
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aminofluorination: transition-metal-free N–F bond insertion into diazocarbonyl compounds†
Gui
Chen
a,
Jinshuai
Song
b,
Yinghua
Yu
a,
Xuesong
Luo
a,
Chunsen
Li
*b and
Xueliang
Huang
*a
aKey Laboratory of Coal to Ethylene Glycol and Its Related Technology, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Yangqiao west road 155#, Fuzhou, Fujian 350002, China. E-mail: huangxl@fjirsm.ac.cn; Web: http://www.fjirsm.cas.cn/yjxt/yyhxyjzs/hxlktz/
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Yangqiao west road 155#, Fuzhou, Fujian 350002, China. E-mail: chunsen.li@fjirsm.ac.cn
First published on 14th December 2015
Abstract
Gem-aminofluorination of diazocarbonyl compounds has been achieved for the first time. This reaction proceeds under mild conditions and does not require any transition-metal promoter or catalyst. Treatment of diazoesters with N-fluorobenzenesulfonimide (NFSI), which serves as both a fluorine and nitrogen source, results in the facile construction of C–N and C–F bonds, providing aminofluorination products in moderate to excellent yields. Kinetic studies and DFT calculations have provided valuable insight into the potential mechanism for this novel N–F bond insertion.
Introduction
Diazoacetate derivatives play an important role in the synthetic community. Over the past century, significant advances have been made towards the generation of carbenoid intermediates triggered by transition-metals. The resulting reactive species can undergo valuable transformations, such as three-membered ring (cyclopropane, cyclopropene) formation, X–H bond insertion (X = C, N, O, S, etc.) and ylide generation (Scheme 1a–c).1 van Vranken, Barluenga, Wang, and others have developed palladium- or copper-catalysed multiple component reactions of diazo compounds, which allow the installation of two separated functional moieties on the carbenic carbon via a single operation (Scheme 1d).2 Despite these important advances, the direct introduction of two functional groups to the same carbon center, namely gem-difunctionalization of donor/acceptor (D/A) carbenes, is still far from well developed.3,4 It is of note that these processes at least involve one C–C bond formation.5 Thus the studies on the gem-difunctionalization of D/A carbenes, which involves two distinct carbon heteroatom bond formations, would greatly enhance the synthetic applications of diazo compounds.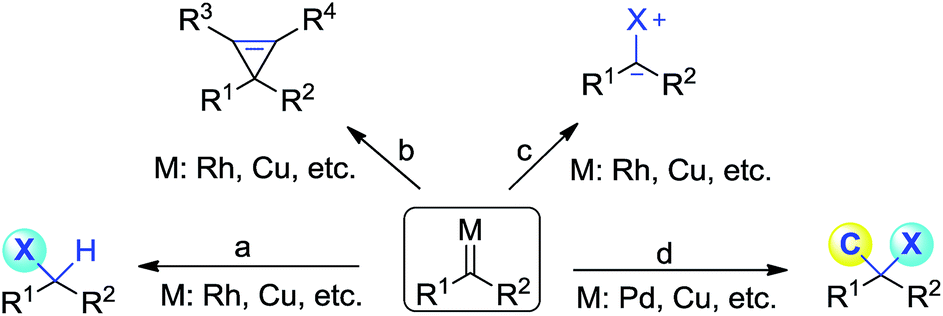 | ||
| Scheme 1 Representative reaction modes of a transition-metal carbenoid. | ||
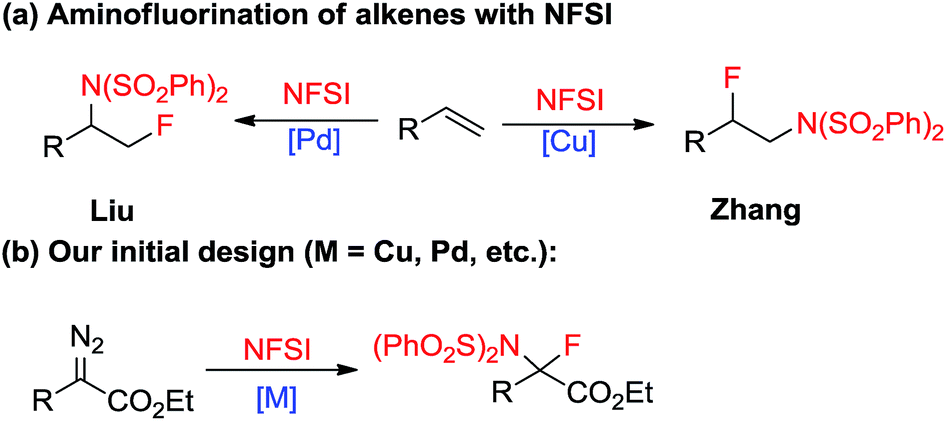
Single substitution of hydrogen with fluorine may alter the chemical and physical properties of a potential drug candidate by blocking undesired metabolism at a specific site.6 In contrast to the relatively large number of reports on the catalytic insertion of N–H bonds to α-diazocarbonyl compounds,7 a simple yet appealing concept for the transition-metal catalysed N–F bond insertion has not been realized thus far. N-Fluorobenzenesulfonimide (NFSI) is inexpensive and shelf stable, and is often employed as a mild electrophilic fluorinating or aminating reagent.8,9 Recently, Liu10 and Zhang11 demonstrated that NFSI could serve as both an amino and fluorine source for the transition-metal-catalysed aminofluorination of alkenes (Scheme 2a). Inspired by these seminal works, we envisioned that NFSI might be an ideal candidate for the transition-metal catalysed N–F bond insertion into D/A carbenes (Scheme 2b). Although procedures for the amination7,12 or fluorination13,14 of diazo compounds are known, to our knowledge, direct aminofluorination of diazo compounds remains unexplored. Herein, we present our primary results on gem-aminofluorination of diazocarbonyl compounds under mild conditions. Kinetic studies and DFT calculations shed light on the mechanism of the current N–F bond insertion.
