
NBS-mediated dinitrogen extrusion of diazoacetamides under catalyst-free conditions: practical access to 3-bromooxindole derivatives†
Xiangbo Wanga,
Kuiyong Donga,
Bin Yanb,
Cheng Zhanga,
Lihua Qiu*a and
Xinfang Xu*ab
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: xinfangxu@suda.edu.cn
bJinghua Anti-cancer Pharmaceutical Engineering Center, Nantong 226407, China
First published on 18th July 2016
Abstract
A synthetically useful transformation arises from NBS-mediated dinitrogen extrusion of N-aryl diazoacetamides under catalyst-free conditions, which gives 3-bromooxindoles in high to excellent yields with high selectivity via a non-carbene process.
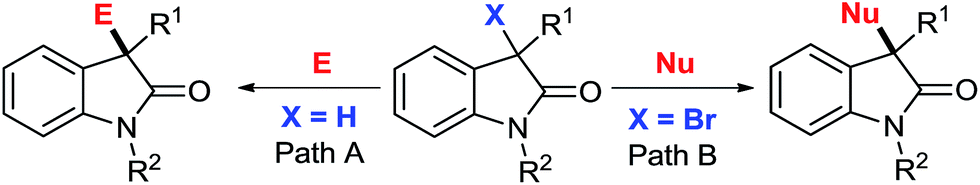
3,3-Disubstituted oxindoles and indoles are pervasive structural motifs in bioactive compounds,1 including Citrinadin A and B,1c,2 Gelsemine,3 Chitosenine,4 Spirotryprostatin A and B5 etc. Over the past several years, effective methods have been developed for 3-position functionalization on existing indole frameworks,6 including nucleophilic addition with oxindole units (Scheme 1, Path A)7 and electrophilic addition with 3-bromooxindoles (Path B).8 Among these reports, examples using 3-bromooxindole as an electrophile for the construction of 3-substituted or 3,3-disubstituted derivatives are limited. The main obstacle in this context is the underdeveloped access to 3-bromooxindole derivatives with diversified functional groups.8,9 Here, we report a practical method for accessing these building blocks under catalyst-free conditions with broad substrate tolerance.10
 | ||
| Scheme 1 General approaches to 3,3-disubstituted oxindoles. | ||
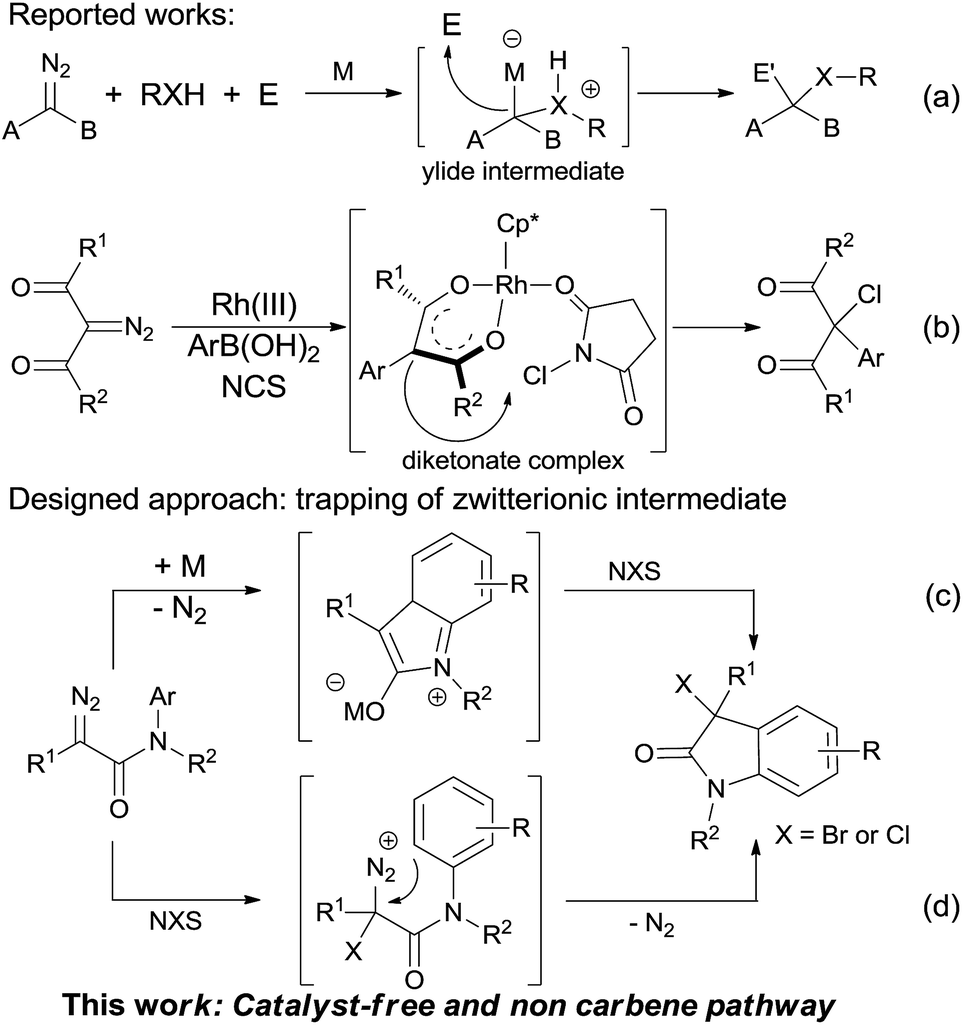
Diazo compound is a useful material in organic synthesis and its typical utility is metal-catalyzed carbene transformation.11 In the last few years, Hu and co-workers have reported a three-component reaction,12 in which various electrophiles, including aldehydes,13 α,β-unsaturated aldehydes/ketones14 and imines,15 are utilized to trap the active ylide intermediate and give the vicinal difunctionalized products in highly selective fashion (Scheme 2a). Recently, Yu's group disclosed a cascade reaction via trapping