
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catechol–TiO2 hybrids for photocatalytic H2 production and photocathode assembly†‡
Katherine L.
Orchard
 ab,
Daisuke
Hojo
b,
Katarzyna P.
Sokol
a,
Meng-Ju
Chan
b,
Naoki
Asao§
b,
Tadafumi
Adschiri
*b and
Erwin
Reisner
*a
ab,
Daisuke
Hojo
b,
Katarzyna P.
Sokol
a,
Meng-Ju
Chan
b,
Naoki
Asao§
b,
Tadafumi
Adschiri
*b and
Erwin
Reisner
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk; Web: http://www-reisner.ch.cam.ac.uk
bWPI Advanced Institute for Materials Research (AIMR), Tohoku University, 2-1-1 Katahira Aoba-ku Sendai, Miyagi 980-8577, Japan. E-mail: ajiri@tagen.tohoku.ac.jp
First published on 9th November 2017
Abstract
Visible-light driven H2 evolution in water is achieved using catechol-photosensitised TiO2 nanoparticles with a molecular nickel catalyst. Layer-by-layer immobilisation of catechol–TiO2 onto tin-doped indium oxide electrodes generates photocathodic currents in the presence of an electron acceptor. This approach represents a new strategy for controlling photocurrent direction in dye-sensitised photoelectrochemical applications.
Immobilising molecular dyes onto wide band-gap metal oxide semiconductors is a well-established strategy for preparing tuneable, visible-light responsive materials from relatively inexpensive components. This approach was pioneered for solar energy conversion in dye-sensitised solar cells (DSCs)1 and is an emerging field in the area of solar fuels synthesis.2 In the majority of dye-sensitised systems, photoexcitation occurs in the dye (HOMO–LUMO transition) followed by fast charge injection into the semiconductor (Type I sensitisation, Fig. 1a). This approach has been effective in H2 production using dye-sensitised photocatalysis (DSP)2a and in forming photoanodes for dye-sensitised photoelectrochemical cells (DSPEC);2b,c however, the translation of DSP systems to photocathodes is more challenging, often relying on low-performance p-type semiconductors (such as NiO)3 or molecular engineering4 to control the direction of electron transfer.
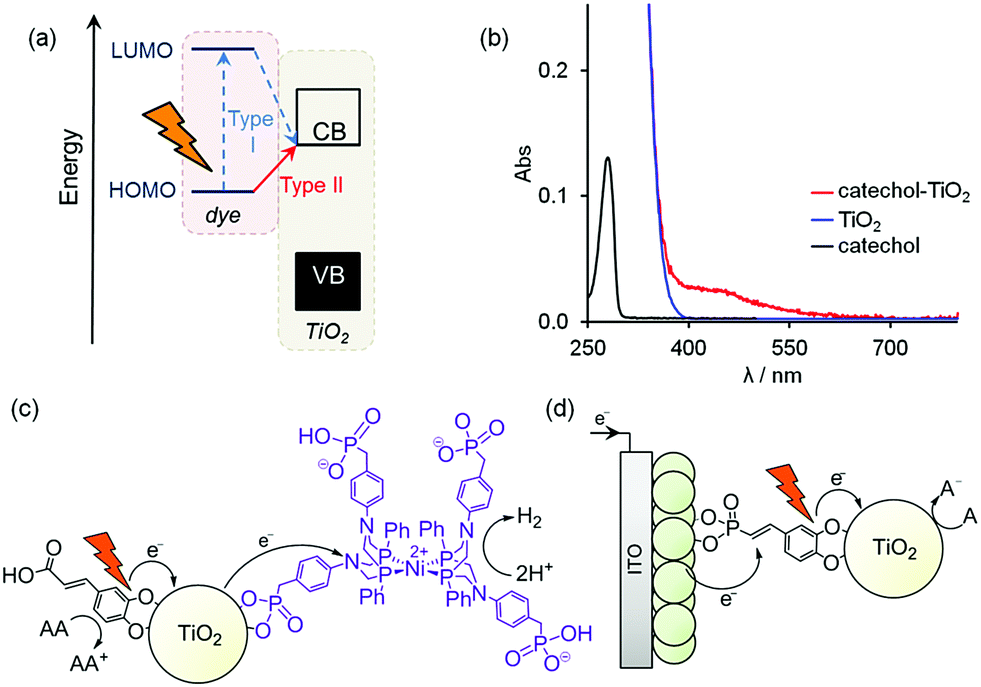 | ||
| Fig. 1 (a) Energy transfer in Type I (two-step) and Type II (one-step) dye-sensitised TiO2 systems; (b) diffuse reflectance absorption spectrum of catechol-modified TiO2 compared to bare TiO2 and the transmission spectrum of catechol in water (catechol shown is DHCA); (c) solar H2 production in DSP system (AA = ascorbic acid); (d) layer-by-layer photocathode incorporating catechol–TiO2 (A = acceptor; catechol shown is DHSP). | ||
