
Iterative catalyst controlled diastereodivergent synthesis of polypropionates†‡
D.
Roke
,
M.
Fañanás-Mastral§
* and
B. L.
Feringa
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborg 4, 9747 AG Groningen, Netherlands. E-mail: B.L.Feringa@rug.nl
First published on 7th July 2016
Abstract
Polypropionate fragments are synthesized using a combination of a copper-catalyzed asymmetric allylic alkylation, ruthenium-catalyzed cross-metathesis and iridium-catalyzed asymmetric allylic etherification. The use of an appropriate chiral ligand for each catalytic reaction allows access to 1,2-hydroxymethyl arrays with good to excellent control over the relative and absolute configuration of the different stereocenters.
Introduction
Polypropionates are important subunits in many polyketides, one of the major classes of natural products featuring a wide range of biological activities (Fig. 1).1,2 In pharmaceutics, polyketides have also found various applications, being used as antibiotics, immunosuppressants, antiparasitics, cholesterol lowering drugs, and antitumor agents.3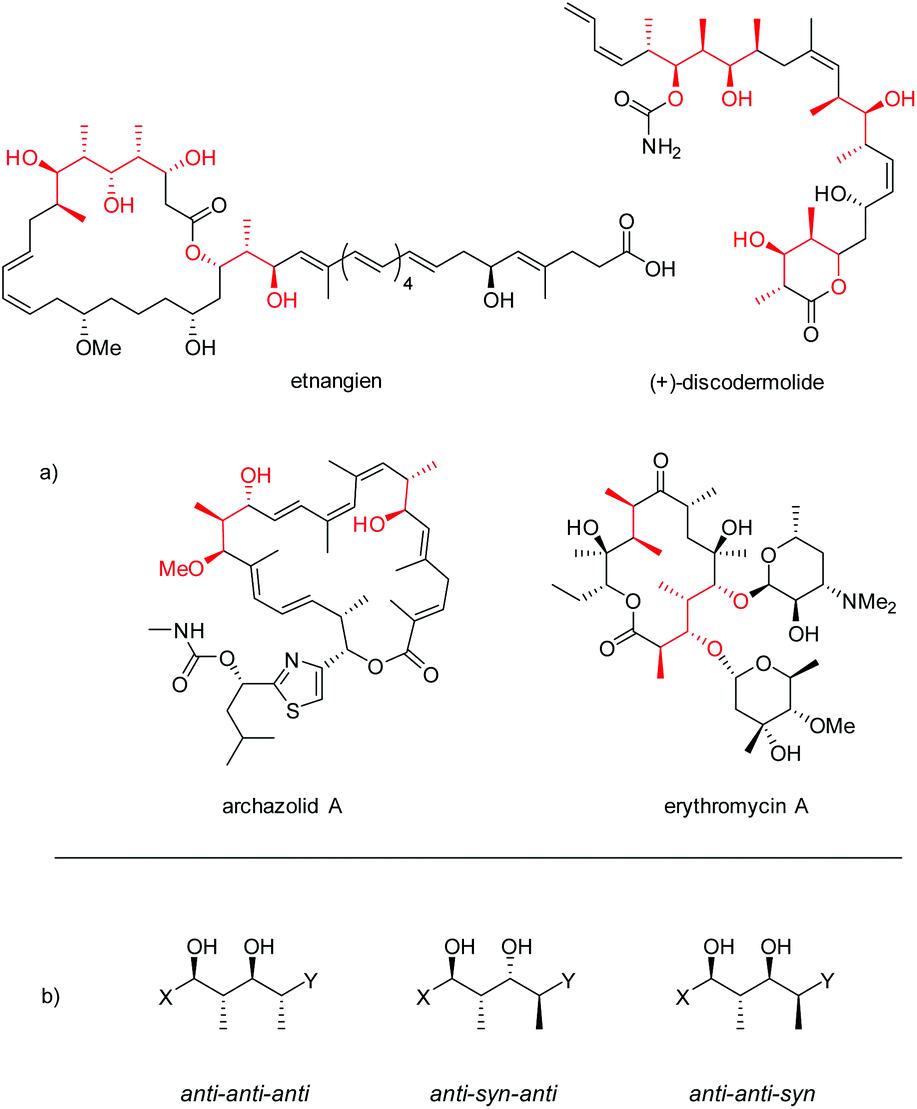 | ||
| Fig. 1 (a) Examples of polyketides bearing polypropionate segments (highlighted in red). (b) Three configurations of polypropionate segments described in this paper. | ||
Polypropionates consist of alternating methyl- and hydroxyl-substituted stereocenters. The combination of these two different motifs leads to a large number of possible distinct stereochemical combinations. The importance of these complex chemical structures has directed a lot of attention towards the diastereoselective and enantioselective synthesis of polypropionates.4 In an attempt to mimic the biosynthetic pathway, the stereoselective aldol reaction is arguably the most extensively studied method.5,6 However, a large number of highly successful alternative approaches has been reported such as crotylations,7 reductive aldol couplings8 and epoxide ring-opening reactions.9 Important methods for the asymmetric synthesis of polypropionates frequently start with stoichiometric amounts of chiral materials, either from natural sources or often in the form of chiral auxiliaries which are to be attached and cleaved off in separate steps.
A growing number of catalytic methods towards polypropionates are emerging,4 and examples include (organo)catalytic aldol couplings,5,10 reductive aldol couplings4,8,11 and catalytic crotylations.12 Although already a variety of useful catalytic asymmetric methods towards chiral propionate units exists, most of these methods have been limited due to the fact that only one 1,2-hydroxymethyl moiety is introduced,10–12 making it difficult to perform these methods iteratively to allow the formation of polypropionates.
The transition metal-catalyzed asymmetric allylic substitution is a widely investigated reaction, which allows the formation of C–C, C–H, C–S, C–N and C–O bonds in a highly enantioselective manner.13 In particular, the SN2′-selective allylic substitution provides a versatile substrate which bears a stereocenter next to a double bond which can be further functionalized.13,14 Copper-catalyzed asymmetric allylic alkylations with organometallic reagents usually proceed with high SN2′ selectivity being a powerful tool for the construction of optically active carbon stereocenters.15 A wide range of chiral ligands has been utilized for the copper-catalyzed asymmetric allylic alkylation, examples include phosphoramidites,16 diphosphines,17 phosphine–phosphite18 and NHC ligands.19 Among these different catalytic systems, the combination of CuBr·SMe2 with Taniaphos L1 has emerged as an excellent catalyst for the introduction of the methyl unit via copper-catalyzed AAA.17
On the other hand different transition metals have been employed for the asymmetric allylic C–O bond formation towards branched allyl ethers and alcohols.20–28 Among these systems Ir/phosphoramidite complexes have emerged as very efficient catalysts for SN2′-selective asymmetric allylic etherification.13d,f,g,23–27 Substitutions with phenoxides23 and alkoxides24a,b and alcohols24c have been shown to provide excellent results. Moreover silanoates25 and caboxylates26 can be introduced and even a direct hydroxylation using carbonates as pronucleophiles has been reported.27
An iterative approach, in which the methyl and hydroxyl substituents are introduced in separate allylic substitution steps, could be a potentially powerful approach towards polypropionates. A related strategy has been used before for the synthesis of 1,2-dihydroxy arrays23e,28 and, albeit non-iteratively, for 1,2-aminoethyl arrays.29 However for polypropionates only a few sequential approaches have been published,30 and to the best of our knowledge a catalytic iterative approach in which alternating methyl and hydroxyl groups are sequentially introduced to arrive at the distinct diastereomers and enantiomers of polypropionates has not been reported.4 A distinct advantage of such a catalytic iterative approach is that it can be performed in a diastereodivergent way, in which the diastereoselectivity is controlled fully by the chiral catalyst.
Herein we present an iterative catalytic method for the synthesis of polypropionates in which stereocenters are sequentially generated using a copper-catalyzed asymmetric allylic alkylation, reported earlier by our group,17f in combination with an iridium-catalyzed asymmetric allylic etherification (Scheme 1).23d,e The combination of subsequent asymmetric allylic substitution reactions allows for the sequential introduction of methyl- and hydroxyl-substituted stereocenters. Control over the distinct configurations was achieved by the use of an appropriate enantiomer of the chiral ligand to obtain the desired stereochemistry. After each new stereocenter that is introduced by a catalytic allylic substitution, a ruthenium-catalyzed cross-metathesis is performed to introduce either an allylic carbonate or allylic bromide to allow the next stereocenter to be introduced.
