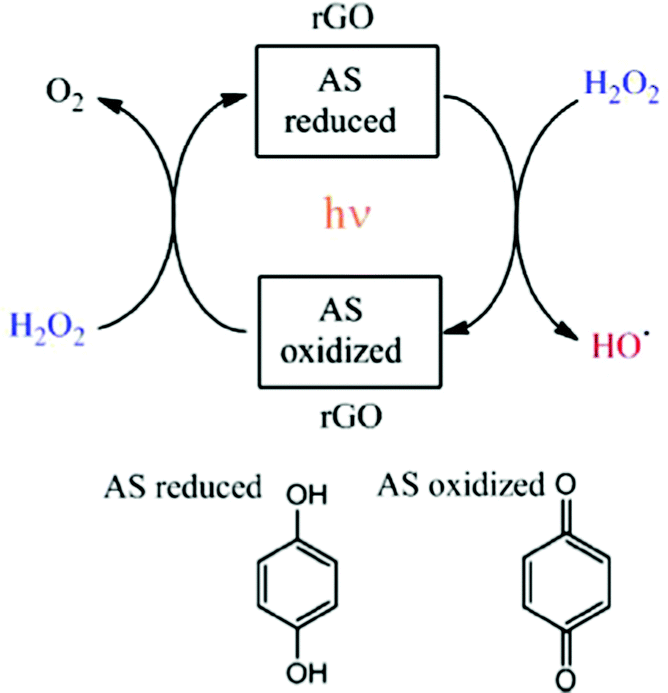 | ||
| Scheme 3 Mechanistic proposal for the generation of hydroxyl radicals using hydroquinone/p-benzoquinone model molecules for the active sites (AS) in rGO. Reproduced from ref. 63 with permission from Wiley, copyright 2017. | ||
The catalytic activity of G to promote the Fenton reaction is a clear example of how it could be possible to employ Gs with adequate composition to replace metals in paradigmatic reactions that have been assumed to be promoted exclusively by metals, similarly to one that will be commented later on the C–C coupling of iodobenzene and benzene. In any case, however, the need for acidic pH values to promote the (photo)Fenton reaction is still a severe limitation for the general implementation of the process.
Recently, rGO has also been employed to catalyze the decomposition of p-hydroxybenzoic acid as a model pollutant in water by ozone.70 It was found that rGO promotes O3 decomposition to O2˙− and 1O2 as evidenced by EPR measurements as well as selective quenching experiments using p-benzoquinone (O2˙− quencher) and sodium azide (selective 1O2 quencher), respectively. The catalytic activity of rGO was higher than that of acidic AC or GO. Catalyst deactivation occurs due to the oxidation of the rGO surface, although a thermal treatment of the used rGO catalyst can restore completely the catalytic activity. Due to the low ID/IG ratio (∼0.84) it was speculated that carbonyl groups on the rGO surface could be the active sites for O3 activation. For this reaction, due to the good catalytic activity observed and the possibility of catalyst reuse, it is important to get more insights into the nature of the active sites on rGO. The use of molecules mimicking the possible active sites, together with theoretical calculations predicting reasonable mechanisms and evaluation of materials with different populations and nature of functional groups would be recommendable to clarify the nature of active sites responsible for the enhanced generation of O2˙− and 1O2 from O3.
Besides (photo)Fenton and ozonation, catalytic oxidation using potassium peroxymonosulfate (PMS; oxone; KHSO5·0.5KHSO4·0.5K2SO4) as an oxidant120 is also a promising AOP for pollutant degradation in water, particularly for neutral solutions. The main advantage of the use of PMS compared to H2O2 is the lower pH dependence of the reaction for the generation of SO4˙− compared to the generation of HO˙ radicals from H2O2.121 In addition, the oxidation potential of sulfate radicals (∼3.1 V) is higher than that of HO˙ radicals (∼2.8 V).71,121,122 Typically, sulfate (eqn (3)) and hydroxyl radicals (eqn (4)) are generated from PMS employing Co(II) or Mn(II) as homogeneous catalysts (eqn (3)). Heterogeneous catalysts for this process include supported or unsupported cobalt oxides123,124 or manganese oxides.125,126
| Co2+ + HSO5− → Co3+ + HO− + SO4˙− | (3) |
| SO4˙− + HO− → SO42− + HO˙ k = 6.5 × 107 M−1 s−1 | (4) |
| Sample | Rate constant (min−1) | I D/IG ratio | O (at%) | CO (%) |
COOH (%) | C–OH/C–O–C (%) | H2Oads (%) | Pore size (nm) | Pore vol (cm3 g−1) | S BET (m2 g−1) |
|---|---|---|---|---|---|---|---|---|---|---|
| GO | 0.001 | 1.22 | 31.35 | 13.7 | 59.42 | 25.08 | 1.80 | 6.7 | 0.04 | 29.9 |
| rGO300 | 0.002 | 1.24 | 12.35 | 24.44 | 20.44 | 43.76 | 11.36 | 8.5 | 0.33 | 118 |
| rGO500 | 0.004 | 1.28 | 7.39 | 16.14 | 27.82 | 42.7 | 13.34 | 8.1 | 0.43 | 173 |
| rGO700 | 0.015 | 1.39 | 5.29 | 19.14 | 21.87 | 46.43 | 12.55 | 8.1 | 0.51 | 203 |
| rGO900 | 0.027 | 1.69 | 2.95 | 21.52 | 21.17 | 43.6 | 13.71 | 8.2 | 0.69 | 286 |
| rGO-CM | 0.005 | 0.94 | 9.16 | 16.30 | 22.02 | 51.69 | 9.99 | — | 140 | |
| rGO-HH | 0.010 | 1.52 | 9.05 | 28.87 | 29.97 | 32.69 | 8.47 | — | 338 | |
| rGO-HT | 0.026 | 1.35 | 9.63 | 32.65 | 26.01 | 30.46 | 7.89 | — | 142 |
 | ||
| Scheme 4 Transformation of GO into rGO by thermal annealing. Note that this is a cartoon highlighting the importance of carbonyl groups as active centers. | ||
DFT calculations showed the higher adsorption energy, electronic transfer and O–O bond length in PMS (SO4–OH) adsorbed on carbon vacancy defects, zigzag or armchair edges compared to adsorption on the G basal plane.59 The role of the oxygen functionalities was studied using three rGO samples having similar oxygen content (9.05–9.63 at%) obtained by either chemical or thermal reduction methods. As commented, GO with a high oxygen content (>35 at%) exhibits negligible catalytic activity. Among the three rGO samples prepared, the one with the highest ketonic content exhibited the highest activity even though its ID/IG ratio, surface area and phenol adsorption were lower. DFT calculations showed higher O–O bond elongation when PMS interacts with oxygen groups. Among them, ketonic groups were presumed to be the main oxygen-functional group responsible for PMS activation. The main feature of the ketonic group is the presence of lone-pair electrons acting as weak Lewis basic sites able to coordinate and participate in electron transfer processes with PMS. It should be mentioned that the stronger oxidation potential of SO52− (pKa1 < 0, pKa2 = 9.9) with an asymmetric structure and negatively charged oxygen atoms makes PMS decomposition easy even under basic conditions, in contrast to the previously commented case of H2O2.
In line with these results, the same authors showed that, besides rGO, PMS activation can be performed by other carbonaceous materials including MWCNTs, pyrolized nanodiamond (ND) and cubic-ordered mesoporous carbon (CMK-3) (Fig. 4).71 Selective HO˙ and SO4˙− quenching experiments using MeOH and theoretical calculations were employed to understand the concomitant occurrence of radical or non-radical reaction mechanisms when using PMS as the oxidant. Importantly, the nature of the active sites present on the carbonaceous materials strongly influences the prevalence of the radical over the non-radical mechanism for PMS activation. In this regard, it was proposed that ketonic groups and sp2 hybridized carbon lattice can donate electrons to PMS leading to the formation of sulfate radicals. It should be, however, commented that ketones and quinones are electron acceptors and not donors. Nevertheless, in a catalytic cycle the process can start with electron donor centers with the structure of allylic alcohols and hydroquinones that would donate electrons and, then, would be converted into ketones. On the other hand, defective edges on pyrolized ND and rGO promote non-radical pathways in which SO52− interacts with the edges and becomes activated for concerted oxygen or electron transfer to the substrate (Fig. 4). Unfortunately, rGO is not a stable catalyst, becoming partially oxidized during the reaction and its catalytic activity decreases to a large extent upon reuse. This contrasts with the already commented stability of G for the Fenton and photoFenton processes and could reflect the higher reactivity of SO4˙− radicals vs. HO˙ radicals toward the attack to the graphenic π system. The used rGO catalyst was subjected to a thermal treatment to regenerate the active sites and remove the excess of oxygen functional groups. However, the catalytic activity could only be recovered partially by these treatments. Therefore, more efforts are still necessary to increase G stability for PMS activation. In addition, the excess of PMS employed with respect to phenol (molar ratio of 31) should probably be decreased to make the process economically more attractive in real applications.
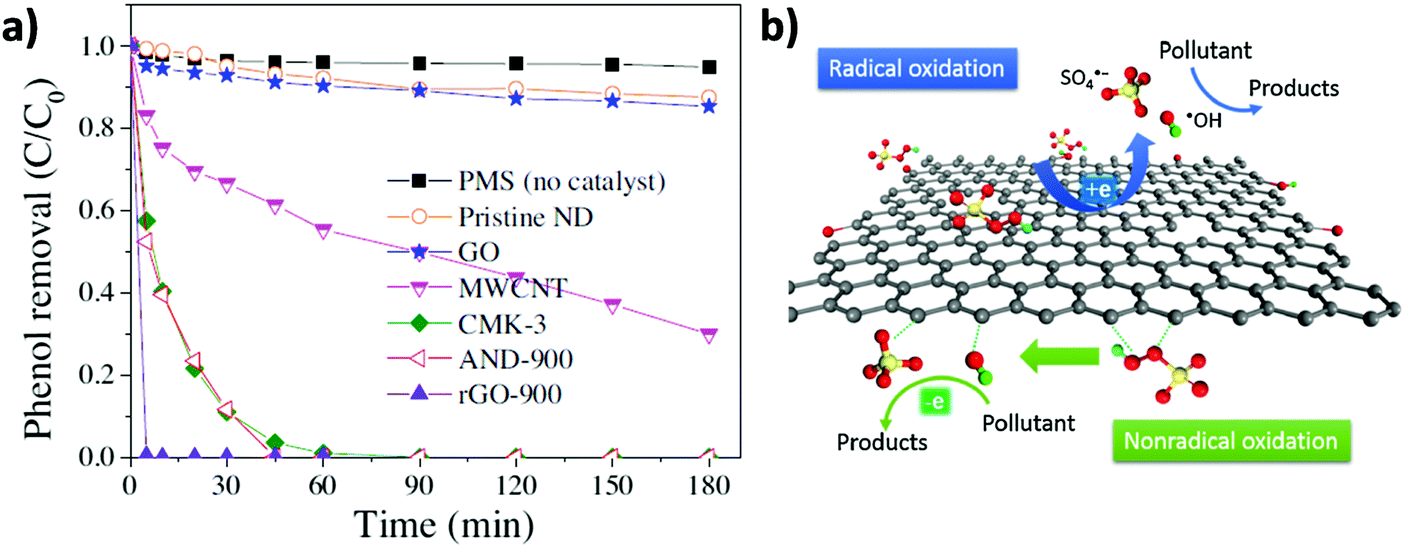 | ||
| Fig. 4 (a) Phenol oxidation by PMS activation on nanocarbons ([Catalyst]0 = 0.1 g L−1, [PMS]0 = 6.5 mM, [T] = 25 °C, and [phenol]0 = 20 mg L−1, pH free). (b) Illustration of radical and non-radical oxidation mechanism using PMS as the oxidant. Reproduced from ref. 71 with permission from Elsevier, copyright 2017. | ||
Similarly to the previously commented use of oxone, catalytic persulfate activation (K2S2O8, PS) (eqn (5)) is considered an alternative to the use of hydrogen peroxide in AOPs to achieve degradation of organic compounds in water.128
| Fe2+ + S2O82− → Fe3+ + SO42− + SO4˙− k = 2.7 × 101 M−1 s−1 | (5) |
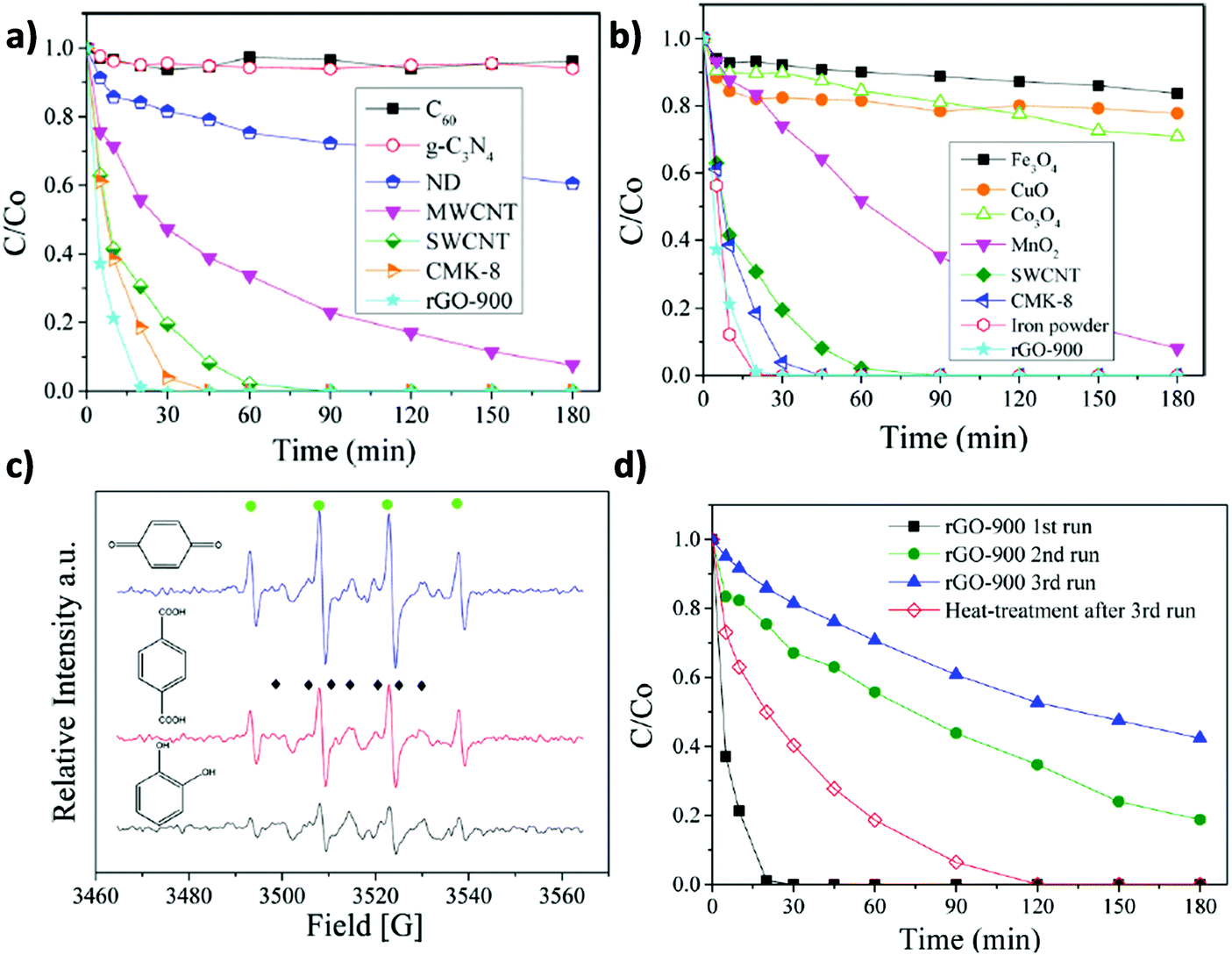 | ||
| Fig. 5 Catalytic phenol degradation by PS using various (a) carbonaceous materials or (b) metal oxides as catalysts; (c) EPR spectra of PS activation using different organocatalysts (●: DMPO-OH). (◆: DMPO-SO4). (d) Reusability experiments for phenol degradation by PS employing rGO-900 as a carbocatalyst. Reaction conditions: catalyst (0.2 g L−1), 25 °C, PS (6.5 mM), phenol (20 mg L−1; 0.21 mM). Reproduced from ref. 72 with permission from American Chemical Society, copyright 2017. | ||
4.3. Benzyl alcohol-type groups
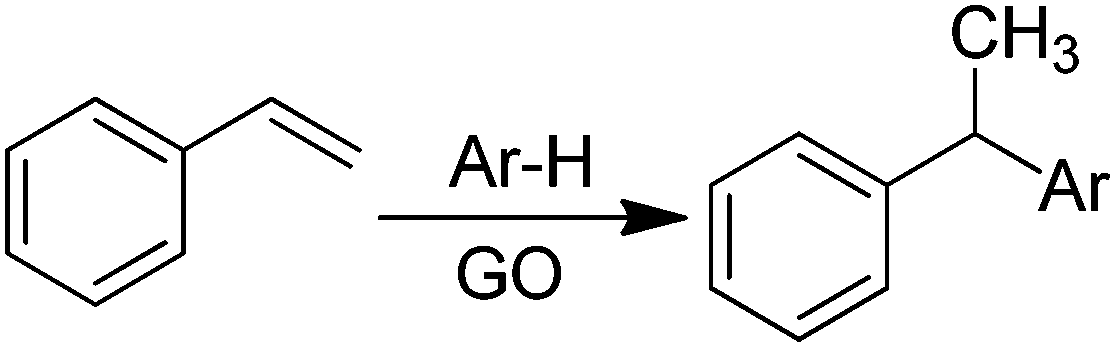
Recently, H2-treated GO in combination with KOtBu has been employed as an efficient heterogeneous catalyst for the direct C–H arylation of benzene by 4-methoxyiodobenzene to form 4-methoxybiphenyl (Scheme 5).73 The interest in this process is that it is related to the classical Suzuki–Miyaura cross coupling reaction that is catalyzed by Pd.129,130 In the present case, it was found that Mn or Fe impurities do not play any role and GO is assumed to be among the first metal-free catalysts for C–C coupling reactions. Reactions with various oxygen containing model compounds and DFT calculations support that negatively charged oxygen atoms are likely to be responsible for the overall transformation. The stabilizing and activating effect of K+ ions is proposed to facilitate the activation of the C–I bond of C6H5–I after adsorption. This hypothesis was further supported by performing a reaction in the presence of 18-crown-6 to trap the K+ ions, observing that the C–H arylation of benzene with 4-iodoanisole is effectively stopped. In addition, the G π system also greatly favors the reaction by adsorbing the aromatic reagents before coupling.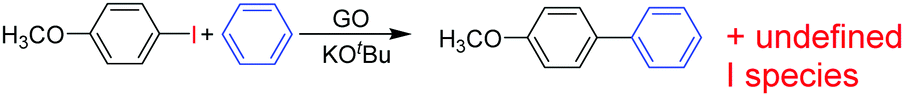 | ||
| Scheme 5 Direct GO-catalyzed C–H arylation of benzene. | ||
To investigate the nature of oxygen species, four catalysts with different oxygen contents were synthesized by controlling the H2 annealing temperature of GO at 300, 500, 700, or 900 °C to obtain GO-300, GO-500, GO-700, and GO-900, respectively. XPS spectra indicated that the peak intensity of the oxygen species decreases from 16.1 to 4.2 wt% as the treatment temperature increases. Catalytic data revealed that the lower the oxygen content in GO, the lower the catalytic activity. A series of model oxygen containing compounds (Fig. 6) showed that those compounds with oxygen functional groups directly attached to the benzene ring gave the desired product in very low yield or not at all. On the other hand, when the oxygen functional groups were attached to a benzylic position, then, the model compounds catalyzed the desired reaction, benzyl alcohol being the most active of the organic molecules able to promote the coupling. Furthermore, it is believed that aside from the π–π interaction stabilization, K+ ions plays a role in the promotion of C–I bond cleavage of iodobenzene. It is, however, unclear how a K+ can stabilize C6H5˙ radicals that were detected and predicted to be the key intermediates. It is also unknown what is the energy cost of C–I bond cleavage by C6H5–I adsorbed on GO.
 | ||
| Fig. 6 A series of model oxygen containing compounds investigated to identify the nature of active sites. The numbers correspond to the yield of the coupling biphenyl under the reaction conditions. Reaction conditions: p-methoxyiodobenzene (0.4 mmol), benzene (4 mL), catalyst (0.3 mmol), KOtBu (1.2 mmol), 120 °C, 2 h. | ||
As an additional comment, it is, however, unusual to expect a high density of benzylic alcohols on Gs that ideally should have exclusively sp2 carbons. Detection and quantification of the population of these –CH2OH groups would be of great interest to confirm their presence and to develop more efficient synthetic protocols to increase their number.