Charge-transfer sensitisation, in which photoexcitation occurs directly from a surface-adsorbed molecule to the semiconductor conduction band (Type II, Fig. 1a), has recently been explored as a low-cost alternative in DSCs5 and solar fuel production.6 Type II sensitisation is known to occur in TiO2 functionalised with phenol and enediol groups, such as catechols.5d These groups bind to Ti4+ ions on the surface of TiO2, forming complexes that absorb visible light due to ligand-to-metal charge-transfer (Fig. 1b).7 This one-step transfer from dye to TiO2 results in very fast electron injection (on the order of fs),5b and, importantly, by avoiding excitation into the dye LUMO the charge-transfer is inherently directional (Fig. 1a and Fig. S1, ESI‡). Type II sensitisation also occurs between carbon nitride and TiO2, and has been used to enhance solar light-driven H2 evolution with hydrogenase,8 and to form photoanodes for photoelectrochemical water oxidation.9
Catechol-containing dyes based on alizarin red have been studied for DSP and for photocurrent switching applications;10 however, these dyes are also Type I sensitisers.5d To our knowledge, the only example of purely Type II visible-light driven H2 evolution uses phenolic resin-sensitised TiO2 nanoparticles, with Pt as the H2 evolution catalyst (HEC) in the presence of a sacrificial electron donor (SED).6b,c
Replacing noble metals with inexpensive catalysts and moving towards SED-free systems are both important goals in achieving sustainable fuel synthesis. Here we take steps towards addressing these challenges by developing a new route for assembling noble metal-free photocathodes. First, we demonstrate that Pt can be replaced by a 3d transition metal catalyst to form an active Type II DSP system (Fig. 1c). We then develop a SED-free photoelectrochemical system, using the directionality of the charge-transfer complex to form photocathodes on a conductive electrode support (Fig. 1d).
