
Cu-catalyzed β-functionalization of saturated ketones with indoles: a one-step synthesis of C3-substituted indoles†
Zhao Yanga,
Chengkou Liub,
Yu Zengb,
Jingming Zhangb,
Zhixiang Wanga,
Zheng Fang*b and
Kai Guo *bc
*bc
aCollege of Engineering China Pharmaceutical University, 24 Tongjiaxiang, Nanjing, 210003, China
bCollege of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, 30 Puzhu South Road, Nanjing, 211816, China. E-mail: guok@njtech.edu.cn; fzcpu@163.com; Fax: +862558139926; Tel: +862558139935
cState Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, 30 Puzhu South Road, Nanjing, 211816, China
First published on 13th September 2016
Abstract
An effective one-pot synthesis of β-indolylketones from saturated ketones and indoles was developed. It is noteworthy that this methodology has a good functional group tolerance (alkyl, nitro, halogen, hydroxyl, methoxyl, carboxyl and ester) and is of great significance to the preparation of many indole heterocycles.
Alkylated indole derivatives, especially at the C3-position (A, Scheme 1), are present in many natural products with a variety of biological activities.1 Moreover, they can serve as versatile synthetic intermediates to prepare more compounds with indole skeletons.1b,c,2–5 They exist in numerous medicinally relevant compounds, alkaloid products and agrochemicals such as spiroindimicin B,1c NITD609,3 SYN351 (ref. 4) or Hapalindol D (Scheme 1).1b,5 Therefore, the synthesis of alkylated indoles has attracted considerable interest. Traditionally, alkylated indoles were synthesized from the conjugated addition of indoles to α,β-unsaturated compounds.6–9 This process can be promoted by Lewis acids,6 protic acids,7 transition-metals8 or organ-catalysts.9 Nevertheless, this application was limited by acidic conditions, expensive reagents and polymerization of the vinyl ketones. Most importantly, α,β-unsaturated compounds are usually costly and commercially unavailable, although some methods of direct dehydrogenation of ketones were developed.10
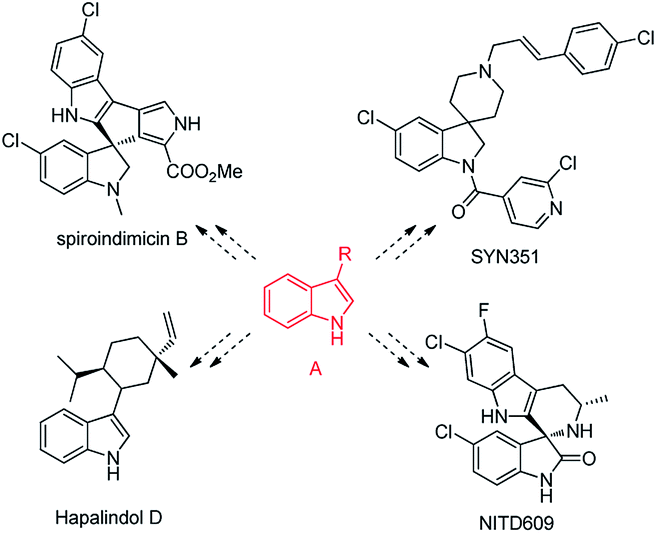 | ||
| Scheme 1 Biologically active molecules with indole skeletons (synthesized from alkylated indole derivatives A). | ||
Recently, some direct β-functionalizations of saturated ketones have been reported.11 Pd-based catalysts were used mostly. During the preparation of this article, Su and his co-workers reported a copper-catalysted β-functionalization of saturated ketones with nitro, oxygen and 1,3-dicarbonyl compounds.12
Herein, we reported a copper-catalyzed one-pot synthesis of β-indolylketones from β-functionalization of saturated ketones with indoles (Scheme 2). To the best of our knowledge, there has been no report on this conversion before.