4.4. Acid sites
Recently, hydrothermally treated GO (HGO) was found to be a new metal free catalyst for the reduction from 4-nitrophenol to 4-aminophenol using NaBH4 as a reducing agent.65 The reaction mechanism using metal hydrides is different from the one discussed earlier for hydrazine activation. While reduction of nitroaromatics by hydrazine requires some oxidation, the reactivity of metal hydrides derives from the polarization of the metal–hydrogen bond that increases with the metal electropositivity. For this reason, it is expected that this nature of the active sites on G should be different for both types of nitroaromatic hydrogenation. Combined experimental and theoretical investigations revealed that using NaBH4 as reducing agent hydroxyl groups and holes are beneficial for promoting the reduction, while in contrast, epoxy and carboxyl groups should not exhibit catalytic activity. In a control experiment, HGO was treated with a concentrated NaOH solution to modify its functional groups. It was observed that the NaOH treated sample showed about one tenth lower catalytic activity compared to the bare HGO catalyst. Assuming that NaOH only neutralizes acid sites, this activity data suggest that the catalytic performance of HGO mainly originates from the acidic O–H groups on the surface present on HGO. Furthermore, XPS results also confirmed the decrease of hydroxyl groups after NaOH treatment and this information that is reasonable with the expected reactivity of NaOH supports the involvement of acidic OH as centers of nitro group reduction.131Recently, a general strategy for alkylation of arenes with styrenes and benzylic alcohols catalyzed by GO has been explored (Scheme 6), exploiting the unique property of Gs as metal-free catalysts to afford valuable diarylalkanes in high yields and excellent regioselectivity.74
 | ||
| Scheme 6 GO catalyzed alkylation of arenes. | ||
In order to elucidate the nature of the active sites on G in these alkylations, extensive characterization of the GO samples before and after reaction was conducted. It was observed that the recovered GO in suspension measured by methylene blue adsorption exhibits a surface area of 367 m2 g−1, indicating the occurrence of partial π-stacking of the GO sheets as compared to the parent GO material (1371 m2 g−1). The increase in the π–π stacking is consistent with a partial reduction of initial GO to rGO during the course of the reaction as was indicated in XPS by a substantial decrease of C–O/CO functional groups on the GO surface from 49.4% to 40.9% with a concomitant increase in the intensity of peaks corresponding to CC bonds (from 37.0% to 52.0%). The partial reduction of GO to rGO during the process points toward the importance of oxygen-containing functionalities anchored on the GO surface such as hydroxyl, epoxide, and carboxylic acid groups as active sites. Based on these observations, it was proposed that the reaction mechanism of the alkylation involves activation of both coupling partners by adsorption on G (Scheme 7). The key step would be activation of the olefin by hydration to an adsorbed alcohol, and a transition state in which the arene nucleophile is prepositioned by π-stacking interactions with the GO sheet for a concerted C–C bond forming step to give the alkylated product and regenerate the catalyst after the release of water. It should be noted, however, that the classical mechanism for arene alkylation requires the presence of strong acid sites, whose presence in this case has not been discussed, particularly, the possibility that the transformation of GO to rGO is accompanied by the removal of sulfate and sulfonic groups. It is also noteworthy that in the proposed reaction mechanism the key step is the reaction of adsorbed reactants with the simultaneous breaking of a strong C–O bond and formation of a C–Ar bond without assistance of any acid sites. It will be discussed below that indeed electron transfer via charge transfer interactions between the substrate and the catalyst can have a similar promotional effect as an acid, i.e. partial charge transfer from the aromatics to the carbocatalyst would leave a positive partial charge on the substrate which then drives the reaction. It is clear that further studies are required to provide some support to this proposed mechanism.
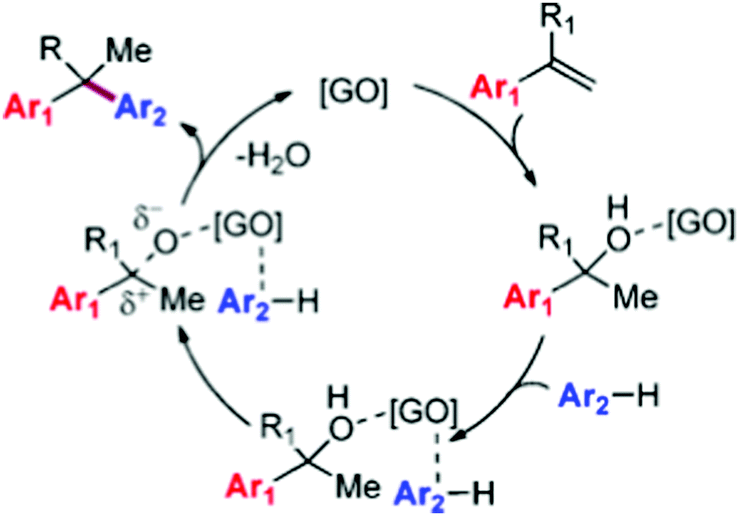 | ||
| Scheme 7 Proposed mechanism for the GO-catalyzed alkylation of arenes. Reproduced from ref. 74 with permission from American Chemical Society, copyright 2017. | ||
An efficient protocol for the formation of 2,3-dihydroquinazolinones has been reported employing GO as a carbocatalyst (Table 3).75 The catalytic activity of GO is higher than that of other catalysts such as PEG–SO3H, rGO, SnO2-QDs (QDs: quantum dots) or nano CuFe2O4. The activity of GO retains more than 90% of the catalytic activity of the fresh sample after five uses. Based on GO characterization and the catalytic activity observed using organic molecules as models of active sites and organocatalysts, carboxylic acid functionalities are the most likely active sites on GO (Table 3).
|
|
||
|---|---|---|
| Time (h) | Yield (%) | |
| Reaction conditions: catalyst (25 mg), anthranilamide (1 mmol), meta-nitrobenzaldehyde (1 mmol), water (3 mL), room temperature. | ||
| Benzoic acid (10 mol%) | 0.25 | 65 |
| Ethylene glycol (10 mol%) | 0.25 | — |
| 1,2-Diphenylethano-1,2-diol (5 mol%) | 0.5 | — |
| H2SO4 (13 mol%) | 0.25 | 25 |
| Styrene oxide (10 mol%) | 0.5 | — |
| Stilbene oxide (10 mol%) | 0.5 | — |
| 2-Iodophenol (10 mol%) | 0.5 | — |
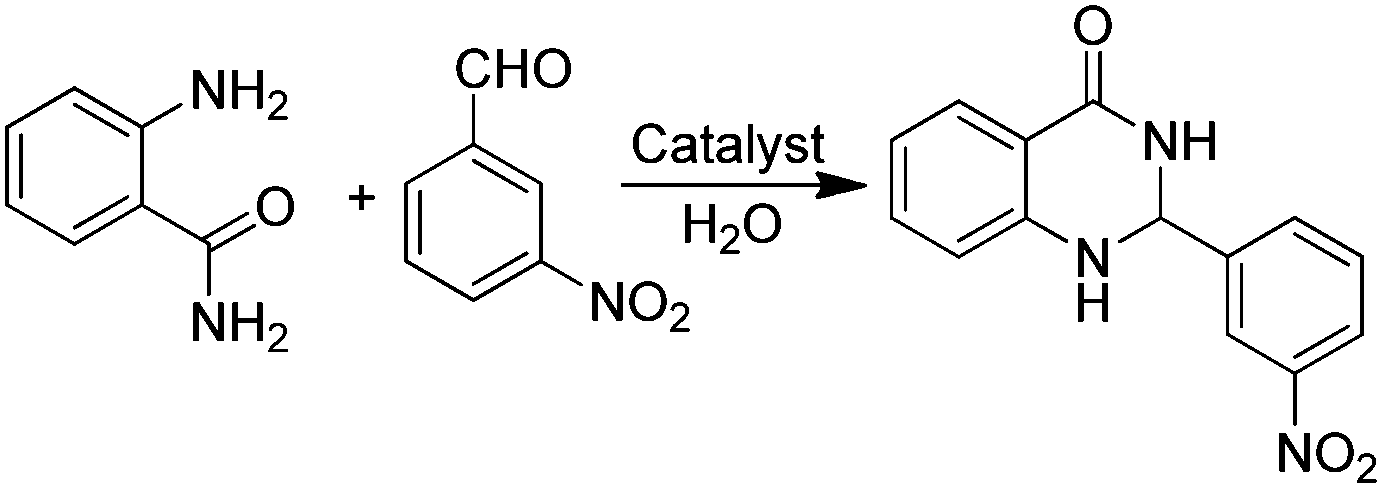
In addition, the presence of oxone as an oxidant in the system allows expanding this cyclocondensation for the selective preparation of quinazolin-4(3H)-one derivatives (Scheme 8). In the present case, the possible role of GO activating oxone was not investigated and the possibility that metal traces present on GO due to the use in Hummer's oxidation of a large excess of KMnO4 could play a role in the catalysis was not addressed.
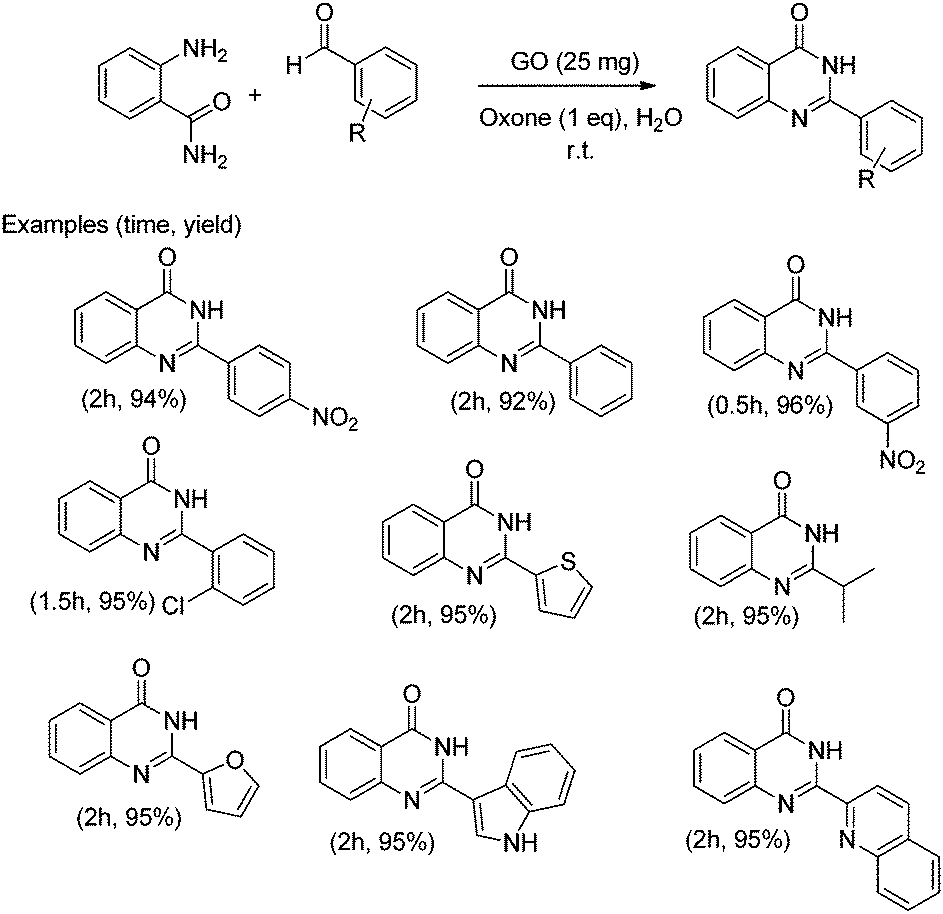 | ||
| Scheme 8 Synthesis of quinazolin-4(3H)-one derivatives. Data adapted from ref. 75. | ||
5. Sulfur functional groups
5.1. Sulfate groups
Active sites on G can also be impurities or adventitious groups introduced in the material during its preparation procedure.14 One of these cases is the catalytic activity of sulfonic or sulfate groups introduced on rGO prepared from GO due to the large excess of H2SO4 employed in graphite oxidation. In this context, Garcia and co-workers have reported the activity of GO as a metal free carbocatalyst for the room-temperature ring opening of epoxides using methanol and other primary alcohols as nucleophiles and solvent (Scheme 9).66 Interestingly, GO at 0.19 wt% in the ring opening of styrene oxide by methanol exhibited 99% conversion with 97% selectivity towards the desired product. Interestingly, the amount of GO used as the catalyst in this reaction is much lower than that used for benzyl alcohol oxidation or alkyne hydration (200 wt%)68 or for hydration of propylene oxide (3.4 wt%).132 It was confirmed that the impurities present in GO introduced during graphite oxidation as a consequence of the harsh treatment conditions are responsible for the catalytic activity of GO. Thus, the catalytic activity decreased when S was partially removed by thermal treatment at 200 °C. Furthermore, the presence of pyridine in the reaction mixture completely stopped the reaction due to neutralization of acid sites present on GO. A comparable activity to GO was observed for H2SO4 and p-toluenesulfonic acid, while glacial acetic acid showed no conversion of styrene oxide, thus, indicating that hydrogen sulfate groups that can be partially removed by methanol washings are probably the main active sites responsible for the catalytic activity. The interaction of the strong acid sites on GO with alcohols was monitored by IR spectroscopy using methanol as a probe molecule showing the presence of these active sites.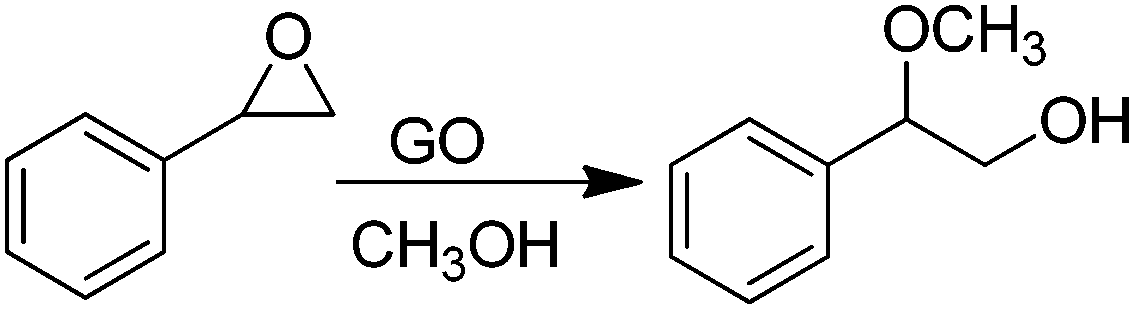 | ||
| Scheme 9 Ring opening of styrene oxide catalyzed by GO. | ||
Room temperature acetalization of benzaldehyde by methanol can also be similarly performed in quantitative yield with complete selectivity using GO as a carbocatalyst (Scheme 10).67 In contrast, the use of other carbonaceous materials, such as graphite or AC (Norit A), highly porous metal–organic frameworks [Fe(BTC) or Cu3(BTC)2 (BTC = 1,3,5-benzenetricarboxylate)], or an acid resin such as Amberlite XAD4 as a catalyst resulted in yields below 14% for acetalization of benzaldehyde to benzaldehyde dimethyl acetal. In the case of other carbonaceous materials, formation of benzoic acid was also observed, showing the lack of acid sites in these materials and a certain oxidation activity. In this reaction, hydrogen sulfate groups on GO whose proportion can be decreased by exhaustive washings with methanol were proposed as being responsible for the observed catalytic activity.
