
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The third orthogonal dynamic covalent bond†
Inés
Lascano
ab,
Kang-Da
Zhang‡
ab,
Robin
Wehlauch
ac,
Karl
Gademann
 ac,
Naomi
Sakai
ab and
Stefan
Matile
*ab
ac,
Naomi
Sakai
ab and
Stefan
Matile
*ab
aNational Centre of Competence in Research (NCCR), Molecular Systems Engineering (MSE), Switzerland Web: http://www.unige.ch/sciences/chiorg/matile/
bDepartment of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: http://www.unige.ch/sciences/chiorg/matile/ Fax: +41 22 379 5123; Tel: +41 22 379 6523
cDepartment of Chemistry, University of Zurich, Zurich, Switzerland
First published on 20th April 2016
Abstract
Orthogonal dynamic covalent bonds are of interest for the construction of functional systems. The orthogonality of disulfide and hydrazone exchange under basic and acidic conditions, respectively, is well established. However, the integration of boronate esters as the third bond has failed so far because they exchanged too easily, especially under hydrazone exchange conditions. In this report, a collection of bioinspired catechols derived from adhesive natural products from cyanobacteria is screened with phenylboronic acids with proximal alcohols (benzoboroxoles), amines and fluorines to identify the least labile boronate esters. Moreover, Kool's 2-aminophenol catalysts are introduced to selectively accelerate hydrazone exchange without disturbing sufficiently inert boronate esters. Based on these results, we identified three different conditions to selectively exchange disulfides, hydrazones and boronate esters, that is to demonstrate the existence of three orthogonal dynamic covalent bonds. Moreover, their compatibility with functional systems is confirmed by successful hydrazone exchange in multicomponent surface architectures in the presence of intact boronate esters and disulfides.
Dynamic covalent bonds are fascinating because they can be either as stable as covalent bonds or as rapidly exchanging as non-covalent bonds, depending on the conditions.1–4 This dual nature makes them ideal tools for the construction of multicomponent functional systems. However, contrary to the routine use of several non-covalent bonds at the same time, dynamic covalent bonds are usually used alone. This could be in part due to the poor orthogonality between different types of dynamic covalent bonds. Orthogonality is defined by the existence of conditions that allow the exchange of one without disturbing the others (Fig. 1). The most popular pair of orthogonal dynamic bonds, hydrazones2 and disulfides3 exchange exclusively under acidic and basic conditions, respectively, and thus enabled the construction and operation of various doubly dynamic functional systems.5–8
 | ||
| Fig. 1 Selective exchange under conditions A, B and C defines orthogonality of disulfides, hydrazones and boronic/boronate esters. | ||
Extending this approach, triply dynamic functional systems have been constructed very recently9,10 with boronate esters,1 hydrazones and disulfides. These studies have shown the feasibility to exchange boronate esters in the presence of intact hydrazones and disulfides9 and to simultaneously form these three types of bonds.10 However, the orthogonality of boronate esters as the third dynamic covalent bond could not be established so far because (a) they are too labile, particularly under acidic conditions, and (b) hydrazone exchange requires strong acids.9,10 To overcome this dilemma, we first considered the use of electron-deficient catechol derivatives of anachelin, a natural product from the cyanobacterium Anabaena cylindrica. The high affinity of these bioinspired catechols to mineral oxides11 suggested that they might also afford more inert boronate esters. To overcome the second obstacle en route to the third orthogonal organic dynamic covalent bond, weakening of the hydrazone bond was not an option because it would destabilize the entire functional system.12 Acceleration of the exchange of inert aryl hydrazones appeared more promising, also because the original aniline catalysts13 have been dramatically improved recently with bifunctional acid/base catalysts.14–16 The anthranilic acids15 and, more interestingly, the 2-aminophenols16 are thought to operate by intramolecular protonation of the OH leaving group via 8- and 7-membered rings, respectively, to accelerate the rate-limiting dehydration during hydrazone formation from aldehydes and hydrazines. Contemplating this elegant catalyst design, we felt that the proposed mechanism could also extend to the activation of the NH leaving group during hydrazone exchange. In the following, we show that indeed the combination of bioinspired catechol adhesives11 and cutting-edge organocatalysts16 was the key to find the third orthogonal dynamic covalent bond.