 | ||
| Scheme 2 Aminofluorination catalysed by transition-metals. | ||
Results and discussion
Optimization studies and substrate scope
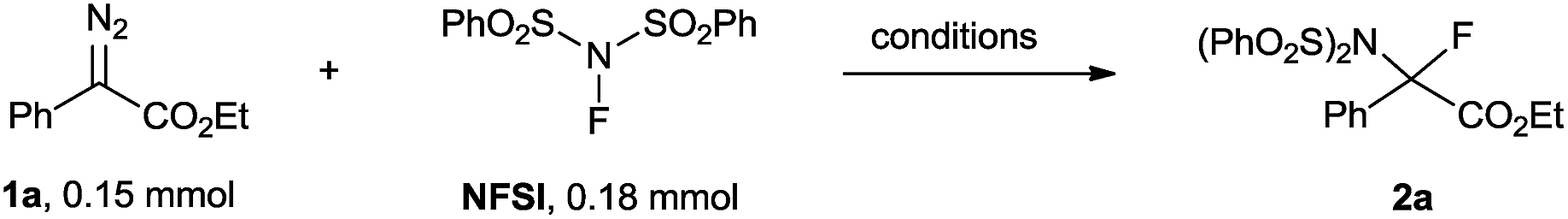
We initiated our studies using the reaction of ethyl diazophenyl acetate 1a (ref. 15) with NFSI using CuBr as a precatalyst and bathocuproine (2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline; BC) as a ligand.11 Gratifyingly, in the presence of 5 mol% CuBr and 6 mol% BC, ethyl diazophenyl acetate 1a was completely consumed after stirring in a reaction medium of 1,2-dichloroethane (DCE) at 60 °C for 21 h, and the desired product 2a was obtained in a 79% NMR yield (Table 1, entry 1). To our surprise, the reaction proceeded equally well in the absence of both CuBr and BC, giving 2a in 89% yield (Table 1, entry 2). The reaction could complete in a comparably short time when the reaction was carried out at an elevated temperature (Table 1, entries 2–4 vs. entry 5). A brief examination of the solvent effects revealed that DCE was still the best choice (Table 1, entries 6–14). Notably, the reaction can also be performed in water, giving 2a in a moderate yield, which indicates an environmentally benign perspective (Table 1, entry 15). Due to the similar polarity between 2a and NFSI, a slight excess of 1a (1.5 eq.) was necessary to make sure that NFSI reached full conversion. In this case, 2a was isolated in a nearly quantitative yield (Table 1, entry 16).|
|
||||
|---|---|---|---|---|
| Entrya | Solvent | Temp (°C) | Time (h) | Yieldb (%) |
| a All reactions were carried out under an atmosphere of argon in 0.15 mmol scale, [1a] = 0.10 M, [NFSI] = 0.12 M. b Yields were determined by 19F NMR, using 1-bromo-4-fluorobenzene as the internal standard. c In the presence of 5 mol% CuCl and 6 mol% BC. d Under air. e The reaction was carried out in 0.3 mmol scale, [1a] = 0.15 M, [NFSI] = 0.10 M. f Isolated yield. | ||||
| 1c | DCE | 60 | 21 | 79 |
| 2 | DCE | 60 | 21 | 89 |
| 3 | DCE | 80 | 7 | 77 |
| 4 | DCE | 100 | 4 | 73 |
| 5 | DCE | RT | 24 | 17 |
| 6 | CH2Cl2 | 60 | 24 | 81 |
| 7 | CHCl3 | 60 | 24 | 70 |
| 8 | Toluene | 60 | 24 | 58 |
| 9 | MeCN | 60 | 24 | 69 |
| 10 | THF | 60 | 24 | 47 |
| 11 | Dioxane | 60 | 24 | 22 |
| 12 | DMF | 60 | 24 | 0 |
| 13 | MeOH | 60 | 24 | 0 |
| 14 | H2O | 60 | 12 | 62 |
| 15d | DCE | 60 | 24 | 82 |
| 16e | DCE | 60 | 48 | 97f |
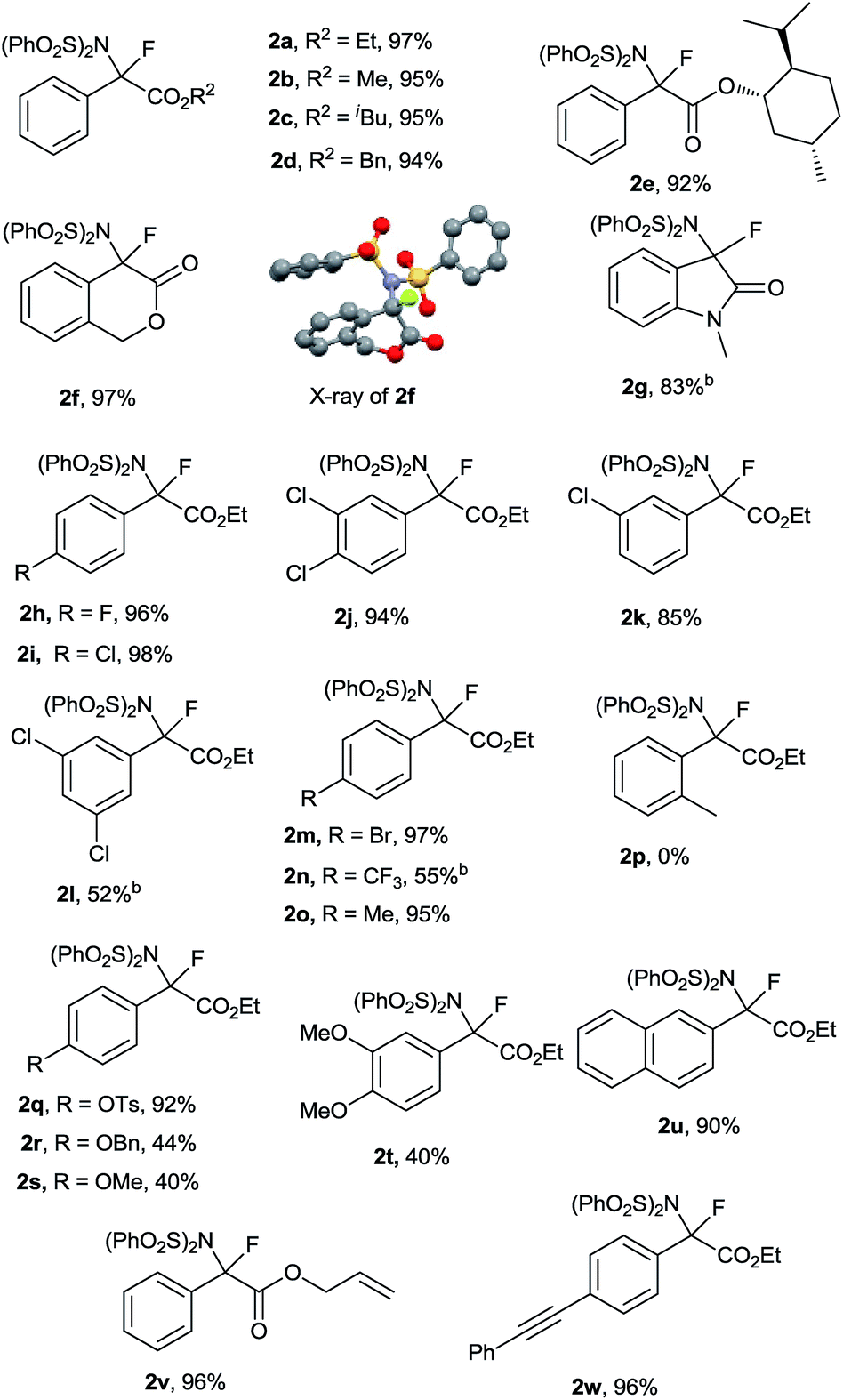