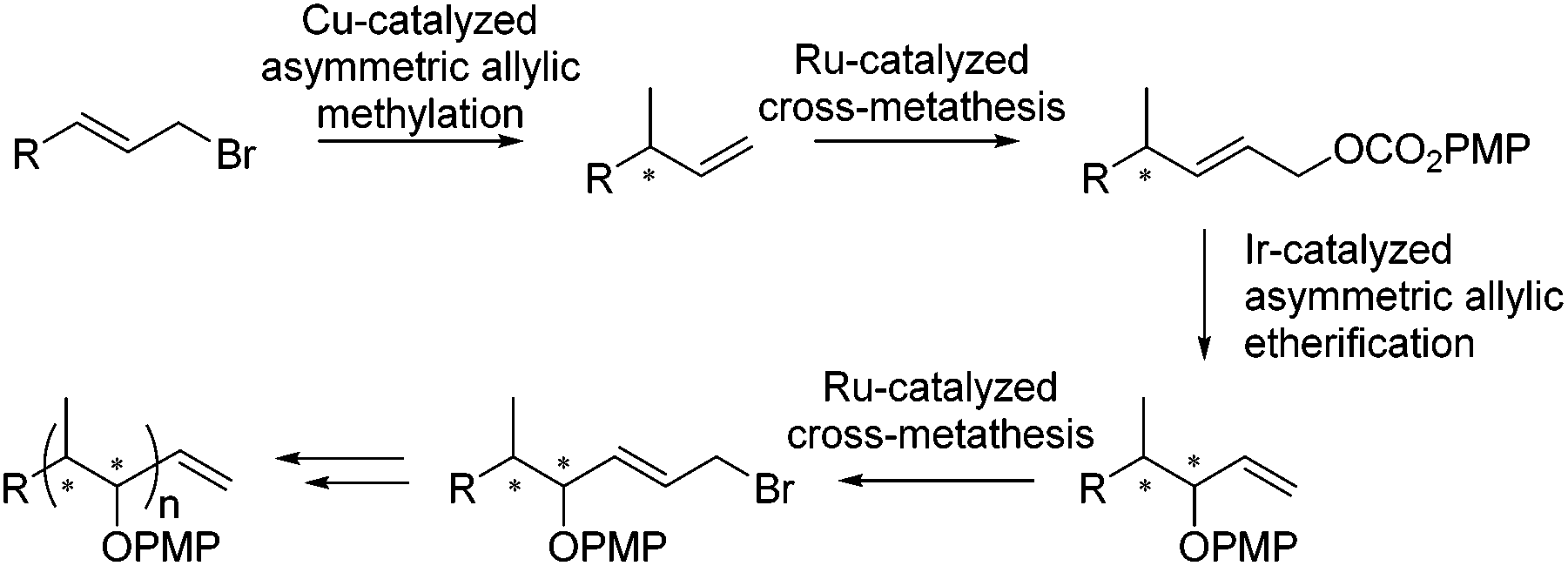 | ||
| Scheme 1 Iterative catalytic approach towards polypropionates. | ||
Results and discussion
Starting with acetal protected allyl bromide 1, the first copper-catalyzed asymmetric allylic alkylation with MeMgBr was carried out using a Cu–Taniaphos catalyst to provide product 2 with high levels of stereocontrol (anti dr > 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; syn dr = 90:10, Scheme 2).17f Both the syn- and anti-isomers of product 2 can be obtained by choosing the appropriate enantiomer of the Taniaphos ligand L1. The pure syn-2b diastereomer was readily obtained by column chromatography. The configuration of the newly formed stereocenter is determined largely by the chiral catalyst, rather than the chirality present in the substrate, indicating a high level of catalyst control. Protecting the adjacent allylic hydroxyl using a dioxolane ring appeared to be essential, since other protecting groups gave rather low conversion and/or regioselectivity.17f
:1; syn dr = 90:10, Scheme 2).17f Both the syn- and anti-isomers of product 2 can be obtained by choosing the appropriate enantiomer of the Taniaphos ligand L1. The pure syn-2b diastereomer was readily obtained by column chromatography. The configuration of the newly formed stereocenter is determined largely by the chiral catalyst, rather than the chirality present in the substrate, indicating a high level of catalyst control. Protecting the adjacent allylic hydroxyl using a dioxolane ring appeared to be essential, since other protecting groups gave rather low conversion and/or regioselectivity.17f
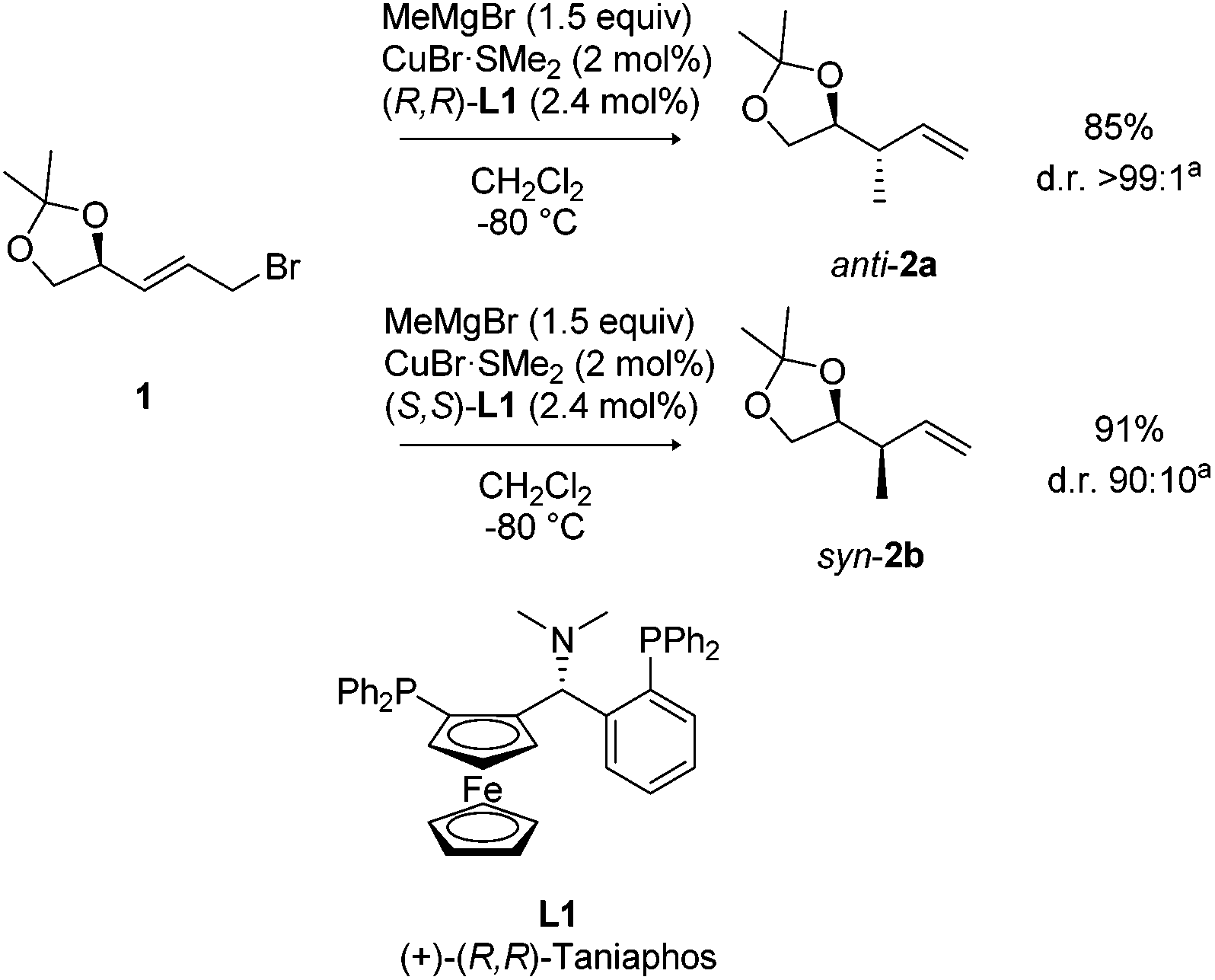 | ||
| Scheme 2 Asymmetric allylic alkylations of allyl bromide 1 towards products 2. aDiastereomeric ratio (dr) determined by GC-MS analysis. | ||
Next, both anti- and syn-products 2 were converted into the corresponding allyl carbonates 4 by a cross-metathesis reaction. Using a Hoveyda–Grubbs 2nd generation catalyst and the readily available (Z)-but-2-ene-1,4-diyl bis(4-methoxyphenyl) dicarbonate (3, Scheme 3), carbonates 4a and 4b were obtained in 69% and 66% yield, respectively.