Catechols 1–9 were chosen for this study (Fig. 2) and the protected anachelin chromophore 511 served as starting point for the design of catechols. Recognizing the power of electron-deficient aromatic units, we prepared new catecholate derivatives 2, 7–9 for subsequent experiments. Phenylboronic acids 10–12 with proximal primary alcohols (benzoboroxoles),17 tertiary amines1c,18 and fluorines7 have been reported previously. The formation of boronate esters was assessed in MeOH/water 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, pH 7.8, at room temperature (Fig. S1 and 2†). Changes in the absorption spectra of catechols, kept constant at low micromolar concentrations, were recorded in response to increasing concentrations of boronic acids. Reverse titration of boronate esters of alizarin red 4 provided access to the KD's of otherwise “invisible” catechols.
:1, pH 7.8, at room temperature (Fig. S1 and 2†). Changes in the absorption spectra of catechols, kept constant at low micromolar concentrations, were recorded in response to increasing concentrations of boronic acids. Reverse titration of boronate esters of alizarin red 4 provided access to the KD's of otherwise “invisible” catechols.
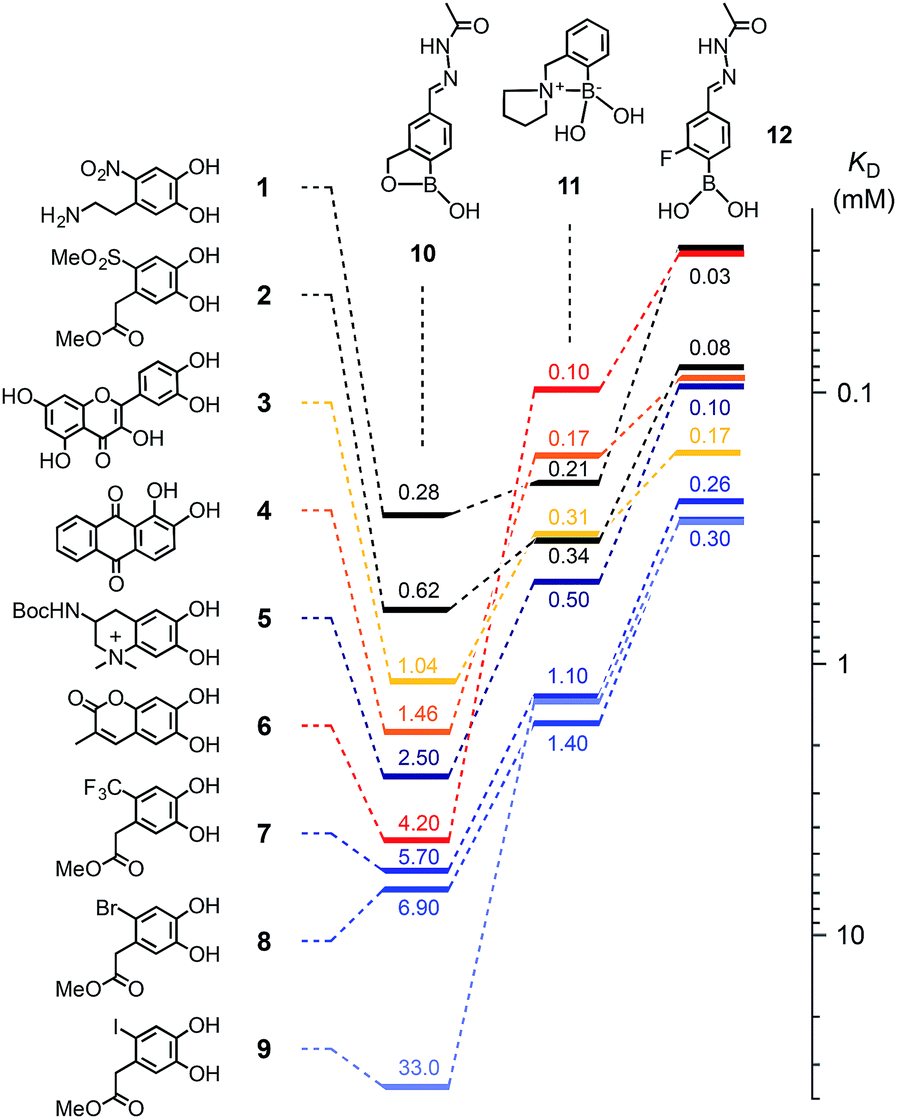 | ||
| Fig. 2 Dissociation constants KD (mM) of boronic esters obtained from catechols 1–9 and boronic acids 10–12 in MeOH/H2O 3:1 (100 mM HEPES, pH 7.8); from changes in absorption upon direct titration (1, 3, 4 and 6) or reverse titration with 4 (2, 5, 7–9). | ||
Compared to the previous best performing catechols 3, 4 and 6, the most distinctive results with the new “bioadhesives” 1, 2, 5 and 7–9 were obtained with benzoboroxoles 10 (Fig. 2, Table S1†). As on oxide surfaces,11 nitrodopamine 1 with a KD = 280 μM was best also with regard to boronate ester formation. It was followed by the similarly electron-deficient methyl sulfone 2 at 620 μM, quercetin 3 and alizarin red 4 as the first positive controls at 1.04 and 1.46 mM, and the close anachelin mimic 5 at 2.50 mM. “Bioadhesives” 2, 5 and particularly 1 also gave top values with the fluorinated boronic acid 12, whereas boronic acid 11 was overall slightly less convincing with anachelin mimics, particularly 1.
