
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Overall water splitting by Pt/g-C3N4 photocatalysts without using sacrificial agents†
Guigang
Zhang
,
Zhi-An
Lan
,
Lihua
Lin
,
Sen
Lin
and
Xinchen
Wang
*
State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou, 350002, China. E-mail: xcwang@fzu.edu.cn; Web: http://www.wanglab.fzu.edu.cn
First published on 27th January 2016
Abstract
We report the direct splitting of pure water by light-excited graphitic carbon nitride (g-C3N4) modified with Pt, PtOx, and CoOx as redox cocatalysts, while pure g-C3N4 is virtually inactive for overall water splitting by photocatalysis. The novelty is in the selective creation of both H2 and O2 cocatalysts on surface active sites of g-C3N4via photodeposition triggering the splitting of water for the simultaneous evolution of H2 and O2 gases in a stoichiometric ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, irradiated with light, without using any sacrificial reagents. The photocatalyst was stable for 510 hours of reaction.
:1, irradiated with light, without using any sacrificial reagents. The photocatalyst was stable for 510 hours of reaction.
Using photocatalysts to produce hydrogen sustainably by water splitting is the “holy grail” in modern science. Over the past 40 years, inorganic semiconductors, such as metal oxides and metal (oxy)nitrides, have been utilized as photocatalysts for hydrogen production.1–8 However, direct water splitting in a wireless powder photocatalytic system to produce gaseous hydrogen and oxygen has not yet been achieved using conjugated polymers (CPs). These materials have already shown great promise for use in organic electronics and photovoltaic devices, such as solar cells, light-emitting diodes, and field-effect transistors, due to their good processability and tuneable electronic structures.9–13
The key challenge to using pristine CPs for direct water splitting is the insufficient hopping charge transport of the chains (usually below 10−4 cm2 V−1 s−1) and a poor stability in water and under light irradiation.12 Increasing the structural dimensions of the CPs (e.g., from 1D chains to 2D architectures) is desirable because the hole mobility is greatly increased (up to 0.1 cm2 V−1 s−1) by the remarkably reduced binding energies of the Frenkel-type excitons and the robust stability of the 2D extended π-conjugated units.14 However, further progress in direct water splitting by CPs will rely on breakthroughs in combining stable CP light transducers with suitable redox cocatalysts (usually noble metals) to promote charge separation and to reduce charge build-up on the polymer surface to prevent photocorrosion. Indeed, the promise of this type of system has been demonstrated by the successful development of 2D graphitic carbon nitride (g-C3N4) polymer and metal-based redox cocatalyst systems for CO2 reduction, organic synthesis and water half-splitting reactions using sacrificial reagents.15–22 In contrast, it is difficult to achieve overall water splitting without using sacrificial reagents because it depends not only on a rational chemical synthesis to tune the textural properties of the polymer but also on a rational design of the composite to control the reaction kinetics on the polymer surface.23–27
Photocatalytic water splitting by a prototypical g-C3N4 polymer was shown to be thermodynamically possible because the C2p and N2p orbital bands straddle the water splitting redox potentials,15–22,28–34 but pure g-C3N4 is typically limited by sluggish kinetics in photocatalyzing overall water splitting due to a lack of surface redox active sites. By optimizing the g-C3N4 bulk and morphological properties and employing suitable redox cocatalysts (e.g., Pt for H2 evolution and Co(OH)2 for oxygen evolution), activities for the water half-splitting reactions (water reduction and oxidation) can be dramatically increased.28–34 Therefore, if the appropriate water redox cocatalysts are simultaneously deposited on g-C3N4, pure water splitting to produce gaseous hydrogen and oxygen could be achieved. However, the rough deposition of cocatalysts by traditional chemical reduction (e.g., H2 and NaBH4) cannot fully amplify the activity. Besides, the densely stacked graphitic layer also causes trouble for charge separation and migration due to a long bulk diffusion distance, resulting in a low photocatalytic quantum efficiency.15 It is advisable to reduce the diffusion distance by rational synthesis of a g-C3N4 nanosheet together with suitable cocatalyst modification to achieve water splitting. Up to now, direct water splitting photocatayzed by g-C3N4 CPs in the absence of sacrificial reagents has never been realized and still remains a significant basic science challenge. Here, we demonstrate that light-excited g-C3N4 CPs can induce a one-step water splitting reaction via a four-electron pathway to generate gaseous H2 and O2 in a stoichiometric molar ratio of 2:1 when their morphology is modified and the reaction kinetics are improved by modification with Pt, PtOx, and CoOxvia photodeposition. The optimal g-C3N4-based nanocomposite had a turnover number of 3.1 moles of H2 and O2 per mole of g-C3N4 photocatalyst for the overall water splitting reaction. The nanocomposite was stable in water and under light irradiation.