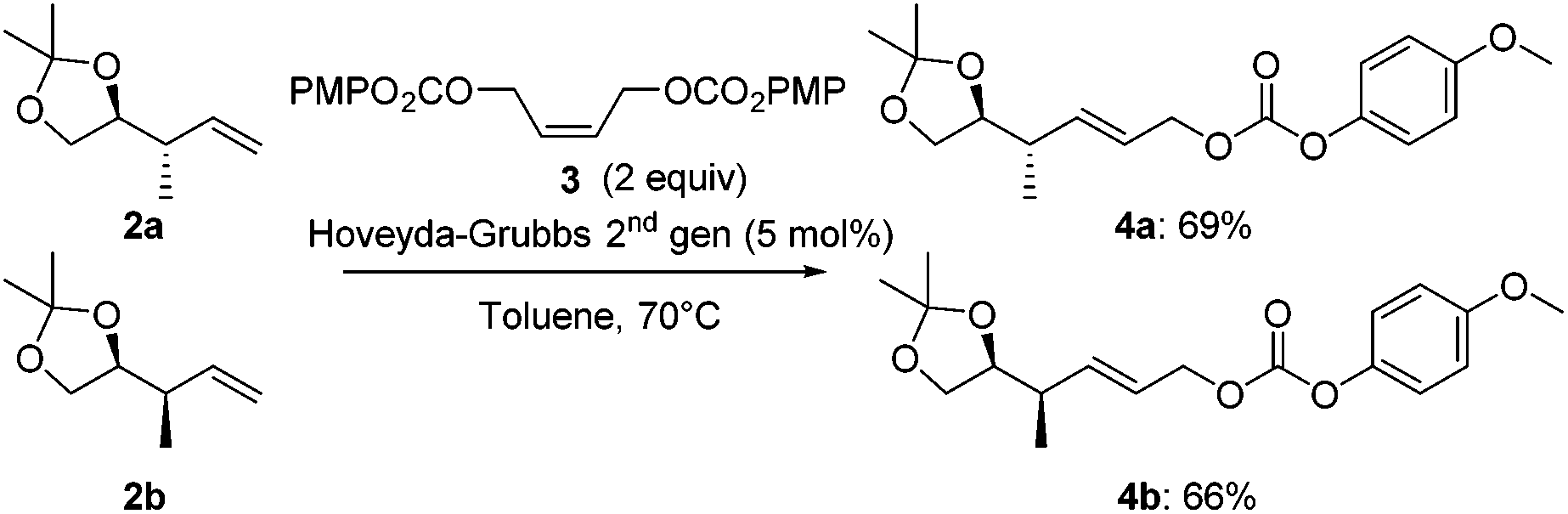 | ||
| Scheme 3 Cross-metathesis of anti- and syn-products 2 towards allyl carbonates 4. | ||
In the next step we explored the iridium-catalyzed asymmetric allylic etherification giving products 5 bearing a 1,3-hydroxy-2-methyl unit, featuring three contiguous stereocenters (Scheme 4). A wide range of different methods for asymmetric allylic alkylations using O-nucleophiles have been reported, often giving rise to allylic ethers with excellent regio- and enantioselectivity.20,21,23–27 Our attention was drawn by one specific method reported by Han and coworkers, in which decarboxylative allylic etherification was performed, to provide branched allylic para-methoxyphenyl ethers (–OPMP).23d,e In contrast to the previous asymmetric allylic etherifications, this method does not require the use of an external nucleophile, leading to a higher atom efficiency. Moreover, the para-methoxyphenyl ethers are generally easily cleaved under oxidative conditions to liberate the free hydroxyl moiety.31
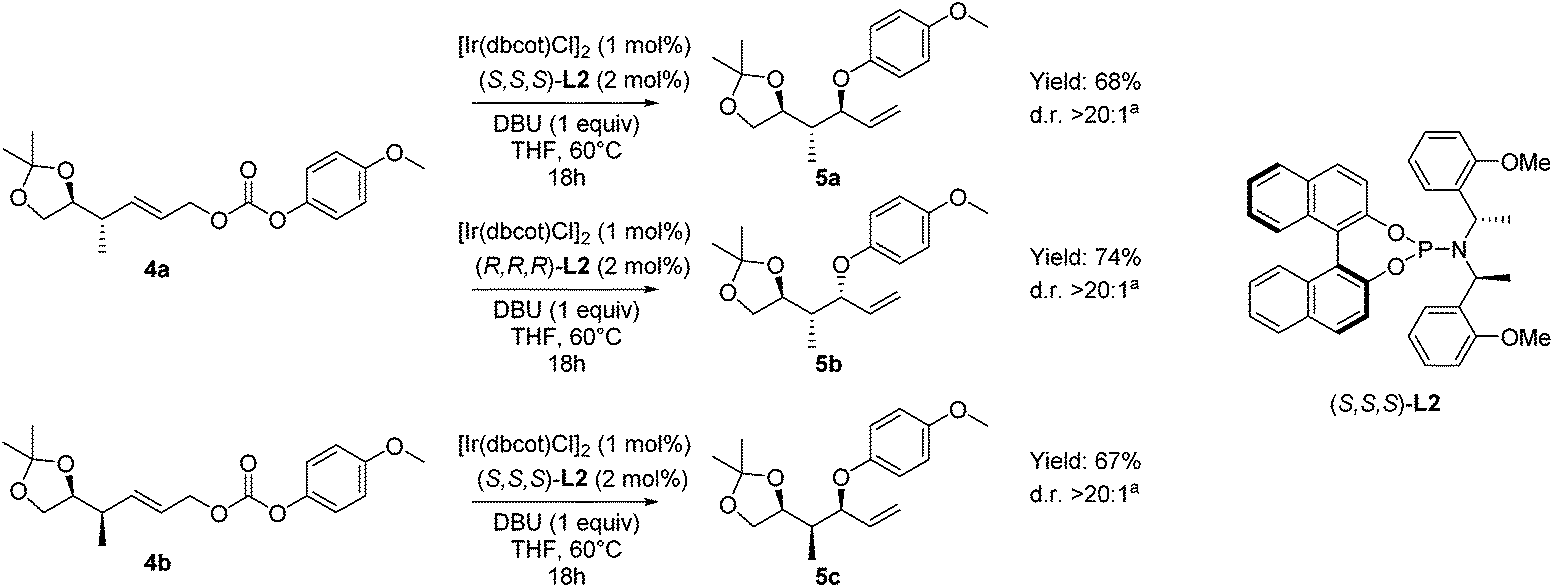 | ||
| Scheme 4 Asymmetric allylic etherifications of allyl carbonates 4 forming allyl ethers 5. aDiastereomeric ratio determined by GC-MS and 1H-NMR analysis of the crude product. | ||
Performing the etherification, using [Ir(dbcot)Cl]232 in the presence of chiral phosphoramidite ligand L2 and DBU as a base in THF (Scheme 4), resulted in high conversion towards the allylic ethers 5. To our delight, when we applied Han's conditions to allylic carbonate 4a with (S,S,S)-L2, anti–anti isomer 5a was obtained with excellent diastereoselectivity. When the opposite enantiomer (R,R,R)-L2 was used, substrate 4a was converted into anti–syn-isomer 5b. A diastereomeric ratio of over 20:1 was found, showing no obvious mismatch effect. Finally, when the asymmetric allylic etherification was performed on substrate 4b using ligand (S,S,S)-L2, syn–syn-isomer 5c was obtained, again with excellent control over diastereoselectivity, with a diastereomeric ratio of over 20:1. Moreover, in all cases, the control over regioselectivity is perfect, as no linear product due to SN2 substitution was observed.
To highlight the potential iteration of our method, allyl bromides 7 were synthesized to perform a second copper-catalyzed asymmetric allylic alkylation. The terminal olefin was converted into the required allylic bromide necessary for the subsequent alkylation using a challenging cross-metathesis reaction (Scheme 5).33 Substrates 5a and 5b were converted into the allylic bromides 7a and 7b, respectively, with (E)-1,4-dibromo-2-butene (6) applying the Hoveyda–Grubbs 2nd generation catalyst. Allylic bromides 7a and 7b were obtained in 81% and 64% yield, respectively.34
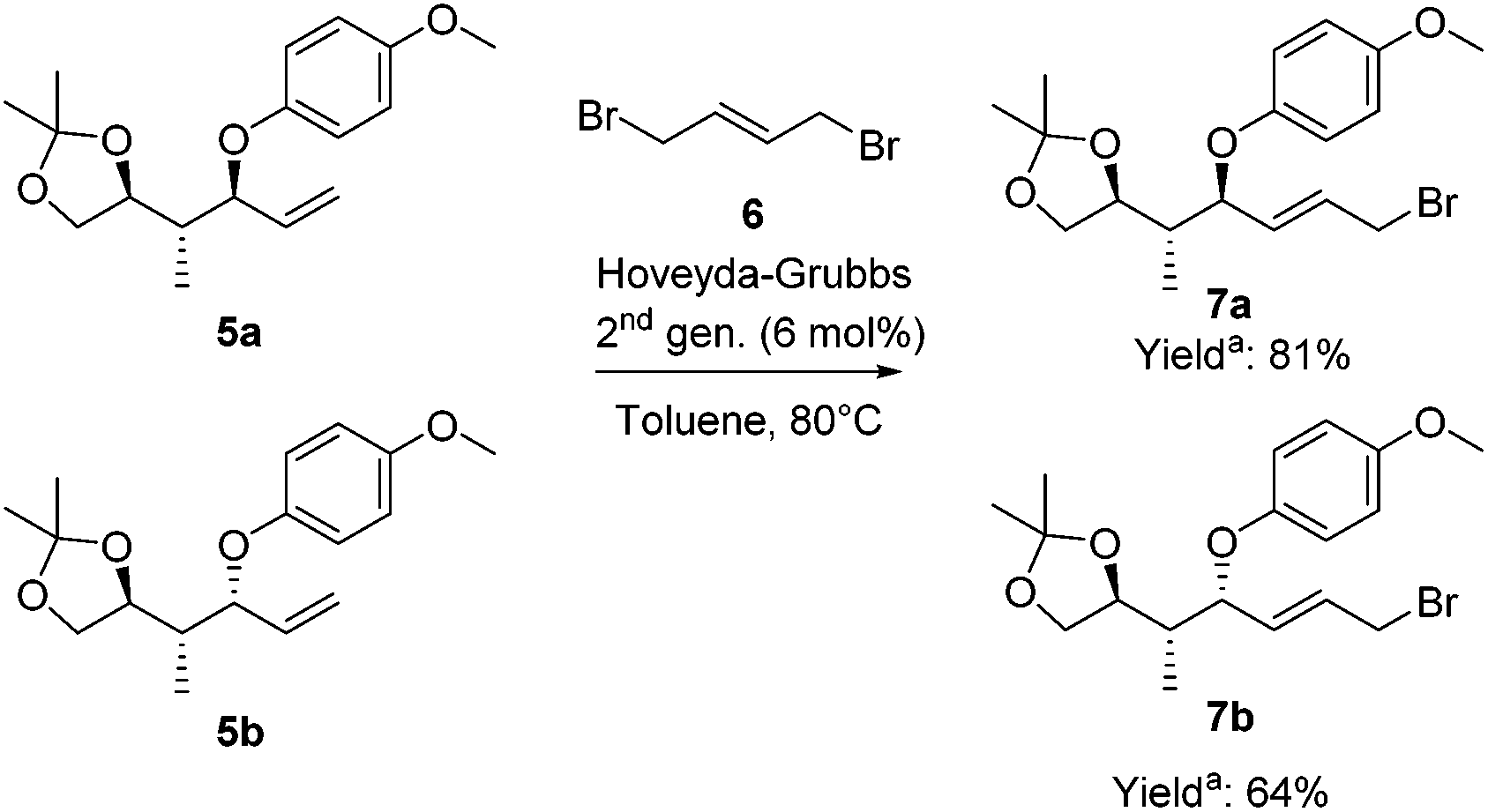 | ||
| Scheme 5 Cross-metathesis reactions to convert the terminal olefins into allylic bromides. aYields are based on the recovered starting material. | ||
A second methyl substituent was introduced via a copper-catalyzed asymmetric allylic alkylation, creating a molecule bearing four consecutive stereocenters (Scheme 6). When we subjected substrates 7 to our previously optimized conditions for the copper-catalyzed asymmetric allylic alkylation of substrate 1, high selectivity was obtained, and rather low conversion was observed. Therefore, a screening of temperatures, copper complexes and ligands was performed on a model substrate (see the ESI‡), showing that none of the tested systems gave better selectivity than our initial system consisting of CuBr·SMe2 and Taniaphos ligand L1. However, using this catalyst system, high conversion could be obtained by raising the temperature to −60 °C. When we subjected allylic bromide 7a to these newly optimized conditions using (R,R)-Taniaphos L1, nearly full conversion and high