The outstanding KD's of the new “bioadhesives” 1 and 2 with benzoboroxole 10 were most important because boronate esters of benzoboroxoles have been identified previously as best in functional systems, presumably because the less electrophilic tetrahedral boronate is intramolecularly stabilized.9 Testifying its inertness, the boronate ester 13, prepared from the best performing anachelin mimic 1 and 10, underwent only marginal exchange with catechol 8 to boronate 14 under acidic hydrazone exchange conditions B (around 10%, see below, Fig. 3a, dB and eB). In contrast, the exchange reached around 35% when replacing nitrodopamine 1 with anachelin 5 (Fig. S20
and eB). In contrast, the exchange reached around 35% when replacing nitrodopamine 1 with anachelin 5 (Fig. S20 and 21†).
and 21†).
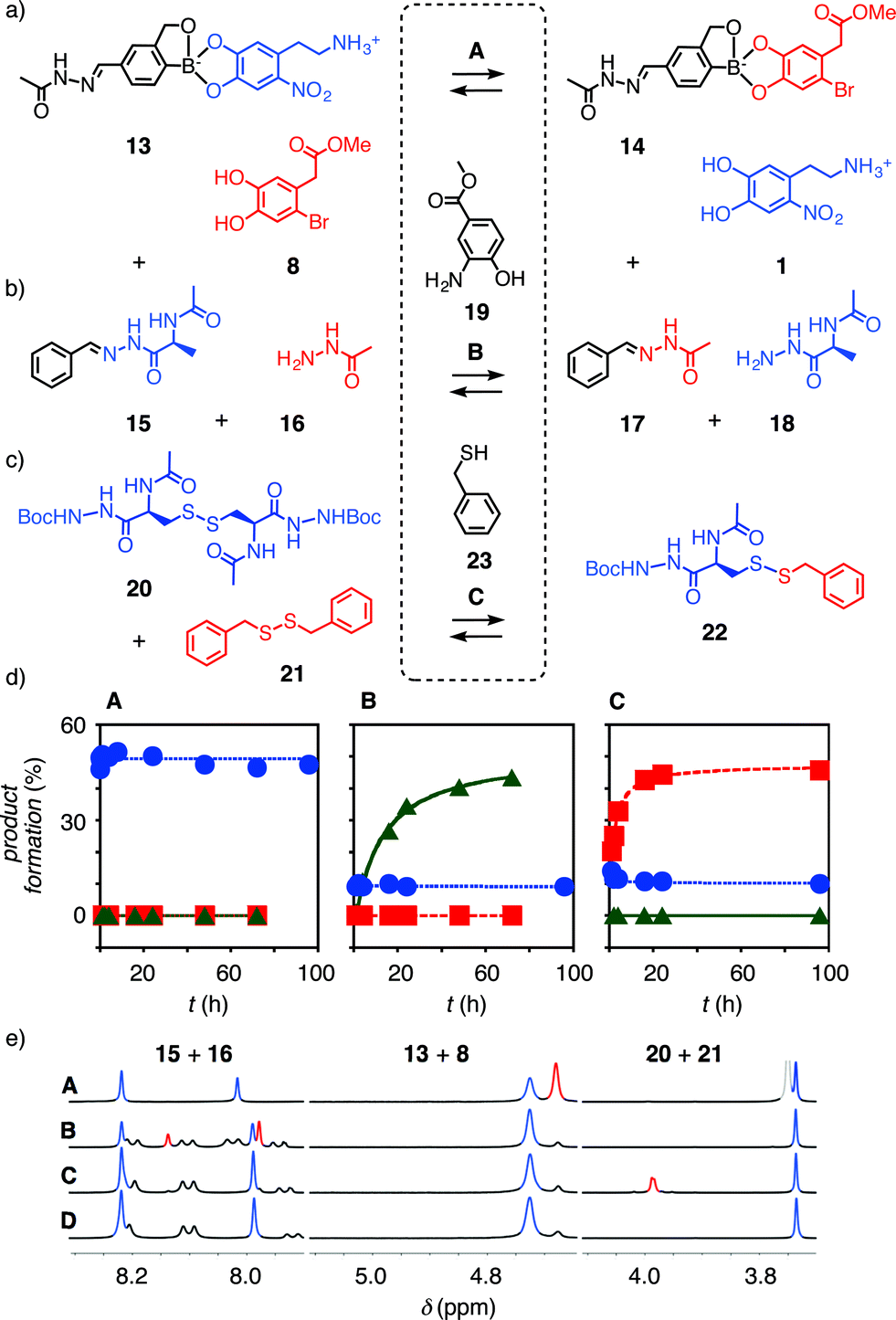 | ||
Fig. 3 (a–c) Exchange reactions used in this study to identify orthogonality under conditions A, B and C. (d) Exchange kinetics for boronate esters ( , 20 mM 13, 22 mM 8) hydrazones ( , 20 mM 13, 22 mM 8) hydrazones ( , 20 mM 15, 20 mM 16) and disulfides ( , 20 mM 15, 20 mM 16) and disulfides ( , 20 mM 20, 20 mM 21) under conditions A (DMSO-d6, 10% D2O, 2% DIPEA), B (1.0 mM 19, 1.5 mM TFA in DMSO-d6), and C (2.0 mM 22, 2.0 mM TEA in DMSO-d6). (e) Original data showing diagnostic peaks in 1H NMR spectra in conditions A–C and D (DMSO-d6) of 15, 20 and 13 (blue peaks) and their exchange products (red peaks). The signals at ∼8.1 ppm disappear in conditions A due to H/D exchange. , 20 mM 20, 20 mM 21) under conditions A (DMSO-d6, 10% D2O, 2% DIPEA), B (1.0 mM 19, 1.5 mM TFA in DMSO-d6), and C (2.0 mM 22, 2.0 mM TEA in DMSO-d6). (e) Original data showing diagnostic peaks in 1H NMR spectra in conditions A–C and D (DMSO-d6) of 15, 20 and 13 (blue peaks) and their exchange products (red peaks). The signals at ∼8.1 ppm disappear in conditions A due to H/D exchange. | ||