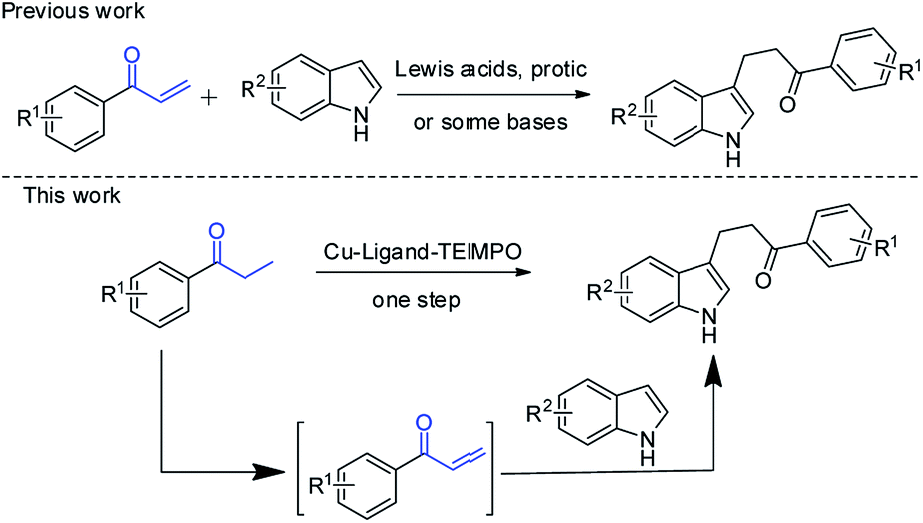 | ||
| Scheme 2 One-pot synthesis of β-indolylketones from direct β-functionalization of saturated ketones with indoles. | ||
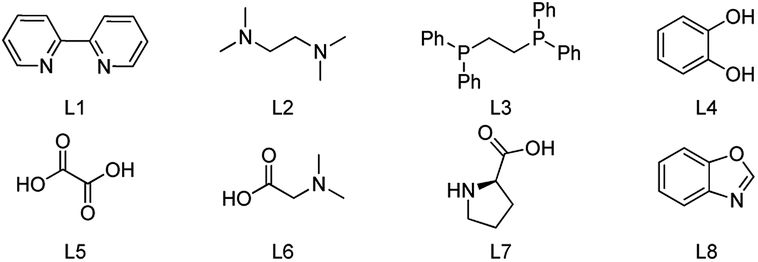
Recently, one-pot preparation of α-amine ketones was developed successfully by our group.13 Then, we have been devoted to synthesis of α-indolylketones from saturated ketones with indoles. Interestingly, reaction of propiophenone with indole in toluene at 100 °C in the presence of Cu(OAc)2 (0.1 equiv.) and TEMPO (0.5 equiv.) afforded the β-indolylketone product in 4% yield, which was more meaningful. So, more efforts were devoted to one-pot preparation of β-indolylketones from β-functionalization of saturated ketones with indoles. The coupling of propiophenone with indole was chosen for initial research shown in Table 1.