selectivities in the allylic alkylation towards product 8a with the anti–anti–anti configuration were obtained. This configuration has shown to be the most challenging stereochemical array to synthesize via conventional methods.4d,35 Remarkably, the increase in temperature from −80 °C to −60 °C compared to the asymmetric allylic alkylation of substrate 1 did not affect the SN2′:SN2 ratio. Studies on copper-catalyzed asymmetric allylic alkylations of related structures bearing a vicinal stereocenter are very scarce. Notably, a recent study from our group showed that low regioselectivity is obtained with related substrates with other protecting groups at the allylic alcohol unit.17f Apart from this, as far as we know only a kinetic resolution of allylic substrates bearing an alkyl substituent at the allylic stereocenter has been published.36
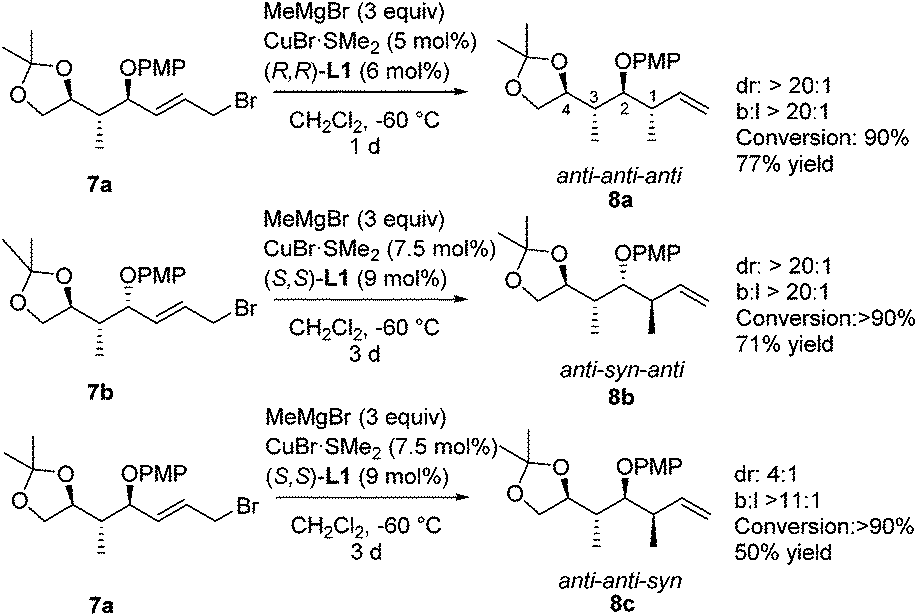 | ||
| Scheme 6 Copper-catalyzed asymmetric allylic alkylation of allylic bromides 7. Branched to linear ratio, diastereomeric ratio and conversion were determined by 1H-NMR analysis of the crude product. For products 8b and 8c, after one day and with 5 mol% of copper, 30% and 50% conversion was obtained, respectively. | ||
Product 8b with the anti–syn–anti-configuration was obtained with excellent selectivities and high conversion, although prolonged reaction times and increased catalyst loading are needed. Even though the configuration of the newly introduced substituent in products 8a and 8b is both anti with respect to the vicinal stereocenter, it is clear that the reaction towards product 8a proceeds faster (1 d versus 3 d) and with a lower catalyst loading (5 mol% versus 7.5 mol%). In this case it seems that the interaction with the more remote stereocenter (1,3-methyl–methyl interaction) has a rather large influence on the reactivity and that the 1,3-syn-configuration in product 8a is preferred over the 1,3-anti-configuration in product 8b. A similar effect was observed in the synthesis of deoxypropionates reported by Hanessian and coworkers and it was hypothesized that syn-pentane interactions37 would be minimized in the CuIII-intermediate when the methyl and CuIII-complex are syn to each other, leading to the syn-product (Fig. 2).38 When these substituents are anti to each other (leading to anti-products), these interactions are maximized. It is however not clear what the effect of the bulky –OPMP substituent is and how this changes the 1,3-interaction.
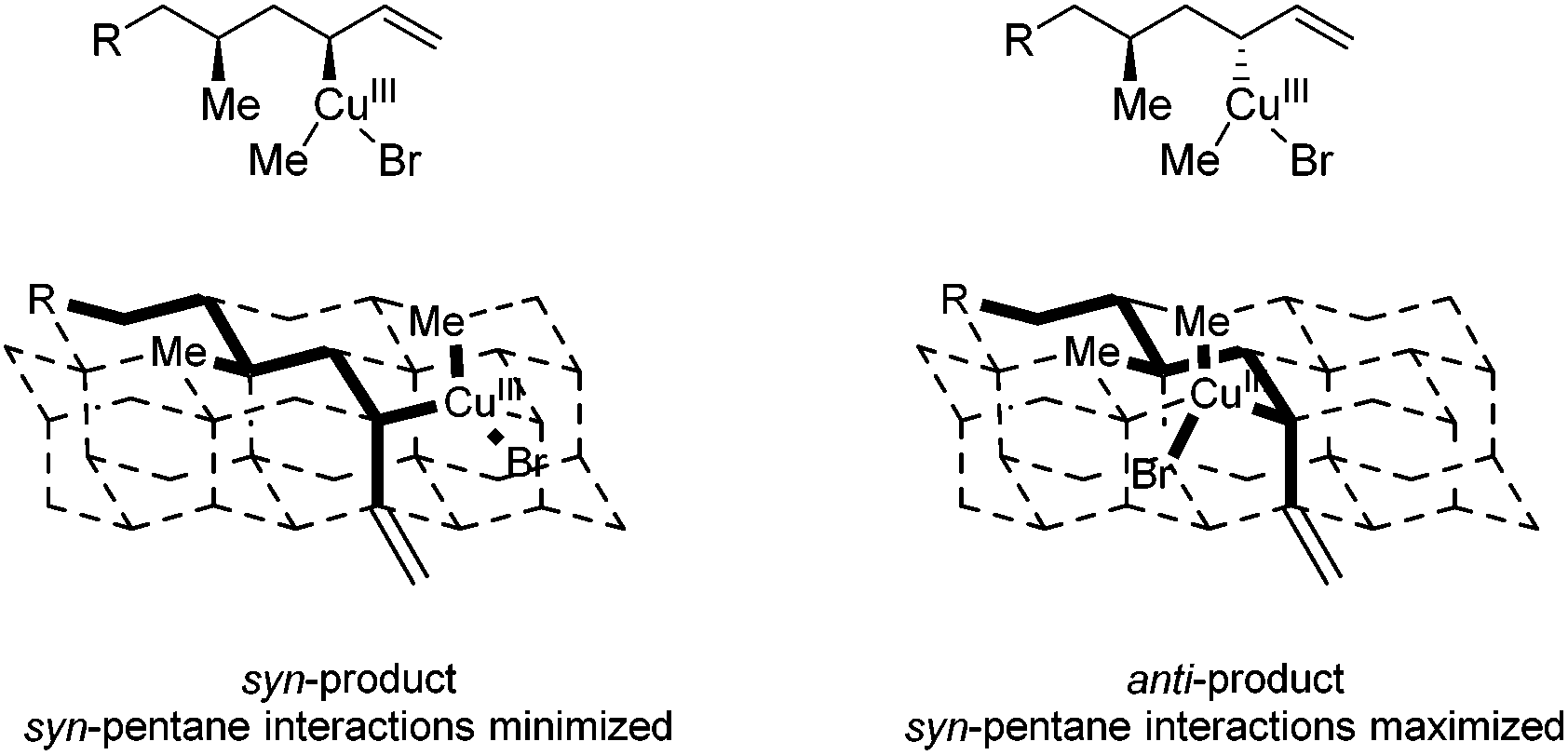 | ||
| Fig. 2 syn-Pentane interactions of 1,3-dimethyl moieties in a diamond lattice. Ligands on copper are not presented for clarification. | ||
The new protocol presented here was also effective for the even more challenging methylation towards the anti–anti–syn-8c stereoisomer. In this case, the methyl unit that had to be introduced is syn with respect to the vicinal stereocenter already present. As shown for substrate 1, a mismatch effect is expected due to unfavorable steric interactions of the substrate with the catalyst when a methyl substituent is introduced with a 1,2-syn-configuration. Moreover, also the 1,3-methyl–methyl interactions are unfavorable, making this diastereomer the most difficult one to access. Despite the highly unfavorable interactions, high regioselectivity (b:l > 11:1) was observed towards product 1,2-syn-8c when we subjected substrate 7a to our reaction conditions, using (S,S)-Taniaphos L1. Although a slightly reduced stereoselectivity was observed in this case, most probably arising from competing substrate control, the anti–anti–syn isomer 8c could still be obtained with good selectivity (dr = 4:1). It is important to note that the small amount of minor stereoisomer was readily removed by column chromatography to provide product 8c as a single enantiomer in 50% yield.