According to the kinetics measurements using 1H NMR spectroscopy, the exchange reaction of boronate ester 13 with the catechol 8 in DMSO-d6 containing 10% D2O and 2% Hünig base, i.e., conditions A, reached the equilibrium rapidly with a t50 < 3 minutes (Table 1, entry 1, Fig. 3a, dA , eA (red peak) and S4†). The result was a mixture of 13 and 14 at a ratio of 1:1.1. Reactivity in pure DMSO-d6 (condition D, Fig. 3e) and the other conditions B and C was low (Fig. 3a, dB
, eA (red peak) and S4†). The result was a mixture of 13 and 14 at a ratio of 1:1.1. Reactivity in pure DMSO-d6 (condition D, Fig. 3e) and the other conditions B and C was low (Fig. 3a, dB , dC
, dC , eB and C, S3–7†). Thus, standard boronate exchange conditions were already compatible with hydrazone and disulfide bonds.
, eB and C, S3–7†). Thus, standard boronate exchange conditions were already compatible with hydrazone and disulfide bonds.
It was difficult to identify the hydrazone exchange conditions compatible with the other two dynamic covalent bonds. Boronate ester 13 hydrolyzed quickly and completely under the acidic hydrazone exchange conditions even with aniline13–15 or with the recent anthranilic acid catalysts,15 because the necessary amounts of acid and catalysts were excessive (Fig. S19†). However, 2-aminophenol 19, acidified by a withdrawing ester in position 4,16 catalyzed hydrazone exchange with a t50 = 11.9 h to reach equilibrium at a ratio 15/17 = 1.3:1 under condition B (Fig. 3b, dB , eB (red peaks), S10†; Table 1, entry 2). Most importantly, the 1H NMR spectrum of boronate ester 13 did not change much under condition B, and disulfides were not affected either (Fig. 3dB
, eB (red peaks), S10†; Table 1, entry 2). Most importantly, the 1H NMR spectrum of boronate ester 13 did not change much under condition B, and disulfides were not affected either (Fig. 3dB ,
,  , eB, S9–13†, Table 1).