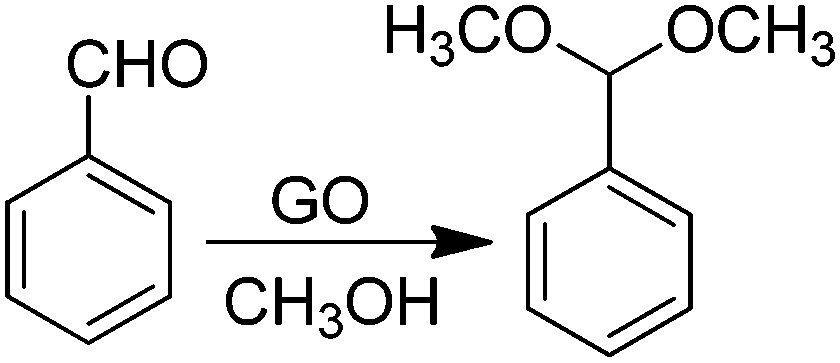 | ||
| Scheme 10 Acetalization of benzaldehyde with methanol using GO as the catalyst. | ||
5.2. Sulfonic groups
GO was reported to be an efficient carbocatalyst for the dehydration of fructose into HMF (Scheme 11) with a yield of 87% at 120 °C after 6 h.76 To gain understanding on the nature of active sites responsible for this key transformation in biomass valorization, a series of control experiments were performed. Glacial acetic acid, p-toluenesulfonic acid, concentrated sulfuric acid and Amberlyst-15 were used as catalysts in their optimum amounts under similar conditions to those used with GO. It was observed that, except for glacial acetic acid, the other acid catalysts could facilely promote the dehydration reaction, suggesting that the active sites of GO are probably the sulfonic groups present as impurities rather than the carboxylic groups. However, the yield of HMF (87%) with GO as the catalyst was clearly higher than that using Amberlyst-15 (71%) that could be considered a structurally related analog for arylsulfonic groups present in GO. It was, therefore, proposed that this difference between these two catalysts arises from the synergy derived from the presence on the GO of a diverse array of oxygen-containing groups, such as hydroxy, epoxy, and carboxyl groups that are absent in Amberlyst-15 containing exclusively sulfonic groups. It was proposed that these other oxygen-containing functionalities present on the surface of GO would also play a role in the dehydration of fructose into HMF. In support of this proposal, the catalytic activity of GO decreased rapidly after GO treatment above 200 °C, while its activity is retained for GO treated below 150 °C. These data are in agreement with the assumption that oxygen-containing groups on GO play an important role in maintaining the high catalytic activity of GO for fructose dehydration. To gain more insights, the affinity of GO and Amberlyst-15 for fructose and HMF was evaluated by adsorption measurements. These experiments indicated that the amount of fructose adsorbed on GO is much higher than that on Amberlyst-15. The higher affinity of GO for fructose was attributed to the presence of surface oxygen-containing groups forming hydrogen bonds with the hydroxy groups of fructose. On the other hand, a larger amount of ether byproduct was identified for Amberlyst-15 as the catalyst derived from the stronger acidity of sulfonic acid groups on this resin. Accordingly, GO would have adequate acidity and strong adsorption towards fructose, and these two factors contribute to its higher catalytic activity and selectivity. | ||
| Scheme 11 GO catalyzed dehydration of fructose to HMF. | ||
Similarly to the previous case of fructose dehydration, GO has been reported as an acid catalyst for the synthesis of polyoxymethylene dimethyl ethers (PODEn) from methanol and trioxymethylene (Scheme 12).77 It was proposed that the active sites are the combination of the sulfonic groups and the hydroxyl and carboxyl groups present on the surface of GO establishing a cooperative synergy. This conclusion was reached by comparing the catalytic activity in the synthesis of PODEn of various modified GO catalysts differing in the oxygenated functional groups in a series of modified GO catalysts. Three types of modified GOs masking selectively one type of oxygen functional group were prepared. Thus, the hydroxyl groups were selectively blocked by silylation with tetramethoxysilane. The carbonyl and carboxyl groups on GO were selectively removed by reduction with NaBH4. Similarly, 84.7% of the SO3H groups on GO were removed by hydrothermal treatment at 150 °C for 12 h. When the hydroxyl and carboxyl groups are selectively masked or removed, the modified GO exhibited much lower activity and selectivity to PODE2–8 than the parent GO under identical conditions. All these control experiments clearly support that the hydroxyl and carboxyl groups contribute to the catalytic activity of GO for the synthesis of PODEn whose main active sites appear to be strong sulfonic acid groups. This proposal was further supported by studying the catalytic activity of a series of model compounds including p-hydroquinone, p-benzoquinone, terephthalic acid and salicylic acid that showed very low catalytic activity in the synthesis of PODEn with dimethoxymethane as the dominant product. In contrast, benzenesulfonic and sulfosalicylic acids as catalysts gave higher trioxymethylene conversions. Also, sulphuric acid as a model catalyst showed 72.2% conversion of trioxymethylene with selectivity to PODE2–8 (19.3%), dimethoxymethane (29.2%), methylformate (0.9%) and formaldehyde (50.6%). These data prove that the carboxyl, hydroxyl and carbonyl groups are catalytically active for the dissociation of trioxymethylene and the formation of dimethoxymethane from formaldehyde and methanol; however, their acidity is probably insufficient for chain propagation to form PODE2–8. Among the model compounds tested the formation of long chain PODE2–8 products could be effectively catalyzed only by the sulfonic groups or sulfonic acid. It should, however, be commented that polymers and high oligomers are only formed at high substrate concentrations, best in the absence of solvents, and besides acid strength the most important parameter controlling polymer growth is monomer concentration.
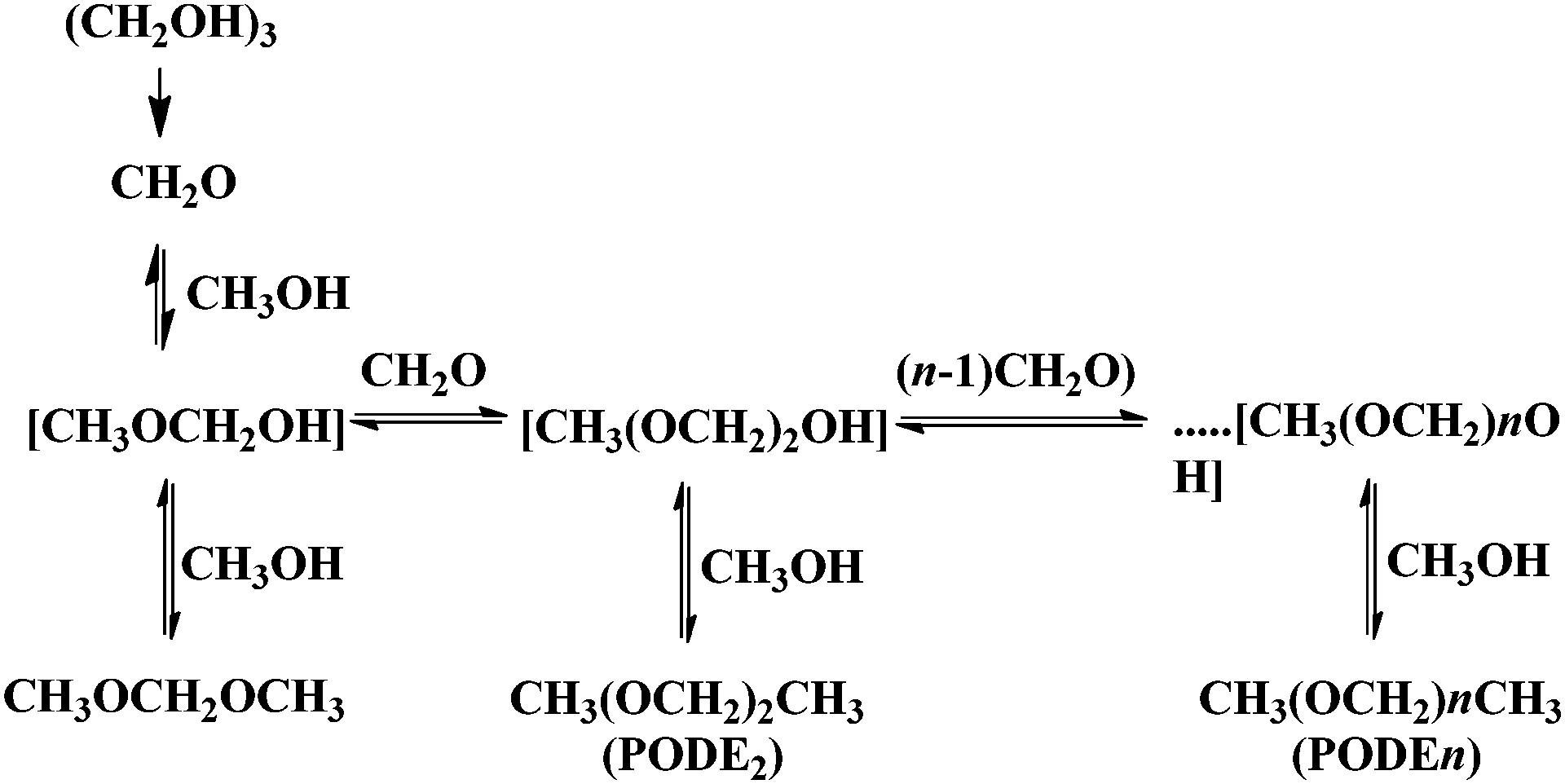 | ||
| Scheme 12 Products obtained by treatment of trioxymethylene in methanol under acid catalysts. | ||
GO was efficient in promoting in high yields ring opening reactions of chiral aromatic epoxides by indoles (Scheme 13) in a regio/enantioselective manner under solvent- and metal-free conditions.78 The Friedel–Crafts products were obtained with yields in the range of 25–80% and enantioselectivity up to 99%. It was believed that the presence of different acid functionalities on the carbocatalyst surface activates the epoxide ring-opening and forms an incipient carbocation that reacts with the nucleophilic carbon of the indole. Furthermore, the high stereoselectivity achieved in all cases with complete inversion of stereochemistry indicates that no free-carbocation is fully developed, the nucleophilic attack by indole taking place regioselectively at the benzylic position as in the SN1-type mechanism. Further studies are required to delineate the exact role of oxygen functionalities on the activity of GO.
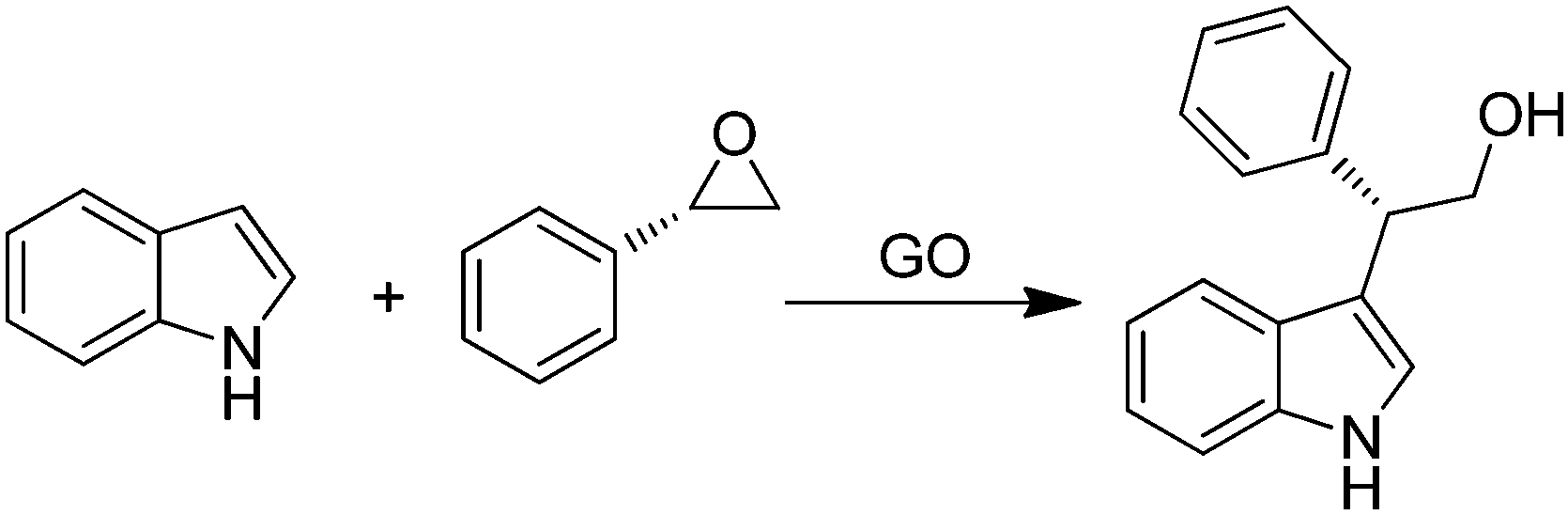 | ||
| Scheme 13 GO catalyzed ring opening reactions of chiral styrene oxide by indole. | ||
6. Carbon vacancies
Loh and co-workers have reported that simple base and acid washings of as-prepared GO (ba-GO) can release and enlarge the defects in GO, thereby enhancing the catalytic activity of treated ba-GO for the oxidative coupling of amines to imines to 98% yield under solvent-free and open-air conditions.64 In contrast, untreated, as-prepared GO exhibited 44% yield under identical conditions. 13C NMR spectrum of ba-GO revealed a significant decrease in the concentration of oxygen functionalities appearing in the spectrum at 71 ppm (C–OH) and 61.9 ppm (C–O–C), which are attributable to hydroxyl and epoxide groups, respectively. This suggests that these oxygenated functional groups are not involved in the catalysis. Nevertheless, in situ FTIR analysis of ba-GO indicated that residual oxygen functional groups, mainly as ketones, epoxides (significantly reduced) and carboxylic acids, still remain in ba-GO.To gain more insights into the nature of active sites, ba-GO was reduced with NaBH4 to decrease the population of ketonic functional groups, followed by subsequent acid treatment to regenerate the carboxyl peak at 1650–1750 cm−1. A catalytic activity of 78% imine yield similar to that of unreduced ba-GO was observed. This result was taken as support for the involvement of carboxylic acid groups in the catalysis. This claim was further supported by performing a control experiment with 1-pyrene carboxylic acid as a molecular analogue of the active sites present in ba-GO. A yield of 95% comparable to that of ba-GO was achieved using 1-pyrene carboxylic acid as the organocatalyst. Scanning tunneling microscopy revealed that GO sheets become highly porous after the base treatment with an average pore area of around 5 nm2. These holes are likely to be created during the harsh oxidation process and would be freed from amorphous acidic carbon debris by the etching process during the base treatment. The available data suggest that the active catalysis sites in ba-GO are likely to be hole defects in the conjugated domain as well as the edges of the hole defect terminated by the carboxylic acids (Scheme 14).
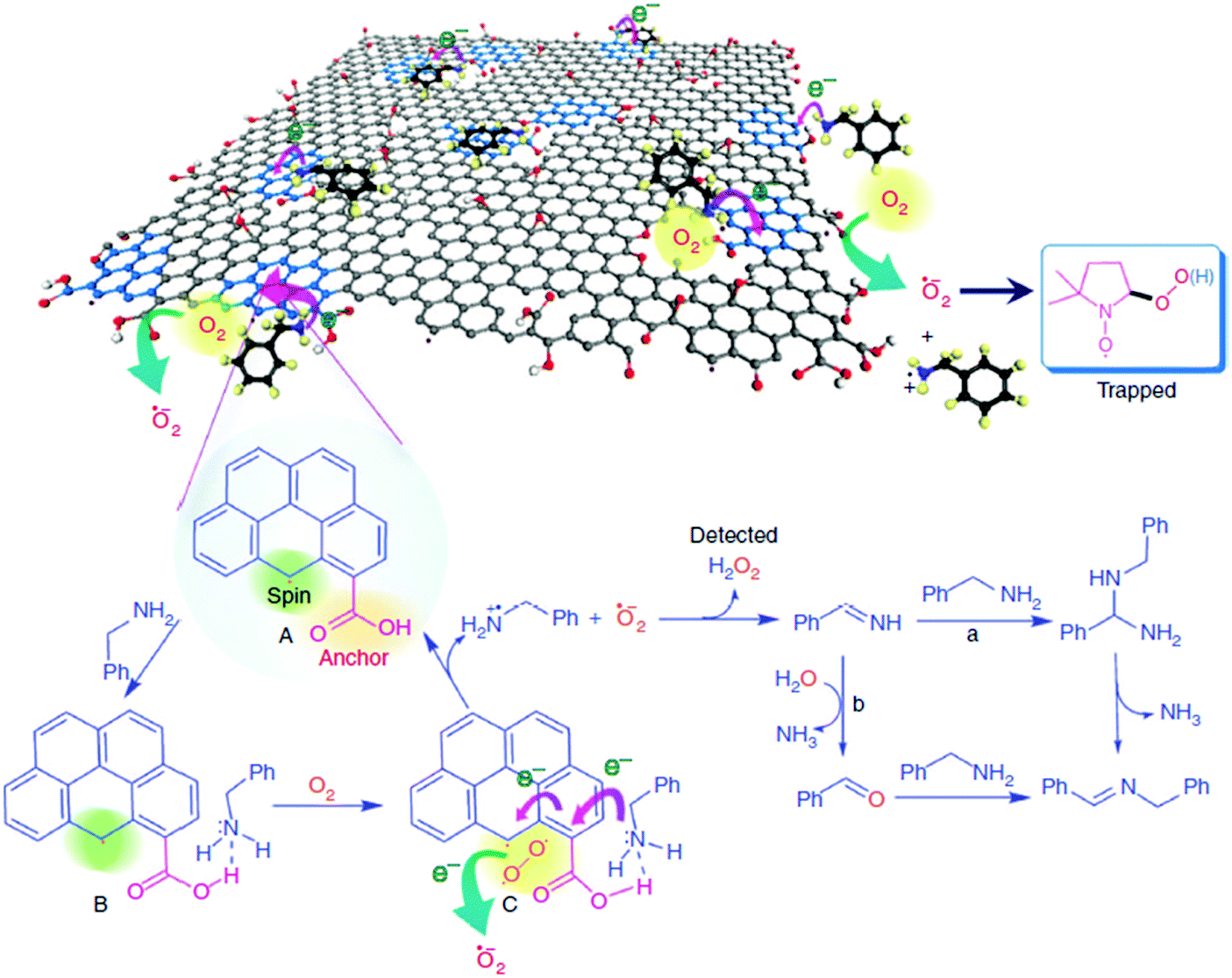 | ||
| Scheme 14 Proposed mechanism of ba-GO catalyzed oxidative coupling of primary amines. Reproduced from ref. 64 with permission from Nature, copyright 2017. | ||
The role of carbon vacancies in the catalytic activity of ba-GO was supported by observing a broad and a sharp signal in ESR with an intensity ratio of about 5:1. Therefore, it was proposed that the observed catalytic ability is related to the localized spins present at the edges of the π-electron system. Furthermore, EPR spectroscopy showed the characteristic spectrum with hyperfine couplings of the adduct corresponding to the superoxide radical being trapped by 5,5-dimethyl-1-pyrroline-N-oxide (DMPO). Also, the formation of H2O2 was detected by using Horseradish peroxidase to promote the enzymatic oxidation of N,N-diethyl-1,4-phenylenediammonium sulfate (DPD). Therefore, these edge sites with unpaired electrons would enhance the kinetics for molecular oxygen activation by a sequence of electron transport and reduction to superoxide radicals.