the diketonate complex with N-chlorosuccinimide (NCS) (Scheme 2b).16 Inspired by these works, a direct 3-halogenated oxindole derivatives formation could be envisioned via a zwitterionic intermediate trapping transformation with halogenating agent (NCS or NBS) (Scheme 2c).15b Here, we report our recent progress on this direction, and the results turned out that the reaction did not go through carbene intermediate; instead, it was a direct electrophilic addition on the carbon of the diazo group followed by an F–C type reaction with dinitrogen extrusion simultaneously to give the final product (Scheme 2d). Besides the numerous reports on metal- or organo-catalysis, photolysis and thermolysis in carbene transformations with diazo compounds,11,17 this report presents the rare example of substrate promoted reaction pathway.
 | ||
| Scheme 2 Designed transformation to 3-bromooxindoles. | ||
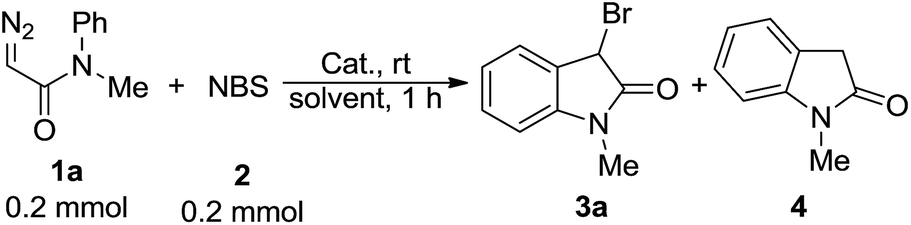
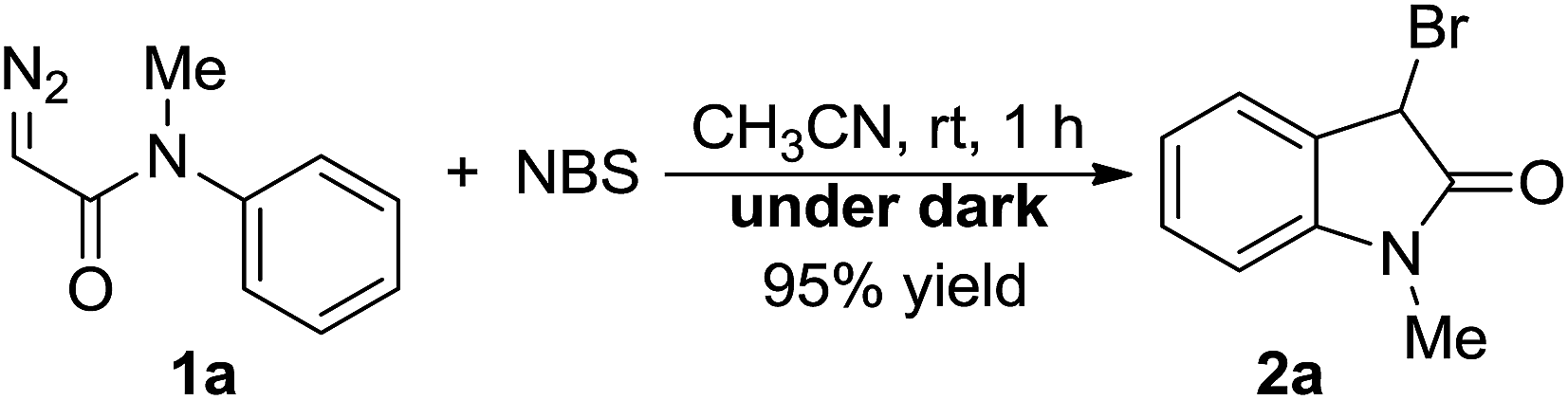
Our study of designed approach to 3-bromooxindoles began by evaluating the model reaction of diazoacetamide 1a with NBS catalyzed by Rh2(OAc)4 in DCM at room temperature, and only 42% of desired product 3a was obtained contaminated with 54% of direct intramolecular aromatic substitution product 4 (Table 1, entry 1). The structure of 3a was confirmed spectroscopically8b and single-crystal X-ray diffraction analysis.18 Next, we examined several of copper-catalysts, including Cu(I) and Cu(II), and all of them worked very well to predominantly give 3 in >90% yields (entries 2–4). Interestingly, when the control reaction was carried out in the absence of catalyst, it gave the brominated product in 82% yield (entry 5), which suggested a non carbene process of this reaction. Various solvents were screened for further optimization of reaction conditions in the absence of catalyst (entries 5–9), and CH3CN turned out to give the higher yields due to it better solubility for NBS (entry 6, in 97%). In addition, the synthetic practicability of the present method was demonstrated through a gram–scale reaction, which generated the 3-bromooxindole in 70% yield combined with another 10% of 3,5-dibromooxindole byproduct (entry 10).