, eB, S9–13†, Table 1).
To complete the series needed to demonstrate the existence of the third orthogonal dynamic covalent bond, disulfide exchange from substrates 20 and 21 to mixed product 22 and back was initiated with traces of thiol 23. Under condition C, equilibrium was reached with t50 = 2.5 h at a ratio of 20/22 = 1:1.6 (Fig. 3c, dC , eC (red peak)), without disturbing hydrazones and boronate esters (Fig. 3dC
, eC (red peak)), without disturbing hydrazones and boronate esters (Fig. 3dC ,
,  , eC, S14–18†; Table 1, entry 3).
, eC, S14–18†; Table 1, entry 3).
Compatibility of the third orthogonal dynamic covalent bond with functional systems was explored with multicomponent surface architecture 24 (Fig. 4). This system was obtained by the well-established SOSIP-TSE method (SOSIP = self-organizing surface-initiated polymerization; TSE = templated stack exchange, Fig. S22†).8,9 Namely, ring opening disulfide exchange polymerization was initiated by the thiolate groups bound on an indium-tin oxide (ITO) surface to yield, after the removal of benzaldehyde protecting group, the architecture 25. Reaction of boroxole aldehyde 10* in DMSO/AcOH with the hydrazides along the central stacks in 25 gave surface architecture 26. Incubation of electrode 26 in a solution of nitrodopamine 1 in DMSO/Hünig base afforded the desired multicomponent architecture 24.
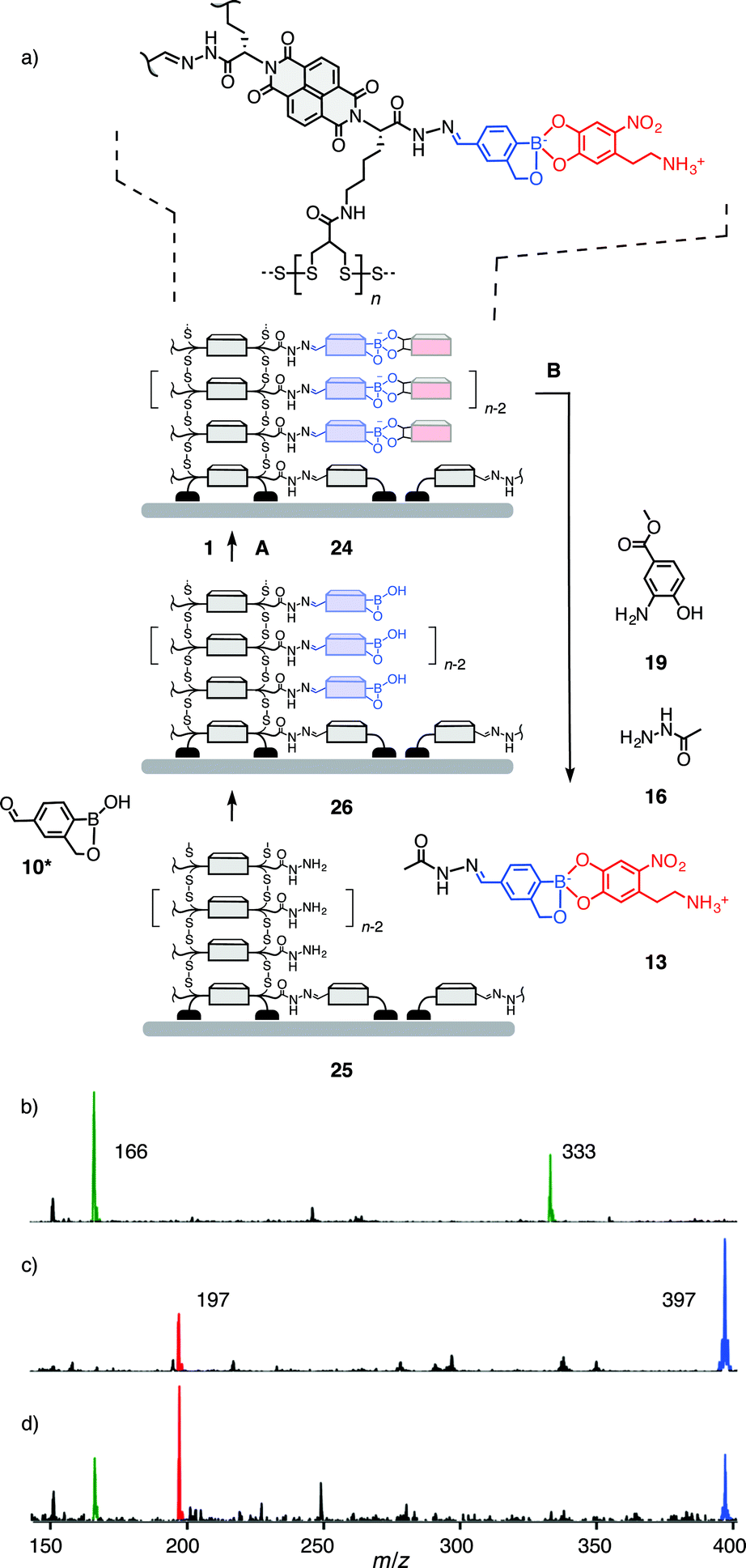 | ||
| Fig. 4 (a) Synthesis of architecture 24 and subsequent recovery of intact boronate ester 13 by orthogonal hydrazone exchange, as evidenced by ESI-MS (negative mode) of (b) catalyst 19 (m/z 166 ([M − H]−), 333 ([2M − H]−)), (c) 13 (m/z 397 ([M − H]−)) and catechol 1 (m/z 197 ([M − H]−)), and (d) the solution obtained from incubation of multicomponent surface architecture 24 with hydrazide 16 and 1.0 mM catalyst 19 in DMSO-d6, 1.5 mM TFA, for 7 h at 40 °C (10: m/z 217 ([M − H]−)). | ||
While the orthogonal formation of three dynamic covalent bonds has been demonstrated for the construction of this system9 and others,10 true orthogonality in functional systems, i.e., selective exchange, cleavage and formation of one bond without disturbing the others, remained to be demonstrated. The so far missing piece of evidence was hydrazone exchange in the presence of intact boronate esters. With the multicomponent surface