To overcome the mismatch effect observed in the allylic alkylation towards product syn-8c which resulted in a decrease in diastereoselectivity, an alternative approach was studied. In previous work, we showed that the presence of the dioxolane ring is essential to obtain high selectivity in an asymmetric allylic alkylation towards 1,2-hydroxymethyl units.17f We envisioned that installing a dioxane ring in our system might also improve the stereoselectivity. A sequence consisting of deprotection–protection steps starting from 5a afforded olefin 9 in 59% overall yield (Scheme 7). Next, a ruthenium-catalyzed cross-metathesis was performed with (E)-1,4-dibromo-2-butene and the Hoveyda–Grubbs 2nd generation catalyst to provide allylic bromide 10.
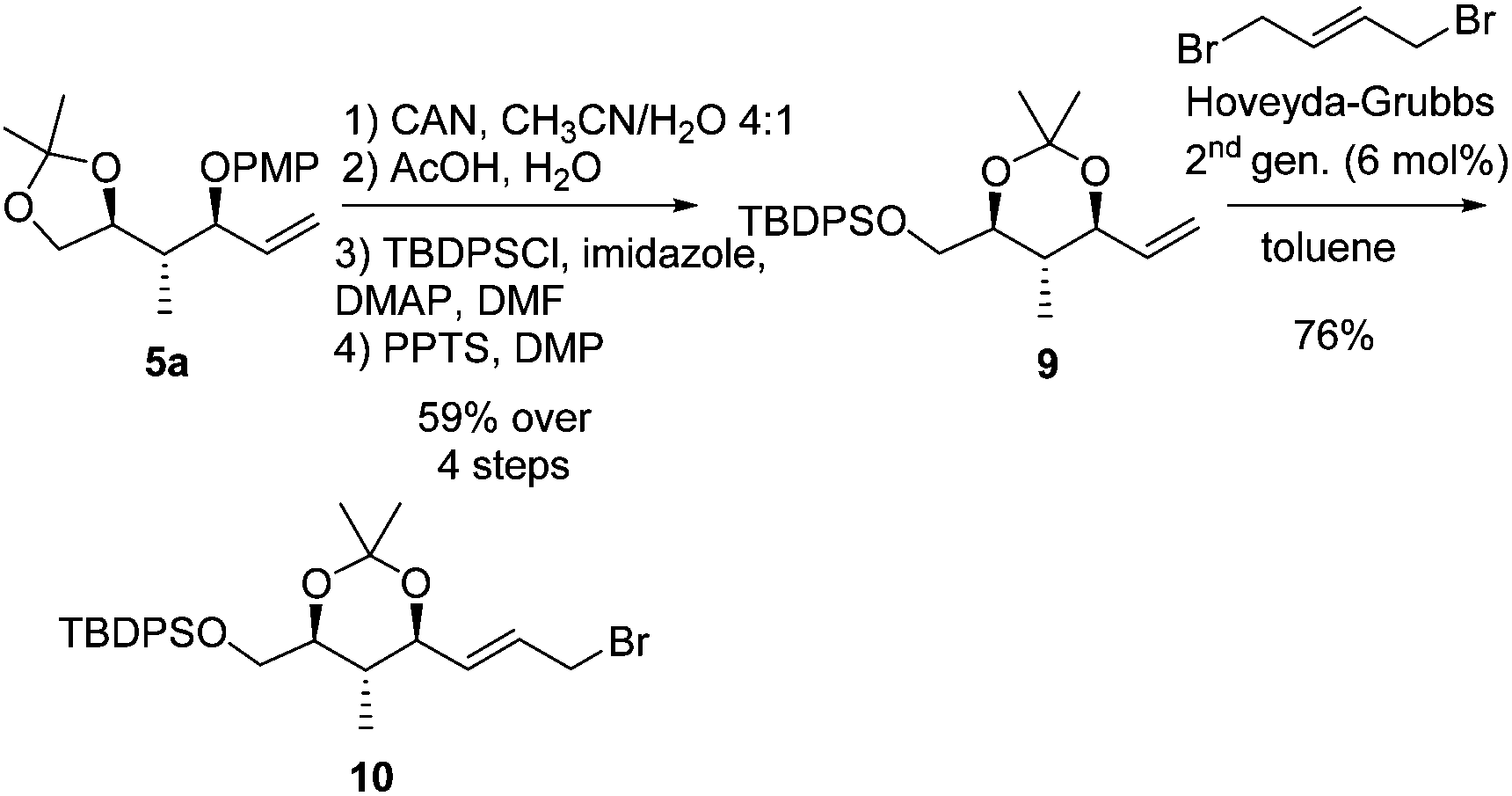 | ||
| Scheme 7 Synthesis of allylic bromide 10. | ||
This adapted strategy was highly rewarding as the copper-catalyzed asymmetric allylic alkylation of allylic bromide 10 shows that excellent stereoselectivity and regioselectivity are obtained towards product 11 (Scheme 8). Product 11 has the same anti–anti–syn configuration as product 8c, but shows a near perfect degree of stereocontrol, likely due to the protection of the hydroxyl moiety as part of the dioxane ring. Although this reaction suffers from rather low conversion, starting materials can be readily recovered and resubjected to the reaction. These results show that the three polypropionate segments bearing anti–anti–anti, anti–syn–anti and anti–anti–syn stereochemical units can be obtained in a catalytic iterative manner with branched linear and diastereomeric ratios of over 95:5.
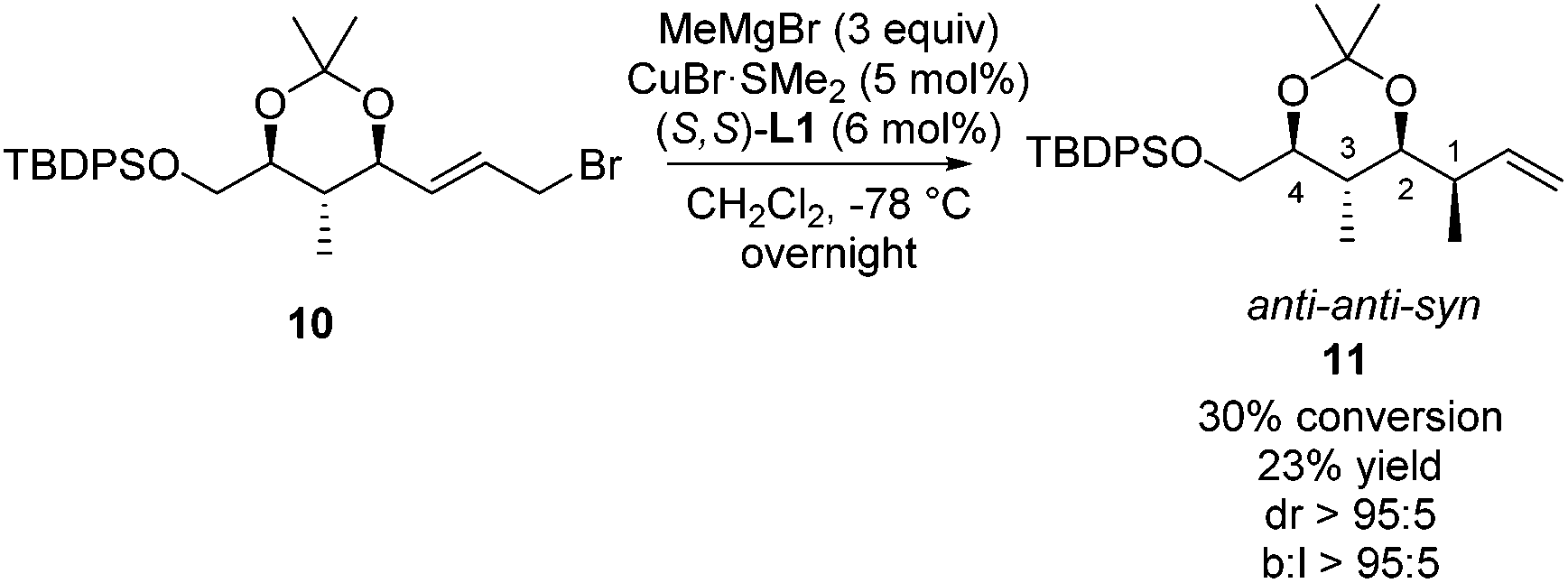 | ||
| Scheme 8 Copper-catalyzed asymmetric allylic alkylation towards product 11. Conversion, diastereomeric ratio and branched to linear ratio are based on the 1H-NMR analysis of the crude product. | ||
Conclusions
In summary, a new method for the iterative catalyst controlled diastereodivergent synthesis of polypropionates is developed by combining a copper-catalyzed asymmetric allylic alkylation with a ruthenium-catalyzed cross-metathesis and an iridium-catalyzed asymmetric allylic etherification. Both methyl- and hydroxyl-substituted stereocenters are introduced with high regio- and diastereoselectivity. In each allylic substitution, despite the presence of (multiple) stereocenters, nearly full control over selectivity by the chiral catalyst is observed and the ability of synthesizing several diastereomers under catalyst control has been demonstrated with the preparation of anti–anti–anti-, anti–syn–anti- and anti–anti–syn-isomers. These synthetically valuable fragments can be accessed with excellent selectivities by the use of an appropriate enantiomer of the ligand in each catalytic reaction. Moreover, remarkably high selectivity towards the branched product was observed in the copper-catalyzed asymmetric allylic alkylation.Experimental section
Typical procedure for cross metathesis of olefins 2 with dicarbonate 3
In a Schlenk tube equipped with a septum and a stirring bar, the Hoveyda–Grubbs 2nd generation catalyst (7.68 μmol, 4.81 mg), dicarbonate 3 (0.256 mmol, 99.4 mg) and olefin 2 (0.256 mmol, 40 mg) were dissolved in dry degassed toluene (2.5 mL) and stirred under a nitrogen atmosphere at 70 °C. After 5 h, a second portion of the Hoveyda–Grubbs 2nd generation catalyst (5.20 μmol, 3.21 mg) and dicarbonate 3 (0.256 mmol, 99.4 mg) were added. After 18 h, water was added and the aqueous layer was extracted 3 times with CH2Cl2. The organic layers were combined and dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography to yield the corresponding carbonate 4.:1) afforded 4a (65.5 mg, 69%) as a colorless oil. [α]20D = −2.4° (c = 1.0 in CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.09 (m, 2H), 6.88 (m, 2H), 5.91 (dd, J = 7.1, 15.6 Hz, 1 H), 5.71 (dt, J = 15.6, 6.5 Hz, 1H), 4.71 (m, 2H), 3.99 (m, 2H), 3.80 (s, 3H), 3.63 (dd, J = 6.1, 13.3 Hz, 1H), 1.41 (s, 1H), 1.36 (s, 1H), 1.03 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 157.4, 153.9, 144.7, 138.4, 123.5, 121.9, 114.4, 109.1, 79.1, 69.1, 67.4, 55.6, 39.5, 26.5, 25.5, 15.6. HRMS (ESI+, m/z): Calcd for C18H25O6 [M + H+]: 337.1641, found: 337.1646.