With the optimized reaction conditions in hand (Table 1, entry 16), the generality and limitations of this metal-free N–F bond insertion were investigated. The results are summarized in Table 2. Most of the α-diazoacetates reacted with NFSI smoothly to give the corresponding products in moderate to excellent yields. When changing the ester group (R2) from methyl to isobutyl, or benzyl, no significant decrease of the product yields was observed (Table 2, 2a–d). When the diazo compound contained a chiral auxiliary, an ester derived from (+)-menthol, a pair of diastereoisomers 2e were obtained in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Interestingly, diazo compounds derived from cyclic esters and amides were proven to be viable substrates. N–F bond insertion of 1f and 1g gave the corresponding products 2f and 2g in 97% and 83% yields, respectively. The structure of 2f was confirmed by X-ray crystallographic analysis.16 The effects of substituents on the phenyl ring were also examined. Both electron-withdrawing and electron-donating groups on the aromatic ring of 1 were tolerated under the reaction conditions. The reaction of diazo acetates bearing mild electron-withdrawing or electron-donating groups (fluoro, chloro, bromo and methyl) at the para position of the phenyl ring gave the corresponding products in excellent yields (Table 2, 2h, 2i, 2m and 2o). Incorporation of one chloro group to the meta position had no obvious impact on the yields of the products (Table 2, 2i and 2k). Interestingly, the yield of 2l bearing two meta chloro substituents was decreased to 52%. While the diazo acetate bearing an ortho substituted group was not compatible to the current conditions (Table 2 and 2p), probably due to the steric hindrance effect. Similarly, strong electron-donating or -withdrawing substituents on the aromatic ring were amenable for the current aminofluorination, albeit giving moderate yields (Table 2, 2n, 2r–t). Of note, vinyl and alkyl moieties remained intact (Table 2, 2v and 2w), indicating that the involvement of a free carbene intermediate is less likely (vide infra). Notably, the reaction could be scaled up to gram scale without sacrificing the yield of 2a (5 mmol scale, 2.39 g 2a was obtained with a 97% yield). It is worthwhile to mention that the current conditions are not applicable to alkyl or heteroaryl acetate derived diazo compounds.17