architecture 24, this translated to the task to release intact boronate ester 13.19 To tackle this challenge, the conditions B elaborated above for selective hydrazone exchange were applied (20 mM hydrazide 16, 1.0 mM catalyst 19, 1.5 mM TFA in DMSO-d6). Because of the small amounts of material present per electrode, the same exchange solution was used for multiple electrodes (10 electrodes for 0.5 mL of solution). 1H NMR of the obtained exchange solution showed the diagnostic peak of the boronate ester 13 at 4.72 ppm and only a small peak of the hydrolysed benzoboroxole 10 at 5.01 ppm (Fig. S24†). Corroborative evidence for the release of intact ester 13 was obtained by ESI-MS (Fig. 4, S25†). These results identify the multicomponent surface architecture 24 as the first functional system that operates with three fully orthogonal organic20 dynamic covalent bonds, to the best of our knowledge. The insights gained here will be important for future developments in the construction of dynamic functional systems, and lead to the tantalizing question of the possible existence of a fourth orthogonal organic dynamic covalent bond.
Acknowledgements
We thank the NMR and the Sciences Mass Spectrometry (SMS) platforms for services, and the University of Geneva, the European Research Council (ERC Advanced Investigator), the Swiss National Centre of Competence in Research (NCCR) Molecular Systems Engineering, the NCCR Chemical Biology and the Swiss NSF for financial support.Notes and references
- (a) X. Wu, Z. Li, X.-X. Chen, J. S. Fossey, T. D. James and Y.-B. Jiang, Chem. Soc. Rev., 2013, 42, 8032–8048 RSC; (b) T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed., 1996, 35, 1910–1922 CrossRef; (c) S. L. Wiskur and E. V. Anslyn, J. Am. Chem. Soc., 2001, 123, 10109–10110 CrossRef CAS PubMed; (d) W. Niu, M. D. Smith and J. J. Lavigne, J. Am. Chem. Soc., 2006, 128, 16466–16467 CrossRef CAS PubMed; (e) M. Li, N. Lin, Z. Huang, L. Du, C. Altier, H. Fang and B. Wang, J. Am. Chem. Soc., 2008, 130, 12636–12638 CrossRef CAS PubMed; (f) G. Zhang, O. Presly, F. White, I. M. Oppel and M. Mastalerz, Angew. Chem., Int. Ed., 2014, 53, 5126–5130 CAS; (g) N. Luisier, K. Schenk and K. Severin, Chem. Commun., 2014, 50, 10233–10236 RSC; (h) B. Akgun and D. G. Hall, Angew. Chem., Int. Ed., 2016, 55, 3909–3913 CrossRef CAS PubMed.