The g-C3N4 polymers used for photocatalytic water splitting were typically prepared by thermally polymerizing urea into heptazine units at 550 °C which pack together like graphitic crystals. This structure was confirmed by X-ray diffraction (XRD), Fourier transform infrared (FT-IR) spectroscopy, and Raman spectroscopy (Fig. S1†).15,35–37 The g-C3N4 optical properties measured by UV-vis diffuse reflection spectroscopy (DRS) were characteristic of a semiconductor; g-C3N4 had an optical absorption edge at 442 nm due to the excitation of electrons from its valence band to its conduction band (Fig. 1a). The conduction band minimum (CBM) and valence band maximum (VBM) of the g-C3N4 semiconductor were determined to be −1.31 V and 1.49 V (vs. NHE, pH = 7), respectively, from electrochemical Mott–Schottky plots (Fig. 1b and c), where an estimated flat potential was directly used as the conduction band potential. Density functional theory (DFT) calculations revealed that the band gap was 2.56 eV with the CBM and VBM located at −1.0137 and 1.5505 V (vs. NHE, pH = 7), respectively, which enables g-C3N4 to act as a redox shuttle for the water splitting reaction (Fig. S2†). This calculated band gap is consistent with the experimental data and further demonstrates that in theory, g-C3N4 could be used to split water.
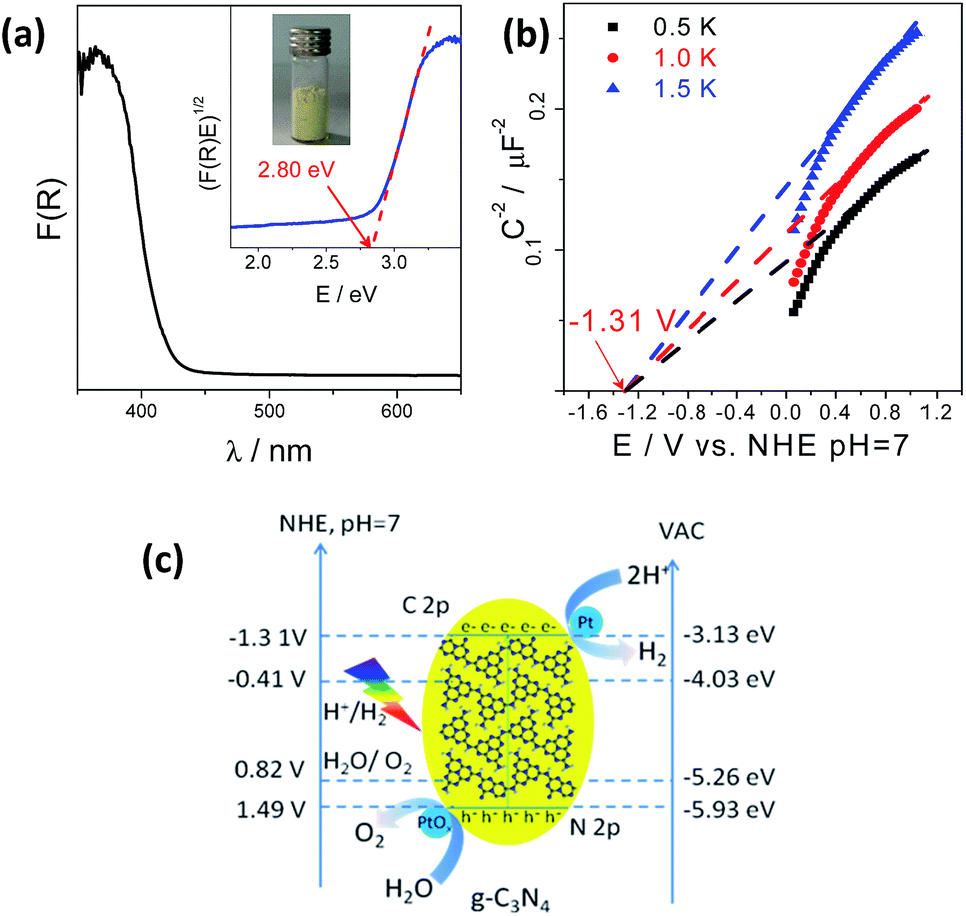 | ||
| Fig. 1 (a) UV-vis DRS spectrum of g-C3N4 polymers; inset: the corresponding Tauc plot. (b) Mott–Schottky plots of the g-C3N4 electrode in 0.2 M Na2SO4, pH = 7. (c) Band structure diagram of g-C3N4 polymers calculated by optical absorption and typical electrochemical Mott–Schottky methods. | ||
First, the effects of g-C3N4 morphology on the photocatalytic activity were investigated. We prepared three types of g-C3N4 using dicyandiamide (DCDA), ammonium thiocyanate (ATC) and urea as precursors. The results showed that after in situ photo-deposition with Pt, the urea-derived g-C3N4 exhibited significant photocatalytic activity for the overall water splitting reaction, while the other samples were inactive for overall water splitting (Table S1†). It should be noted that all pure g-C3N4 polymers showed no activity for overall water splitting in the absence of cocatalysts, implying that surface kinetic control using Pt species was indispensable to achieve overall water splitting by g-C3N4 based photocatalysts. N2 sorption measurements revealed that the DCDA- and ATC-derived g-C3N4 samples had smaller surface areas than the urea-derived samples (ca. 10 m2 g−1vs. 61 m2 g−1). However, mpg-C3N4 with a surface area of ca. 67 m2 g−1 also exhibited no water splitting activity. This indicated that surface area was not the major factor in controlling the water splitting activity and the splitting of water on densely stacked g-C3N4 polymers was indeed very difficult to achieve. To better understand the real mechanism of water splitting on the soft surface of the CPs, we characterized the morphology of the above different polymers. TEM images of DCDA- and ATC-derived g-C3N4 and mpg-C3N4 samples revealed densely stacked polymer layers, which were very different from the silk-like thin nanosheets of the urea-derived one (Fig. S3†). The fast evolution of O in the form of CO2 or CO could accelerate the deamination rate. Thus, the texture, morphology and electronic properties of the CNU