The carboxylic groups at the edges of defects, along with the localized unpaired electrons, work synergistically to trap molecular oxygen and the amine molecules. In this regard, while pyrene did not give any oxidation, 1-pyrene carboxylic acid was able to promote aerobic oxidation of benzylamine. It should be, however, commented that while 1-pyrenecarboxylic acid as a model molecule of the active site contains a carboxylic acid at the periphery of a large π-conjugated system, the dangling bonds responsible for the ESR signals and responsible for molecular oxygen activation are not present in the model molecule.
As a continuation of the concept of an active site constituted by a carboxylic acid group and a radical, the use of GO with TEMPO as a cocatalyst has been reported for the selective oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-diformylfuran (DFF) with 100% HMF conversion with 99.6% selectivity to DFF using 80 wt% GO loading at 1 atm air pressure.79 In order to identify the nature of the active center in this catalytic system for selective HMF oxidation, a series of model catalysts were tested with various oxygen functional groups like hydroxyl, carbonyl, anhydride, and carboxyl groups with TEMPO as the cocatalyst. The activity of various carboxylic acids in the tested substrates increased in the order: acetic acid ∼ hexanoic acid < benzoic acid < 1-pyrene carboxylic acid ≪ GO. The data with model compounds demonstrate that the carboxylic acid–TEMPO system has always intrinsic activity for the aerobic oxidation of HMF. Furthermore, the large π-conjugation system appears to have a strong synergistic effect with the connected carboxylic acid group leading to an enhanced catalytic activity. The role of holes as active sites was also proposed. It was believed that these holes are created during the harsh oxidation process of graphite, and the hole defects are enlarged during thermal exfoliation by partial removal of oxygen groups in the form of CO and CO2.
These findings suggest that the active catalytic sites in GO are likely to be holes surrounded by conjugated domains with the edges of the hole defects terminated by carboxylic acid groups. It is interesting to comment that ESR spectroscopy can be used to address the location of the dangling bonds, either at internal holes or at the periphery. Thus ESR spectrum of ultrasonically exfoliated GO resulted in a sharper peak with a line width close to that expected for σ dangling bond spins in nanoporous carbon materials (∼0.1 mT), whereas thermally exfoliated GO showed ESR peaks that have a line width similar to that of localized spins originating from the edge of G (∼1 mT), indicating the existence of unpaired spins originating from the edges of G.64
In the GO/TEMPO system, the TEMPO acts as the active site losing an electron in the presence of oxygen and takes the oxoammonium form. The role of GO is to act as an electron transfer mediator from the TEMPO to oxygen (Scheme 15). Accordingly, it can be inferred that the enhanced catalytic activity of the thermally exfoliated GO in the GO/TEMPO system compared to a series of molecular analogues and ultrasonically exfoliated GO should reflect a more efficient electron relay from TEMPO to oxygen. Thus, it is proposed that the enhanced activity of the thermally exfoliated GO in the oxidation originates from the synergistic effect of carboxylic acid groups and unpaired electrons at the edges. Scheme 15 summarizes the proposed reaction pathway for molecular O2 activation in the presence of GO/TEMPO.
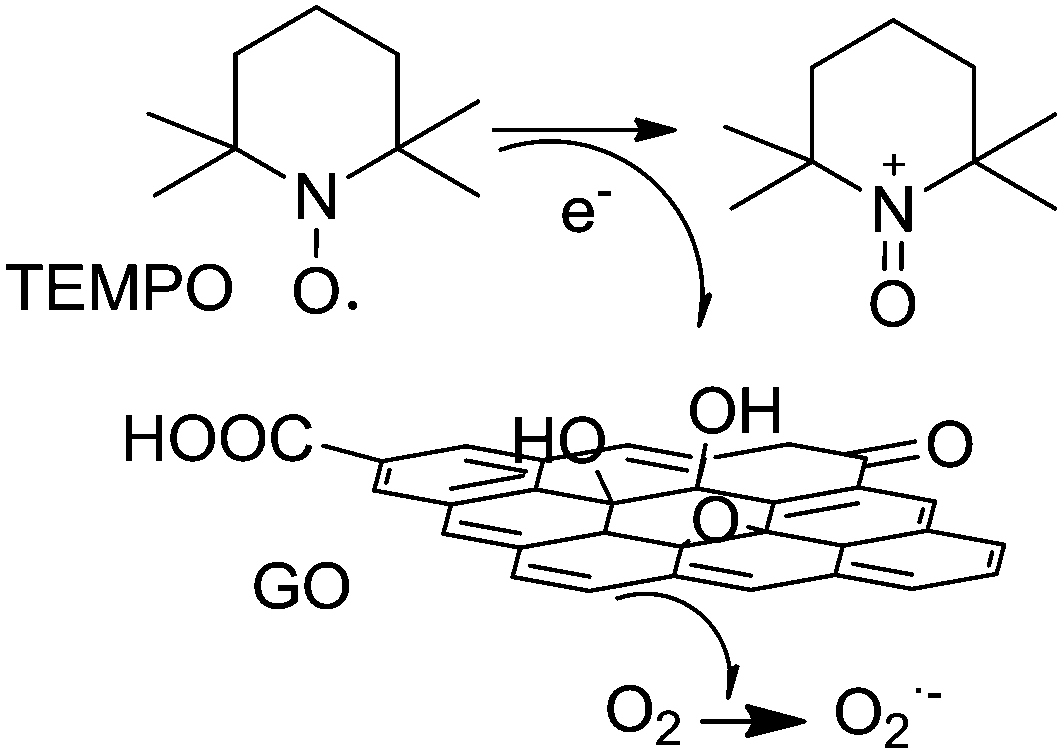 | ||
| Scheme 15 Proposed reaction pathway for molecular O2 activation using GO/TEMPO as the catalyst. Adapted from ref. 79. | ||
7. Edges
The importance of dangling bonds at the edges of Gs and rGO has been already commented in the previous examples of Schemes 14 and 15. Generally speaking, the catalytic activity of edges derives from the presence of dangling bonds and they are suitable to promote radical chain oxidations.Another possibility of edges acting as catalytic sites is to activate covalent bonds by interacting with them. One of these examples is the previously commented activation of SO52− through a non-radical pathway. Similarly, a combination of experimental and theoretical calculations has been used to rationalize the catalytic activity of G nanofibers for hydrogen release from sodium alanate (NaAlH4).80 It was found that G nanofibers considerably improve hydrogen release from NaAlH4, which is of significance for the implementation of NaAlH4 for environmentally friendly and effective hydrogen storage and release. DFT revealed that carbon atoms at sheet edges, regardless whether zigzag or armchair, can weaken Al–H bonds in sodium alanate. This effect is believed to be due to a combination of NaAlH4 destabilization and dissociation product stabilization (Scheme 16).
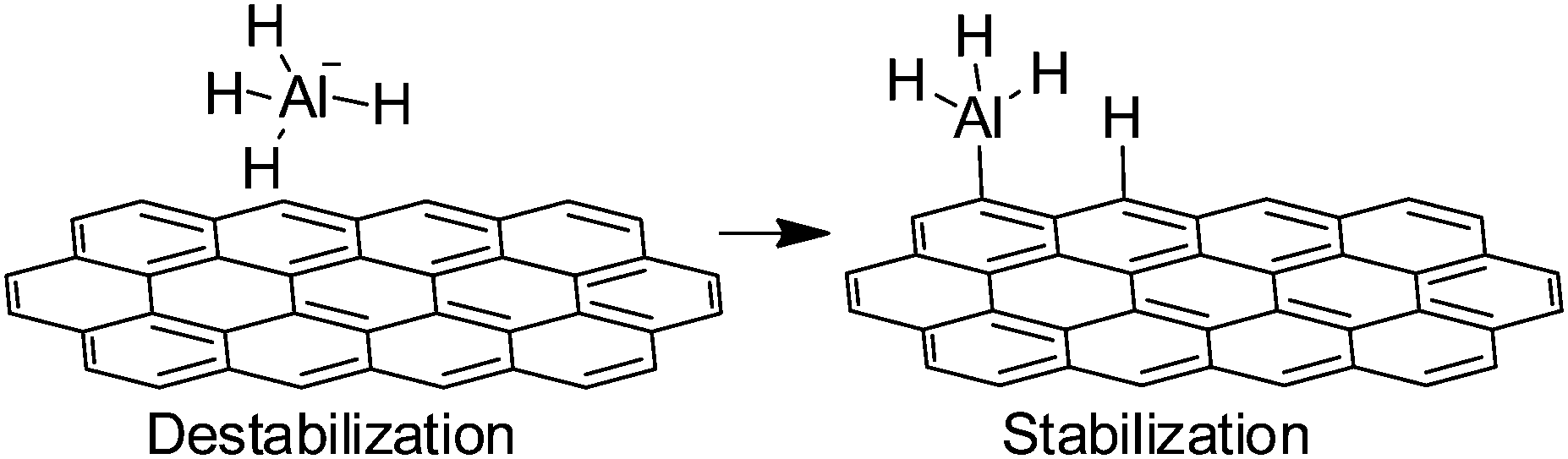 | ||
| Scheme 16 Proposal for the mechanism of NaAlH4 decomposition by activation at the edges of G. | ||
One interesting issue to be considered is that edges can have zigzag and armchair configurations and some theoretical models suggest that the interaction of these two edges with small molecules like H2 should be different. Therefore, it would be important to provide some experimental data to confirm the difference in catalytic activity of zigzag vs. armchair periphery.
8. Frustrated Lewis pairs
A series of G-based catalysts have been reported as metal-free carbocatalysts for hydrogenation of carbon–carbon multiple bonds. In one of these reports the selective hydrogenation of acetylene in the presence of a large excess of ethylene was reported.81 The selective conversion of acetylene can be promoted by a G catalyst obtained from pyrolysis of alginate at 900 °C followed by exfoliation of the graphitic carbon residue. The activity of this material increases with temperature, reaching an optimal value of 99% of acetylene conversion with only 21% conversion of ethylene at 120 °C. Hydrogenations are examples of reactions catalyzed by noble or transition metals and, therefore, the finding that G is a catalyst for this selective hydrogenation represents an important step towards replacement of metal catalysts. Under the optimized reaction conditions, GO was able to achieve 50.6% of acetylene conversion to ethylene with no measurable ethylene hydrogenation at 150 °C, while rGO reached complete acetylene conversion with only 5% ethylene hydrogenation at 130 °C. On the other hand, a series of heteroatom doped Gs exhibited significantly lower activity than G or rGO, a fact that indicates that dopant elements are not the main active sites for hydrogenation under the reaction conditions. However, the observed influence of doping on the selectivity of acetylene vs. ethylene constitutes a notable example of how the activity and selectivity of a carbocatalyst can be controlled. Again, one of the possible influences of doping is to favor charge-transfer interactions between doped G and the substrate. XP spectra indicated no changes on the used G and rGO catalysts with respect to the fresh samples, while in contrast, GO is converted into rGO. Furthermore, XPS analysis of the spent N-doped G showed the removal of nitrogen atom from the sample, indicating the occurrence of denitrogenation of N-doped G under the reaction conditions.To explain the catalytic activity of G and rGO for hydrogenation an analogy with organocatalysis was suggested. It has been well documented in the literature that there are discrete organic molecules comprising Lewis acid–base pairs rigidly separated at a short distance without undergoing neutralization (‘frustrated Lewis acid–base pairs’) that can activate hydrogen and act as metal-free hydrogenation catalysts.133–136 It was proposed that the activation of H2 on G takes place similarly to how these discrete organic molecules activate H2 in frustrated Lewis acid–base pairs resulting in the simultaneous formation of H+-like and H-like sites (Scheme 17). This claim was further supported by preadsorbing D2 on G, followed by evacuation and subsequent heating on a H2 stream, whereby the detection of H–D by mass spectrometry confirmed H2 activation by observation of isotopic H–D scrambling, presumably through the involvement of frustrated Lewis acid–base pairs. This was further supported by control experiments where acetylene conversion was found to depend on the presence of CO2 (increasing conversion by 33%) or NH3 (decreasing conversion by 9%) in the stream. These effects were reversible and the catalytic activity of G was recovered to the initial state in the absence of CO2 or NH3, when these gases were removed from the stream. This clearly demonstrates that under the reaction conditions, the interaction of acidic (CO2) and basic (NH3) molecules with the active sites of G results in changes in the catalytic activity. This interaction was not irreversible at the reaction temperature.
 | ||
| Scheme 17 Proposed mechanism for the activity of G materials as hydrogenation catalysts based on the presence of frustrated Lewis acid–base sites on G. Triangle and star represent the acid and basic sites, respectively, on the G sheet at a certain distance suitable to react with H2 molecules (as black spheres). These active hydrogen atoms on the G surface will be subsequently transferred to the alkene. Reproduced from ref. 81 with permission from Nature, copyright 2017. | ||
rGO and G obtained by pyrolysis of alginate at 900 °C under an inert atmosphere have also been reported as metal-free catalysts for the reduction of nitro to the corresponding amino compounds by hydrogen as a reducing agent.82 As previously commented for the selective hydrogenation of acetylene, doped Gs obtained by pyrolysis and exfoliation of suitable precursors including N and S-doped Gs were found to be unstable under hydrogenation conditions, undergoing complete or partial hydrogenative removal of the dopant element.
To put into context the catalytic activity of G for hydrogenation of nitro groups it is worth mentioning that supported Au NPs exhibited a preferential selectivity higher than 90% at high substrate conversions for the selective hydrogenation of nitro groups with respect to CC double bonds.137,138 In contrast to these observations, G was found much less selective, the selectivity depending on the nature of the substrate. Reduction of 4-nitrostyrene using G as a catalyst gives rise to 4-aminostyrene at low conversions, whereas at higher conversions the highest selectivity is observed for 4-aminoethylbenzene, corresponding to the hydrogenation of both the nitro and CC double bond.82 As commented earlier, the proposed active sites for hydrogen activation on G are frustrated Lewis acid–base pairs that are acid and basic centers separated by less than 4 Å, but without collapsing by mutual neutralization. This claim was further supported by performing two additional tests in which 4-nitrostyrene and 1-nitrocyclohexene were submitted to hydrogenation using G as the catalyst, with the addition of a minute amount of acetic acid or ethyl acetate. Addition of 134 μmol of acetic acid results in no hydrogenation of 4-nitrostyrene and decreased the conversion of 1-nitrocyclohexene from 86 to 9%. In contrast, addition of ethyl acetate in the same amount (134 μmol) had much less effect on the process, showing that the quenching effect of acetic acid has to do with its acidic character, leading to neutralization of basic centers that are involved as active sites.
Very recently, theoretical models have supported that a G bilayer and G ribbon having boron and nitrogen as dopant elements should be effective frustrated Lewis pair catalysts for activation of molecular hydrogen.83 Analysis of the structures along the reaction path suggested that the hydrogen molecule should be heterolytically dissociated and that the transition state structure should be close to that of the product. The activation is asynchronous as one of the hydrogen atoms interacts initially with the active site followed by the other one. According to this model, it would be very important to ensure that the synthesized doped carbon catalysts have separated boron and nitrogen dopants in different G sheets in order for them to act as frustrated Lewis pair catalysts. This model where B and N atoms are located in different G sheets can explain why experimental results using a codoped (B,N)G catalyst showed lower activity in the hydrogenation of acetylene. By separating the N and B atoms in different G layers, the combination of the two should have a clear synergistic effect. This already points to the discussion that Gs are more than molecules, but packed solid state structures, and that a catalytic site can extend over more than one layer, but involve the electronic environment of a number of separated units. In particular, simultaneous acid–base interactions, charge or bond frustration (due to the fact that graphitic stacking with about 0.34 nm is much longer than any chemical bond) or bipolar charge-transfer activation work much better “over the layers”.
9. Beyond chemical functionality as the active site: solid state effects in catalysis
Adsorption of substrates and reagents on G is generally claimed as one of the reasons for the high activity observed for the compared G catalysts.14 Particularly, π–π interactions of G with condensed polycyclic aromatic compounds are known to be very strong and intermolecular complexes have been reported between G and pyrenes, among other aromatic compounds.139In other cases, this preassociation of substrates with the G basal plane has been proposed as the cause of a synergy with other active sites resulting in high activity of G as metal-free catalysts.14
The above comments lead to the interesting discussion on how much heterogeneous carbocatalysts can really be described by the techniques developed for molecules and how much collective properties of an electronically coupled solid state material play a role. It is clear that substrate binding via charge transfer interactions, electron transfer, or so called π–π-interactions will depend on the electron density of the carbocatalyst, and the higher the difference in electron density, the stronger the binding and the activation/deactivation towards a specific chemical reaction. A remarkable clear case was reported for related graphitic carbon nitrides in Friedel–Crafts-catalysis.84 Here, electron transfer from benzene to G does the same role as a solid acid catalyst, and benzene could be successfully activated to be alkylated by methanol or substituted by cyano groups using urea. Similar arguments hold true for the catalyzed trimerization of nitriles85 which was also shown to be driven by charge transfer interactions of nitriles with the graphitic catalyst. It is clear that such “diffuse” effects due to charge transfer without the existence of a precise center do overlap with many of the sites described above.