For the DSP system, a range of charge-transfer dye-sensitised TiO2 powders were prepared by mixing simple catechol derivatives (Fig. 2a) with commercial P25 TiO2 nanoparticles (average crystallite diameter 21 nm, 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of anatase:rutile). The resulting coloured powders exhibited broad light absorption over the visible range (Fig. S2, ESI‡). The dye loadings ranged from 75–172 nmol (mg TiO2)−1 (Table S1, ESI‡). These loadings correspond to an estimated footprint of 0.5–1.1 nm2 molecule−1, consistent with reported ranges for sub-monolayer to monolayer coverage of catechols on TiO2.11
:2 mixture of anatase:rutile). The resulting coloured powders exhibited broad light absorption over the visible range (Fig. S2, ESI‡). The dye loadings ranged from 75–172 nmol (mg TiO2)−1 (Table S1, ESI‡). These loadings correspond to an estimated footprint of 0.5–1.1 nm2 molecule−1, consistent with reported ranges for sub-monolayer to monolayer coverage of catechols on TiO2.11
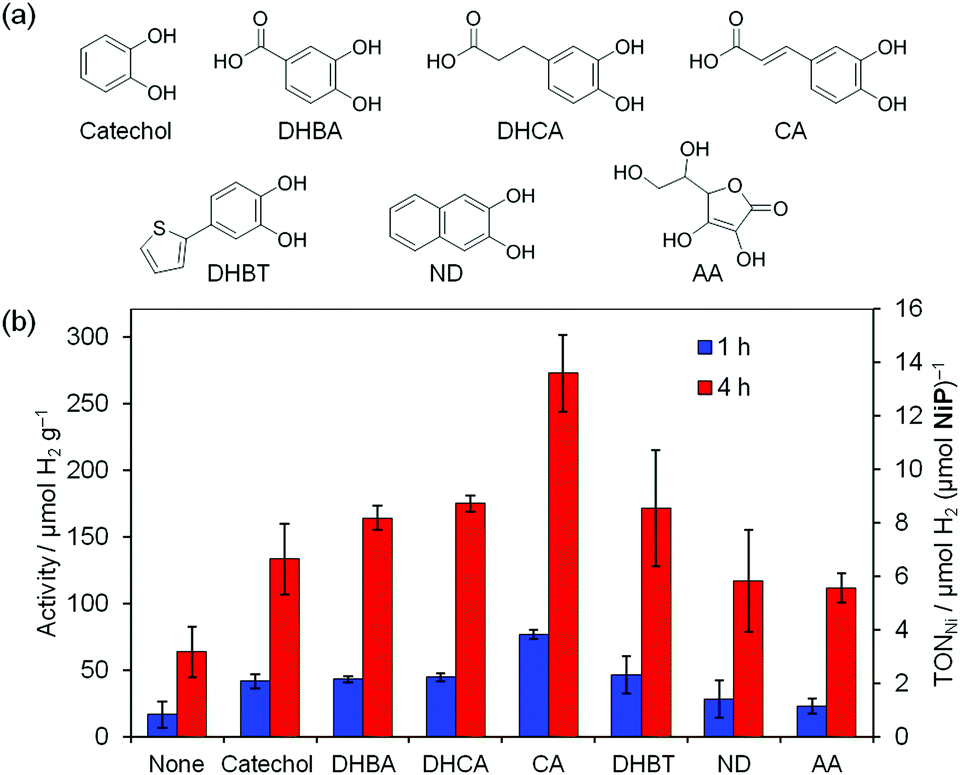 | ||
| Fig. 2 (a) Chemical structures of charge-transfer dyes used in this study (catechol = 1,2-dihydroxybenzene, DHBA = 3,4-dihydroxybenzoic acid, DHCA = 3,4-dihydroxyhydrocinnamic acid, CA = caffeic acid, DHBT = 2-(3,4-dihydroxybenzene)thiophene, ND = 2,3-naphthalenediol, AA = ascorbic acid); (b) comparison of visible-light driven H2 evolution with charge-transfer dye-functionalised P25 TiO2 powders in the presence of a molecular Ni catalyst, NiP (0.1 M AA, pH 4.5, 2.5 mg powder, 50 nmol NiP, 2.25 mL, λ > 420 nm, AM 1.5 G, 100 mW cm−2, 25 °C). | ||
Photocatalysis solutions were prepared by suspending the dye-P25 powders (2.5 mg) in aqueous SED solution (ascorbic acid, AA, 0.1 M, 2.25 mL). We selected the HEC [Ni(PPh2NC6H4CH2P(O)(OH)22)2]Br2, NiP (Fig. 1c), as it has previously been shown to be highly active in aqueous DSP12 and DSPEC.3a,4bNiP (50 nmol) was added to the dye-P25 suspension as a solution in methanol (1 mM) before the photoreactors were purged with N2 and irradiated with simulated solar light (λ > 420 nm, AM 1.5 G, 100 mW cm−2). The formation of the hybrid DSP system (Fig. 1c) is supported by FTIR and UV-vis spectroscopy (Fig. S3, ESI‡), with an estimated NiP loading of 38 nmol mg−1 for caffeic acid (CA)-P25 (calculated using the difference in absorption at 450 nm of a NiP solution before and after exposure to TiO2). No H2 was detected in control experiments without AA, TiO2 or NiP, or with NiCl2 in place of NiP.