:1. Interestingly, diazo compounds derived from cyclic esters and amides were proven to be viable substrates. N–F bond insertion of 1f and 1g gave the corresponding products 2f and 2g in 97% and 83% yields, respectively. The structure of 2f was confirmed by X-ray crystallographic analysis.16 The effects of substituents on the phenyl ring were also examined. Both electron-withdrawing and electron-donating groups on the aromatic ring of 1 were tolerated under the reaction conditions. The reaction of diazo acetates bearing mild electron-withdrawing or electron-donating groups (fluoro, chloro, bromo and methyl) at the para position of the phenyl ring gave the corresponding products in excellent yields (Table 2, 2h, 2i, 2m and 2o). Incorporation of one chloro group to the meta position had no obvious impact on the yields of the products (Table 2, 2i and 2k). Interestingly, the yield of 2l bearing two meta chloro substituents was decreased to 52%. While the diazo acetate bearing an ortho substituted group was not compatible to the current conditions (Table 2 and 2p), probably due to the steric hindrance effect. Similarly, strong electron-donating or -withdrawing substituents on the aromatic ring were amenable for the current aminofluorination, albeit giving moderate yields (Table 2, 2n, 2r–t). Of note, vinyl and alkyl moieties remained intact (Table 2, 2v and 2w), indicating that the involvement of a free carbene intermediate is less likely (vide infra). Notably, the reaction could be scaled up to gram scale without sacrificing the yield of 2a (5 mmol scale, 2.39 g 2a was obtained with a 97% yield). It is worthwhile to mention that the current conditions are not applicable to alkyl or heteroaryl acetate derived diazo compounds.17

Mechanistic studies
Having uncovered an efficient method for N–F bond insertion, we sought to gain more insight into the reaction mechanism. Thus a series of additional experiments were subsequently carried out. Performing the reaction under irradiation by UV light, 2a was obtained in a much lower yield (Scheme 3a). Furthermore, evolution of N2 was significantly slow in the absence of NFSI under otherwise identical conditions (Fig. S1†). These experiments also suggest that a pathway via a free carbene intermediate is unfavourable. Addition of the radical scavenger 2,6-di-tert-butyl-4-methylphenol (BHT) had no obvious influence on the reaction efficiency (Scheme 3b), which indicates that a mechanism that involves free radical species is also less likely. Additionally, considering the diazo carbon atom is mildly nucleophilic and might be trapped by the electrophilic fluorine atom, a subsequent displacement of dinitrogen by the imide moiety of NFSI would give 2a. With this consideration in mind, the following control experiment was carried out immediately. The combination of selectfluor, a reactive electrophilic fluorinating reagent, with tetrapropyl ammonium benzenesulfonimide did afford 2a in 8% yield (Scheme 3c).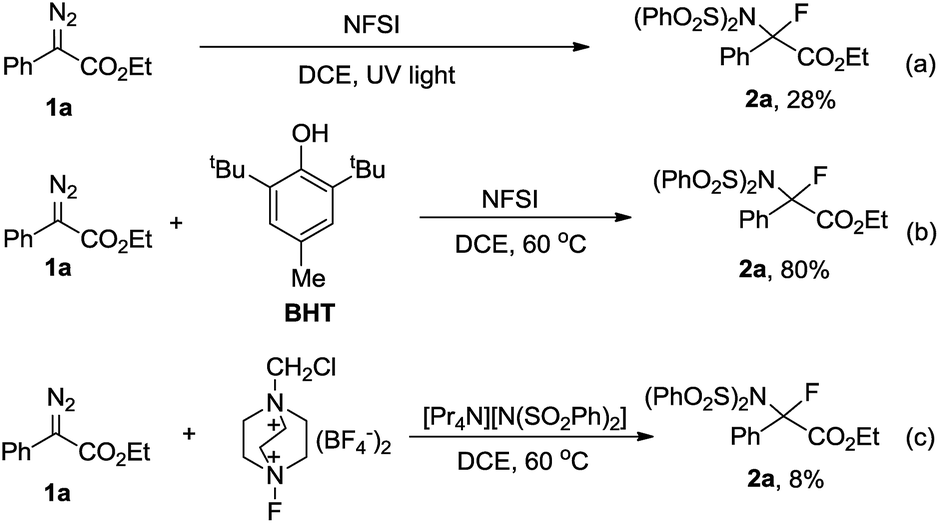 | ||