samples were optimized, and contributed to creating the active Pt/g-C3N4 photocatalysts for overall water splitting. Evidently, accelerated charge separation and migration on the nanosheets can be obtained in comparison with densely stacked graphitic layers, which is elucidated well by the corresponding large decrease of PL emission intensity (Fig. S4†). The nanosheet structure can also be certified by an AFM experiment. As shown in Fig. 2a, the thickness of the nanosheet is determined as ∼2 nm. One can now easily conclude that the ultrathin 2D geometry of urea-derived g-C3N4 is crucial for achieving overall water splitting as demonstrated by the fact that g-C3N4 samples prepared from urea at different temperatures all have remarkable water splitting activities (Fig. S5†) due to their similar thin nanosheet structures (Fig. S6†). The CNU samples prepared at 550 °C showed optimum activities. This is because when the temperature is lower than 550 °C, the heptazine cycle doesn't completely form, while partial decomposition occurs when the temperature is higher than 550 °C. Both of these two aspects may generate inactive CNU samples. DCDA- and ATC-derived g-C3N4 and mpg-C3N4 samples revealed densely stacked polymer layers, and the deposition rate of Pt nanoparticles on the surface of the polymer was very slow, in the absence of organic sacrificial agents to react with the holes. Optimization of the deposition technique of Pt is needed to enhance the overall water splitting activities of this bulky g-C3N4.
 | ||
| Fig. 2 (a) AFM image of the g-C3N4 polymers with Pt deposited in situ. (b) TEM image of the g-C3N4 polymers with Pt deposited in situ. (c) HR-TEM image of the g-C3N4 polymers with Pt deposited in situ. (d) STEM images of the g-C3N4 polymers with Pt deposited in situ. Scale bar for a, b, c and d is 50 nm, 100 nm, 2 nm and 50 nm, respectively. | ||
We then investigated the effect of cocatalyst loading techniques on the photocatalytic water splitting activity. Three different cocatalyst loading techniques, in situ photodeposition, and H2 and NaBH4 reduction, were developed to decorate the g-C3N4 nanosheets. As shown in Fig. S7,† evident water splitting activity was generated for photodeposition of Pt on the surface of the g-C3N4 nanosheets, while only very slow H2 and no O2 evolution were found for both H2 and NaBH4 reduction modified ones. In the first case, when g-C3N4 was irradiated with light, photoexcited charge carriers were generated and then immediately migrated to the surface of the g-C3N4 nanosheets without recombination. The surface adsorbed Pt4+ was then reduced in situ by the excited electrons and deposited on the active sites, which can efficiently promote the water splitting. For the other investigated techniques, Pt4+ was reduced by H2 or NaBH4 and then randomly deposited on the surface, resulting in poor activities. The selective photodeposition of Pt on thin g-C3N4 nanosheets resulted in a uniform dispersion of ultrafine Pt nanoparticles (∼1–2 nm) with a (111) crystal lattice spacing of ∼0.23 nm (Fig. 2b and c). The homogeneous deposition of Pt can be further proved by STEM imaging (Fig. 2d). However, serious particle accumulation occurred when the Pt cocatalysts were deposited by H2 and NaBH4 reduction (Fig. S8†), which was the major hindrance which led to decreased water splitting activity.