AA can also sensitise TiO2 through charge-transfer interactions, resulting in small amounts of H2 evolution in the absence of additional catechol during visible light irradiation (Fig. 2b, “none”); however, all ex situ functionalised powders enhanced the activity compared to bare P25 (Fig. 2b and Fig. S4a, ESI‡). The highest activity was obtained with CA (Fig. 2), with an activity with respect to the powder of 273 ± 29 μmol H2 g−1 after 4 h. The activity per hour of 73 ± 3 μmol H2 (g TiO2)−1 h−1 was maintained over the first 4 h, equivalent to a turnover frequency (TOF) per Ni catalyst of 3.6 ± 0.2 μmol H2 (μmol NiP)−1 h−1 and TOF per catechol of 0.6 ± 0.2 μmol H2 (μmol CA)−1 h−1. This result is comparable to that reported for the phenolic resin system with Pt (105 μmol g−1 h−1).6b The TOFNi is an order of magnitude lower than previously reported for the equivalent system with a Ru tris(bipyridine) dye, RuP,12 but RuP can directly reduce NiP in solution, whereas the catechol derivatives cannot. The lower extinction coefficient and faster recombination rate of the charge-transfer band relative to that of metal-based dyes (ps13vs. ns–μs2a) are also expected to contribute to the difference in activity.
The high activity of CA relative to the unconjugated analogue 3,4-dihydroxyhydrocinnamic acid (DHCA) is consistent with the red-shift in the absorption spectrum and increase in both the catechol binding constant and the electron injection efficiency associated with greater conjugation.5b The activity per dye molecule also reflects this trend for 2-(3,4-dihydroxybenzene)thiophene (DHBT; Fig. S4b, ESI‡); however, the lower loading of DHBT on TiO2 compared to CA (Table S1, ESI‡) resulted in a lower activity per gram (Fig. 2b). Ring conjugation (2,3-naphthalenediol, ND) did not enhance activity (Fig. 2b and Fig. S4b, ESI‡), which is likely due to the comparatively low visible light absorption of ND-P25 (Fig. S1, ESI‡).
The turnover number per Ni (TONNi) increased with decreasing NiP loading (Fig. S5a, ESI‡) and increasing CA-P25 powder loading (Fig. S6a, ESI‡). However, the corresponding activity per gram decreased with decreasing NiP loading (Fig. S5b, ESI‡) and reached a maximum at a CA-P25 loading of 1.25 mg (Fig. S6b, ESI‡). Since increasing the powder loading increases both the dye loading and the particle:Ni ratio, it can be inferred that the H2 production rate is limited by the generation and supply of electrons to NiP. This conclusion is supported by the decrease in activity of the system on decreasing the light intensity (Fig. S7, ESI‡). The external quantum efficiency (EQE) of the most active system (5 mg CA-P25, 50 nmol NiP) was measured to be 0.09 ± 0.01%.
Since light absorption is related to the number of catechol–TiO2 surface interactions, light absorption per g can be enhanced by using smaller nanoparticles to increase the surface area. We synthesised TiO2 nanoparticles by a previously reported method (pure anatase, average diameter ∼7 nm, AN7–TiO2, see Fig. S8, ESI‡ for TEM)14 and tested both these and commercial pure anatase nanoparticles (10 nm, AN10–TiO2, Fig. S7, ESI‡) under the standard DSP conditions (CA–TiO2, 2.5 mg, 50 nmol NiP). However, despite the increase in catechol loading (505 nmol (mg TiO2)−1; Table S1, ESI‡) and light absorption (Fig. S9, ESI‡) compared to P25, these particles displayed less photocatalytic activity (Fig. S10, ESI‡). The lower performance was not found to be due to a difference in binding of NiP to the particles (48 and 51 nmol mg−1 for AN10–TiO2 and AN7–TiO2, respectively). However, it is likely that the smaller, pure anatase particles suffer from less efficient charge separation compared to the larger, mixed-phase (anatase/rutile) P25.15 Pure rutile phase TiO2 nanoparticles (R–TiO2, 10–30 nm) displayed lower activity than P25, AN10, and AN7–TiO2 (4.9 ± 0.3 after 3 h, Table S2 and Fig. S10, ESI‡).