| Scheme 3 Preliminary mechanistic studies. | ||
Kinetic studies for the reaction of 1a with NFSI were further performed to get a deeper understanding of the reaction mechanism. The reaction was found to be first-order in both 1a and NFSI (Fig. S2–S5†). The activation parameters ΔH‡ = 17.1 kcal mol−1 and ΔS‡ = −13.0 cal mol−1 K−1 were obtained from Eyring plots by varying the temperature from 313 to 353 K (Fig. 1A, top). The negative ΔS‡ value may suggest the generation of a bimolecular transition state involving NFSI and 1a. Similar kinetic behaviours were observed for the reactions of various para-substituted diazo phenyl acetates 1 with NFSI. A fairly linear Hammett correlation between log(kX/kH) and σ+ was obtained with a reaction constant of ρ = −0.81 (Fig. 1B, bottom). The small negative ρ value suggests that the transition state is weakly polarized with a positive charge at the reaction center.18,19
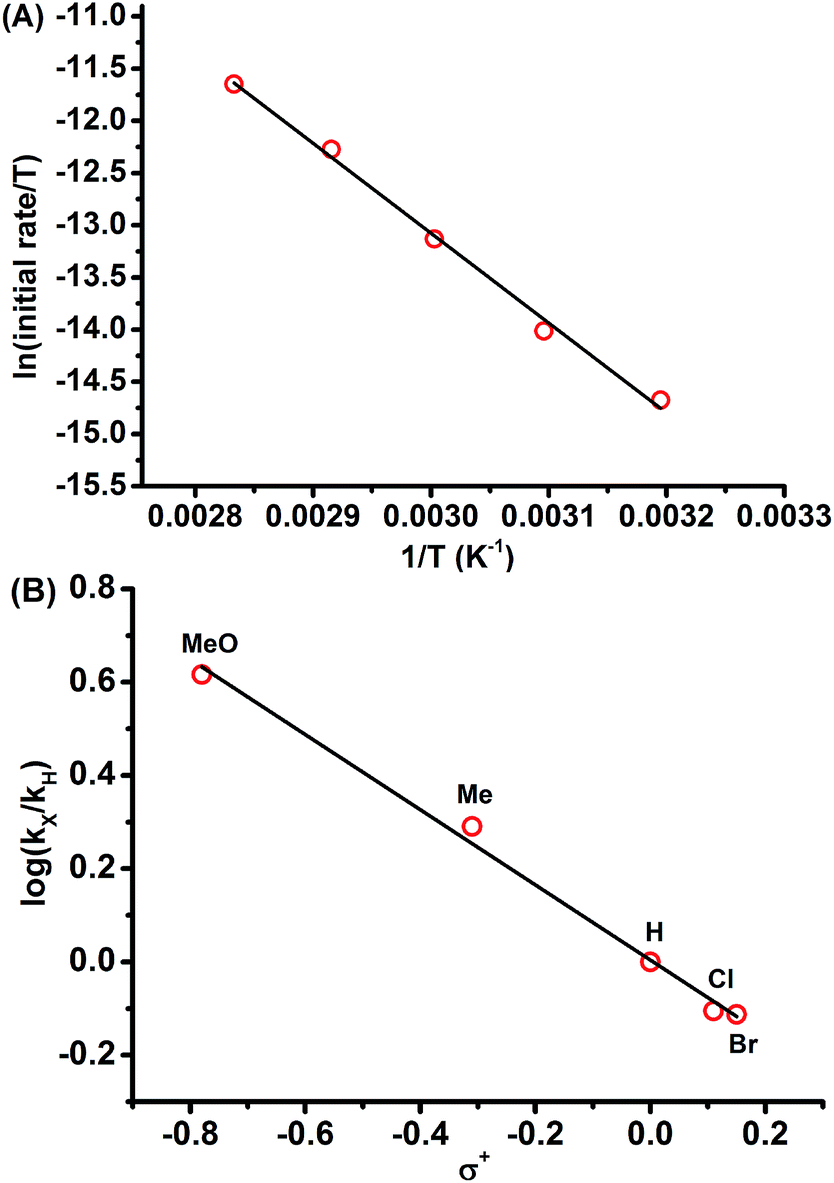 | ||
| Fig. 1 (A) (top) Plot of ln(initial rate/T) vs. 1/T for the reaction between 1a and NFSI in DCE, [1a] = 0.15 M, [NFSI] = 0.1 M, slope = −8.61 × 103, y-intercept = 1.27 × 10, r2 = 0.996. (B) (bottom) Hammett plot of log(kX/kH) vs. σ+ for the reaction of NFSI with para substituted diazo phenyl acetates 1 in DCE at 50 °C, [1] = 0.15 M, [NFSI] = 0.1 M, slope = −0.81, y-intercept = 0.4 × 10−2, r2 = 0.995. | ||
DFT calculations were carried out to provide more details on the potential mechanism (Scheme 4).20 The reaction of 1a with NFSI encounters an activation enthalpy of 16.2 kcal mol−1 with a slight loss of entropy (ΔS‡ = −9.0 cal mol−1 K−1), which matches well with the experimental values (vide supra). According to the downhill energy profile, the following steps are rather facile. The weakly polarized TS further collapses to an ion pair Int1 with an energy change of −41.8 kcal mol−1. Releasing N2 from Int1 has been proven to be an exothermic process. Finally, an SN1 like reaction takes place to give 2a, with a total enthalpy change of −84.8 kcal mol−1. It is of note, that the formation of a procarbonium ion through the reaction of diazo carbon with Cl+, Br+ and I+ has been reported recently.21 However, in our case, the replacement of NFSI by NXS (X = Cl, Br, I) results in no formation of the aminohalogenated product. It is well known that diazo compounds are not stable in the presence of a strong acid. Indeed, addition of strong Brønsted acids (CCl3CO2H or CF3CO2H) led to the rapid decomposition of 1a. However, the addition of acetic acid had no influence on the current N–F bond insertion.22,23