:1) afforded 4b (136.9 mg, 66%) as a colorless oil. [α]20D = +15.2° (c = 1.0 in CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.09 (m, 2H), 6.88 (m, 2H), 5.74 (m, 2H), 4.67 (d, J = 5.3 Hz, 2H), 3.94 (m, 2H), 3.79 (s, 3H), 3.64 (m, 1H), 2.39 (m, 1H), 1.41 (s, 1H), 1.35 (s, 1H), 1.11 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 157.4, 153.9, 144.7, 137.8, 124.0, 121.8, 114.4, 109.1, 79.1, 68.9, 67.4, 55.6, 40.3, 26.7, 25.4, 16.2. HRMS (ESI+, m/z): Calcd for C18H25O6 [M + H+]: 337.1643, found: 337.1646.
Typical procedure for iridium-catalyzed allylic etherification of carbonates 4
In a Schlenk tube equipped with a septum and a stirring bar, [Ir(dbcot)Cl]2 (4 μmol, 3.46 mg) and L2 (8 μmol, 5.0 mg) were stirred for 10 min in THF (0.5 mL) under a nitrogen atmosphere until a homogenous orange solution was obtained. DBU (0.2 mmol, 30 μmol) was added and the color changed to light yellow. Carbonate 4 (0.2 mmol, 67.3 mg) in THF (1.0 mL) was added and the mixture was stirred overnight at 60 °C. Water (1.5 ml) was added and the aqueous layer was extracted with Et2O (3 × 1.5 ml). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography to yield compounds 5.:1) afforded 5a (197.9 mg, 68%) as a colorless oil. [α]20D = −2.4° (c = 1.0 in CHCl3). 1H NMR (400 MHz, CDCl3): δ 6.89–6.83 (m, 2H), 6.83–6.77 (m, 2H), 5.86 (ddd, J = 6.3, 10.7, 17.2 Hz, 1H), 5.31 (dd, J = 8.8, 14.0 Hz, 2H), 4.79 (dd, J = 4.8, 5.7 Hz, 1H), 4.06–3.98 (m, 2H), 3.76 (s, 3H), 3.72–3.65 (m, 1H), 2.27–2.15 (m, 1H), 1.41 (s, 3H), 1.36 (s, 3H), 0.93 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 153.8, 152.1, 134.8, 118.1, 117.1, 114.4, 108.6, 80.0, 76.6, 67.6, 55.7, 40.6, 26.7, 25.7, 10.1. HRMS (ESI+, m/z): Calcd for C17H25O4 [M + H+]: 293.1745, found: 293.1747.
:1) afforded 5b (217.2 mg, 74%) as a pale yellow oil. [α]20D = −2.8° (c = 1.0 in CHCl3). 1H NMR (400 MHz, CDCl3): δ 6.91–6.76 (m, 4H), 5.84 (ddd, J = 17.3, 10.7, 5.3 Hz, 1H), 5.27–5.20 (m, 2H), 4.85–4.81 (m, 1H), 4.17 (td, J = 7.9, 6.1 Hz, 1H), 4.02 (dd, J = 8.0, 6.0 Hz, 1H), 3.76 (s, 3H), 3.67 (t, J = 7.9 Hz, 1H), 1.95–1.86 (m, 1H), 1.41 (s, 3H), 1.32 (s, 3H), 0.96 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 153.9, 152.8, 136.8, 117.4, 116.6, 114.4, 108.5, 79.4, 77.0, 67.7, 55.7, 42.1, 26.7, 25.7, 8.7. HRMS (ESI+, m/z): Calcd for C17H25O4 [M + H+]: 293.1747, found: 293.1748.
:1) afforded 5c (38.9 mg, 67%) as a colorless oil. [α]20D = +7.2° (c = 1.0 in CHCl3). 1H NMR (400 MHz, CDCl3): δ 6.84–6.77 (m, 4H), 5.89–5.79 (m, 1H), 5.29–5.22 (m, 2H), 4.50 (t, J = 5.1 Hz, 1H), 4.13 (dd, J = 13.8, 6.8 Hz, 1H), 4.04 (dd, J = 8.1, 6.0 Hz, 1H), 3.76 (t, J = 8 Hz, 1H), 3.75 (s, 3H), 2.03–1.95 (m, 1H), 1.37 (s, 3H), 1.35 (s, 3H), 1.13 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 153.9, 152.1, 135.7, 117.7, 117.0, 114.5, 105.3, 80.9, 77.1, 68.0, 55.6, 41.1, 26.6, 25.7, 11.4. HRMS (ESI+, m/z): Calcd for C17H25O4 [M + H+]: 293.1747, found: 293.1748.
Typical procedure for cross-metathesis of olefins 5 with (E)-1,4-dibromobut-2-ene
In a Schlenk tube equipped with a septum and a stirring bar, the Hoveyda–Grubbs 2nd generation catalyst (0.015 mmol, 9.40 mg), (E)-1,4-dibromobut-2-ene (2.5 mmol, 577.3 mg) and olefin 5 (0.5 mmol, 146.2 mg) were dissolved in toluene (5 mL) and the mixture was stirred under a nitrogen atmosphere at 80 °C. After 5 h, a second portion of the Hoveyda–Grubbs 2nd generation catalyst (0.010 mmol, 6.27 mg) was added. After 18 h, water (4 ml) was added and the aqueous layer was extracted with CH2Cl2 (3 × 3 ml). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography to yield the corresponding allyl bromides 7.:1) afforded 7a (130.6 mg, 56%) as a pale yellow oil. [α]20D = +9.0° (c = 1.0 in CHCl3). 1H NMR (400 MHz, CDCl3): δ 6.86–6.78 (m, 4H), 6.00 (dt, J = 15.1, 7.5 Hz, 1H), 5.83 (dd, J = 15.4, 6.3 Hz, 1H), 4.87–4.83 (m, 1H), 4.04 (dd, J = 7.8, 6.0 Hz, 1H), 3.99–3.92 (m, 2H), 3.76 (s, 3H), 3.65 (dd, J = 9.9, 5.5 Hz, 1H), 2.26–2.17 (m, 1H), 1.41 (s, 3H), 1.37 (s, 3H), 0.92 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 154.0, 151.8, 132.0, 129.6, 117.1, 114.5, 108.8, 78.4, 76.6, 67.9, 55.7, 41.1, 31.8, 26.8, 25.7, 10.1. HRMS (ESI+, m/z): Calcd for C18H26BrO4 [M + H+]: 385.1009, found: 385.1002.
:1) afforded 7b (114.9 mg, 54%) as a pale yellow oil. [α]20D = −22.0° (c = 1.0 in CHCl3). 1H NMR (400 MHz, CDCl3): δ 6.89–6.77 (m, 1H), 5.90 (dt, J = 7.3, 6.8 Hz, 1H), 5.79 (dd, J = 15.4, 5.0 Hz, 1H), 4.89–4.86 (m, 1H), 4.14 (dt, J = 14.1, 7.1 Hz, 1H), 4.02 (dd, J = 8.0, 6.0 Hz, 1H), 3.95 (d, J = 7.3 Hz, 1H), 3.76 (s, 3H), 3.65 (t, J = 7.8 Hz, 1H), 1.93–1.84 (m, 1H), 1.41 (s, 3H), 1.31 (s, 3H), 0.95 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 154.1, 152.5, 134.1, 128.2, 117.4, 114.5, 108.6, 78.0, 76.9, 67.8, 55.7, 42.2, 31.9, 26.9, 25.7, 8.9. HRMS (ESI+, m/z): Calcd for C18H26BrO4 [M + H+]: 385.1009, found: 385.1002.