Having established that catechol–TiO2 hybrids are effective light absorbers for (UV-filtered) solar H2 evolution with NiP, we sought to transfer the DSP system onto photoelectrodes. We designed a layer-by-layer route to immobilise the TiO2 nanoparticles onto a degenerately doped n-type metal oxide support (indium tin oxide, ITO) in a directed, sequential fashion (Fig. 1d). In order to maximise the deposition of the TiO2 nanoparticles in our system, we used hierarchical inverse opal mesoporous ITO electrodes (IO–ITO; Fig. S11a, ESI‡). IO–ITO is an effective support for nanoscale adsorbates such as enzymes (e.g. Photosystem II, 10.5 × 20.5 × 11.0 nm3), owing to their open, macroporous structure (750 nm pores with 150 nm interconnecting chambers) and high surface area mesoporous scaffold (formed from <50 nm ITO nanoparticles).16 Mesoporous ITO (mesoITO) electrodes (1 μm thickness, Fig. S11b, ESI‡) were also prepared for comparison.17
By studying the attachment of DHCA to IO–ITO in the presence or absence of one equivalent of propylphosphonic acid by UV-vis spectroscopy, we found that ITO binds preferentially to phosphonate groups over catechol groups (83% of DHCA is displaced; Fig. S12a, ESI‡). We therefore synthesised (E)-(3,4-dihydroxystyryl)phosphonic acid (DHSP; see Fig. 1d and ESI‡ for synthetic details) as a bifunctional, phosphonated catechol that is expected to bind to ITO in the correct orientation for subsequent formation of the charge-transfer interaction with TiO2. In contrast to ITO, TiO2 has an approximately equal binding affinity for catechol and phosphonic acid groups (45% of DHCA was displaced in the presence of propylphosphonic acid; Fig. S12b, ESI‡). Therefore, formation of the charge-transfer complex was only effective if DHSP was anchored to ITO before adding TiO2; ex situ-prepared DHSP-sensitised P25 powders did not absorb appreciably in the visible-light region.
DHSP was anchored onto ITO by incubating the electrodes in an aqueous stock solution (1 mM, 2–8 °C, 16 h). Cyclic voltammetry (CV) of DHSP-functionalised flat ITO shows a quasi-reversible wave (E1/2 = 0.84 V vs. RHE), which can be assigned to the catechol/semiquinone redox couple (Fig. S13, ESI‡). By monitoring the change in absorbance of a stock solution of DHSP (1 mM) at 280 nm after incubation with IO–ITO electrodes, the loading of DHSP was estimated to be 64 ± 11 nmol cm−2, which matches the loading estimated from the charge passed during CV (55.5 nmol cm−2, see ESI‡).
The final charge-transfer complexes were formed by depositing TiO2 nanoparticles onto the DHSP-functionalised IO–ITO. In contrast to the photocatalysis results, the electrodes with the highest photocurrent in the presence of an acceptor (O2) were those prepared with AN7–TiO2 nanoparticles (Fig. S14, ESI‡). The nanoparticles were deposited by immersion of the IO–ITO|DHSP electrodes in a stock solution of oleic acid-capped AN7–TiO2,18 followed by treatment with NEt3 (10 vol% in ethanol) to remove surface oleic acid, as confirmed by FTIR spectroscopy (Fig. S15, ESI‡). The formation of the charge-transfer complex was evidenced by a change in colour of the electrodes from pale yellow/green to peach (Fig. S16, ESI‡). The loading of TiO2 was measured to be 212.4 μg cm−2 for 12 μm thick IO–ITO (Ti content; Inductively Coupled Plasma-Optical Emission Spectroscopy, ICP-OES), which corresponds to approximately 83 nmol of nanoparticles per cm2.
Scanning electron microscopy (SEM) of AN7–TiO2 on DHSP-functionalised flat ITO showed that an even monolayer of particles is formed (Fig. S17, ESI‡), and energy-dispersive X-ray spectroscopy (EDX) of the fully assembled IO–ITO|DHSP|AN7–TiO2 electrodes showed that the particles are evenly distributed throughout the IO–ITO structure (Ti content of 11.8, 11.4, and 10.7 at% for the top, middle, and bottom part of the IO–ITO structure respectively; Fig. S18a, ESI‡). In contrast, EDX analysis of equivalent electrodes prepared with P25 TiO2 showed that these nanoparticles do not penetrate beyond the surface (10–12 at% Ti detected only on the top surface and none at lower layers; Fig. S18b, ESI‡). The difference in particle distribution, and subsequent performance, is attributed to the better size match of the macro- and mesopores of the IO–ITO structure to the small, well-dispersed AN7–TiO2 compared to the larger, aggregated P25 particles (close to micron-sized aggregates of 20 nm particles; see Fig. S8, ESI‡ for TEM). AN10–TiO2 nanoparticles also form aggregates (Fig. S8, ESI‡), resulting in low performance (Fig. S14, ESI‡).