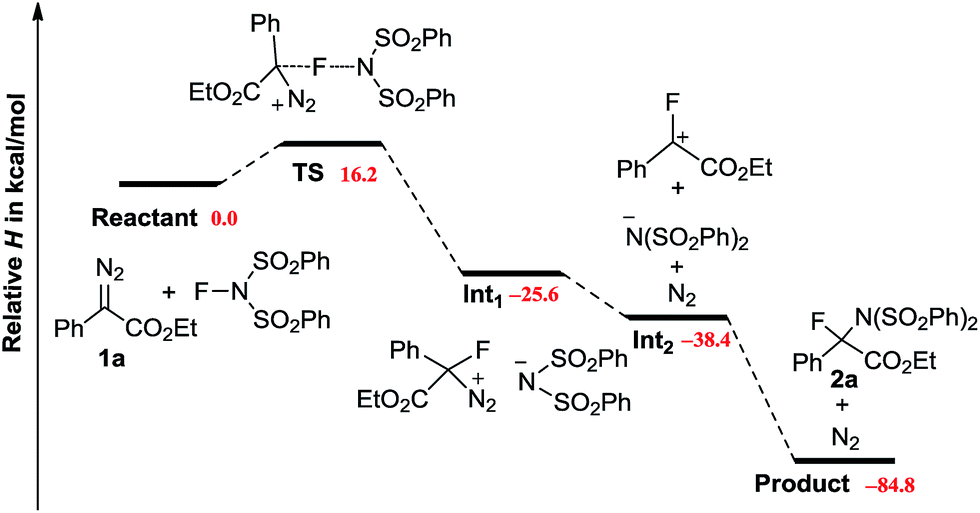 | ||
| Scheme 4 Mechanistic rationale supported by DFT calculations. | ||
Conclusions
In conclusion, we have developed an unprecedented N–F bond insertion into diazocarbonyl compounds. The current method represents a facile approach to construct C–N and C–F bonds on the same carbon without any transition-metal as a catalyst or promoter. Mechanistic studies, including kinetic experiments and DFT calculations, revealed that a reaction sequence of electrophilic activation of 1 by NFSI, followed by an SN1 like displacement of dinitrogen by benzenesulfonimide is preferred. Further study on the asymmetric variant as well as the difunctionalization of the metal carbene is ongoing in our laboratory.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 21402197, 21502190) and Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences.Notes and references
- For selected reviews, see: (a) M. P. Doyle, Chem. Rev., 1986, 86, 919 CrossRef CAS; (b) T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS; (c) A. Padwa and D. J. Austin, Angew. Chem., Int. Ed., 1994, 33, 1797 CrossRef; (d) A. Padwa and M. D. Weingarten, Chem. Rev., 1996, 96, 223 CrossRef CAS PubMed; (e) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed; (f) M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides, John Wiley & Sons, Inc., New York, NY, 1998 Search PubMed; (g) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS PubMed; (h) Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS; (i) Y. Zhang and J. Wang, Chem. Commun., 2009, 45, 5350 RSC; (j) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (k) H. M. L. Davies and Y. Lian, Acc. Chem. Res., 2012, 45, 923 CrossRef CAS PubMed; (l) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed; (m) X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427 CrossRef CAS PubMed; (n) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed.
- (a) K. L. Greenman, D. S. Carter and D. L. van Vranken, Tetrahedron, 2001, 57, 5219 CrossRef CAS ; For selected reviews, see: ; (b) J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 7486 CrossRef CAS PubMed; (c) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS; (d) Y. Zhang and J. Wang, Eur. J. Org. Chem., 2011, 1015 CrossRef; (e) Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586 CrossRef CAS.