|
|||
|---|---|---|---|
| Entry | Catalyst | Ligand | Yieldb (%) |
| a Reaction condition: 1a (3 mmol), 2a (1 mmol), catalyst (0.1 mmol), ligand (0.1 mmol) and TEMPO (2,2,6,6-tetramethylpiperidinooxy, 1 mmol) in toluene were heated at 100 °C for 12 h under Ar (Ar balloon).b Yield of isolated product.c In the open air.d In the absence of TEMPO.e 0.2 equiv. TEMPO was involved under O2. | |||
| 1 | Cu(CH3COO)2 | 9 | |
| 2 | Cu(CH3COO)2 | L1 | 82 |
| 3 | Cu(CH3COO)2 | L2 | 85 |
| 4 | Cu(CH3COO)2 | L3 | 5 |
| 5 | Cu(CH3COO)2 | L4 | 82 |
| 6 | Cu(CH3COO)2 | L5 | Trace |
| 7 | Cu(CH3COO)2 | L6 | 7 |
| 8 | Cu(CH3COO)2 | L7 | 45 |
| 9 | Cu(CH3COO)2 | L8 | 73 |
| 10 | CuI | L2 | 5 |
| 11 | CuBr | L2 | 32 |
| 12 | Cu(CH3COO)2·H2O | L2 | 76 |
| 13 | Pd(CH3COO)2 | L2 | 22 |
| 14 | NiBr2 | L2 | Trace |
| 15 | Ni(CH3COO)2 | L2 | Trace |
| 16 | Cu(CH3COCHCOCH3)2 | L2 | 55 |
| 17 | Cu(CF3COO)2·H2O | L2 | 5 |
| 18 | CoCl2 | L2 | NO |
| 19 | RuClPh3P | L2 | 3 |
| 20 | FeCl3 | L2 | 4 |
| 22 | L2 | NO | |
| 23c | Cu(CH3COO)2 | L2 | 79 |
| 24d | Cu(CH3COO)2 | L2 | NO |
| 25e | Cu(CH3COO)2 | L2 | Trace |
 |
|||
To our delight, increasing the amount of TEMPO (0.5 equiv. to 1 equiv.) resulted in a slight increase of the yield (4% to 9%, Table 1, entry 1). In order to increase the catalyst stability or solubility or prevent aggregation of the metal, a series of ligands were examined (Table 1, entries 2–9). To our delight, the obvious increase of the yield could be observed when L1, L2 or L4 was involved (Table 1, entries 2, 3 and 5). L2 may be the best choice (Table 1, entry 3). Furthermore, numerous metal complexes were tested (Table 1, entries 10–20). Obviously, Cu-based catalysts showed relatively better catalytic activity (Table 1, entries 10–12, 16 and 17). And, the most effective catalyst was Cu(OAc)2 (Table 1, entry 3). Other metal complexes such as Pd, Ni, Co, Ru and Fe were less effective (Table 1, entries 13–15 and 18–20). Moreover, the reaction failed to give the desired product in the absence of metal complex. A slight decrease of the yield was obtained when the reaction was occurred under air instead of Ar atmosphere (Table 1, entries 3 and 23). The instability of the metal catalyst under the air (O2 and water were existed) may be the reason. It was found that the reaction did not proceed in the absence of TEMPO, which indicated that it was necessary in this process (Table 1, entry 24). Only trace amount of the desired product was detected when catalytic amount of TEMPO was involved under O2, which indicated that the cyclic utilization of TEMPO under O2 may be failed. Afterwards, the solvent and temperature were screened shown in Tables S1 and S2 (ESI†). Obviously, only a little or trace amount of corresponding product was obtained when DMF and NMP were involved (Table S1,† entries 2 and 3). It is noteworthy that benzene-type solvents shown similar activity (Table S1,† entry 4). A moderate yield was obtained (65%) with more reaction time consumed when DMSO was used as the solvent (Table S1,† entry 4). p-Xylene was chosen for the following research because higher temperature could be tested. Notably, more reaction time was necessary when the temperature was decreased (Table S2†). 120 °C was chosen because the conversion was finished in the shortest time. Interestingly, almost no desired product was got when the temperature was decreased to 80 °C. The optimized reaction conditions were obtained after the screening of the reaction parameters above: 1a (3 mmol), 2a (1 mmol), Cu(OAc)2 (0.1 mmol), L2 (0.1 mmol) and TEMPO (1 mmol) in p-xylene (3 mL) at 120 °C under Ar for 4 h.
Following the optimized reaction conditions, a series of indoles and ketones were examined to explore the scope of this reaction shown in Scheme 3.