In the presence of the electron acceptor O2, cathodic photocurrents were obtained under visible light irradiation (λ > 400 nm, 100 mW cm−2) with IO–ITO|DHSP|AN7–TiO2 electrodes below the redox potential of DHSP (Eapplied = 0.3 V vs. RHE, Fig. 3a). Initial current spikes were observed in the photocurrent curves of these electrodes (Fig. 3a), which likely occur due to rapid polarisation and charge accumulation at the electrode.19 A switch to anodic photocurrent was observed at potentials where DHSP is oxidised (Eapplied > ∼0.7 V vs. RHE, Fig. S19, ESI‡). The photocurrent is attributed to the formation of the charge-transfer complex since little photocurrent is obtained in the absence of the catechol and/or TiO2 (Fig. 3a). The magnitude of the photocurrent scales with the thickness of the ITO scaffold, reflecting the increase in loading of the dye–TiO2 complex with increasing ITO surface area (Fig. 3b). The photocathodic currents obtained with 12 μm thick IO–ITO (70 μA cm−2) are comparable to those obtained with Ru-based Type I dyes in the presence of an acceptor,20 demonstrating the promise of employing metal-free Type II dyes.
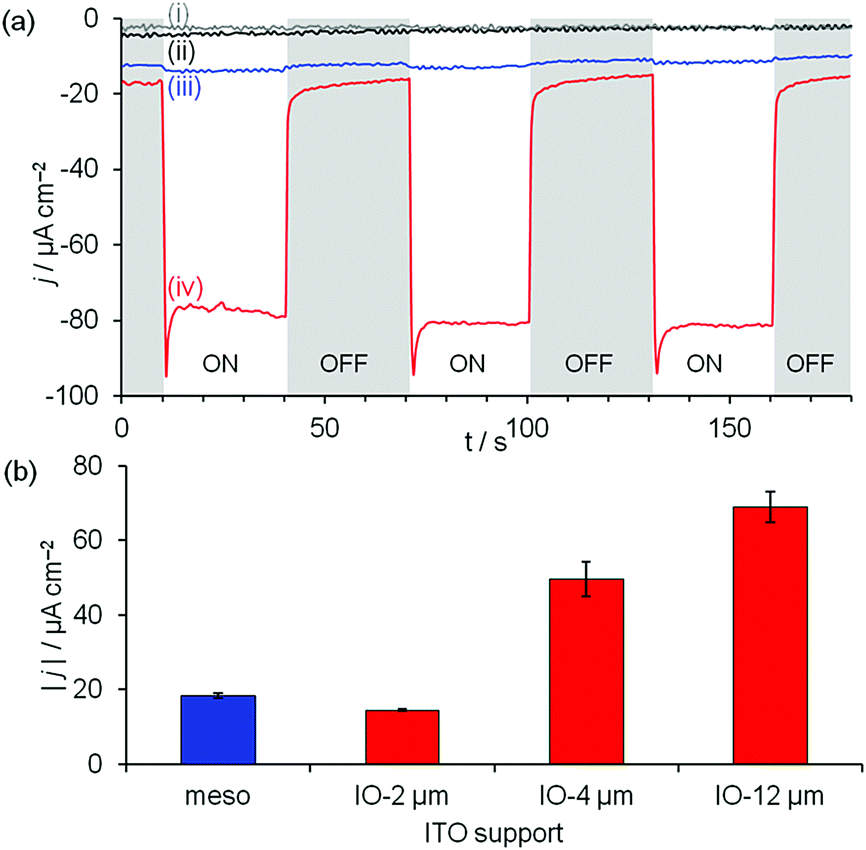 | ||
| Fig. 3 (a) Chronoamperograms under chopped irradiation of (i) IO–ITO|DHSP, (ii) IO–ITO, (iii) IO–ITO|AN7–TiO2 and (iv) IO–ITO|DHSP|AN7–TiO2 electrodes. (b) Effect of ITO morphology and thickness on visible-light photocurrent of ITO|DHSP|AN7–TiO2 electrodes. Due to the hierarchical structure consisting of macro- and mesopores, the effective surface area of IO-2 μm ITO is lower than 1 μm mesoITO. Conditions: Na2SO4 (0.1 M), pH 4.5, 0.3 V vs. RHE, 100 mW cm−2, λ > 400 nm, room temperature, under air. | ||
In the absence of O2 (N2 purged solution), small cathodic currents were maintained for the bare IO–ITO|DHSP|AN7–TiO2 electrodes (Fig. S20, ESI‡). Although NiP could be immobilised onto the electrodes (maximum loading of 29 ± 3 nmol per cm2; UV-vis spectroscopy), photo-driven H2 evolution was not achieved with these electrodes. It is possible that the phosphonate groups in NiP may displace either some or all of the DHSP groups from the surface of TiO2. NiP can also bind directly to ITO, by-passing the dye–TiO2 construct. Therefore, although the layer-by-layer design successfully generates a directional, dye-sensitised electrode, further catalyst design is required in order to fully translate the DSP system to DSPEC.