Another useful concept from solid state physics with impact in catalysis is the “flat band potential” of electrons within an electronically coupled system, which corresponds to the HOMO in molecular orbital theory.140 The flat band potential is a collective property, i.e. the accessibility of electrons at a specific site depends on the electron acceptor and electron donor properties of the complete conjugated system.140–142 This is especially important for the heteroatom doping discussed below, as for instance nitrogen is known to make the HOMO more positive, i.e. more electron poor and thereby more “acidic”/electron accepting. Nitrogen-doped carbon made by appropriate techniques is thereby “noble”, has a metallic cluster, and cannot be oxidized in air even up to 700 °C:86 it is a quantum-mechanical effect that electron density cannot be discontinuous, but “bends” into any other medium. This means that even acidic groups bound to such carbocatalysts will sense the electron density, and these acid sites on doped G will behave in a way as molecular acids having electron pulling units: a more noble support will increase the acid strength. In that sense, controlling the electron density within a carbon framework can be regarded as the heterogeneous version of controlling electron density by ligand substitution or by modifying an organic environment:143 it is now in the hands of the chemist to fine-tune catalytic activity and selectivity.
In the case of graphitic materials, it has been reported that preadsorption can drive the selectivity of a reaction towards a given stereoisomer that is different from the one observed in homogeneous catalysis. Thus, recently, the Mukaiyama–Michael addition of 2-(trimethylsiloxy)furan (TMSOF) and β-nitrostyrene (Scheme 18) has been reported using graphite oxide/graphene oxide-based catalysts.87 Lowering the graphite oxide loading from 12 to 0.2 wt% results in good yields in all cases, while the selectivity in favor of the anti diastereoisomer increases continuously. For instance, 0.2 wt% graphite oxide exhibited 85% yield after 6 h at room temperature with a syn/anti ratio of 25/75. Furthermore, the use of 0.2 wt% exfoliated graphite oxide further improved the yield to 90% after 6 h at room temperature and also an unprecedented anti selectivity up to a diastereoisomeric ratio of 23/77 (syn/anti) was observed.
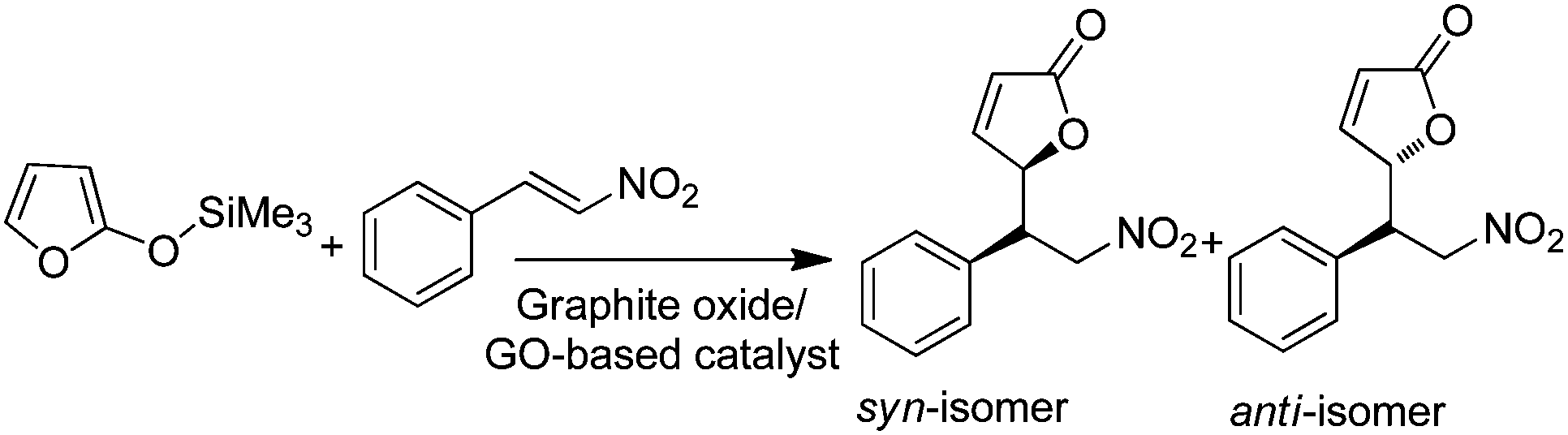 | ||
| Scheme 18 Mukaiyama–Michael addition between TMSOF and β-nitrostyrene with a graphite based catalyst. | ||
DFT calculations were performed to shed light on the origin of this diastereoselectivity. The G model for DFT calculations was constructed by considering 54 C atoms consisting of 19 fused six-membered rings. The geometry of the transition states shown in Fig. 7 indicates that the β-nitrostyrene molecule establishes a π-stacking interaction with the G layer. Due to this interaction, TMSOF can attack the activated β-nitrostyrene only from the top. The main difference between the two transition states is in the relative orientation of TMSOF, which according to the calculations establishes a series of dispersive interactions with the C atoms of the G in the anti-transition state (see the short distances in Fig. 7A), because the SiMe3 group protrudes away from the reactants. In contrast, in the syn transition state, the predicted geometry shows that the SiMe3 group is basically oriented above the –NO2 group of β-nitrostyrene, and only weak interactions can occur with the G layer, as indicated by a long distance of ≥5 Å (see Fig. 7B). On the other hand, in the case of the uncatalyzed reaction, a favorable electrostatic interaction between the positively charged SiMe3 group and the negatively charged nitro group is the main force that drives the formation of the syn product. In contrast to the results attained with graphitic materials in the reaction catalyzed by BINOL–phosphoric acid, the geometrical constraint imposed by the phosphoric group activating the addition matches better the geometry of the syn compared to the anti transition state.
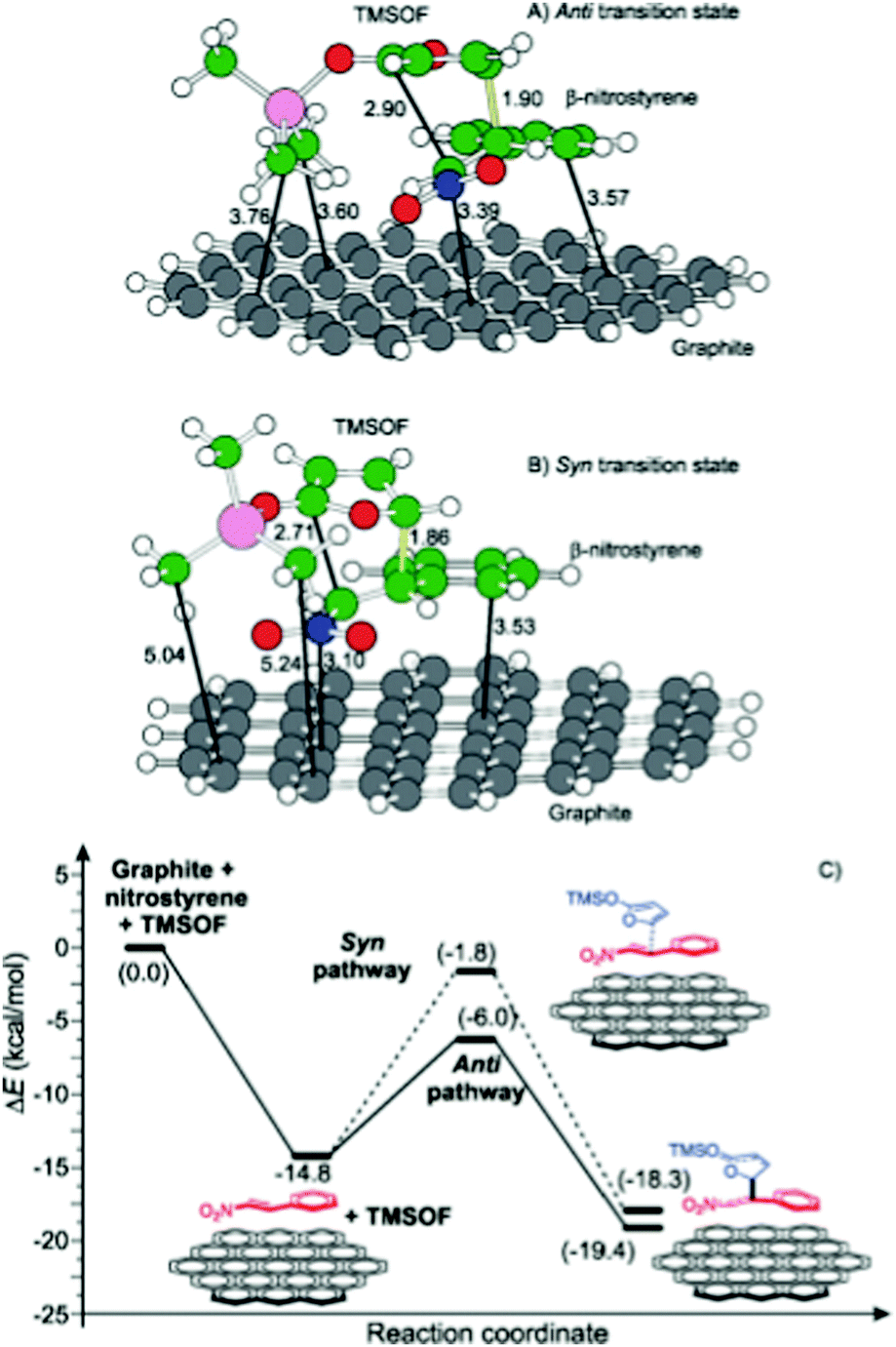 | ||
| Fig. 7 Geometry of the anti and syn transition states for the C–C bond-forming step in the presence of G (A and B). Distances are in angstroms. The C–C bond is colored yellow. (C) Energy profile up to the intermediate corresponding to the complete formation of the C–C bond. Reproduced from ref. 87 with permission from American Chemical Society, copyright 2017. | ||
10. The heterojunction and dyad concepts in catalysis by Gs
A related concept coming from solid state chemistry is the concept of heterojunction.144 This happens when two carbocatalysts of different character are in tight contact with each other, e.g. GO@G or N-doped G@G.145,146 As the electron density cannot discontinuously jump, surface charges build up which in the case of atomic or thin layers can extend over the whole sample. This means that by the right synthesis or processing, one and the same graphitic system can be brought to possess different electronegativities, due to this non-covalent neighbourhood effect. This concept lies behind the unusual selective oxidation of saturated hydrocarbons by dioxygen, using a C@C3N4 heterojunction.88 These secondary phases do not necessarily have to be large and they can even be well defined very electron rich or very electron poor aromatic molecules, such as HAT in Fig. 8. | ||
| Fig. 8 Chemical structures of HAT molecules. | ||
The classical molecular orbital description of this interaction is then, of course, like a charge transfer complex, where electron density is sucked into the π*-orbital of HAT, thus leaving a stronger oxidizing carbocatalyst as the support phase. If molecular entities are involved, this activation was termed “dyadic”.147 Notice that this mechanism of dioxygen activation is different from and complementary to that occurring when oxygen functional groups are responsible for the oxidation.
11. Dopant elements as active sites
As a follow up of the previous discussion on the influence of heteroatoms on the collective properties of G such as electron density and flat band potential, the following sections will summarize those reports in which heteroatoms have been proposed as discrete active sites in reactions promoted by G-based catalysts. It should be noted that although one may think of many other heteroatoms, either as dopants or codopants, the available experimental studies on the catalytic activity of these heteroatoms is at the moment still limited, particularly as metal-free catalysts and many other combinations are expected to be reported in the near future.11.1. N-Doping
Selective aerobic oxidation of benzyl alcohol has been achieved by engineering of active sites on G by N-doping through a high temperature (800–1000 °C) nitridation.89 Among the three types of nitrogen atoms present on the G lattice, pyridinic N, pyrrolic N, and graphitic N, the graphitic sp2 N atoms were established to be the catalytically active centers for this aerobic oxidation. The intensity of the N1s signal can be used to determine the N content that decreases as the nitridation temperature increases in the order NG-800 (4.16 at%) > NG-900 (3.48 at%) > NG-1000 (1.71 at%). The presence of N heteroatoms on G promotes benzyl alcohol oxidation with 100% selectivity of aldehyde by NG-900 that exhibited a ∼4-fold enhancement in activity upon increasing the reaction temperature from 313 to 343 K for benzyl, p-nitrobenzyl, p-fluorobenzyl, p-methylbenzyl and p-methoxybenzyl alcohols, thus, indicating the wide scope of N-doped G materials as catalysts for the aerobic oxidation of alcohols. However, the NG-900 catalyst was catalytically inactive for 1-phenylethanol presumably due to the steric encumbrance around the hydroxyl group. The spatial constraints on the reactive sites are apparently one of the important factors determining the value of the pre-exponential constant of the kinetic equation for aerobic alcohol oxidation over multilayer N-doped Gs as catalysts.NG–T and G samples prepared by high temperature exfoliation are believed to react with methanol to produce methoxyl radicals which was trapped by DMPO allowing recording in the sextet peaks corresponding to DMPO–CH3O˙ adducts.148 It is proposed that the formation of CH3O˙ is correlated closely with the presence of electron-deficient defects on the NG–T surface. It is interesting to note that NG-900 with the lowest density of defects (ID/IG 1.51) generates a larger amount of DMPO–CH3O˙ compared with NG-800 (ID/IG 1.58) and G (ID/IG 1.56), suggesting that not all types of N atoms are able to activate methanol to form the methoxyl radical. Addition of benzyl alcohol decreased the EPR line intensity of DMPO–CH3O˙, indicating that benzyl alcohol may be activated by the electron-deficient defects and the N dopant to form the C6H5CH2O˙ radical, which competes with the generation of CH3O˙. Apart from N atoms as catalytically active sites, the contribution of other sites like defects, including edges and carbon vacancies cannot be excluded.149 Adsorption of molecular oxygen over the graphitic N atoms to form a sp2 N–O2 adduct transition state seems to be the elementary step leading to O2 activation. The activated oxygen shows high chemical reactivity towards primary alcohols since it can abstract α-H atoms of alcohol to finally form water in several steps. No H2O2 was detected during the catalytic reaction.
In a series of studies, the effect of G-doping or co-doping with various heteroatoms such as N, B, S and P was also studied for PMS activation. For comparison, an undoped rGO sample prepared by thermal reduction of GO at 350 °C under nitrogen was also tested. It was observed that N-doped rGO resulted in the most active catalyst for PMS activation of the various materials tested including analogous undoped rGO, doped B-rGO and P-rGO, as well as co-doped NB–rGO or NP–rGO, although the dopant content varied from sample to sample.90 In the case of co-doped BN–rGO and PN–rGO samples their lower activity was rationalized considering that co-doping in the same benzene ring with an electron rich and an electron deficient atom could hardly break the inertness of G. The study suggests that N doping facilitates the electron transfer from the G sheet to PMS leading to the formation of SO4˙− and HO˙ as observed by EPR measurements. This is, of course, related to the more positive HOMO position of the N-doped carbon, resulting in a stronger oxidation power, as discussed above. Unfortunately, the catalytic activity of N-doped G decreases upon reuse and could only partly be recovered by thermal treatment. As already commented, this deactivation could be caused by self-oxidation of the catalyst throughout the course of PMS decomposition, and it would have been interesting to show that it is possible to regain the activity of the fresh catalyst by some reduction process.
In a related work, the importance of the preparation method of N doped G and the proposal of graphitic-N as active sites were again suggested.91 The most active N-doped G sample (NG-700), obtained by reaction of GO and melamine and subsequent pyrolysis at 700 °C, showed an enhancement of catalytic activity with respect to rGO-700 of 80 times (Fig. 9). In addition, NG-700 exhibits much higher activity than that of other samples prepared from GO and ammonium nitrate at 350 °C. It is important to note the high N content achieved using melamine as the precursor (NG-700, 9.68 at%; 54.41% pyridinic, 23.09% pyrrolic and 22.49% graphitic) compared to the use of ammonium nitrate (5.61 at%),150 annealing of GO with NH3 (3–5 at%)151 and CVD (5.0 at%)152 among others. Interestingly, the NG-700 sample is even more active than the benchmark heterogeneous Co3O4 catalyst and more active than SWCNTs, N doped CNT and G nanosheets (GNs) (Fig. 9).
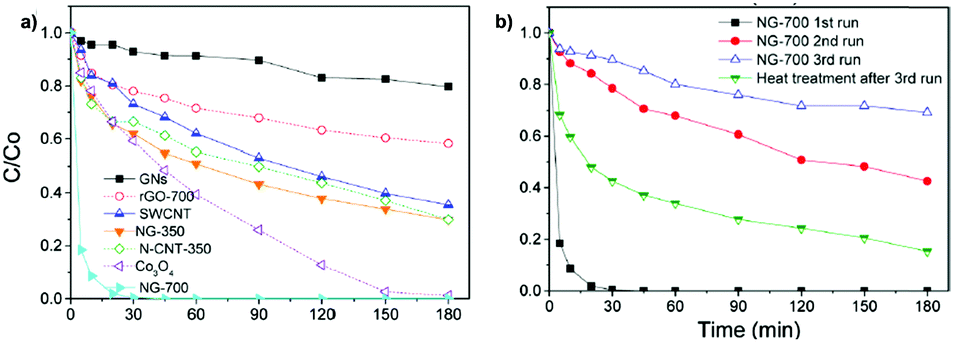 | ||
| Fig. 9 (a) Phenol degradation by PMS using different catalysts. (b) Stability and recyclability of NG-700. Reaction conditions: catalyst (0.1 g L−1), PMS (6.5 mM), phenol (20 mg L−1, 0.21 mM), 25 °C, pH not indicated. Reproduced from ref. 91 with permission from American Chemical Society, copyright 2017. | ||
In this study the proposed mechanism for PMS activation by N–G involved transfer of delocalized electrons from the zigzag edges of G to PMS.91 Importantly, theoretical calculations at the DFT level revealed that the presence of graphitic N enhances the adsorption of PMS and the electron transfer from G to PMS compared to pristine G. These calculations also predict that the activity of pyridinic or pyrrolic N should not be significant compared to that of graphitic. Furthermore, EPR measurements using DMPO as a spin trap as well as radical quenching experiments using ethanol and tert-butanol provide direct and indirect evidence in support of the generation of HO˙ and SO4˙− radicals. Unfortunately, as in all the previous G-based catalysts commented the NG-700 catalyst deactivates upon reuse and thermal annealing of the used catalyst to diminish the presence of oxygenated functional groups can hardly recover part of the catalytic activity. XPS showed that the used catalyst has a higher oxygen (13.74 vs.3.11 at%) content and lower N-doping (1.56 vs. 9.68 wt%) with respect to the fresh material giving hints that deactivation should originate from the introduction of oxygenated functional groups and N loss.