We also investigated the chemical composition and valence state of the Pt species. As shown in Fig. 3a and b, electron energy loss spectroscopy (EELS) and XRD analysis confirmed the existence of a Pt (111) plane.38 Besides, no evident structure variation occurred after modification with the Pt cocatalysts, implying a robust stability of the g-C3N4 CPs.39–41 Three pairs of XPS peaks corresponding to Pt0, Pt2+, and Pt4+ with binding energy at 72.13, 74.26 and 78.17 eV, respectively, were measured (Fig. 3c). Pt0 was effective for H2 evolution while PtOx were able to promote O2 evolution.42 However, two pairs of XPS peaks were deconvoluted for a NaBH4 reduction modified one (Fig. S9†), indicating the complete reduction of Pt4+ into Pt2+ and Pt0. To confirm that PtOx were active for the promotion of a water oxidation reaction, we evaluated the photocatalytic water oxidation activities of the as-prepared PtOx/g-C3N4. As shown in Fig. S10,† this material showed enhanced activity for water oxidation in comparison with the pure one, emphasizing the positive role of PtOx in improving the water oxidation rate. In addition, the water splitting rates and evolved H2/O2 gas ratio (Fig. S11†) could be finely tuned by simply adjusting the total loading from 0.2 to 5 wt% due to the change of the ratio of Pt and PtOx intensities (Fig. S12 and Table S2†) and the alteration of particle size (Fig. S13†). The creation of metal/polymer surface junctions promotes the interfacial redox reaction which can be confirmed by a rapidly decreased PL intensity (Fig. 3d). The optimum activity was achieved at a Pt loading content of 3 wt%. When Pt or PtOx were singly deposited on the g-C3N4 nanosheets, the sample exhibited very poor activity in both cases, which once again highlighted that the simultaneous creation of both H2 and O2 evolution cocatalysts on the active sites was indeed essential for triggering the overall splitting of water.
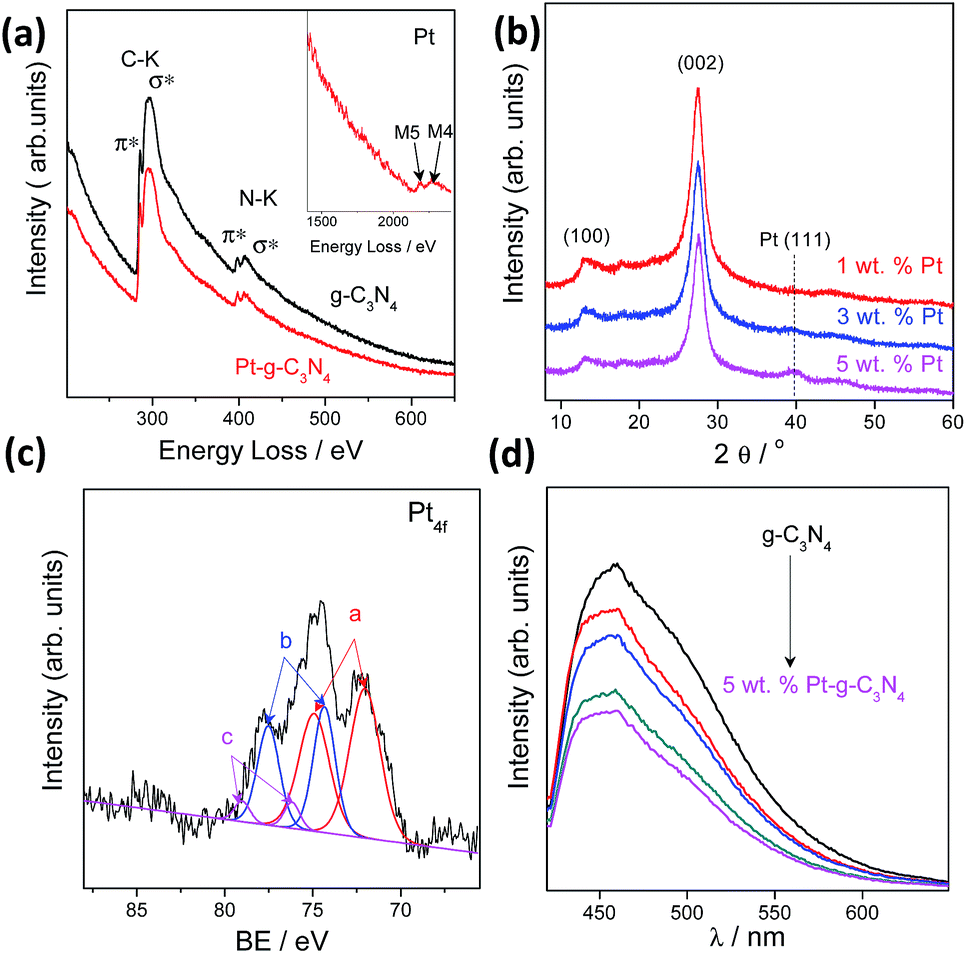 | ||
| Fig. 3 (a) EELS of the g-C3N4 polymers with Pt deposited in situ. (b) XRD of the g-C3N4 polymers with Pt deposited in situ. (c) High resolution of XPS analysis of Pt4f. (d) PL spectra of the g-C3N4 polymers with Pt deposited in situ. | ||
The g-C3N4 nanosheets modified by other noble metals (e.g., Rh, Ru, or Au) via in situ photodeposition all just showed trace H2 and no O2 evolution (Fig. S14†), implying the importance of Pt for water splitting. The pH value and amount of polymer powders used for water splitting were also optimized (Fig. S15 and S16†). The optimum water splitting rate was obtained for samples prepared by photodepositing 3 wt% Pt on 0.2 g of g-C3N4 nanosheets under neutral conditions. We then evaluated their stability for long term reaction.