- For recent selected examples, see: (a) W.-W. Chan, S.-F. Lo, Z. Zhao and W.-Y. Yu, J. Am. Chem. Soc., 2012, 134, 13565 CrossRef CAS PubMed; (b) T. K. Hyster, K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2013, 135, 5364 CrossRef CAS PubMed; (c) Y. Xia, Z. Liu, Z. Liu, R. Ge, F. Ye, M. Hossain, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2014, 136, 3013 CrossRef CAS PubMed; (d) A. Yada, S. Fujita and M. Murakami, J. Am. Chem. Soc., 2014, 136, 7217 CrossRef CAS PubMed; (e) F.-N. Ng, Y.-F. Lau, Z. Zhou and W.-Y. Yu, Org. Lett., 2015, 17, 1676 CrossRef CAS PubMed.
- For selected examples on Lewis acid promoted insertion to carbonyl compounds, see: (a) T. Hashimoto, Y. Naganawa and K. Maruoka, J. Am. Chem. Soc., 2009, 131, 6614 CrossRef CAS PubMed; (b) D. C. Moebius and J. S. Kingsbury, J. Am. Chem. Soc., 2009, 131, 878 CrossRef CAS PubMed; (c) W. Li, X. Liu, F. Tan, X. Hao, J. Zheng, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2013, 52, 10883 CrossRef CAS PubMedW. Li, F. Tan, X. Hao, G. Wang, Y. Tang, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 1608 CrossRef CAS PubMed.
- (a) R. Kudirka and D. L. van Vranken, J. Org. Chem., 2008, 73, 3585 CrossRef CAS PubMed; (b) R. Kudirka, S. K. J. Devine, C. S. Adams and D. L. van Vranken, Angew. Chem., Int. Ed., 2009, 48, 3677 CrossRef CAS PubMed; (c) P.-X. Zhou, Z.-Z. Zhou, Z.-S. Chen, Y.-Y. Ye, L.-B. Zhao, Y.-F. Yang, X.-F. Xia, J.-Y. Luo and Y.-M. Liang, Chem. Commun., 2013, 49, 561 RSC. For a recent example on Pd(0)-catalysed carbene insertion of Sn–Sn and Si–Si bonds, see: (d) Z. Liu, H. Tan, T. Fu, Y. Xia, D. Qiu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2015, 137, 12800 CrossRef CAS PubMed.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed.
- For recent selected examples, see: (a) S. Bachmann, D. Fielenbach and K. A. Jørgensen, Org. Biomol. Chem., 2004, 2, 3044 RSC; (b) B. Liu, S.-F. Zhu, W. Zhang, C. Chen and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 5834 CrossRef CAS PubMed; (c) E. C. Lee and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 12066 CrossRef CAS PubMed; (d) Z. Hou, J. Wang, P. He, J. Wang, B. Qin, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2010, 49, 4763 CrossRef CAS PubMed; (e) B. Xu, S.-F. Zhu, X.-L. Xie, J.-J. Shen and Q.-L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 11483 CrossRef CAS PubMed; (f) S.-F. Zhu, B. Xu, G.-P. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2012, 134, 436 CrossRef CAS PubMed; (g) Y. Zhu, X. Liu, S. Dong, Y. Zhou, W. Li, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2014, 53, 1636 CrossRef CAS PubMed.