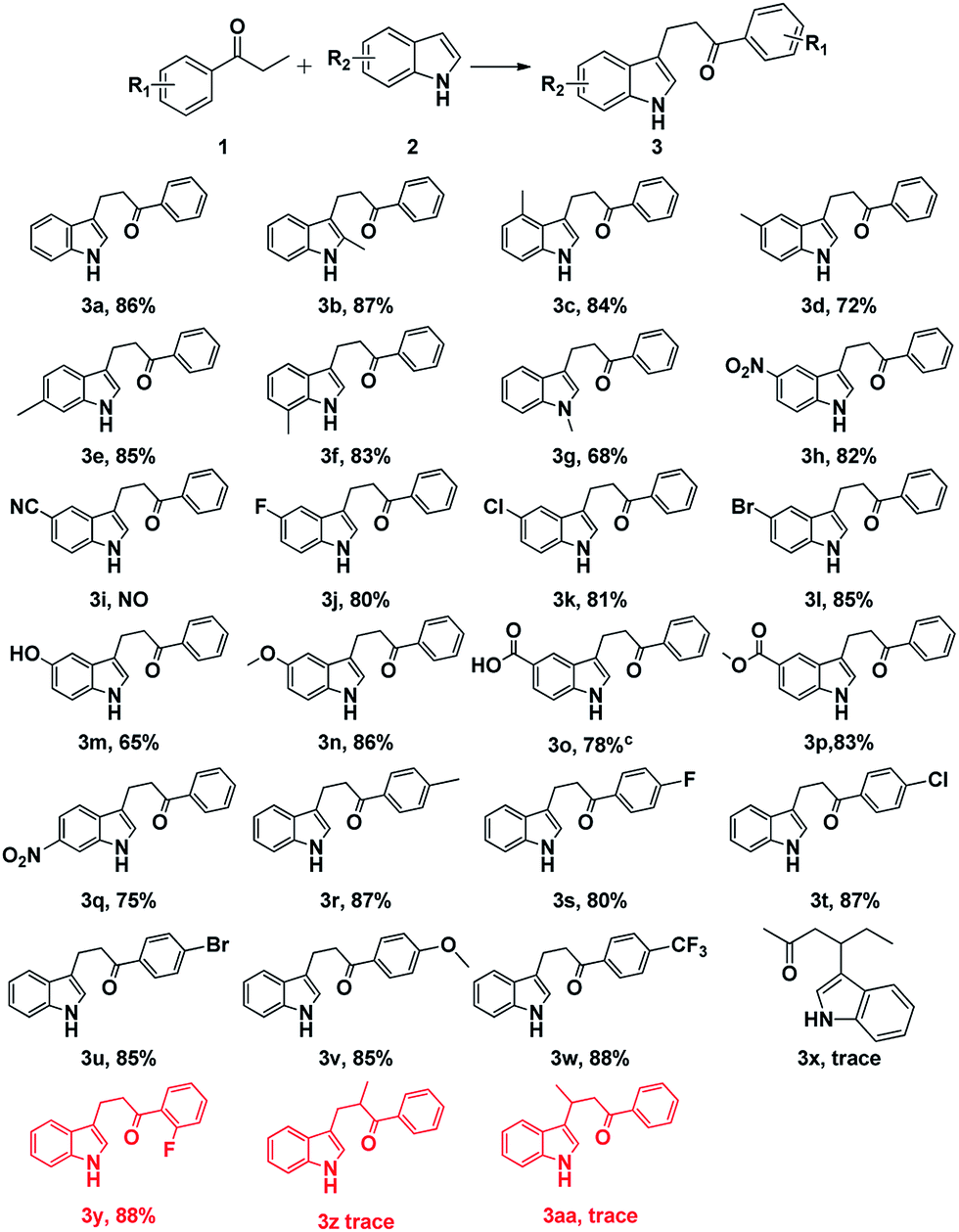 | ||
| Scheme 3 Scope of indoles and ketonea,b. aReaction condition: 1a (3 mmol), 2a (1 mmol), Cu(oAc)2 (0.1 mmol), L2 (0.1 mmol) and TEMPO (1 mmol) in p-xylene (3 mL) at 120 °C under Ar for 4–8 h. bYield of isolated product. cDMSO and p-xylene ((1.5 + 1.5) mL) were used. | ||
As for the substituted indoles, variety of indoles with electron-donating and electron-withdrawing substituents were transformed into the corresponding products in good to moderate yields (Scheme 3, 3a–h and 3j–3w). Notably, indoles bearing strong electron-withdrawing group on the ring such as nitro could react efficiently to give the desired yields (Scheme 3, 3a–3h, 3h and 3q). It was noteworthy that this methodology had a good functional group tolerance, including alkyl, nitro, halogen, hydroxyl, methoxyl, carboxyl and ester (Scheme 3, 3a–3h and 3j–3w). Good functional group tolerance made this methodology more useful in the synthesis of complex natural products and some drug candidates. Meanwhile, the desired products were produced in excellent yields when halo-substituted indoles were involved (Scheme 3, 3j–3l and 3s–3u), which could be further functionalized by cross-coupling reaction.14 Moreover, the effect of substituents at different positions on the ring of indoles could be ignored (Scheme 3, 3b–3g). Then, 2′-fluoropropiophenone, 1-phenyl-2-methyl-1-propanone and butyrophenone were tested under the optimized conditions. Fortunately, a good yield (88%) was obtained when 2′-fluoropropiophenone was involved. However, 1-phenyl-2-methyl-1-propanone and butyrophenone failed to give the desired products. It indicated that steric hindrance on the ring could be ignored. Nevertheless, steric hindrance on alpha-carbon or beta-carbon affects the reaction process dramatically (Scheme 3, 3y–3aa). Nevertheless, the reaction of 5-cyanoindole with propiophenone failed to give the desired product, because cyano was unstable under this reaction conditions. As for substituted ketones, varies functional groups were examined. Good group tolerance was obtained. Unfortunately, only trace amount of desired product was detected when aliphatic ketone was involved (Scheme 3, 3x).