As shown in Fig. S17,† the optimized Pt/g-C3N4 showed good water splitting stabilities under both UV and visible light irradiation for 580 hours of continuous reaction. It should be noted that N2 gas was evolved along with H2 and O2 at the initial stage of the reaction. This arises from the self-oxidation of the surface un-condensed amino groups (–NH) by excited holes.43–45 As the reaction proceeded, after 80 hours almost no N2 evolution was observed, suggesting a complete consumption of the –NH groups. When the Xe lamp was turned off, the amounts of the evolved gases quickly diminished in just four hours (Fig. S18†), indicating a fast occurrence of the backward reaction of water splitting on the Pt species (H2 and O2 recombination for water formation). Thus, to further enhance the overall water splitting activity of the system, an efficient restraint of the backward reaction via rational structural design of the cocatalysts (e.g., core/shell nanostructure) should be considered.
The addition of cobalt species for in situ formation of cobalt-based cocatalysts can also sufficiently promote the water oxidation selectivity and efficiency of metal-free semiconductors such as g-C3N4 and h-BCN.43–47 As expected, the simultaneous evolution of H2 and O2 gases in a stoichiometric ratio of 2:1 by Pt–Co/g-C3N4 under UV (λ > 300 nm) (12.2 and 6.3 μmol h−1) (Fig. 4a) and visible light irradiation (λ > 420 nm) (1.2 and 0.6 μmol h−1) (Fig. 4b) was significantly enhanced after 1 wt% CoOx were further modified for use as O2 evolution cocatalysts, which can be determined by XPS analysis (Fig. S19†). The slightly decreased activity in each run of reaction may be attributed to the stacked samples on the inner side of the reactor (Fig. S20†). Furthermore, no obvious deactivation was observed after 510 hours of reaction (Fig. S21†), demonstrating the robust resistance of the composites to water and light corrosion at the soft interface. The total amount of gaseous H2 and O2 collected reached ∼6.2 mmol, which corresponded to turnover numbers (TON) of 3.1 and 111.3 based on g-C3N4 and Pt, respectively. The apparent quantum yield (AQY) for the overall water splitting reaction was calculated to be 0.3% at 405 nm (Fig. S22†) and was monitored by an on-line gas chromatograph (Fig. S23†). This is lower than the value of 2.5% of (Ga1−xZnx) (N1−xOx) inorganic photocatalysts. However, it is a remarkable first observation that photocatalytic overall water splitting can occur on the surface of an organic/polymer semiconductor via a 4-electron pathway. Optimization of the system to further improve the efficiency is ongoing in our lab.