|
||||
|---|---|---|---|---|
| Entry | Cat (X mol%) | Solvent | 3ab (%) | 4b (%) |
| a Reactions were carried out at room temperature on a 0.2 mmol scale in 2.0 mL solvent with corresponding catalyst for 1 h.b Isolated yield.c Reaction carried out in 7.0 mmol scale and 1.1 g of 3a was isolated combined with 10% corresponding 3,5-dibromooxindole byproduct. TBME = tert-butyl methyl ether; DMB = 2,2-dimethylbutane. | ||||
| 1 | Rh2(OAc)4 (1.0%) | DCM | 42 | 54 |
| 2 | Cu(CH3CN)4PF6 (5.0%) | DCM | 90 | — |
| 3 | CuI (5.0%) | DCM | 93 | — |
| 4 | Cu(OTf)2 (5.0%) | DCM | 97 | — |
| 5 | — | DCM | 82 | — |
| 6 | — | CH3CN | 97 | — |
| 7 | — | TBME | 80 | — |
| 8 | — | Toluene | 73 | — |
| 9 | — | DMB | 62 | — |
| 10c | — | CH3CN | 70 | — |
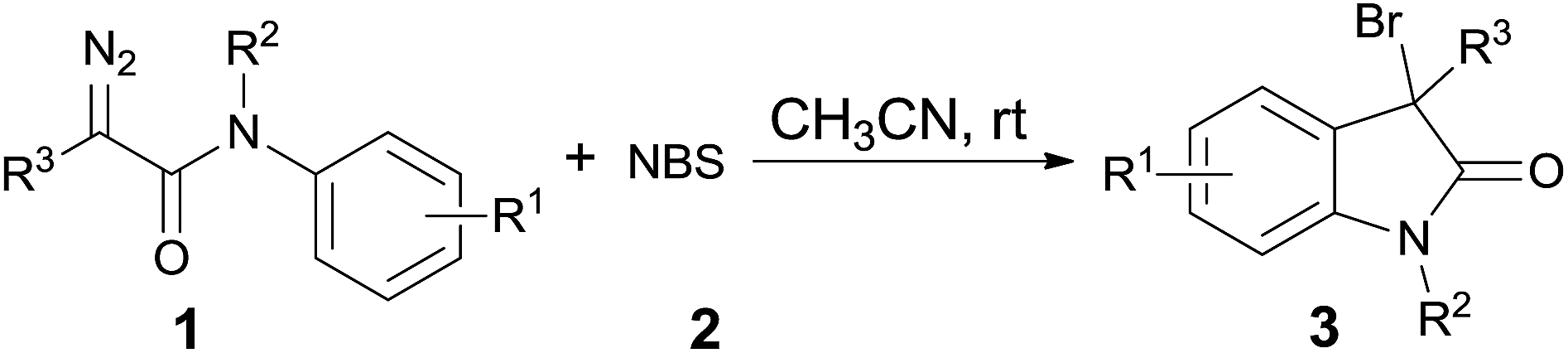
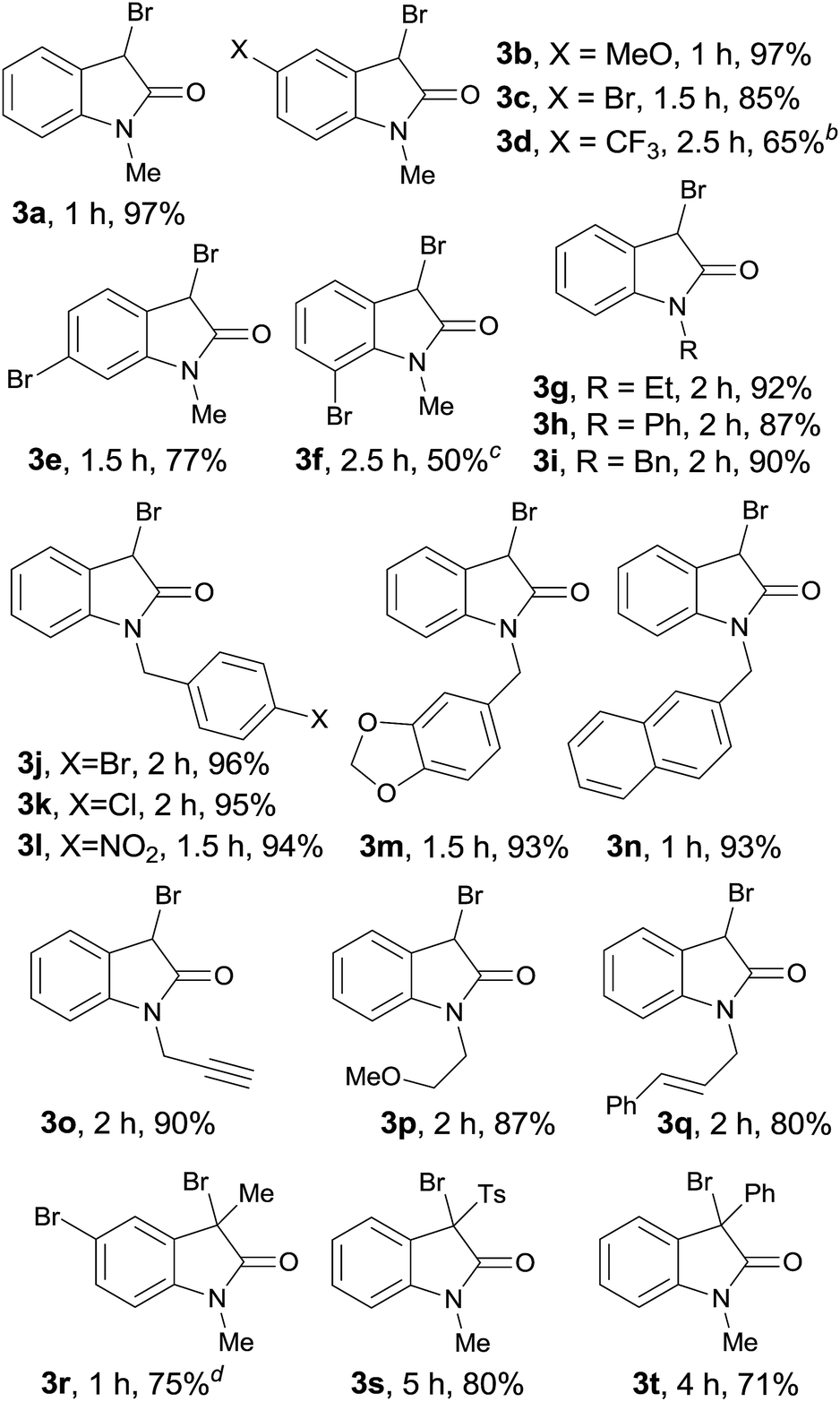
After identifying the optimized reaction conditions, we evaluated the substrate scope of this method. As shown in Table 2, all the tested diazoacetamides were tolerated in these reaction conditions to give moderate to high yields. Substrates carrying neutral or electronic rich groups on the aryl ring showed higher reactivity compared to the ones with electronic deficient substitutions (3a, 3b vs. 3c, 3d). Ortho- or meta-position substituted compounds were both effective and the F–C type addition occurred at the position with less steric hindrance to give the corresponding products 3e and 3f in 70% and 50% yields, respectively. Due to steric effect, using excess amount of NBS was necessary to ensure the full conversion of 1f; however, considerable amount of multi-brominated byproduct 3f′ was also obtained under these conditions. To extensively explore the substrates scope, diazoacetamides 1 with various R2 groups were investigated and it did not make any obvious differences in the yields. It is worth mentioning that the reaction was highly selective. Diversified substitutions or functional groups were tolerated under these mild catalyst-free reaction conditions, including compounds with benzyl group (3i–3n, >90% yields), propargyl group (3o, 90% yield), ether group (3p, 87% yield) and allyl group (3q, 80% yield), which were all perfect reacting partners in typical carbene transformations and they were all untouched in this reaction. What's more, α-substituted diazoacetamides 1, either with methyl, phenyl, or electronic deficient Ts groups, all performed well and yielded the 3,3-disubstituted oxindoles with >70% yields (3r–3t).