In line with the above commented results, N-doping of rGO (∼9 wt% N) improves the catalytic activity with respect to rGO also for catalytic wet air oxidation (∼160 °C at 7 bar O2) and catalytic ozonation (room temperature and atmospheric pressure).92 The N content was assigned to pyridinic, pyrrolic and quaternary N atoms based on XPS. Further studies, however, about the nature of the N-atoms that are acting as catalytic sites in these oxidation processes are still needed.
N-Doped G has also been reported as a metal-free catalyst for the reduction of 4-nitrophenol to 4-aminophenol by NaBH4.93 N–G exhibited a pseudo zero order kinetics, that was found different from pseudo first order reactions catalyzed by metal NPs.153 From characterization by in situ FT-IR spectroscopy and theoretical studies, it was suggested that 4-nitrophenol tends to interact with N–G via the O atom of the hydroxyl group. Also, evidence was obtained in favor to consider adsorption of 4-nitrophenol as the decisive elementary step responsible for the pseudo zero order kinetics. Theoretical studies, on the other hand, suggested that only the carbon atoms bonded to N atoms are activated and exhibit more favorable charge density than the rest of the C atoms on the G sheet. All the four kinds of N atoms (pyridinic, pyrrolic, graphitic and amine) present on G have good adsorption capability for 4-nitrophenol and the nitro groups of the adsorbed 4-nitrophenol become always activated by adsorption.
11.2. (S,N)-Doping
The presence of two or more dopant elements enlarges the potential to introduce active sites on G and the possibility of tuning the catalytic activity of this material.23,90 Continuing with activation of PMS, it has been observed that co-doping with S (0.69%) and N (8.15%) causes a synergism for this process (Fig. 10).94 The amount of residual O surely present on rGO, its variation in the different samples and its possible influence on the catalytic activity were though not considered. The low S content with respect to N was attributed to the larger difference in atomic diameter, 1.03 and 0.71 Å, respectively, with respect to the carbon atoms (0.75 Å) that should make S atom more difficult to accommodate in the G lattice. Raman spectroscopy showed that the NS–rGO sample exhibits the highest disorder and larger population of defective sites interrupting the graphitic sp2 carbon domains and disturbing the electronic density distribution of G sheets. In particular, the obtained ID/IG ratios were NS–rGO (1.30) > N–rGO (1.23) > rGO (1.20) > GO (1.18). Based on XPS, the N content mainly distributed in pyridine-like (39.8%), pyrrole-like (51.6) and quaternary or graphitic (8.6%). The sulfur content was assigned to C–S–C and C–SOX–C. DFT calculations of models for undoped G, S–G, N–G, S–N–G and S–S–N–G were carried out to obtain some insights into the effect of dopants on the electronic density of bonded carbon atoms on a G model (Fig. 10). In the case of S-doped G insignificant charge transfer was found, while N doping induces a positive charge density on the adjacent carbons (C1, C2 and C3). Simultaneous co-doping with N and S of G sheet further increases the charge density of the C2 carbon atom from 0.31 to 0.48 (Fig. 10). Thus, a good correlation between the disruption of electronic density of pristine G by the dopant elements and the experimental catalytic activity for PMS decomposition was observed. Interestingly, an experimental optimal S loading was observed for the catalytic activity of SN–rGO. The decrease of catalytic activity when an excessive S doping content is present was justified based on DFT calculations by observing the decrease of the charge density in the adjacent carbon atoms (C1, C2 and C3) when considering a S–S–N–G model with respect to a S–N–G model (Fig. 10). EPR measurements showed the formation of SO4˙− and HO˙ radicals by NS–rGO decomposition of PMS. Unfortunately, SN–rGO is deactivated upon use and its catalytic activity could not be recovered. Further studies are, therefore, still needed to increase the stability of codoped NS–rGO catalysts to allow reusability of the material. | ||
| Fig. 10 (a) Temporal profile of phenol disappearance by PMS activation promoted by a series of doped G catalysts as indicated in the plot; (b) model of co-doped G used for theoretical calculations; (c) electrostatic potential mapping from the charge density matrix for (a) undoped model G, (b) S–G, (c) N–G, (d) S–N–G and (e) S–S–N–G. Reproduced from ref. 94 with permission from Wiley, copyright 2017. | ||
11.3. (B,N)-Doping
Nitrogen and boron co-doped G (B,N–G) was prepared by a two-step method and its catalytic activity was tested for acetylene hydrochlorination.95 XPS and Raman spectroscopy strongly supported that N and B atoms covalently dope the G sheets. Under the same reaction conditions, the initial acetylene conversion for GO, B–G, N–G, and B,N–G catalysts follows the order B,N–G > N–G > B–G > GO. These results show the benefits of co-doping on G sheet increasing the catalytic activity. Thus, the estimated turnover frequency (TOF) values for N–G and B,N–G catalysts were 8.33 × 10−3 min−1 and 3.32 × 10−2 min−1, respectively. These TOF values are higher than that of the SiC@N–C composite reported in an earlier precedent (∼5.63 × 10−3 min−1),154 and also higher than that of the C3N4 material (1.42 × 10−2 min−1).155 To put these data into context, it is worth noting that the TOF values for the two most active catalysts, namely, Au/AC and Hg/AC, were 5.38 and 0.22 min−1, respectively.156 Hence, the catalytic performance of B,N–G is significant, but lower, compared to Hg/AC and and Au/AC catalysts.Both experimental and theoretical studies suggest that the carbon atoms bonded to N species are the active sites. The enhanced activity of B,N–G (10.96% N; 15. 47% B) may be due to its higher N content compared with N–G (5.47% N). Also, the presence of B atoms should change the electronic density of the N atom and, therefore, should influence HCl adsorption on the N active sites. It should be noted that adsorption of HCl on the G-catalyst is the rate-determining step of acetylene hydrochlorination. In the matrix of B,N–G, carbon atoms act as the adsorbing site for C2H2, and the nitrogen atom provides the catalytic active site for HCl adsorption. Therefore, the ensemble of carbon atoms bonded to N atoms and the N atoms appear to be the active sites. In contrast to the role of N atoms, the presence of oxygen atoms on N–G and B,N–G catalysts decreases the adsorption of HCl. DFT calculations anticipate that a higher HCl adsorption value should occur for B,N–G due to the combined interaction between pyridinic N and B atoms compared to N–G wherein all three types of N are present. In agreement with this theoretical prediction, it was measured that the presence of B increases dramatically the adsorption energy. TPD analysis showed that the binding strength of HCl with various G-materials increases in the following order of GO < N–G < B,N–G. One of the major drawbacks of the B,N–G catalyst is that acetylene conversion decreases from 94.89% to 61.88% within 4 h on stream, indicating its poor catalytic stability during acetylene hydrochlorination. This decay in activity was ascribed to the formation of coke around the active sites.
11.4. P-Doping
Besides N, P atoms in the corresponding P-doped Gs can act as catalytic centers in aerobic oxidations.90 In this context, a simple and efficient approach was reported to synthesize P-doped graphitic porous carbon (PGc) materials by heating phytic acid (see the structure in Scheme 19) in a domestic microwave oven (1100 W) for 40 s.96 Using this preparation methodology, it is possible to control coordination of P atoms. The catalytic activity of the resulting P-doped porous carbon was compared with that of a sample of N-doped G in the aerobic oxidation of benzyl alcohol. This P-doped carbon has a very unique structure consisting of a porous carbon monolith sandwiched by two highly wrinkled G-like sheets (Scheme 19). The wrinkled structure was proposed to result from P doping and the larger diameter of P atoms compared to C atoms which induce local geometrical stress in the carbon network around each P atom. XPS measurements indicated that the porous P-doped carbon contains 4.9 atomic% P in agreement with the presence of P in the carbon material. | ||
| Scheme 19 Synthesis of porous P-doped graphitic carbon from phytic acid. Reproduced from ref. 96 with permission from American Chemical Society, copyright 2017. | ||
This porous P-doped carbon resulted in benzyl alcohol conversion of 33.4% at 60 °C with >99% selectivity to benzaldehyde, which is approximately three times higher than the conversion achieved with a sample N-doped G under similar reaction conditions.89 Interestingly, the P-doped carbon was active in the aerobic oxidation of 1-phenylethanol reaching 46.5% conversion with complete selectivity to acetophenone at 80 °C, while in contrast, N-doped carbon catalysts were inactive.89 This may be due to the unique “protruding out” structure of P atoms on the G matrix, which is different than that of planar disposition of N atoms, relaxing the geometrical constraint around the active site N in comparison to P. It was shown that the formation of a large amount of H2O2 byproduct seems to be unavoidable when using noble metal based catalysts for selective oxidation of alcohols to aldehydes by molecular oxygen.157 However, using P-doped G as the catalyst no detectable H2O2 is generated which is another specific feature of this P-doped material compared to transition metal catalysts.158,159 Since P has the same number of valence electrons as N, it is believed that the mechanism of O2 activation could be similar to that of N-doped G, involving the formation of peroxo-like species on the heteroatom.160 Both theoretical and experimental studies have demonstrated that P-doped G activates molecular oxygen in an electrochemical oxygen reduction reaction and this electrochemical oxidation is proposed to be related to the oxygen activation occurring in benzyl alcohol oxidation.161–163 An alcohol conversion of 18% after 48 h of reaction at 80 °C was observed when the reaction is carried out under N2. This conversion value is slightly lower than when the reaction is carried out in the presence of O2 (23% conversion). This oxidation of benzyl alcohol under a N2 atmosphere proves that some functional groups on P-doped graphitic carbon can directly promote alcohol oxidation in a stoichiometric way without the requirement of external O2. This result is compatible with the proposal that O2 does not directly react with the substrate, but however it is needed to regenerate the functional groups/active site present on the P-doped porous carbon involved in the catalytic oxidation. This mechanism wherein the active sites on P-doped carbon oxidize the benzylic alcohol is completely different from that proposed for GO, N-doped, and N, B co-doped carbon catalysts.68,89,164,165 Addition of butylated hydroxytoluene (BHT), as a free radical quencher, to a reaction mixture of benzyl alcohol in the presence of P-doped carbon as a catalyst under the same conditions does not alter conversion and selectivity, supporting that the reaction using P-doped carbon as the catalyst does not proceed via a free radical mechanism. This result is again contrasting with the GO catalyzed aerobic oxidation of alcohols, wherein a significant decrease from 24 to 5% was observed in the conversion upon addition of BHT.68 XPS analysis indicated that P-doped porous carbon contains mainly PO, instead of CO, whereas C–O–C groups are below the detection limit, and the majority of –OH present on it are directly bonded to P with abundant P–OH functionalities. Further, the atomic O/P ratio of ∼4 measured by XPS for P-doped carbon suggests that some P atoms possibly connect with two or more oxygen containing functionalities, such as OH groups. A series of control experiments using PGc materials with a similar oxygen content but different P–OH populations suggest that the P–OH functional groups are likely to play an important role in the oxidation promoted by P-doped carbon. On the other hand, the benzyl alcohol oxidation reaction was also performed in the presence of molecules containing PO and P–OH functional groups, such as phytic acid and phosphorous acid, observing no activity. These data suggest the importance of the graphitic regions on the P-doped material establishing π–π interactions with aromatic substrates. Based on these data, a possible reaction mechanism is proposed in Scheme 20. In the first step, condensation between the alcohol and PO moieties on P-doped carbon would take place, and an ether intermediate would be formed. The condensation is likely facilitated by the interaction of the alcohol with the surface by π–π interactions with the graphitic domains and by hydrogen bonding with the polar P–OH groups. In the second step of the reaction, a rate determining H transfer would take place, possibly through a cyclic transition state releasing simultaneously the aromatic aldehyde and a water molecule. Then, the generated P(III) groups on P-doped carbon should react with molecular oxygen to regenerate the P(V) centers for further reaction, thus completing the catalytic cycle (Scheme 20). This mechanistic proposal was further supported by analyzing the spent P-doped catalyst by FT-IR spectroscopy. The peaks at 1166 cm−1 (PO), 1035 cm−1 (P–O/CO), and 900 cm−1 (shoulder, P–OH) have similar intensities as the fresh PGc catalyst. These unchanged intensities were taken as an indication that the catalytic sites are largely regenerated during the reaction in the presence of O2.
 | ||
| Scheme 20 Proposed mechanism for aerobic oxidation of benzyl alcohol by P-doped carbon. Reproduced from ref. 96 with permission from American Chemical Society, copyright 2017. | ||
11.5 Dopant elements in electrochemical reactions
Due to the electrical conductivity G and related materials they have been widely used in the preparation of electrodes either in the combination of metals or even in the absence of metals.166–169 In most of the electrochemical reactions the key issue is to perform the transformation with electrochemical potentials as close as the thermodynamic value. However this is not possible and in order to have adequate reaction rates and current densities it is necessary to work under an overpotential to increase the kinetics of the process. It is known that this overpotential depends on the nature of the electrode and this is generally termed an electrocatalyst when the overpotential is reasonably low. Doped Gs are among the most widely reported metal-free electrocatalysts and it is known that this electrocatalytic activity is related to the presence of dopant elements.95 Herein we will limit to comment three of the currently most studied electrochemical reactions that are of large importance in the development of renewable energy sources and, particularly, in the conversion of power to chemicals (hydrogen evolution reaction or HER and oxygen evolution reaction or OER) and on the operation of fuel cells that corresponds to the reverse case, i.e. chemicals to power (oxygen reduction reaction or ORR) (eqn (6)–(9)).95,170–174 In these three processes metals either as bulk or in the form of nanoparticles on various supports are the preferred electrocatalysts, platinum being the most efficient. However, for the sake of sustainability and affordability there is much current interest in replacing precious metals by carbon-based electrocatalysts, since this could facilitate the introduction of electrolysis and fuel cells on a large scale.| ORR O2 + 4H+ + 4e− → 2H2O | (6) |
| O2 + 2H+ + 2e− → H2O2 | (7) |
| OER 2H2O → O2 + 4H+ + 4e− | (8) |
| HER 4H+ + 4e− → 2H2 | (9) |
Nitrogen doping on G has led to one of the most efficient metal-free ORR electrocatalysts and there has been numerous studies describing the preparation of N-doped G in various manners for their use as ORR electrocatalysts.95,172,177 Of the various types of the N atoms experimental data indicate that graphitic N atoms act as the most efficient type although there has been some controversy about the influence of pyridinic N atoms.97,98,172 Theoretical studies at the DFT level have provided much insight into the reaction mechanism and the nature of the active site in this process.97,98 These calculations indicate that the carbon atoms bonded to graphitic N are electron deficient and act as active sites by increasing oxygen adsorption and favoring its reduction by electron transfer. In the catalytic cycle graphitic N can be interconverted into pyridinic N atoms and for this reason some of the experimental data have failed to provide conclusive evidence in support of the nature of the active site.97 Theoretical calculations also indicate that pyridinic N atoms do exert an influence on the selectivity of the four electrons versus two electron reductions. In fact ORR can lead to the formation of H2O and also to H2O2 and it seems that N atoms also depending on its nature drive the process toward H2O2. Scheme 21 illustrates one of the reaction mechanisms that have been proposed.97
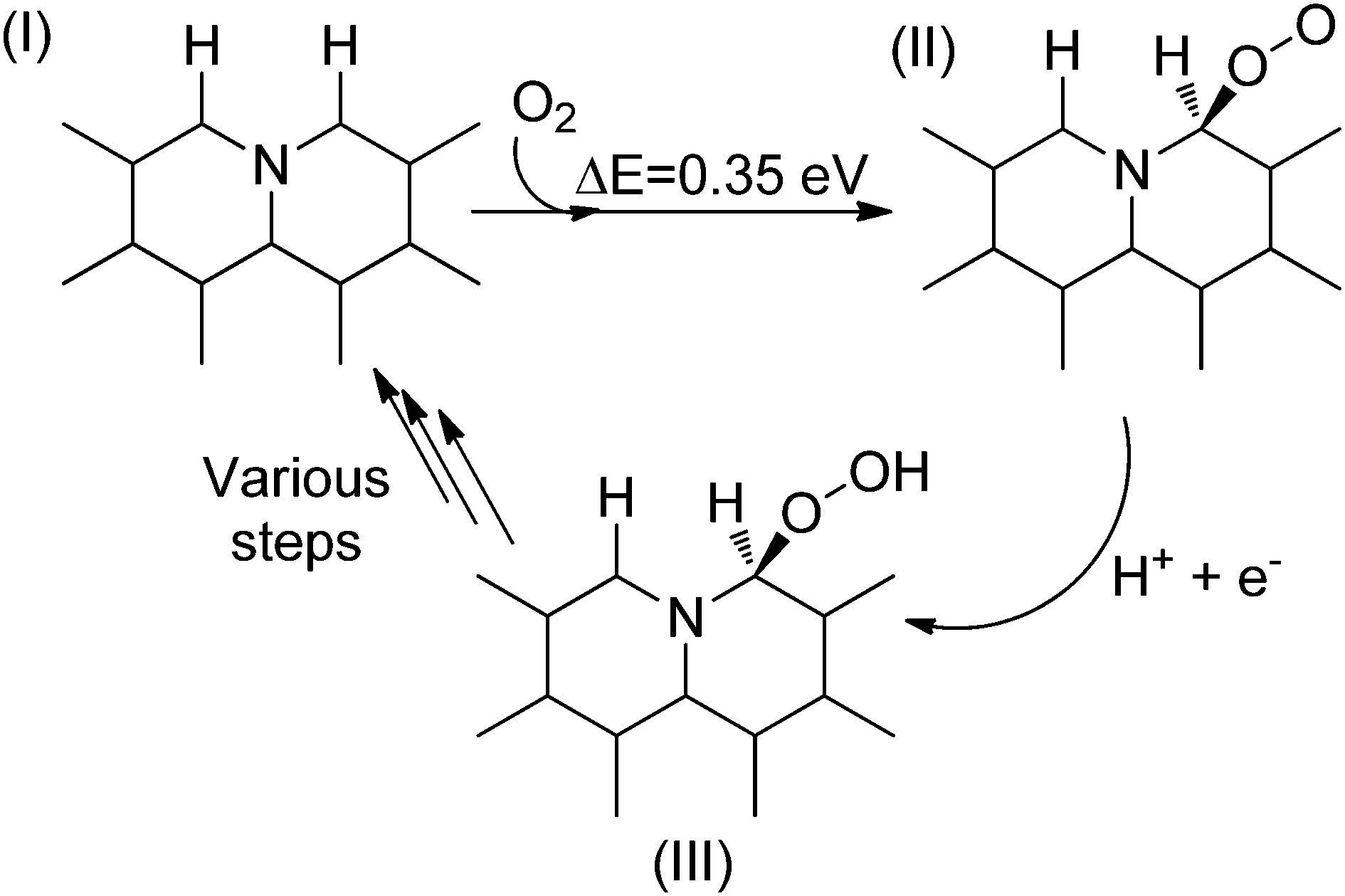 | ||
| Scheme 21 Proposed ORR catalytic cycle considering graphitic nitrogen as the initial active site. | ||
Besides N, B and other heteroatoms can also introduce electrocatalytic activity on G for ORR.172,178–181 Similarly to the cases that have been commented for N it seems that the electrocatalytic activity derives from the influence of the heteroatom (mostly graphitic) on the electron density of the adjacent carbon atoms. By modulating this electron density and also the spin density, oxygen adsorption is favored around these sites and as a result electron transfer is favored.