An obvious phenomenon was observed that α,β-unsaturated ketones was detected and confirmed by comparison with standard product by TLC among the reaction process. This result indicated that α,β-unsaturated ketones were formed and acted as the significant intermediates.12 Then nucleophilic 1,2-addition between α,β-unsaturated ketones and C3–H of indoles occurred to generate the desired product.
In conclusion, one-pot synthesis of β-indolylketones from β-functionalization of saturated ketones with indoles was developed firstly. β-Indolylketone is a very useful intermediate in the context of complex compounds with indole skeletons. A series of substrates containing strong electron-withdrawing and electron-donating groups could be transformed into the corresponding products in good yields. Moreover, many functional groups, particularly some sensitive groups such as methoxyl, ester, hydroxyl and halogen, were stable under the optimized conditions. Further optimization of the reaction condition and scope of the reaction was ongoing in our laboratory.
Acknowledgements
The research has been supported by National Key Basic Research Program of China (973 Program) 2012CB721104 and 2012CB725204; the National Natural Science Foundation of China (Grant No. U1463201 and 21402240); Jiangsu Province Natural Science Fund (Grant No. BK20150031 and BK20130913).Notes and references
- (a) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed; (b) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS PubMed; (c) W. Zhang, Z. Liu, S. Li, T. Yang, Q. Zhang, L. Ma, X. Tian, H. Zhang, C. Huang, S. Zhang, J. Ju, Y. Shen and C. Zhang, Org. Lett., 2012, 14, 3364 CrossRef CAS PubMed; (d) J. J. Jackson, H. Kobayashi, S. D. Steffens and A. Zakarian, Angew. Chem., Int. Ed., 2015, 54, 9971 CrossRef CAS PubMed.