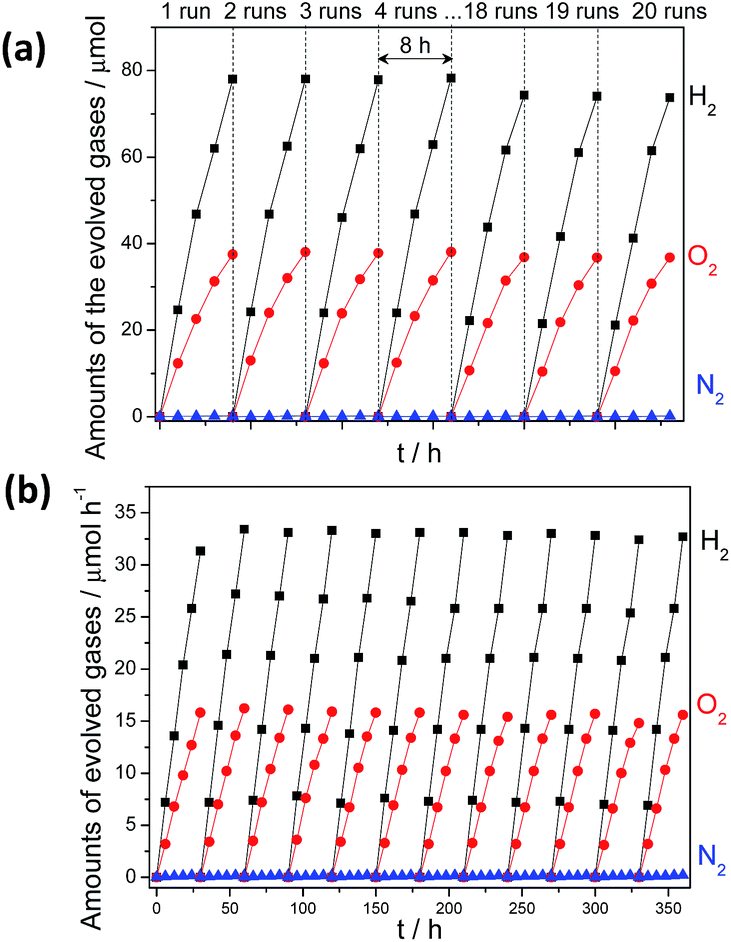 | ||
| Fig. 4 Time course of water splitting activities of 3 wt% Pt, PtOx and 1 wt% CoOx Co-modified g-C3N4 polymers under (a) UV-vis (λ > 300 nm) irradiation and (b) visible light (λ > 420 nm) irradiation. | ||
Conclusions
The discovery of Pt/g-C3N4 CPs that can split pure water without the use of sacrificial reagents establishes a new chemical paradigm for exploiting clean, renewable solar energy using organic semiconductor light-energy transducers. Ongoing efforts are focused on modifying the electronic and textural structures of g-C3N4 CPs and coupling them to low-cost kinetic promoters to facilitate photocatalytic cascade processes for water splitting and CO2 fixation that are relevant to sustainable energy production via artificial photosynthesis.48–50Acknowledgements
This work is financially supported by the National Basic Research Program of China (2013CB632405), and the National Natural Science Foundation of China (21425309).Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed
- N. Lewis and D. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed
- M. Jacobson, W. Colella and D. Golden, Science, 2005, 308, 1901–1905 CrossRef CAS PubMed
- H. Yang, C. Sun, S. Qiao, J. Zou, G. Liu, S. Smith, H. Cheng and G. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625–627 CrossRef CAS PubMed
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed
- A. Paracchino, V. Laporte, K. Sivula, M. Gratzel and E. Himsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed
- J. Burroughes, D. Bradley, A. Brown, R. Marks, K. Mackay, R. Friend, P. Burns and A. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS
- N. Sariciftci, L. Smilowitz, A. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CAS
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. Macdonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner and W. Zhang, Nat. Mater., 2006, 5, 328–333 CrossRef CAS PubMed
- W. Huynh, J. Dittmer and A. Alivisatos, Science, 2002, 295, 2425 CrossRef CAS PubMed
- A. Slater and A. Cooper, Science, 2015, 348, 988–997 CrossRef CAS PubMed
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. Langeveld-Voss, A. Spiering, R. Janssen, E. Meijer and P. Herwig, Nature, 1999, 401, 685–688 CrossRef CAS
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- D. Zheng, C. Pang and X. Wang, Chem. Commun., 2015, 51, 17467–17470 RSC
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11001–11005 CrossRef CAS PubMed
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed
- F. Goettmann, A. Thomas and M. Antonietti, Angew. Chem., Int. Ed., 2007, 46, 2717–2720 CrossRef CAS PubMed
- R. Sprick, J. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS PubMed
- G. Liu, T. Wang, H. Zhang, X. Meng, D. Hao, K. Chang, P. Li, T. Kako and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 13561–13565 CrossRef CAS PubMed