The possibility of applying the developed reaction conditions to the formation of other 3-halogenated oxindoles could be expected (eqn (1)), and the corresponding chlorooxindoles 5a and
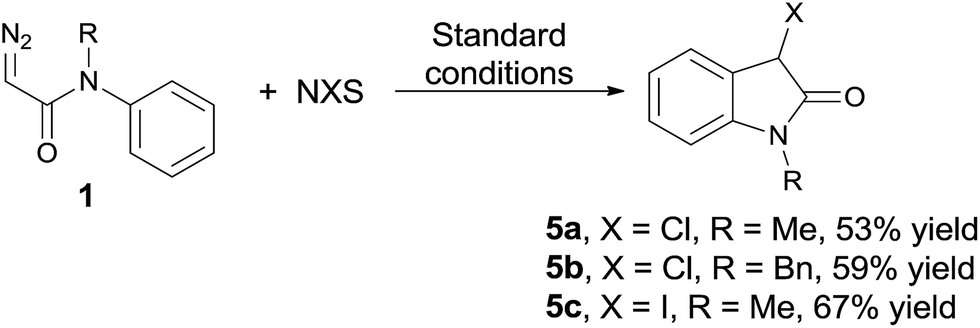 | (1) |
To gain insight into the mechanism details, a few control reactions were carried out. Reaction pathways through metal carbene or carbene intermediates were first excluded; otherwise byproducts could be envisioned from 1i–1q with most possibilities. The other possibility of via intermediate 4 could be ruled out, which only gave the aryl brominated product 6 under standard conditions (eqn (2)). Photolysis was also
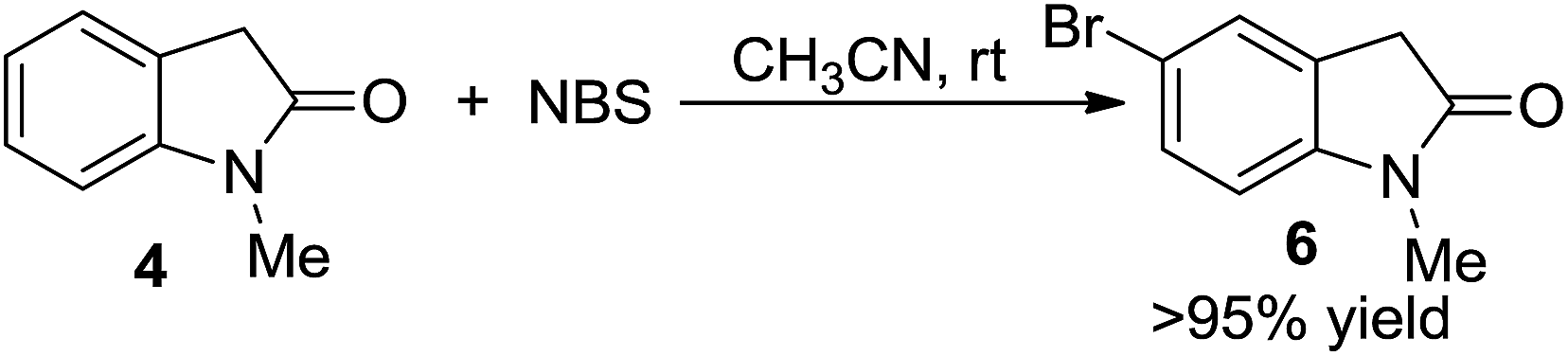 | (2) |
 | (3) |
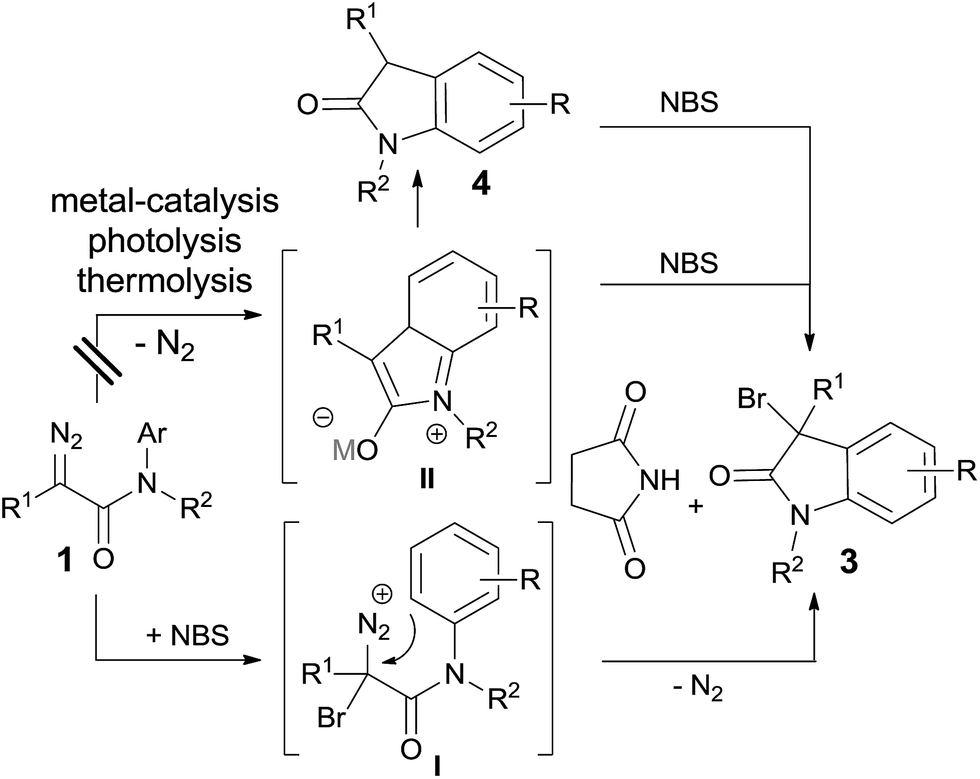 | ||
| Scheme 3 Proposed reaction pathway. | ||
The generated 3-bromooxindoles is an important synthon in the synthesis of bioactive indole and oxindole derivatives.9,10 Here, two general protocols were carried out for the further derivation of the obtained products (Scheme 4). The 3-position of 3c could be easily brominated to give 3,3,5-tribromooxindole 7 in 78% yield. Dimerized product 8 could be obtained under basic condition with 85% yields from 3a.
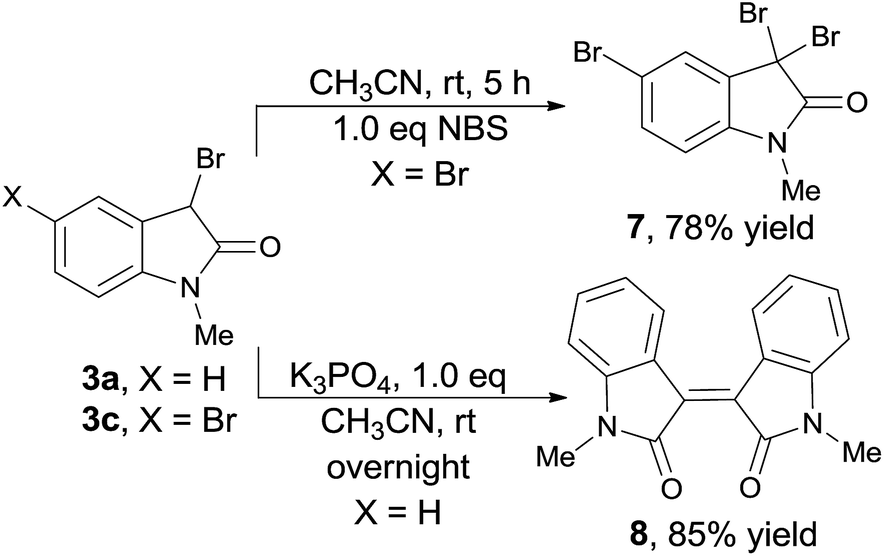 | ||
| Scheme 4 Derivation. | ||
Conclusions