- (a) J.-A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed; (b) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1 CrossRef CAS PubMed; (c) T. Liang, C. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (d) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (e) P. A. Champagne, J. Desroches, J. D. Hamel, M. Vandamme and J. F. Paquin, Chem. Rev., 2015, 115, 9073 CrossRef CAS PubMed; (f) P. Chen and G. Liu, Eur. J. Org. Chem., 2015, 4295 CrossRef CAS.
- Y. Li and Q. Zhang, Synthesis, 2015, 159 Search PubMed.
- S. Qiu, T. Xu, J. Zhou, Y. Guo and G. Liu, J. Am. Chem. Soc., 2010, 132, 2856 CrossRef CAS PubMed.
- H. Zhang, Y. Song, J. Zhao, J. Zhang and Q. Zhang, Angew. Chem., Int. Ed., 2014, 53, 11079 CrossRef CAS PubMed.
- For selected examples on metal-free N–H insertion of diazoesters, see: (a) S. S. So and A. E. Mattson, J. Am. Chem. Soc., 2012, 134, 8798 CrossRef CAS PubMed; (b) S. R. Hansen, J. E. Spangler, J. H. Hansen and H. M. L. Davies, Org. Lett., 2012, 14, 4626 CrossRef CAS PubMed; (c) D. M. Nickerson, V. V. Angeles, T. J. Auvil, S. S. So and A. E. Mattson, Chem. Commun., 2013, 49, 4289 RSC.
- (a) G. A. Olah and J. Welch, Synthesis, 1974, 896 CrossRef CAS; (b) R. Pasceri, H. E. Bartrum, C. J. Hayes and C. J. Moody, Chem. Commun., 2012, 48, 12077 RSC; (c) A. K. Yadav, V. P. Srivastava and L. D. S. Yadav, Chem. Commun., 2013, 49, 2154 RSC; (d) J. Tao, R. Tran and G. K. Murphy, J. Am. Chem. Soc., 2013, 135, 16312 CrossRef CAS PubMed.
- It should be mentioned that slightly before the submission of this manuscript, an elegant work on fluorohalogenation of diazos from Katzenellenbogen and co-workers was published online, see: N. Yasui, C. G. Mayne and J. A. Katzenellenbogen, Org. Lett., 2015, 17, 5540 CrossRef CAS PubMed.
- (a) G. Maas, Angew. Chem., Int. Ed., 2009, 48, 8186 CrossRef CAS PubMed; (b) F. Ye, C. Wang, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 11625 CrossRef CAS PubMed.
- ESI†.
- See ESI† for the details.
- J. Wang, F. Liang and B. Chen, J. Org. Chem., 1998, 63, 8589 CrossRef CAS.
- W.-L. Man, J. Xie, P.-K. Lo, W. W.-Y. Lam, S.-M. Yiu, K.-C. Lau and T.-C. Lau, Angew. Chem., Int. Ed., 2014, 53, 8463 CrossRef CAS PubMed.
- See ESI† for the details on DFT calculation.
- H. Mao, Z. Tang, H. Hu, Y. Cheng, W.-H. Zheng and C. Zhu, Chem. Commun., 2014, 50, 9773 RSC.
- Details on the reactions in presence of Brønsted acids, see:
. - H. Peng, Z. Yuan, H.-Y. Wang, Y.-L. Guo and G. Liu, Chem. Sci., 2013, 4, 3172 RSC.
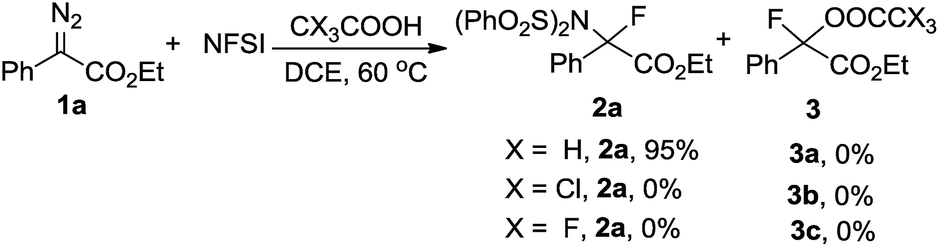
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, and data for new compounds. CCDC 1417241. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04237b |
| This journal is © The Royal Society of Chemistry 2016 |