The present review is limited to the use of Gs and related materials as metal-free catalysts. In many cases it is considered that since the major elements of Gs are non-metals the possible metal impurities that could be present do not play any role. However, it has been frequently observed that impurities of highly active transition metals even at the ppm level can contribute to a significant extent to the observed catalytic activity.182,183 This is the case of ORR where experimental data have been provided in support that metal impurities, even if not intentionally added to the sample, do play a major role in the electrocatalytic activity of Gs and carbon-based catalysts.184 Chemical analyses have shown that iron, copper, and manganese among other metals are present in graphite samples as major metal impurities and when G is obtained from graphite these metal impurities can remain present in the carbon material, either as charge balancing cations or even as small metal nanoparticles.130,185 Even more, most of the studies on the electrocatalytic activity of doped Gs use GO as the starting material, obtained from graphite by using a large excess of permanganate. In this case manganese can be present in the GO sample at the level of hundreds of ppms accompanied by other metal impurities as determined by exhaustive chemical analysis. In other cases the G sample can have other origins but the absence of metals should not be taken for granted without a detailed chemical analysis because the metals can be present either in the precursors of the Gs or during manipulation of the sample. This problem of metal impurities as the major contributors of the catalytic activity, even though not intentionally added to the sample, is a current issue in catalysis and has led to controversies both in homogeneous and heterogeneous catalysis.185 For instance it has been found that during the synthesis of zeolites by extended hydrothermal treatments at temperatures about 150 °C, iron can be incorporated into the structure coming from the stainless steel used in the autoclaves.186 In another example, the catalytic activity initially attributed to iron for C–C cross-coupling was later shown to be due to copper impurities present in iron salts with low purity. Similarly, for ORR catalytic activity Pumera and co-workers have shown in a series of papers that metal impurities can be responsible for a large part of the observed activity.184,187–189 By careful purification of the G sample by thermal treatment with chlorine resulting in the formation of volatile metal chlorides as confirmed by chemical analysis it was found that most of the ORR electrocatalytic activity of Gs is lost.187 Accordingly, caution should be taken when attributing the catalytic activity of Gs to certain sites. However, as commented earlier, theory and experimental data suggest at the moment that doping, particularly with graphitic N atoms, can promote ORR, although a certain contribution of metals could overlap with the observed activity.
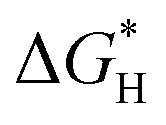 close to zero since H2 desorption will then be favorable. Accordingly, the performance of a series of electrocatalysts should exhibit a volcano shape with respect to
close to zero since H2 desorption will then be favorable. Accordingly, the performance of a series of electrocatalysts should exhibit a volcano shape with respect to 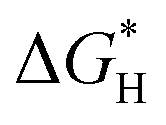 , decreasing the reaction rate as adsorption of a hydrogen atom becomes more favorable or disfavorable. This descriptor correlates well with the experimental data of the electrocatalytic activity of a series of doped Gs. It was found that co-doping with two elements with different electronegativities and, in particular B–N, S–N and P–N, results in materials with high electrocatalytic activity. Specifically, for the case of N and P co-doping Fig. 11 shows the current density as a function of the potential for single N or P doped Gs compared with the performance of the co-doped material showing that the latter exhibits much higher electrocatalytic activity.99 This electrocatalytic activity of N,P doped G corresponds, according to DFT calculations for p-type pyridinic N, to a
, decreasing the reaction rate as adsorption of a hydrogen atom becomes more favorable or disfavorable. This descriptor correlates well with the experimental data of the electrocatalytic activity of a series of doped Gs. It was found that co-doping with two elements with different electronegativities and, in particular B–N, S–N and P–N, results in materials with high electrocatalytic activity. Specifically, for the case of N and P co-doping Fig. 11 shows the current density as a function of the potential for single N or P doped Gs compared with the performance of the co-doped material showing that the latter exhibits much higher electrocatalytic activity.99 This electrocatalytic activity of N,P doped G corresponds, according to DFT calculations for p-type pyridinic N, to a  of 0.08 eV while the calculated
of 0.08 eV while the calculated 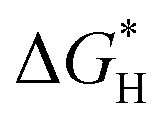 for pyridinic N-doped G is about 0.7 eV and that of P–G is about 0.2 eV. It is probably also that this effect occurs by polarization of the adjacent carbon atoms.
for pyridinic N-doped G is about 0.7 eV and that of P–G is about 0.2 eV. It is probably also that this effect occurs by polarization of the adjacent carbon atoms.
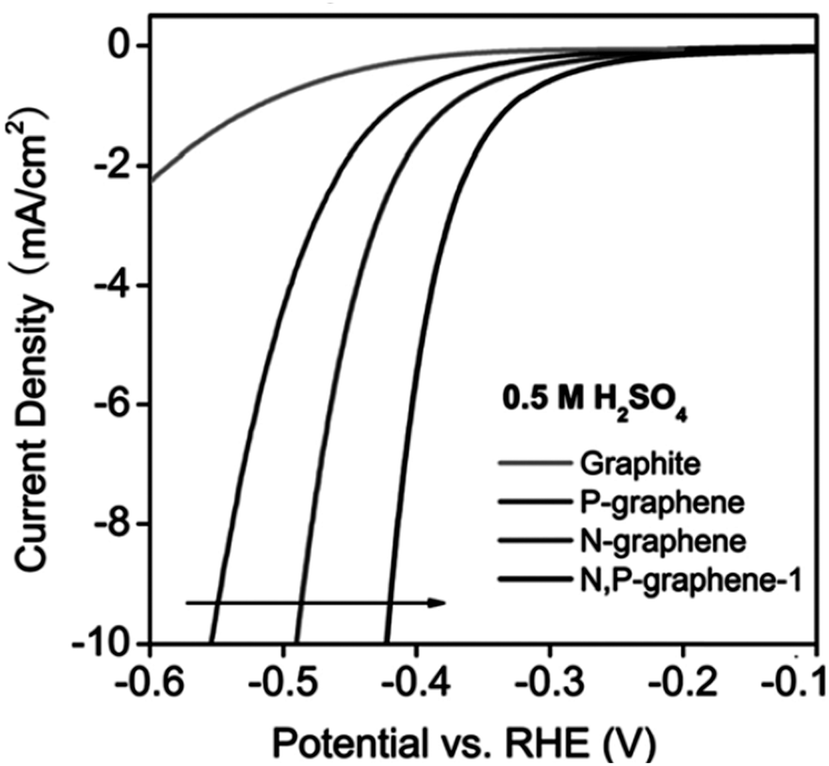 | ||
| Fig. 11 HER polarization curves of P and/or N doped graphene electrocatalysts. Reproduced from ref. 99 with permission from American Chemical Society, copyright 2017. | ||
Co-doping and the use of 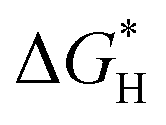 as a descriptor of the electrocatalytic activity also apply to the case of other carbon nanoforms and even to the combination of Gs with other materials. Thus, for instance, the combination of N-doped G with g-C3N4 gives a HER electrocatalyst with a performance closer to that of platinum. It seems that hydrogen adsorption on g-C3N4 is too strong and in N–G too weak and the combination of the two components in a material leads to an average
as a descriptor of the electrocatalytic activity also apply to the case of other carbon nanoforms and even to the combination of Gs with other materials. Thus, for instance, the combination of N-doped G with g-C3N4 gives a HER electrocatalyst with a performance closer to that of platinum. It seems that hydrogen adsorption on g-C3N4 is too strong and in N–G too weak and the combination of the two components in a material leads to an average 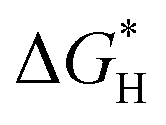 that is close to zero and this results in a high electrocatalytic activity.
that is close to zero and this results in a high electrocatalytic activity.
12. Summary of the current state of the art and future developments
There is meanwhile broad evidence that Gs are metal-free catalysts for a large diversity of different organic reactions, well beyond radical chain aerobic oxidations, and including some reaction types in which transition metals were considered paradigmatic catalysts, such as the Fenton oxidation, hydrogenations, and C–C couplings. Considering the research interest that is currently attracted by carbon catalysis, it is expected that many other reaction types will be described in the near future as being catalyzed by Gs in the absence of metals. The target is to establish the scope and limitations of Gs as carbocatalysts.From the description of reports in the area, it is clear that the most powerful methodology to assess the nature of the active sites is the combination of theoretical calculations on models with instrumental in situ techniques to follow the reaction mechanism and the course of the reaction, while the classic catalytic methodology is based on comparison of the catalytic activity of different samples with well characterized composition and properties. The current approaches tend to attribute the catalytic activity to discrete substructures present on G such as certain functional groups and vacancies, while the importance of the collective properties of Gs as materials is usually underestimated. This is well understandable, taking into account the difficulty to address the issue of the impact of the electron density of G in the catalysis using organic models or even models in calculations. The unique property of Gs is to offer a joint, delocalized electron pool which can be modified even by distant atoms. These collective properties offer certainly additional tools that have just started to be explored in catalysis.
Besides delimiting the reactions that can be catalyzed by Gs, another aspect of interest is to prepare more active materials and to establish valid comparison of the catalytic activity of Gs for these reactions with that of benchmark metal catalysts. In this area, the knowledge of the active sites should serve as a powerful tool to direct carbon synthesis. The long term goal should be to convincingly show that the graphenes are the catalysts of choice for certain industrially important reactions. In this regard, rather than petrochemistry or other industrial processes that have well established catalysts and technologies, Gs offer promising potential in novel areas currently under development and, particularly, for biomass transformation and environmental remediation. In these areas, Gs offer as advantages their compatibility with biomass feedstocks or polluted wastes and the fact that Gs can be considered renewable catalysts that do not need to be recovered after the reaction and can be processed with the final residue, e.g. simply burned. Since novel reactions are seeking suitable catalysts to be commercially implemented, there is in these cases a distinct possibility of competing with the use of transition and precious metals to be used as catalysts at the industrial scale. Certainly, implementation of Gs as catalysts for large scale processes will serve to consolidate these materials by showing that they can be the most suitable option for at least some processes.
Acknowledgements
Financial support by the Spanish Ministry of Economy and Competitiveness (CTQ 2015-69153-CO2-1, CTQ2014-53292-R, Severo Ochoa, Grapas), Generalitat Valenciana (Prometeo 2013-014) and the European Community by fondos FEDER (CTQ2014-53292-R) is gratefully acknowledged. MA wants to thank the German Excellence Cluster Unicat for continued support. SN thanks financial support from the Fundación Ramón Areces (XVIII Concurso Nacional para la Adjudicación de Ayudas a la Investigación en Ciencias de la Vida y de la Materia, 2016). ADM thanks the University Grants Commission (UGC), New Delhi, for the award of an Assistant Professorship under its Faculty Recharge Programme. ADM also thanks the Department of Science and Technology, India, for the financial support through Extra Mural Research Funding (EMR/2016/006500). Open Access funding provided by the Max Planck Society.References
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Coord. Chem. Rev., 2016, 312, 99–148 CrossRef CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS PubMed.
- L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- E. A. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768–2813 CrossRef CAS PubMed.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 12663–12676 CrossRef CAS PubMed.
- P. Manickam-Periyaraman, S. M. Espinosa, J. C. Espinosa, S. Subramanian, M. Álvaro and H. García, J. Environ. Chem. Eng., 2016, 4, 4485–4493 CrossRef CAS.
- S. Navalón and H. García, Nanomaterials, 2016, 6, 123–125 CrossRef PubMed.
- J. C. Espinosa, S. Navalón, M. Álvaro and H. García, Catal. Sci. Technol., 2016, 6, 7077–7085 CAS.
- J. C. Espinosa, S. Navalon, M. Álvaro and H. García, ChemCatChem, 2015, 7, 2682–2688 CrossRef CAS.
- S. Navalon, D. Sempere, M. Alvaro and H. Garcia, ACS Appl. Mater. Interfaces, 2013, 5, 7160–7169 CAS.
- D. Sempere, S. Navalon, M. Dančíková, M. Alvaro and H. Garcia, Appl. Catal., B, 2013, 142–143, 259–267 CrossRef CAS.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed.
- Y. Zhai, Z. Zhu and S. Dong, ChemCatChem, 2015, 7, 2806–2815 CrossRef CAS.
- A. Primo, V. Parvulescu and H. Garcia, J. Phys. Chem. Lett., 2017, 8, 264–278 CrossRef CAS PubMed.
- K. Sakthivel, W. Notz, T. Bui and C. F. Barbas, J. Am. Chem. Soc., 2001, 123, 5260–5267 CrossRef CAS PubMed.
- P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289–296 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Mueller, R. Schloegl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- D. S. Su and G. Centi, J. Energy Chem., 2013, 22, 151–173 CrossRef CAS.
- J. Albero and H. Garcia, J. Mol. Catal. A: Chem., 2015, 408, 296–309 CrossRef CAS.
- A. Bhatnagar, W. Hogland, M. Marques and M. Sillanpaa, Chem. Eng. J., 2013, 219, 499–511 CrossRef CAS.
- A. G. Goncalves, J. F. Figueiredo, J. J. M. Orfao and M. F. R. Pereira, Carbon, 2010, 48, 4369–4381 CrossRef CAS.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed.
- J. F. Blandez, S. Navalon, M. Alvaro and H. Garcia, ChemCatChem, 2015, 7, 3020–3026 CrossRef CAS.
- K. Bao, F. Li, H. Liu, Z. Wang, Q. Shen, J. Wang and W. Zhang, Sci. Rep., 2015, 5, 10360–10366 CrossRef PubMed.
- P. C. C. Faria, J. J. M. Órfão and M. F. R. Pereira, Appl. Catal., B, 2008, 79, 237–243 CrossRef CAS.
- E. C. Larsen and J. H. Walton, J. Phys. Chem., 1940, 44, 70–85 CrossRef CAS.
- J.-K. Jeon, H. Kim, Y.-K. Park, C. H. F. Peden and D. H. Kim, Chem. Eng. J., 2011, 174, 242–248 CrossRef CAS.
- K. Tsuji and I. Shiraishi, Fuel, 1997, 76, 549–553 CrossRef CAS.
- C.-H. Yoon, J. Korean Soc. Envrion. Eng., 2010, 32, 201–208 Search PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2013, 42, 3222–3233 RSC.
- A. Ciesielski and P. Samorì, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288–5301 RSC.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- D. K. James and J. M. Tour, Acc. Chem. Res., 2013, 46, 2307–2318 CrossRef CAS PubMed.
- X. Jia, J. Campos-Delgado, M. Terrones, V. Meunier and M. S. Dresselhaus, Nanoscale, 2011, 3, 86–95 RSC.
- T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2012, 57, 1061–1105 CrossRef CAS.
- K. P. Loh, Q. Bao, P. K. Ang and J. Yang, J. Mater. Chem., 2010, 20, 2277–2289 RSC.
- J. Malig, N. Jux and D. M. Guldi, Acc. Chem. Res., 2013, 46, 53–64 CrossRef CAS PubMed.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS PubMed.
- L. Rodríguez-Pérez, M. A. Herranz and N. Martín, Chem. Commun., 2013, 49, 3721–3735 RSC.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CrossRef CAS.
- C. Soldano, A. Mahmood and E. Dujardin, Carbon, 2010, 48, 2127–2150 CrossRef CAS.
- J. P. Collman, Acc. Chem. Res., 1968, 1, 136–143 CrossRef CAS.
- J. Halpern, Acc. Chem. Res., 1970, 3, 386–392 CrossRef CAS.
- B. F. Machado and P. Serp, Catal. Sci. Technol., 2012, 2, 54–75 CAS.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- I. S. Flyagina, K. J. Hughes, D. C. Mielczarek, D. B. Ingham and M. Pourkashanian, Fuel Cells, 2016, 16, 568–576 CrossRef CAS.
- O. Vaughan, Nat. Nanotechnol., 2016 DOI:10.1038/nnano.2016.36.
- D. W. Boukhvalov, RSC Adv., 2013, 3, 7150–7159 RSC.
- D. W. Boukhvalov, D. R. Dreyer, C. W. Bielawski and Y.-W. Son, ChemCatChem, 2012, 4, 1844–1849 CrossRef CAS.
- J. C. Espinosa, S. Navalón, A. Primo, M. Moral, J. Fernández Sanz, M. Álvaro and H. García, Chem. – Eur. J., 2015, 21, 11966–11971 CrossRef CAS PubMed.
- X. Duan, H. Sun, Z. Ao, L. Zhou, G. Wang and S. Wang, Carbon, 2016, 107, 371–378 CrossRef CAS.
- W. Qi, W. Liu, B. Zhang, X. Gu, X. Guo and D. Su, Angew. Chem., Int. Ed., 2013, 14224–14228 CrossRef CAS PubMed.
- S. Wu, G. Wen, X. Liu, B. Zhong and D. S. Su, ChemCatChem, 2014, 6, 1558–1561 CrossRef CAS.