Typical procedure for asymmetric allylic alkylations of allylic bromides 7
In a Schlenk tube equipped with a stirring bar and a septum, CuBr·SMe2 (0.010 mmol, 2.06 mg) and the corresponding ligand (0.012 mmol) were dissolved in CH2Cl2 (0.8 ml) and the mixture was stirred under a nitrogen atmosphere at room temperature for 15 min. The mixture was cooled to −60 °C and MeMgBr (0.2 ml, 3 M solution in Et2O) was added dropwise. Allyl bromide 7 (0.2 mmol, 77.1 mg) was then added dropwise as a solution in CH2Cl2 (0.32 ml) over 1 h using a syringe pump. Once the addition was complete the resulting mixture was further stirred at −60 °C for 3 days. The reaction was quenched by addition of MeOH (0.2 ml) and the mixture was allowed to reach rt. Then, saturated aqueous NH4Cl solution (2 ml) was added to the mixture. The organic phase was separated, and the resulting aqueous layer was extracted with CH2Cl2 (3 × 2 ml). The organic layers were combined and dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography to yield the corresponding products 8.:1) afforded 8a (48.9 mg, 77%) as a colorless oil. [α]20D = +23.4° (c = 1.0 in CHCl3). 1H NMR (400 MHz, CDCl3): δ 6.88–6.77 (m, 4H), 6.01–5.91 (m, 1H), 5.07 (d, J = 16.9 Hz, 1H), 5.01 (dd, J = 10.3, 1.0 Hz, 1H), 4.31 (dd, J = 13.9, 6.3 Hz, 1H), 4.08 (t, J = 5.2 Hz, 1H), 3.89 (dd, J = 8.1, 6.3 Hz, 1H), 3.76 (s, 3H), 3.63 (t, J = 7.9 Hz, 1H), 2.73–2.63 (m, 1H), 2.34–2.24 (m, 1H), 1.40 (s, 3H), 1.34 (s, 3H), 1.11 (d, J = 6.9 Hz, 3H), 0.92 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 153.6, 153.5, 140.7, 116.6, 114.7, 114.6 (2×), 108.2, 83.5, 76.1, 66.6, 56.7, 40.3, 38.2, 26.5, 25.4, 17.9, 11.3. HRMS (ESI+, m/z): Calcd for C19H28O4Na [M + Na+]: 343.1880, found: 343.1880.
:1) afforded 8b (48.9 mg, 77%) as a colorless oil. [α]20D = +2.2° (c = 1.0 in CHCl3). 1H NMR (400 MHz, CDCl3): δ 6.98–6.92 (m, 2H), 6.79–6.74 (m, 2H), 5.80 (ddd, J = 17.6, 10.3, 7.5 Hz, 1H), 5.06 (dd, J = 17.2, 1.2 Hz, 1H), 4.93 (d, J = 10.3 Hz, 1H), 4.34 (dd, J = 8.3, 1.8 Hz, 1H), 3.99–3.91 (m, 2H), 3.75 (s, 3H), 3.58–3.52 (m, 1H), 2.63–2.53 (m, 1H), 2.00–1.91 (m, 1H), 1.40 (s, 3H), 1.24 (s, 3H), 1.02 (d, J = 6.9 Hz, 3H), 0.94 (d, J = 6.9, 3H). 13C NMR (101 MHz, CDCl3): δ 154.3, 153.4, 141.4, 117.2, 114.5, 114.3, 108.6, 81.8, 77.2, 68.5, 55.7, 40.8, 39.3, 27.0, 25.6, 16.6, 9.1. HRMS (ESI+, m/z): Calcd for C19H28O4Na [M + Na+]: 343.1880, found: 343.1879.
:1) afforded 8c (17.9 mg, 50%) as a colorless oil. [α]20D = −2.4° (c = 1.0 in CHCl3). 1H NMR (400 MHz, CDCl3): δ 6.90–6.83 (m, 2H), 6.82–6.77 (m, 2H), 5.82 (ddd, J = 17.4, 10.2, 8.6 Hz, 1H), 5.12 (dd, J = 17.6, 1.6 Hz, 1H), 5.03 (dd, J = 10.3, 1.7 Hz, 1H), 4.30 (q, J = 7.1 Hz, 1H), 4.14 (dd, J = 8.2, 3.4 Hz, 1H), 3.95 (dd, J = 8.1, 6.1 Hz, 1H), 3.76 (s, 2H), 3.58 (t, J = 8.0 Hz, 1H), 2.69 (h, J = 7.2 Hz, 1H), 2.25 (td, J = 7.0, 3.4 Hz, 1H), 1.40 (s, 3H), 1.36 (s, 3H), 1.09 (d, J = 6.7 Hz, 3H), 0.92 (d, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 153.6, 153.4, 141.1, 116.5, 115.0, 114.6, 108.2, 82.8, 76.1, 67.4, 55.7, 41.0, 38.6, 26.5, 25.6, 17.7, 11.3. HRMS (ESI+, m/z): Calcd for C19H28O4Na [M + Na+]: 343.1880, found: 343.1881.
:1) to afford syn-11a (23%, 10.5 mg) as a colorless oil. [α]20D = +6.8° (c = 0.5 in CHCl3) 1H NMR (400 MHz, CDCl3) δ 7.76–7.67 (m, 4H), 7.44–7.33 (m, 6H), 5.85 (ddd, J = 17.0, 10.5, 9.1 Hz, 1H), 5.04–4.92 (m, 2H), 3.79–3.70 (m, 2H), 3.54–3.48 (m, 1H), 3.36 (dd, J = 10.4, 2.1 Hz, 1H), 2.46–2.36 (m, 1H), 1.67 (ddd, J = 16.9, 6.6, 3.8 Hz, 1H), 1.38 (s, 6H), 1.06 (d, J = 6.1 Hz, 3H), 1.05 (s, 9H), 0.70 (d, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 139.9, 135.8, 129.4, 127.5, 114.7, 112.9, 97.6, 75.4, 65.7, 39.5, 32.0, 29.9, 26.8, 19.4, 19.3, 18.0, 11.5. HRMS (ESI+, m/z): Calcd for C28H40O3SiNa [M + Na+]: 475.2639, found: 475.2630.
Acknowledgements
Financial support from The Netherlands Organization for Scientific Research (NWO-CW), the National Research School of Catalysis (NRSC-C) and the Ministry of Education, Culture and Science (Gravitation Program 024.001.035) is gratefully acknowledged.Notes and references
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688 CrossRef CAS PubMed.
- A. M. P. Koskinen and K. Karisalmi, Chem. Soc. Rev., 2005, 34, 677 RSC.
- K. J. Weissman and R. Müller, Nat. Prod. Rep., 2010, 27, 1276 RSC.
- (a) J. Li and D. Menche, Synthesis, 2009, 2293 CAS; (b) D. Herkommer, B. Schmalzbauer and D. Menche, Nat. Prod. Rep., 2014, 31, 456 RSC; (c) A. R. Dechert-Schmitt, D. C. Schmitt, X. Gao, T. Itoh and M. J. Krische, Nat. Prod. Rep., 2014, 31, 504 RSC; (d) R. W. Hoffmann, G. Dahmann and M. W. Andersen, Synthesis, 1994, 629 CrossRef CAS.
- (a) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC; (b) B. Schetter and R. Mahrwald, Angew. Chem., Int. Ed., 2006, 45, 7506 CrossRef CAS PubMed.
- (a) D. A. Evans, J. S. Tedrow, J. T. Shaw and C. W. Downey, J. Am. Chem. Soc., 2002, 124, 392 CrossRef CAS PubMed; (b) M. A. Walker and C. H. Heathcock, J. Org. Chem., 1991, 56, 5747 CrossRef CAS.
- (a) R. W. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 555 CrossRef; (b) W. R. Roush and R. L. Halterman, J. Am. Chem. Soc., 1986, 108, 294 CrossRef CAS; (c) H. C. Brown, K. S. Bhat and R. S. Randad, J. Org. Chem., 1989, 54, 1570 CrossRef CAS; (d) R. O. Duthaler, A. Hafner and M. Riediker, Pure Appl. Chem., 1990, 62, 631 CrossRef CAS; (e) J. Otera, Modern Carbonyl Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed; (f) B. M. Hackman, P. J. Lombardi and J. L. Leighton, Org. Lett., 2004, 6, 4375 CrossRef CAS PubMed.
- H. Nishiyama and T. Shiomi, in Metal Catalyzed Reductive C–C Bond Formation, ed. M. J. Krische, Springer-Verlag, Berlin Heidelberg, 2007, pp. 105–137 Search PubMed.
- C. Bonini and G. Righi, Synthesis, 1994, 225 CrossRef CAS.
- (a) A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 6798 CrossRef CAS PubMed; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed; (c) For a recent catalytic iterative cross-aldol method see: L. Lin, K. Yamamoto, H. Mitsunuma, Y. Kanzaki, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2015, 137, 15418 CrossRef CAS PubMed.
- S. J. Taylor, M. O. Duffey and J. P. J. Morken, J. Am. Chem. Soc., 2000, 122, 4528 CrossRef CAS.
- (a) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS PubMed; (b) D. G. Hall, Synlett, 2007, 1644 CrossRef CAS; (c) I. S. Kim, M. Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6340 CrossRef CAS PubMed.
- (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (c) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258 CrossRef CAS PubMed; (d) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461 CrossRef CAS PubMed; (e) U. Kazmaier, Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, Springer-Verlag, Berlin, Heidelberg, 2012 Search PubMed; (f) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 CrossRef CAS PubMed; (g) S. Förster, G. Helmchen and U. Kazmaier, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley, New Jersey, 2010, pp. 497–641 Search PubMed.
- For representative examples, see: (a) A. W. Van Zijl, F. López, A. J. Minnaard and B. L. Feringa, J. Org. Chem., 2007, 72, 2558 CrossRef CAS PubMed; (b) Y. Shi, B. Jung, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2015, 137, 8948 CrossRef CAS PubMed.
- (a) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824 CrossRef CAS PubMed; (b) H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2005, 44, 4435 CrossRef CAS PubMed; (c) A. H. Hoveyda, A. W. Hird and M. A. Kacprzynski, Chem. Commun., 2004, 16, 1779 RSC; (d) A. Alexakis, J. E. Bäckvall, N. Krause, O. Pàmies and M. Diéguez, Chem. Rev., 2008, 108, 2796 CrossRef CAS PubMed; (e) C. A. Falciola and A. Alexakis, Eur. J. Org. Chem., 2008, 3765 CrossRef CAS; (f) J. B. Langlois and A. Alexakis, Top. Organomet. Chem., 2012, 38, 235 CrossRef; (g) O. Baslé, A. Denicourt-Nowicki, C. Crévisy and M. Mauduit, in Copper-Catalyzed Asymmetric Synthesis, ed. A. Alexakis, N. Krause and S. Woodward, Wiley-VCH, Weinheim, 2014, ch. 4 Search PubMed.