In this work, a practical catalyst-free reaction for the synthesis of 3-bromooxindole derivatives with N-aryl diazoacetamides and NBS has been developed.22 The reaction initiates from the electrophilic addition of bromonium ion with the carbon on the diazo group followed by intramolecular F–C type addition with the aryl ring, and releasing a N2 synchronously to give the final products in high yield. In addition, this non carbene process shows high selectivity and tolerates diversified substitutions and functional groups, which are competitive reactive sites in typical carbene transformations and are untouched in this process.Acknowledgements
We thank the grants from the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD); and Natural Science Foundation of China Jiangsu province (SBK20150315 and 15KJD150004).Notes and references
- For reviews: (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed; (b) J. Yang, X. Z. Wearing, P. W. Quesne, J. R. Deschamps and J. M. Cook, J. Nat. Prod., 2008, 71, 1431 CrossRef CAS PubMed; (c) Z. Bian, C. C. Marvin, M. Pettersson and S. F. Martin, J. Am. Chem. Soc., 2014, 136, 14184 CrossRef CAS PubMed.
- (a) M. Tsuda, Y. Kasai, K. Komatsu, T. Sone, M. Tanaka, Y. Mikami and J. Kobayashi, Org. Lett., 2004, 6, 3087 CrossRef CAS PubMed; (b) Z. Bian, C. C. Marvin and S. F. Martin, J. Am. Chem. Soc., 2013, 135, 10886 CrossRef CAS PubMed.
- (a) H. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 36 CrossRef CAS; (b) T. Fukuyama and G. Liu, J. Am. Chem. Soc., 1996, 118, 7426 CrossRef CAS.