- (a) R. K. Nandi, R. Guillot, C. Kouklovsky and G. Vincent, Org. Lett., 2016, 18, 1716 CrossRef CAS PubMed; (b) W. Xu, W. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 9546 CrossRef CAS PubMed; (c) M. J. James, J. D. Cuthbertson, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2015, 54, 7640 CrossRef CAS PubMed; (d) Y. Zheng, J. Cleaveland, D. Richardson and Y. Yuan, Org. Lett., 2015, 17, 4240 CrossRef CAS PubMed; (e) M. J. James, R. E. Clubley, K. Y. Palate, T. J. Procter, A. C. Wyton, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Org. Lett., 2015, 17, 4372 CrossRef CAS PubMed; (f) T. D. Montgomery, A. E. Nibbs, Y. Zhu and V. H. Rawal, Org. Lett., 2014, 16, 3480 CrossRef CAS PubMed; (g) S. Chowdhury, M. Chafeev, S. Liu, J. Sun, V. Raina, R. Chui, W. Young, R. Kwan, J. Fu and J. A. Cadieux, Bioorg. Med. Chem. Lett., 2011, 21, 3676 CrossRef CAS PubMed.
- M. Rottmann, C. McNamara, B. K. S. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. Gonzalez-Paez, S. B. Lakshminaraya-na, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H.-P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175 CrossRef CAS PubMed.
- A. Sluder, S. Shah, J. Cassayre, R. Clover, P. Maienfisch, L.-P. Molleyres, E. A. Hirst, A. J. Flemming, M. Shi, P. Cutler, C. Stanger, R. S. Roberts, D. J. Hughes, T. Flury, M. P. Robinson, E. Hillesheim, T. Pitterna, F. Cederbaum, P. A. Worthington, A. J. Crossthwaite, J. D. Windass, R. A. Currie and F. G. P. Earley, PLoS One, 2012, 7, e34712 CAS.
- P. Harrington and M. A. Kerr, Can. J. Chem., 1998, 76, 1256 CrossRef CAS.
- (a) D. Liang, X. Li, W. Zhang, Y. Li, M. Zhang and P. Cheng, Tetrahedron Lett., 2016, 57, 1027 CrossRef CAS; (b) W. Wang, X. Liu, W. Cao, J. Wang, L. Lin and X. Feng, Chem.–Eur. J., 2010, 16, 1664 CrossRef CAS PubMed; (c) T. Tsubogo, Y. Kano, Y. Yamashita and S. Kobayashi, Chem.–Asian J., 2010, 5, 1974 CrossRef CAS PubMed; (d) M. Bandini, M. Fagioli, M. Garavelli, A. Melloni, V. Trigari and A. Umani-Ronchi, J. Org. Chem., 2004, 69, 7511 CrossRef CAS PubMed; (e) N. Yamagiwa, H. B. Qin, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 13419 CrossRef CAS PubMed; (f) M. Bandini, P. G. Cozzi, M. Giacomini, P. Melchiorre, S. Selva and A. Umani-Ronchi, J. Org. Chem., 2002, 67, 3700 CrossRef CAS PubMed; (g) T. P. Loh, J. Pei and M. Lin, Chem. Commun., 1996, 32, 2315 RSC.
- Z. Iqbal, A. H. Jackson and K. R. N. Rao, Tetrahedron Lett., 1988, 29, 2577 CrossRef CAS.
- (a) S. S. Mohapatra, P. Mukhi, A. Mohanty, S. Pal, A. O. Sahoo, D. Das and S. Roy, Tetrahedron Lett., 2015, 56, 5709 CrossRef CAS; (b) S.-S. Li, H. Lin, X.-M. Zhang and L. Dong, Org. Biomol. Chem., 2015, 13, 1254 RSC; (c) D. Das, S. Pratihar and S. Roy, J. Org. Chem., 2013, 78, 2430 CrossRef CAS PubMed; (d) G. Blay, J. Cano, L. Cardona, I. Fernandez, M. Carmen Munoz, J. R. Pedro and C. Vila, J. Org. Chem., 2012, 77, 10545 CrossRef CAS PubMed; (e) A. L. Gottumukkala, J. G. de Vries and A. J. Minnaard, Chem.–Eur. J., 2011, 17, 3091 CrossRef CAS PubMed; (f) A. Badoiu, G. Bernardinelli, C. Besnard and E. P. Kuendig, Org. Biomol. Chem., 2010, 8, 193 RSC; (g) A. Peng, R. Rosenblatt and K. Nolin, Tetrahedron Lett., 2012, 53, 2712 CrossRef CAS; (h) T. B. Poulsen and K. A. Jorgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS PubMed.