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096–4099 CrossRef CAS PubMed
- S. Khan, M. Al-Shahry and W. Ingler, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed
- Z. Yi, J. Ye, N. Kikugawa, T. Kako, S. Ouyang, H. Stuart-Williams, H. Yang, J. Cao, W. Luo and Z. Li, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed
- X. Chen, L. Liu, Y. Yu and S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS PubMed
- Y. Jun, J. Park, S. Lee, A. Thomas, W. Hong and G. Stucky, Angew. Chem., Int. Ed., 2013, 52, 11083–11087 CrossRef CAS PubMed
- Y. Zheng, L. Lin, X. Ye, F. Guo and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 11926–11930 CrossRef CAS PubMed
- P. Niu, L. Yin, Y. Yang, G. Liu and H. Cheng, Adv. Mater., 2014, 26, 8046–8052 CrossRef CAS PubMed
- J. Zhang, M. Zhang, L. Lin and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 6297–6301 CrossRef CAS PubMed
- Y. Hou, A. Laursen, J. Zhang, G. Zhang, Y. Zhu, X. Wang, S. Dahl and I. Chorkendorff, Angew. Chem., Int. Ed., 2013, 52, 3621–3625 CrossRef CAS PubMed
- G. Zhang, S. Zang and X. Wang, ACS Catal., 2015, 5, 941–947 CrossRef CAS
- D. Martin, P. Reardon, S. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568–19245 CrossRef CAS PubMed
- M. Shalom, S. Inal, C. Fettkenhauer, D. Neher and M. Antonietti, J. Am. Chem. Soc., 2013, 135, 7118–7121 CrossRef CAS PubMed
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. Muller, R. Schlogl and J. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC
- Y. Zheng, Y. Jiao, Y. Zhu, L. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Qiao, Nat. Commun., 2014, 5, 3783–3791 Search PubMed
- Q. Liang, Z. Li, X. Yu, Z. Huang, F. Kang and Q. Yang, Adv. Mater., 2015, 27, 4634–4639 CrossRef CAS PubMed
- Y. Zhao, F. Zhao, X. Wang, C. Xu, Z. Zhang, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2014, 53, 13934–13939 CrossRef CAS PubMed
- J. Hong, X. Xia, Y. Wang and R. Xu, J. Mater. Chem., 2012, 22, 15006–15012 RSC
- J. Kiwi and M. Graetzel, Angew. Chem., Int. Ed. Engl., 1978, 17, 860–861 CrossRef
- J. Zhang, M. Grzelczak, Y. Hou, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Sci., 2012, 3, 443–446 RSC
- G. Zhang, C. Huang and X. Wang, Small, 2015, 11, 1215–1221 CrossRef CAS PubMed
- G. Zhang, S. Zang, Z. Lan, C. Huang, G. Li and X. Wang, J. Mater. Chem. A, 2015, 3, 17946–17950 CAS
- M. Zhang, Z. Luo, M. Zhou, C. Huang and X. Wang, Sci. China Mater., 2015, 58, 867–876 CrossRef
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 7698–7704 CrossRef PubMed
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 CAS
- J. Qin, S. Wang, H. Ren, Y. Hou and X. Wang, Appl. Catal., B, 2015, 179, 1–8 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Characterization and experimental detail. See DOI: 10.1039/c5sc04572j |
| This journal is © The Royal Society of Chemistry 2016 |