- (a) G. O. Fonsecaa, Z. Wang, O. A. Namjoshi, J. R. Deschamps and J. M. Cook, Tetrahedron Lett., 2015, 56, 3052 CrossRef; (b) S. Sakai, N. Aimi, K. Yamaguchi, E. Yamanaka and J. Haginiwa, J. Chem. Soc., Perkin Trans. 1, 1982, 1257 RSC.
- (a) P. R. Sebahar and R. M. Williams, J. Am. Chem. Soc., 2000, 122, 5666 CrossRef CAS; (b) C. B. Cui, H. Kakeya and H. Osada, J. Antibiot., 1996, 49, 832 CrossRef CAS PubMed; (c) S. Edmondson, S. J. Danishefsky, L. Sepp-Lorenzino and N. Rosen, J. Am. Chem. Soc., 1999, 121, 2147 CrossRef CAS.
- For reviews: (a) B. M. Trost and M. K. Brennan, Synthesis, 2009, 3003 CrossRef CAS; (b) D. J. Cheng, Y. Ishihara, B. Tan and C. F. Barbas III, ACS Catal., 2014, 4, 743 CrossRef CAS; (c) Z. Cao and J. Zhou, Org. Chem. Front., 2015, 2, 849 RSC; (d) H. Wu, H. Huang, C. Ren, L. Liu, D. Wang and C.-J. Li, Chem.–Eur. J., 2015, 21, 16744 CrossRef CAS PubMed.
- Selected recent examples: (a) B. Cui, Y. You, J. Zhao, J. Zuo, Z. Wu, X. Xu, X. Zhang and W. Yuan, Chem. Commun., 2015, 51, 757 RSC; (b) R. He, C. Ding and K. Maruoka, Angew. Chem., Int. Ed., 2009, 48, 4559 CrossRef CAS PubMed; (c) P. Zhou, Y. Cai, L. Lin, X. Lian, Y. Xia, X. Liu and X. Feng, Adv. Synth. Catal., 2015, 357(4), 695 CrossRef CAS; (d) X. Li, B. Zhang, Z. Xi, S. Luo and J. Cheng, Adv. Synth. Catal., 2010, 352, 416 CrossRef CAS; (e) L. Shen, W. Jia and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 585 CrossRef CAS PubMed.
- Selected recent examples: (a) J. Li, T. Du, G. Zhang and Y. Peng, Chem. Commun., 2013, 49, 1330 RSC; (b) Y. Zou, W. Guo, F. Liu, L. Lu, J. Chen and W. Xiao, Green Chem., 2014, 16, 3787 RSC; (c) J. Zuo, Z. Wu, J. Zhao, M. Zhou, X. Xu, X. Zhang and W. Yuan, J. Org. Chem., 2015, 80, 634 CrossRef CAS PubMed; (d) P. Zheng, Q. Ouyang, S. Niu, L. Shuai, Y. Yuan, K. Jiang, T. Liu and Y. Chen, J. Am. Chem. Soc., 2015, 137, 9390 CrossRef CAS PubMed.
- (a) Y. Cai, X. Liu, P. Zhou, Y. Kuang, L. Lin and X. Feng, Chem. Commun., 2013, 49, 8054 RSC; (b) G. Hernández-Torres, B. Tan and C. F. Barbas, Org. Lett., 2012, 14, 1858 CrossRef PubMed; (c) H. Hamamoto, H. Umemoto, M. Umemoto, C. Ohta, M. Dohshita and Y. Miki, Synlett, 2010, 17, 2593 Search PubMed.
- Recent works under catalyst-free conditions: (a) M. Selva, S. Guidi and M. Noè, Green Chem., 2015, 17, 1008 RSC; (b) S. Li, X. Li, Q. Li, Q. Yuan, X. Shi and Q. Xu, Green Chem., 2015, 17, 3260 RSC; (c) Y. Yang, L. Tang, S. Zhang, X. Guo, Z. Zha and Z. Wang, Green Chem., 2014, 16, 4106 RSC; (d) C. Zhang, L. Zhang and N. Jiao, Green Chem., 2012, 14, 3273 RSC; (e) L. Li, W. Liu, H. Zeng, X. Mu, G. Cosa, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2015, 137, 8328 CrossRef CAS PubMed; (f) W. Liu, L. Li and C.-J. Li, Nat. Commun., 2015, 6, 6526 CrossRef CAS PubMed; (g) M. Liao and J. Wang, Green Chem., 2007, 9, 184 RSC.
- For review: (a) M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley & Sons, New York, 1998 Search PubMed; (b) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS PubMed; (c) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2009, 110, 704 CrossRef PubMed; (d) H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899 RSC; (e) S. F. Zhu and Q. L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed; (f) Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586 CrossRef CAS.