In conclusion, we have demonstrated that catechol–TiO2 complexes are effective photosensitisers for driving sacrificial H2 evolution with an earth-abundant molecular catalyst, forming a fully noble metal-free Type II DSP system. Using a layer-by-layer method, we have transferred the dye-sensitised TiO2 to an electrode to form a Type II photocathode. The excellent size match between the hierarchical IO–ITO support and the TiO2 nanoparticles is critical for high dye–TiO2 loading. The obtained photocurrents with a sacrificial electron acceptor are comparable to those obtained with Type I dyes, indicating that this is a promising means to control photocurrent direction and allow the use of n-type metal oxide supports (such as ITO) in DSPEC applications. This work represents an important first step in translating functional DSP systems to DSPEC; however, challenges in coupling these electrodes to a H2 evolution catalyst will need to be overcome in order to apply them for sustainable solar fuels synthesis.
We gratefully acknowledge financial support by the EPSRC and the World Premier International Research Center Initiative, MEXT, Japan. We also thank Dr Manuela Gross and Dr Benjamin Martindale for supplying NiP, Mr Alan Dickerson for ICP-OES measurements, and Dr Janina Willkomm and Mr Charles Creissen for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- (a) J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC; (b) J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC; (c) L. Alibabaei, H. Luo, R. L. House, P. G. Hoertz, R. Lopez and T. J. Meyer, J. Mater. Chem. A, 2013, 1, 4133–4145 RSC.
- (a) M. A. Gross, C. E. Creissen, K. L. Orchard and E. Reisner, Chem. Sci., 2016, 7, 5537–5546 RSC; (b) L. J. Antila, P. Ghamgosar, S. Maji, H. Tian, S. Ott and L. Hammarström, ACS Energy Lett., 2016, 1, 1106–1111 CrossRef CAS; (c) L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988–990 RSC; (d) F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS PubMed; (e) Z. Ji, M. He, Z. Huang, U. Ozkan and Y. Wu, J. Am. Chem. Soc., 2013, 135, 11696–11699 CrossRef CAS PubMed.
- (a) Z. Huang, M. He, M. Yu, K. Click, D. Beauchamp and Y. Wu, Angew. Chem., Int. Ed., 2015, 54, 6857–6861 CrossRef CAS PubMed; (b) B. Shan, A. K. Das, S. Marquard, B. H. Farnum, D. Wang, R. M. Bullock and T. J. Meyer, Energy Environ. Sci., 2016, 9, 3693–3697 RSC.
- (a) Y. Ooyama, M. Kanda, K. Uenaka and J. Ohshita, ChemPhysChem, 2015, 16, 3049–3057 CrossRef CAS PubMed; (b) E. L. Tae, S. H. Lee, J. K. Lee, S. S. Yoo, E. J. Kang and K. B. Yoon, J. Phys. Chem. B, 2005, 109, 22513–22522 CrossRef CAS PubMed; (c) B.-K. An, W. Hu, P. L. Burn and P. Meredith, J. Phys. Chem. C, 2010, 114, 17964–17974 CrossRef CAS; (d) Y. Ooyama and Y. Harima, ChemPhysChem, 2012, 13, 4032–4080 CrossRef CAS PubMed.