- S. Wu, G. Wen, J. Wang, J. Rong, B. Zong, R. Schlogl and D. S. Su, Catal. Sci. Technol., 2014, 4, 4183–4187 CAS.
- J. C. Espinosa, S. Navalón, M. Álvaro and H. García, ChemCatChem, 2016, 8, 2642–2648 CrossRef CAS.
- C. Su, M. Acik, K. Takai, J. Lu, S. J. Hao, Y. Zheng, P. Wu, Q. Bao, T. Enoki, Y. J. Chabal and K. P. Loh, Nat. Commun., 2012, 3, 1298–1306 CrossRef PubMed.
- X.-K. Kong, Q.-W. Chen and Z.-Y. Lun, J. Mater. Chem. A, 2014, 2, 610–613 CAS.
- A. Dhakshinamoorthy, M. Alvaro, P. Concepción, V. Fornés and H. Garcia, Chem. Commun., 2012, 48, 5443–5445 RSC.
- A. Dhakshinamoorthy, M. Alvaro, M. Puche, V. Fornes and H. Garcia, ChemCatChem, 2012, 4, 2026–2030 CrossRef CAS.
- D. R. Dreyer, H. P. Jia and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 6813–6816 CAS.
- W. Cen, M. Hou, J. Liu, S. Yuan, Y. Liu and Y. Chu, RSC Adv., 2015, 5, 22802–22810 RSC.
- Y. Wang, Y. Xie, H. Sun, J. Xiao, H. Cao and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 9710–9720 CAS.
- X. Duan, Z. Ao, L. Zhou, H. Sun, G. Wang and S. Wang, Appl. Catal., B, 2016, 188, 98–105 CrossRef CAS.
- X. Duan, H. Sun, J. Kang, Y. Wang, S. Indrawirawan and S. Wang, ACS Catal., 2015, 5, 4629–4636 CrossRef CAS.
- Y. Gao, P. Tang, H. Zhou, W. Zhang, H. Yang, N. Yan, G. Hu, D. Mei, J. Wang and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 3124–3128 CrossRef CAS PubMed.
- F. Hu, M. Patel, F. Luo, C. Flach, R. Mendelsohn, E. Garfunkel, H. He and M. Szostak, J. Am. Chem. Soc., 2015, 137, 14473–14480 CrossRef CAS PubMed.
- N. Kausar, I. Roy, D. Chattopadhyay and A. R. Das, RSC Adv., 2016, 6, 22320–22330 RSC.
- H. Wang, Q. Kong, Y. Wang, T. Deng, C. Chen, X. Hou and Y. Zhu, ChemCatChem, 2014, 6, 728–732 CrossRef CAS.
- R. Wang, Z. Wu, Z. Qin, C. Chen, H. Zhu, J. Wu, G. Chen, W. Fan and J. Wang, Catal. Sci. Technol., 2016, 6, 993–997 CAS.
- M. R. Acocella, M. Mauro and G. Guerra, ChemSusChem, 2014, 7, 3279–3283 CrossRef CAS PubMed.
- G. Lv, H. Wang, Y. Yang, T. Deng, C. Chen, Y. Zhu and X. Hou, ACS Catal., 2015, 5, 5636–5646 CrossRef CAS.
- Z. Qian, M. Sterlin Leo Hudson, H. Raghubanshi, R. H. Scheicher, B. Pathak, C. M. Araujo, A. Blomqvist, B. Johansson, O. N. Srivastava and R. Ahuja, J. Phys. Chem. C, 2012, 116, 10861–10866 CAS.
- A. Primo, F. Neatu, M. Florea, V. Parvulescu and H. Garcia, Nat. Commun., 2014, 5, 5291–5299 CrossRef CAS PubMed.
- M.-M. Trandafir, M. Florea, F. Neatu, A. Primo, V. I. Parvulescu and H. Garcia, ChemSusChem, 2016, 9, 1565–1569 CrossRef CAS PubMed.
- X. Y. Sun, B. Li, T. F. Liu, J. Song and D. S. Su, Phys. Chem. Chem. Phys., 2016, 18, 11120–11124 RSC.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467–4471 CrossRef CAS PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, New J. Chem., 2007, 31, 1455–1460 RSC.
- F. Tim-Patrick, T. Arne, J. Yuan and M. Antonietti, Adv. Mater., 2013, 25, 5838–5854 CrossRef PubMed.
- M. R. Acocella, M. Mauro, L. Falivene, L. Cavallo and G. Guerra, ACS Catal., 2014, 4, 492–496 CrossRef CAS.
- X.-H. Li, J.-S. Chen, X. Wang and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 8074–8077 CrossRef CAS PubMed.
- J. Long, X. Xie, J. Xu, Q. Gu, L. Chen and X. Wang, ACS Catal., 2012, 2, 622–631 CrossRef CAS.
- X. Duan, S. Indrawirawan, H. Sun and S. Wang, Catal. Today, 2015, 249, 184–191 CrossRef CAS.
- X. Duan, Z. Ao, H. Sun, S. Indrawirawan, Y. Wang, J. Kang, F. Liang, Z. H. Zhu and S. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 4169–4178 CAS.
- R. P. Rocha, A. G. Goncçalves, L. M. Pastrana-Martínez, B. C. Bordoni, O. S. G. P. Soares, J. J. M. Órfão, J. L. Faria, J. L. Figueiredo, A. M. T. Silva and M. F. R. Pereira, Catal. Today, 2015, 249, 192–198 CrossRef CAS.
- X.-K. Kong, Z.-Y. Sun, M. Chen, C.-I. Chen and Q.-W. Chen, Energy Environ. Sci., 2013, 6, 3260–3266 CAS.
- X. Duan, K. O’Donnell, H. Sun, Y. Wang and S. Wang, Small, 2015, 11, 3036–3044 CrossRef CAS PubMed.
- B. Dai, K. Chen, Y. Wang, L. Kang and M. Zhu, ACS Catal., 2015, 5, 2541–2547 CrossRef CAS.
- M. A. Patel, F. Luo, M. R. Khoshi, E. Rabie, Q. Zhang, C. R. Flach, R. Mendelsohn, E. Garfunkel, M. Szostak and H. He, ACS Nano, 2016, 10, 2305–2315 CrossRef CAS PubMed.
- H. Kim, K. Lee, S. Ihl Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505–17510 RSC.
- J. Vazquez-Arenas, A. Galano, D. U. Lee, D. Higgins, A. Guevara-García and Z. Chen, J. Mater. Chem. A, 2016, 4, 976–990 CAS.
- Y. Zheng, Y. Jiao, L. H. Hua Li, T. Xing, Y. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2014, 8, 5290–5296 CrossRef CAS PubMed.
- B. Li and D. Su, Chem. – Eur. J., 2014, 20, 7890–7894 CrossRef CAS PubMed.
- P. Lara and K. Philippot, Catal. Sci. Technol., 2014, 4, 2445–2465 CAS.
- B. H. Han, D. H. Shin and S. Y. Cho, Tetrahedron Lett., 1985, 26, 6233–6234 CrossRef CAS.
- B. J. Li and Z. Xu, J. Am. Chem. Soc., 2009, 131, 16380–16382 CrossRef CAS PubMed.
- Y. J. Gao, D. Ma, C. L. Wang, J. Guan and X. H. Bao, Chem. Commun., 2011, 47, 2432–2434 RSC.
- Y. Imada, H. Iida, T. Kitagawa and T. Naota, Chem. – Eur. J., 2011, 17, 5908–5920 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, S. Navalon, D. Sempere, M. Alvaro and H. Garcia, Chem. Commun., 2013, 49, 2359–2361 RSC.
- W. M. N. Ratnayake, J. S. Grossert and R. G. Ackman, J. Am. Oil Chem. Soc., 1990, 67, 940–946 CrossRef CAS.
- A. Corma, P. Concepcion and P. Serna, Angew. Chem., Int. Ed., 2007, 46, 7266–7269 CrossRef CAS PubMed.
- M. Pera-Titus, V. García-Molina, M. A. Baños, J. Giménez and S. Esplugas, Appl. Catal., B, 2004, 47, 219–256 CrossRef CAS.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS.
- E. Neyens and J. Baeyens, J. Hazard. Mater., 2003, 98, 33–50 CrossRef CAS PubMed.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- E. Lam, J. H. Chong, E. Majid, Y. Liu, S. Hrapovic, A. C. W. Leung and J. H. T. Luong, Carbon, 2012, 50, 1033–1043 CrossRef CAS.
- S. R. Pouran, A. R. A. Aziz and V. M. A. W. Daud, J. Ind. Eng. Chem., 2015, 21, 53–69 CrossRef.
- A. Dhakshinamoorthy, S. Navalon, M. Alvaro and H. Garcia, ChemSusChem, 2012, 5, 46–64 CrossRef CAS PubMed.
- M. C. Pereira, L. C. A. Oliveira and E. Murad, Clay Miner., 2012, 47, 285–302 CrossRef CAS.
- J. Herney-Ramirez, M. A. Vicente and L. M. Madeira, Appl. Catal., B, 2010, 98, 10–26 CrossRef CAS.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ChemSusChem, 2011, 4, 1712–1730 CrossRef CAS PubMed.
- P. Wu, P. Du, H. Zhang and C. Cai, Phys. Chem. Chem. Phys., 2013, 15, 6920–6928 RSC.
- H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3371 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- S. Navalon, M. Alvaro and H. Garcia, Appl. Catal., B, 2010, 99, 1–26 CrossRef CAS.
- G. P. Anipsitakis, E. Stathatos and D. D. Dionysiou, J. Phys. Chem. B, 2005, 109, 13052–13055 CrossRef CAS PubMed.
- Q. Yang, H. Choi, Y. Chen and D. D. Dionysiou, Appl. Catal., B, 2008, 77, 300–307 CrossRef CAS.
- E. Saputra, S. Muhammad, H. Sun, H. M. Ang, M. O. Tadé and S. Wang, Environ. Sci. Technol., 2013, 47, 5882–5887 CrossRef CAS PubMed.
- Y. Wang, H. Sun, H. M. Ang, M. O. Tadé and S. Wang, Appl. Catal., B, 2015, 164, 159–167 CrossRef CAS.
- H. Sun, S. Liu, G. Zhou, H. M. Ang, M. O. Tade and S. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 5466–5471 CAS.
- A. De Luca, X. He, D. D. Dionysiou, R. F. Dantas and S. Esplugas, Chem. Eng. J., 2017, 318, 206–213 CrossRef CAS.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- S. Navalón, M. Álvaro and H. García, ChemCatChem, 2013, 5, 3460–3480 CrossRef.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- L. Liu, J. Sun, L. Zhu, X. Meng, C. Qi and F.-S. Xiao, J. Mater. Chem., 2012, 5495–5502 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535–1539 CAS.
- D. W. Stephan, Dalton Trans., 2009, 3129–3136 RSC.
- P. A. Chase, T. Jurca and D. W. Stephan, Chem. Commun., 2008, 1701–1703 RSC.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS PubMed.
- P. Serna and A. Corma, ACS Catal., 2015, 5, 7114–7121 CrossRef CAS.
- S. Sabater, J. A. Mata and E. Peris, Organometallics, 2015, 34, 1186–1190 CrossRef CAS.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- M. Y. Han, B. Oezyilmaz, Y. Zhang and P. Kim, Phys. Rev. Lett., 2007, 98, 206805 CrossRef PubMed.
- T. Ohta, A. Bostwick, T. Seyller, K. Horn and E. Rotenberg, Science, 2006, 313, 951–954 CrossRef CAS PubMed.
- L. Xin-Hao and M. Antonietti, Chem. Soc. Rev., 2013, 42, 6593–6604 RSC.
- X.-H. Li and M. Antonietti, Chem. Soc. Rev., 2013, 42, 6593–6604 RSC.
- W. Wang, J. C. Yu, D. Xia, P. K. Wong and Y. Li, Environ. Sci. Technol., 2013, 47, 8724–8732 CrossRef CAS PubMed.
- Y. Zhang, T. Mori, L. Niu and J. Ye, Energy Environ. Sci., 2011, 4, 4517–4552 CAS.
- M. Shalom, M. Guttentag, C. Fettkenhauer, S. Inal, D. Neher, A. Llobet and M. Antonietti, Chem. Mater., 2014, 26, 5812–5818 CrossRef CAS.
- F. Chen, Y. Xie, J. He and J. Zhao, J. Photochem. Photobiol., A, 2001, 138, 139–146 CrossRef CAS.
- H.-P. Jia, D. R. Dreyer and C. W. Bielawski, Tetrahedron Lett., 2011, 67, 4431–4434 CrossRef CAS.
- H. Q. Sun, Y. X. Wang, S. Z. Liu, L. Ge, L. Wang, Z. H. Zhu and S. B. Wang, Chem. Commun., 2013, 49, 9914–9916 RSC.
- X. L. Li, H. L. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. J. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- C. D. Wang, Y. A. Zhou, L. F. He, T. W. Ng, G. Hong, Q. H. Wu, F. Gao, C. S. Lee and W. J. Zhang, Nanoscale, 2013, 5, 600–605 RSC.
- H. Li, J. Liao, Y. Du, T. You, W. Liao and L. Wen, Chem. Commun., 2013, 49, 1768–1770 RSC.
- X. Li, X. Pan, L. Yu, P. Ren, X. Wu, L. Sun, F. Jiao and X. Bao, Nat. Commun., 2014, 5, 3688–3694 CAS.
- X. Li, Y. Wang, L. Kang, M. Zhu and B. Dai, J. Catal., 2014, 311, 288–294 CrossRef CAS.
- M. Conte, A. F. Carley, C. Heirene, D. J. Willock, P. Johnston, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 250, 231–239 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- H. L. Liu, Y. L. Liu, Y. W. Li, Z. Y. Tang and H. F. Jiang, J. Phys. Chem. C, 2010, 114, 13362–13369 CAS.
- J. Zhu, P. C. Wang and M. Lu, Appl. Catal., A, 2014, 477, 125–131 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- C. Zhang, N. Mahmood, H. Yin, F. Liu and Y. Hou, Adv. Mater., 2013, 25, 4932–4937 CrossRef CAS PubMed.
- D. S. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. S. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed.
- Z. W. Liu, F. Peng, H. J. Wang, H. Yu, W. X. Zheng and J. Yang, Angew. Chem., Int. Ed., 2011, 50, 3257–3261 CrossRef PubMed.
- A. Dhakshinamoorthy, A. Primo, P. Concepcion, M. Alvaro and H. Garcia, Chem. – Eur. J., 2013, 19, 7547–7554 CrossRef CAS PubMed.
- Y. Gao, G. Hu, J. Zhong, Z. Shi, Y. Zhu, D. S. Su, J. Wang, X. Bao and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 2109–2113 CrossRef CAS PubMed.
- K. Ellmer, Nat. Photonics, 2012, 6, 809–817 CrossRef CAS.
- D. S. Hecht, L. Hu and G. Irvin, Adv. Mater., 2011, 23, 1482–1513 CrossRef CAS PubMed.
- X. Huang, Z. Zeng, Z. Fan, J. Liu and H. Zhang, Adv. Mater., 2012, 24, 5979–6004 CrossRef CAS PubMed.
- Z.-S. Wu, G. Zhou, L.-C. Yin, W. Ren, F. Li and H.-M. Cheng, Nano Energy, 2012, 1, 107–131 CrossRef CAS.
- C. Hu and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 11736–11758 CrossRef CAS PubMed.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- J. Zhang and L. Dai, ACS Catal., 2015, 5, 7244–7253 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- W. Zhou, J. Jia, J. Lu, L. Jang, D. Hou, G. Li and S. Chen, Nano Energy, 2016, 28, 29–43 CrossRef CAS.
- D. Higgins, P. Zamani, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 357–390 CAS.
- Z. Yang, H. Nie, X. Chen, X. Chen and S. Huang, J. Power Sources, 2013, 236, 238–249 CrossRef CAS.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CAS.
- Z. Jin, H. Nie, Z. Yang, J. Zhang, Z. Liu, X. Xu and S. Huang, Nanoscale, 2012, 4, 6455–6460 RSC.
- R. Li, Z. Wei and X. Gou, ACS Catal., 2015, 5, 4133–4142 CrossRef CAS.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. Chen and S. Huang, ACS Nano, 2012, 6, 205–211 CrossRef CAS PubMed.
- X. Yao, H. Nie, Z. Yang, X. Zhou, Z. Liu and S. Huang, Chem. Commun., 2012, 48, 1027–1029 RSC.
- S. Dzwigaj, J. Janas, W. Rojek, L. Stievano, F. E. Wagner, F. Averseng, M. Che, J. Janas, W. Rojek, L. Stievano, F. E. Wagner, F. Averseng and M. Che, Appl. Catal., B, 2009, 86, 45–52 CrossRef CAS.
- J. Pérez-Ramírez and A. Gallardo-Llamas, Appl. Catal., A, 2005, 279, 117–123 CrossRef.
- L. Wang, A. Ambrosi and M. Pumera, Angew. Chem., Int. Ed., 2013, 52, 13818–13821 CrossRef CAS PubMed.
- C. H. An Wong, Z. ek Sofer, M. Kubešová, J. Kučera, S. Matejková and M. Pumera, PNAS, 2014, 38, 13774–13779 CrossRef PubMed.
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896–910 CrossRef CAS PubMed.
- A. Ambrosi, C. K. Chua, B. Khezri, Z. Sofer, R. D. Webster and M. Pumera, PNAS, 2012, 39, 12899–12904 CrossRef PubMed.
- J. Masa, A. Zhao, W. Xia, M. Muhler and W. Schuhmann, Electrochim. Acta, 2014, 128, 271–278 CrossRef CAS.
- J. Masa, A. Zhao, W. Xia, Z. Sun, B. Mei, M. Muhler and W. Schuhmann, Electrochem. Commun., 2013, 34, 113–116 CrossRef CAS.
- Z. Y. Lin, G. H. Waller, Y. Liu, M. L. Liu and C. P. Wong, Carbon, 2013, 53, 130–136 CrossRef CAS.
- L. Wang, F. X. Yin and C. X. Yao, Int. J. Hydrogen Energy, 2014, 39, 15913–15919 CrossRef CAS.
Footnote |
| † Both authors should be considered first authors. |
| This journal is © The Royal Society of Chemistry 2017 |