- (a) J. Wang, H. Li, L. Zu, W. Jiang, H. Xie, W. Duan and W. Wang, J. Am. Chem. Soc., 2006, 128, 12652 CrossRef CAS PubMed; (b) L. Dai, S.-X. Wang and F.-E. Chen, Adv. Synth. Catal., 2010, 352, 2137 CrossRef CAS; (c) G. Bartoli, M. Bosco, A. Carlone, F. Pesciaioli, L. Sambri and P. Melchiorre, Org. Lett., 2007, 9, 1403 CrossRef CAS PubMed; (d) W. Chen, W. Du, L. Yue, R. Li, Y. Wu, L.-S. Ding and Y.-C. Chen, Org. Biomol. Chem., 2007, 5, 816 RSC; (e) D. P. Li, Y. C. Guo, Y. Ding and W. J. Xiao, Chem. Commun., 2006, 42, 799 RSC.
- (a) Y. Chen, A. Turlik and T. R. Newhouse, J. Am. Chem. Soc., 2016, 138, 1166 CrossRef CAS PubMed; (b) Y. Chen, J. P. Romaire and T. R. Newhouse, J. Am. Chem. Soc., 2015, 137, 5875 CrossRef CAS PubMed; (c) T. Diao, D. Pun and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8205 CrossRef CAS PubMed; (d) M. A. Bigi and M. C. White, J. Am. Chem. Soc., 2013, 135, 7831 CrossRef CAS PubMed; (e) T. Diao, T. J. Wadzinski and S. S. Stahl, Chem. Sci., 2012, 3, 887 RSC; (f) W. Gao, Z. He, Y. Qian, J. Zhao and Y. Huang, Chem. Sci., 2012, 3, 883 RSC; (g) T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566 CrossRef CAS PubMed.
- (a) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed; (b) J. L. Jeffrey, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 8404 CrossRef CAS PubMed; (c) Z. Huang, Q. P. Sam and G. Dong, Chem. Sci., 2015, 6, 5491–5498 RSC; (d) M. Okada, T. Fukuyama, K. Yamada, I. Ryu, D. Ravelli and M. Fagnoni, Chem. Sci., 2014, 5, 2893 RSC; (e) J. A. Terrett, M. D. Clift and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 6858 CrossRef CAS PubMed; (f) F. R. Petronijevic, M. Nappi and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 18323 CrossRef CAS PubMed; (g) Z. Huang and G. Dong, J. Am. Chem. Soc., 2013, 135, 17747 CrossRef CAS PubMed.
- X. Jie, Y. Shang, X. Zhang and W. Su, J. Am. Chem. Soc., 2016, 138, 5623 CrossRef CAS PubMed.
- C.-K. Liu, Z. Fang, Z. Yang, Q.-W. Li, S.-Y. Guo and K. Guo, RSC Adv., 2016, 6, 25167 RSC.
- (a) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS PubMed; (b) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2008, 47, 3096 CrossRef CAS PubMed; (c) S. Zheng, C. Yu and Z. Shen, Org. Lett., 2012, 14, 3644 CrossRef CAS PubMed; (d) X. A. Xie, G. R. Cai and D. W. Ma, Org. Lett., 2005, 7, 4693 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional material information, experimental details and characterization data for the compounds. See DOI: 10.1039/c6ra19000f |
| This journal is © The Royal Society of Chemistry 2016 |