- (a) O. Ramström, S. Lohmann, T. Bunyapaiboonsri and J.-M. Lehn, Chem.–Eur. J., 2004, 10, 1711–1715 CrossRef PubMed; (b) J. M. Poolman, J. Boekhoven, A. Besselink, A. G. L. Olive, J. H. van Esch and R. Eelkema, Nat. Protoc., 2014, 9, 977–988 CrossRef CAS PubMed; (c) T. Takeuchi, J. Montenegro, A. Hennig and S. Matile, Chem. Sci., 2011, 2, 303–307 RSC; (d) X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC; (e) C. S. Mahon, M. A. Fascione, C. Sakonsinsiri, T. E. McAllister, W. Bruce Turnbull and D. A. Fulton, Org. Biomol. Chem., 2015, 13, 2756–2761 RSC.
- (a) H. Y. Au-Yeung, F. B. L. Cougnon, S. Otto, G. D. Pantoş and J. K. M. Sanders, Chem. Sci., 2010, 1, 567–574 RSC; (b) E.-K. Bang, M. Lista, G. Sforazzini, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1752–1763 RSC; (c) N. K. P. Samuel, M. Singh, K. Yamaguchi and S. L. Regen, J. Am. Chem. Soc., 1985, 107, 42–47 CrossRef CAS; (d) R. Singh and G. M. Whitesides, J. Am. Chem. Soc., 1990, 112, 1190–1197 CrossRef CAS; (e) A. G. Torres and M. J. Gait, Trends Biotechnol., 2012, 30, 185–190 CrossRef CAS PubMed; (f) D. Basak, R. Kumar and S. Ghosh, Macromol. Rapid Commun., 2014, 35, 1340–1344 CrossRef CAS PubMed; (g) D. Oupický and J. Li, Macromol. Biosci., 2014, 14, 908–922 CrossRef PubMed; (h) L. Monnereau, M. Nieger, T. Muller and S. Bräse, Adv. Funct. Mater., 2014, 24, 1054–1058 CrossRef CAS; (i) C. Wang, M. R. Krause and S. L. Regen, J. Am. Chem. Soc., 2015, 137, 664–666 CrossRef CAS PubMed; (j) M. Lista, J. Areephong, N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 15228–15231 CrossRef CAS PubMed; (k) G. Gasparini, E.-K. Bang, G. Molinard, D. V Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed; (l) P.-T. Skowron, M. Dumartin, E. Jeamet, F. Perret, C. Gourlaouen, A. Baudouin, B. Fenet, J.-V. Naubron, F. Fotiadu, L. Vial and J. Leclaire, J. Org. Chem., 2016, 81, 654–661 CrossRef CAS PubMed.
- (a) J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed; (b) J.-M. Lehn, Top. Curr. Chem., 2011, 322, 1–32 CrossRef; (c) E. Moulin, G. Cormos and N. Giuseppone, Chem. Soc. Rev., 2012, 41, 1031–1049 RSC; (d) Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC; (e) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC; (f) A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC; (g) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC.
- (a) H. H. Jo, R. Edupuganti, L. You, K. N. Dalby and E. V Anslyn, Chem. Sci., 2015, 6, 158–164 RSC; (b) A. G. Campaña, D. A. Leigh and U. Lewandowska, J. Am. Chem. Soc., 2013, 135, 8639–8645 CrossRef PubMed; (c) J. F. Teichert, D. Mazunin and J. W. Bode, J. Am. Chem. Soc., 2013, 135, 11314–11321 CrossRef CAS PubMed; (d) K. D. Okochi, Y. Jin and W. Zhang, Chem. Commun., 2013, 49, 4418–4420 RSC; (e) B. Icli, E. Solari, B. Kilbas, R. Scopelliti and K. Severin, Chem.–Eur. J., 2012, 18, 14867–14874 CrossRef CAS PubMed; (f) M. J. Barrell, A. G. Campaña, M. von Delius, E. M. Geertsema and D. A. Leigh, Angew. Chem., Int. Ed., 2011, 50, 285–290 CrossRef CAS PubMed; (g) Z. Rodriguez-Docampo and S. Otto, Chem. Commun., 2008, 42, 5301–5303 RSC; (h) A. G. Orrillo, A. M. Escalante and R. L. E. Furlan, Chem. Commun., 2008, 42, 5298–5300 RSC; (i) N. Christinat, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2008, 47, 1848–1852 CrossRef CAS PubMed; (j) Y. Pérez-Fuertes, A. M. Kelly, A. L. Johnson, S. Arimori, S. D. Bull and T. D. James, Org. Lett., 2006, 8, 609–612 CrossRef PubMed; (k) H. Hayashi, A. Sobczuk, A. Bolag, N. Sakai and S. Matile, Chem. Sci., 2014, 5, 4610–4614 RSC.