- Review: (a) D. Xing and W. Hu, Tetrahedron Lett., 2014, 55, 777 CrossRef CAS; (b) X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427 CrossRef CAS PubMed.
- (a) D. Xing, C. Jing, X. Li, H. Qiu and W. Hu, Org. Lett., 2013, 15, 3578 CrossRef CAS PubMed; (b) C. Jing, T. Shi, D. Xing, X. Guo and W. Hu, Green Chem., 2013, 15, 620 RSC.
- (a) C. Ma, D. Xing, C. Zhai, J. Che, S. Liu, J. Wang and W. Hu, Org. Lett., 2013, 15, 6140 CrossRef CAS PubMed; (b) C. Jing, D. Xing, Y. Qian, T. Shi, Y. Zhao and W. Hu, Angew. Chem., Int. Ed., 2013, 52, 9289 CrossRef CAS PubMed; (c) X. Guan, L. Yang and W. Hu, Angew. Chem., Int. Ed., 2010, 49, 2190 CrossRef CAS PubMed.
- (a) D. Zhang, H. Qiu, L. Jiang, F. Lv, C. Ma and W. Hu, Angew. Chem., Int. Ed., 2013, 52, 13356 CrossRef CAS PubMed; (b) Q. Huang, M. Li, L. Jiang, F. Lv, L. Zan, C. Zhai, M. P. Doyle and W. Hu, Nat. Chem., 2012, 4, 733 CrossRef PubMed; (c) W. Hu, X. Xu, J. Zhou, W. Liu, H. Huang, J. Hu, L. Yang and L. Gong, J. Am. Chem. Soc., 2008, 130, 7782 CrossRef CAS PubMed; (d) C. Jing, D. Xing and W. Hu, Org. Lett., 2015, 17, 4336 CrossRef CAS PubMed; (e) X. Xu, Y. Qian, L. Yang and W. Hu, Chem. Commun., 2011, 47, 797 RSC.
- F. Ng, Y. Lau, Z. Zhou and W. Yu, Org. Lett., 2015, 17, 1676 CrossRef CAS PubMed.
- Selected recent examples on organo-catalysis: (a) E. Emer, J. Twilton, M. Tredwell, S. Calderwood, T. L. Collier, B. L. M. Taillefer and V. Gouverneur, Org. Lett., 2014, 16, 6004 CrossRef CAS PubMed; (b) L. Huang and W. D. Wulff, J. Am. Chem. Soc., 2011, 133, 8892 CrossRef CAS PubMed; (c) M. Wu, W. He, X. Liu and B. Tan, Angew. Chem., Int. Ed., 2015, 54, 9409 CrossRef CAS PubMed; (d) T. Hashimoto, H. Nakatsu, K. Yamamoto and K. Maruoka, J. Am. Chem. Soc., 2011, 133, 9730 CrossRef CAS PubMed; on photolysis: (e) C. Wentrup, H. Bibas, A. Kuhn, U. Mitschke and M. C. McMills, J. Org. Chem., 2013, 78, 10705 CrossRef CAS PubMed; and on thermolysis: (f) Y. Deng, C. Jing and M. P. Doyle, Chem. Commun., 2015, 51, 12924 RSC; (g) S. R. Ovalles, J. H. Hansen and H. M. L. Davies, Org. Lett., 2011, 13, 4284 CrossRef CAS PubMed; (h) Z. Huang, C. Wang, E. Tokunaga, Y. Sumii and N. Shibata, Org. Lett., 2015, 17, 5610 CrossRef CAS PubMed; (i) S. R. Hansen, J. E. Spangler, J. H. Hansen and H. M. L. Davies, Org. Lett., 2012, 14, 4626 CrossRef CAS PubMed.
- CCDC 1425225 contains the supplementary crystallographic data for 3a.
- T. Wang, D. L. Hoon and Y. Lu, Chem. Commun., 2015, 51, 10186 RSC.
- (a) Å. Kaupang and T. Bonge-Hansen, Beilstein J. Org. Chem., 2013, 9, 1407 CrossRef PubMed; (b) H. T. Bonge, B. Pintea and T. Hansen, Org. Biomol. Chem., 2008, 6, 3670 RSC; (c) H. T. Bonge and T. Hansen, J. Org. Chem., 2010, 75, 2309 CrossRef CAS PubMed.
- H. Mao, Z. Tang, H. Hu, Y. Cheng, W. Zheng and C. Zhu, Chem. Commun., 2014, 50, 9773 RSC.
- Similar transformation for 3-halooxindole synthesis was reported during this manuscript was under reviewing by Hu's group: C. Ma, D. Xing and W. Hu, Org. Lett., 2016, 18, 3134 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1425225. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra16868j |
| This journal is © The Royal Society of Chemistry 2016 |