- (a) Z. Tachan, I. Hod and A. Zaban, Adv. Energy Mater., 2014, 4, 1301249 CrossRef; (b) G. Zhang and W. Choi, Chem. Commun., 2012, 48, 10621–10623 RSC; (c) G. Zhang, C. Kim and W. Choi, Catal. Today, 2017, 281(Part 1), 109–116 CrossRef CAS.
- P. Persson, R. Bergström and S. Lunell, J. Phys. Chem. B, 2000, 104, 10348–10351 CrossRef CAS.
- C. A. Caputo, L. Wang, R. Beranek and E. Reisner, Chem. Sci., 2015, 6, 5690–5694 RSC.
- M. Bledowski, L. Wang, A. Ramakrishnan and R. Beranek, J. Mater. Res., 2013, 28, 411–417 CrossRef CAS.
- (a) Y. Di Iorio, H. B. Rodríguez, E. San Román and M. A. Grela, J. Phys. Chem. C, 2010, 114, 11515–11521 CrossRef CAS; (b) Q. Li, Y. Che, H. Ji, C. Chen, H. Zhu, W. Ma and J. Zhao, Phys. Chem. Chem. Phys., 2014, 16, 6550–6554 RSC.
- (a) P. Z. Araujo, P. J. Morando and M. A. Blesa, Langmuir, 2005, 21, 3470–3474 CrossRef CAS PubMed; (b) M. Rodenstein, S. Zürcher, S. G. P. Tosatti and N. D. Spencer, Langmuir, 2010, 26, 16211–16220 CrossRef CAS PubMed.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS PubMed.
- Y. Wang, K. Hang, N. A. Anderson and T. Lian, J. Phys. Chem. B, 2003, 107, 9434–9440 CrossRef CAS.
- E. T. Hwang, K. Sheikh, K. L. Orchard, D. Hojo, V. Radu, C.-Y. Lee, E. Ainsworth, C. Lockwood, M. A. Gross, T. Adschiri, E. Reisner, J. N. Butt and L. J. C. Jeuken, Adv. Funct. Mater., 2015, 25, 2308–2315 CrossRef CAS PubMed.
- (a) G. Li, C. P. Richter, R. L. Milot, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree, G. W. Brudvig and V. S. Batista, Dalton Trans., 2009, 10078–10085 RSC; (b) J. T. Carneiro, T. J. Savenije, J. A. Moulijn and G. Mul, J. Phys. Chem. C, 2011, 115, 2211–2217 CrossRef CAS.
- D. Mersch, C.-Y. Lee, J. Z. Zhang, K. Brinkert, J. C. Fontecilla-Camps, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2015, 137, 8541–8549 CrossRef CAS PubMed.
- M. Kato, T. Cardona, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2013, 135, 10610–10613 CrossRef CAS PubMed.
- D. Hojo, T. Togashi, D. Iwasa, T. Arita, K. Minami, S. Takami and T. Adschiri, Chem. Mater., 2010, 22, 1862–1869 CrossRef CAS.
- Y. Zhao, J. R. Swierk, J. D. Megiatto Jr., B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 CrossRef CAS PubMed.
- W. Hamd, M. Chavarot-Kerlidou, J. Fize, G. Muller, A. Leyris, M. Matheron, E. Courtin, M. Fontecave, C. Sanchez, V. Artero and C. Laberty-Robert, J. Mater. Chem. A, 2013, 1, 8217–8225 CAS.
Footnotes |
| † Additional data related to this publication are available at the University of Cambridge data repository (https://doi.org/10.17863/CAM.13111). |
| ‡ Electronic supplementary information (ESI) available: Additional figures, tables, and experimental details. See DOI: 10.1039/c7cc05094a |
| § Current address: Faculty of Textile Science and Technology, Shinshu University, Ueda 386-8567, Japan. |
| This journal is © The Royal Society of Chemistry 2017 |