- S. Hagihara, H. Tanaka and S. Matile, J. Am. Chem. Soc., 2008, 130, 5656–5657 CrossRef CAS PubMed.
- N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 18542–18545 CrossRef CAS PubMed.
- (a) K.-D. Zhang and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 8980–8983 CrossRef CAS PubMed; (b) K.-D. Zhang, N. Sakai and S. Matile, Org. Biomol. Chem., 2015, 13, 8687–8694 RSC.
- L. Rocard, A. Berezin, F. De Leo and D. Bonifazi, Angew. Chem., Int. Ed., 2015, 54, 15739–15743 CrossRef CAS PubMed.
- (a) K. Gademann, Acc. Chem. Res., 2015, 48, 731–739 CrossRef CAS PubMed; (b) S. Zürcher, D. Wäckerlin, Y. Bethuel, B. Malisova, M. Textor, S. Tosatti and K. Gademann, J. Am. Chem. Soc., 2006, 128, 1064–1065 CrossRef PubMed; (c) B. Malisova, S. Tosatti, M. Textor, K. Gademann and S. Zürcher, Langmuir, 2010, 26, 4018–4026 CrossRef CAS PubMed.
- (a) R. Nguyen and I. Huc, Chem. Commun., 2003, 39, 942–943 RSC; (b) S. R. Beeren, M. Pittelkow and J. K. M. Sanders, Chem. Commun., 2011, 47, 7359–7361 RSC.
- (a) E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 826–831 CrossRef CAS; (b) A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS PubMed.
- (a) M. Wendeler, L. Grinberg, X. Wang, P. E. Dawson and M. Baca, Bioconjugate Chem., 2014, 25, 93–101 CrossRef CAS PubMed; (b) M. Rashidian, M. M. Mahmoodi, R. Shah, J. K. Dozier, C. R. Wagner and M. D. Distefano, Bioconjugate Chem., 2013, 24, 333–342 CrossRef CAS PubMed.
- (a) E. T. Kool, D. Park and P. Crisalli, J. Am. Chem. Soc., 2013, 135, 17663–17666 CrossRef CAS PubMed; (b) E. T. Kool, P. Crisalli and K. M. Chan, Org. Lett., 2014, 16, 1454–1457 CrossRef CAS PubMed.
- D. Larsen, M. Pittelkow, S. Karmakar and E. T. Kool, Org. Lett., 2015, 17, 274–277 CrossRef CAS PubMed.
- (a) M. Bérubé, M. Dowlut and D. G. Hall, J. Org. Chem., 2008, 73, 6471–6479 CrossRef PubMed; (b) G. A. Ellis, M. J. Palte and R. T. Raines, J. Am. Chem. Soc., 2012, 134, 3631–3634 CrossRef CAS PubMed.
- M. Li, S. Ishihara, M. Akada, M. Liao, L. Sang, J. P. Hill, V. Krishnan, Y. Ma and K. Ariga, J. Am. Chem. Soc., 2011, 133, 7348–7351 CrossRef CAS PubMed.
- Intact poly(disulfide)s during hydrazone exchange, also under hasher conditions, is the basis of all functional studies realized so far with these systems.5,6k,8,9.
- In this study, inorganic coordination chemistry is not considered. Compare, for example: R. J. Sarma, S. Otto and J. R. Nitschke, Chem.–Eur. J., 2007, 13, 9542–9546 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed procedures and results for all reported experiments. See DOI: 10.1039/c6sc01133k |
| ‡ Present address: Department of Chemistry, Zhejiang Normal University, Jinhua, China. |
| This journal is © The Royal Society of Chemistry 2016 |