- (a) K. Tissot-Croset, D. Polet and A. Alexakis, Angew. Chem., Int. Ed., 2004, 43, 2426 CrossRef CAS PubMed; (b) K. Tissot-Croset and A. Alexakis, Tetrahedron Lett., 2004, 45, 7375 CrossRef CAS; (c) C. A. Falciola, K. Tissot-Croset and A. Alexakis, Angew. Chem., Int. Ed., 2006, 45, 5995 CrossRef CAS PubMed; (d) C. A. Falciola and A. Alexakis, Angew. Chem., Int. Ed., 2007, 46, 2619 CrossRef CAS PubMed; (e) M. Fañanás-Mastral and B. L. Feringa, J. Am. Chem. Soc., 2010, 132, 13152 CrossRef PubMed; (f) J. B. Langlois and A. Alexakis, Angew. Chem., Int. Ed., 2011, 50, 1877 CrossRef CAS PubMed; (g) M. Fañanás-Mastral, M. Pérez, P. H. Bos, A. Rudolph, S. R. Harutyunyan and B. L. Feringa, Angew. Chem., Int. Ed., 2012, 51, 1922 CrossRef PubMed; (h) M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, J. Am. Chem. Soc., 2012, 134, 4108 CrossRef CAS PubMed; (i) V. Hornillos, M. Pérez, M. Fañanás-Mastral and B. L. Feringa, J. Am. Chem. Soc., 2013, 135, 2140 CrossRef CAS PubMed; (j) M. Fañanás-Mastral, R. Vitale, M. Pérez and B. L. Feringa, Chem. – Eur. J., 2015, 21, 4209 CrossRef PubMed.
- (a) F. López, A. W. van Zijl, A. J. Minnaard and B. L. Feringa, Chem. Commun., 2006, 409 RSC; (b) K. Geurts, S. P. Fletcher and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 15572 CrossRef CAS PubMed; (c) J. F. Teichert, S. Zhang, A. W. van Zijl, J. W. Slaa, A. J. Minnaard and B. L. Feringa, Org. Lett., 2010, 12, 4658 CrossRef CAS PubMed; (d) T. den Hartog, B. Macia, A. J. Minnaard and B. L. Feringa, Adv. Synth. Catal., 2010, 352, 999 CrossRef CAS; (e) M. Pérez, M. Fañanás-Mastral, P. H. Bos, H. Rudolph, S. R. Harutyunyan and B. L. Feringa, Nat. Chem., 2011, 3, 337 CrossRef PubMed; (f) M. Fañanás-Mastral, B. ter Horst, A. J. Minnaard and B. L. Feringa, Chem. Commun., 2011, 47, 5843 RSC; (g) Y. Huang, M. Fañanás-Mastral, A. J. Minnaard and B. L. Feringa, Chem. Commun., 2013, 49, 3309 RSC; (h) B. Mao, M. Fañanás-Mastral, M. Lutz and B. L. Feringa, Chem. – Eur. J., 2013, 19, 761 CrossRef CAS PubMed; (i) N. C. van der Molen, T. D. Tiemersma-Wegman, M. Fañanás-Mastral and B. L. Feringa, J. Org. Chem., 2015, 80, 4981 CrossRef CAS PubMed.
- (a) W. Lölsberg, S. Ye and H. G. Schmalz, Adv. Synth. Catal., 2010, 352, 2023 CrossRef; (b) W. Lölsberg, S. Werle, J. M. Neudörfl and H. G. Schmalz, Org. Lett., 2012, 14, 5996 CrossRef PubMed.
- (a) S. Tominaga, Y. Oi, T. Kato, D. K. An and S. Okamoto, Tetrahedron Lett., 2004, 45, 5585 CrossRef CAS; (b) J. J. Van Veldhuizen, J. E. Campbell, R. E. Giudici and A. H. Hoveyda, J. Am. Chem. Soc., 2005, 126, 11130 Search PubMed; (c) H. Seo, D. Hirsch-Weil, K. A. Abboud and S. Hong, J. Org. Chem., 2008, 73, 1983 CrossRef CAS PubMed; (d) K. B. Selim, Y. Matsumoto, Y. Yamamoto, K. Yamada and K. Tomioka, Angew. Chem., Int. Ed., 2009, 48, 8733 CrossRef CAS PubMed; (e) F. Gao, Y. Lee, K. Mandai and A. H. Hoveyda, Angew. Chem., Int. Ed., 2010, 49, 8370 CrossRef CAS PubMed; (f) R. Shintani, K. Takatsu, M. Takeda and T. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 8656 CrossRef CAS PubMed; (g) M. Magrez, Y. Le Guen, O. Baslé, C. Crévisy and M. Mauduit, Chem. – Eur. J., 2013, 19, 1199 CrossRef CAS PubMed; (h) A. Harada, Y. Makida, T. Sato, H. Ohmiya and M. Sawamura, J. Am. Chem. Soc., 2014, 136, 13932 CrossRef CAS PubMed.
- Pd-catalyzed asymmetric allylic etherification: (a) B. M. Trost, J. L. Gunzner, O. Dirat and Y. H. Rhee, J. Am. Chem. Soc., 2002, 124, 10396 CrossRef CAS PubMed; (b) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2000, 122, 11262 CrossRef CAS; (c) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1998, 120, 815 CrossRef CAS.
- Ru-catalyzed asymmetric allylic etherification: (a) B. M. Trost, M. Rao and A. P. Dieskau, J. Am. Chem. Soc., 2013, 135, 18697 CrossRef CAS PubMed; (b) N. Kanbayashi and K. Onitsuka, Angew. Chem., Int. Ed., 2011, 50, 5197 CrossRef CAS PubMed; (c) K. Onitsuka, H. Okuda and H. Sasai, Angew. Chem., Int. Ed., 2008, 47, 1454 CrossRef CAS PubMed; (d) K. Takii, N. Kanbayashi and K. Onitsuka, Chem. Commun., 2012, 48, 3872 RSC; (e) M. D. Mbaye, J. L. Renaud, B. Demerseman and C. Bruneau, Chem. Commun., 2004, 1870 RSC.
- Cu-catalyzed allylic borylation and subsequent oxidation: (a) H. Ito, S. Ito, Y. Sasaki, K. Matsuura and M. Sawamura, J. Am. Chem. Soc., 2007, 129, 14856 CrossRef CAS PubMed; (b) A. Guzman-Martinez and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 10634 CrossRef CAS PubMed; (c) J. K. Park, H. H. Lackey, B. A. Ondrusek and D. T. McQuade, J. Am. Chem. Soc., 2011, 133, 2410 CrossRef CAS PubMed.
- (a) F. López, T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 3426 CrossRef PubMed; (b) A. Leitner, C. Shu and J. F. Hartwig, Org. Lett., 2005, 7, 1093 CrossRef CAS PubMed; (c) C. Welter, A. Dahnz, B. Brunner, S. Streiff, P. Dübon and G. Helmchen, Org. Lett., 2005, 7, 1239 CrossRef CAS PubMed; (d) D. Kim, S. Reddy, O. V. Singh, J. S. Lee, S. B. Kong and H. Han, Org. Lett., 2013, 15, 512 CrossRef CAS PubMed; (e) D. Kim, J. S. Lee, S. B. Kong and H. Han, Angew. Chem., Int. Ed., 2013, 52, 4203 CrossRef CAS PubMed.
- (a) C. Shu and J. F. Hartwig, Angew. Chem., Int. Ed., 2004, 43, 4794 CrossRef CAS PubMed; (b) S. Ueno and J. F. Hartwig, Angew. Chem., Int. Ed., 2008, 47, 1928 CrossRef CAS PubMed; (c) M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5568 CrossRef CAS PubMed.
- I. Lyothier, C. Defieber and E. M. Carreira, Angew. Chem., Int. Ed., 2006, 45, 6204 CrossRef CAS PubMed.
- J. Qu, L. Roßberg and G. Helmchen, J. Am. Chem. Soc., 2014, 136, 1272 CrossRef CAS PubMed.
- M. Gärtner, S. Mader, K. Seehafer and G. Helmchen, J. Am. Chem. Soc., 2011, 133, 2072 CrossRef PubMed.
- J. K. Park and D. T. McQuade, Angew. Chem., Int. Ed., 2012, 51, 2717 CrossRef CAS PubMed.
- P. Tosatti, A. J. Campbell, D. House, A. Nelson and S. P. Marsden, J. Org. Chem., 2011, 76, 5495 CrossRef CAS PubMed.
- (a) S. Hanessian, Y. Gai and W. Wang, Tetrahedron Lett., 1996, 37, 7473 CrossRef CAS; (b) S. Hanessian, W. Wang, Y. Gai and E. Olivier, J. Am. Chem. Soc., 1997, 119, 10034 CrossRef CAS; (c) A. Whitehead, J. P. McParland and P. R. Hanson, Org. Lett., 2006, 8, 5025 CrossRef CAS PubMed.
- T. Fukuyama, A. A. Laird and L. M. Hotchkiss, Tetrahedron Lett., 1985, 26, 6291 CrossRef CAS.
- (a) S. Spiess, C. Welter, G. Franck, J. T. Taquet and G. Helmchen, Angew. Chem., Int. Ed., 2008, 47, 7652 CrossRef CAS PubMed; (b) K.-Y. Ye, H. He, W.-B. Liu, L.-X. Dai, G. Helmchen and S.-L. You, J. Am. Chem. Soc., 2011, 133, 19006 CrossRef CAS PubMed.
- A. W. van Zijl, A. J. Minnaard and B. L. Feringa, J. Org. Chem., 2008, 73, 5651 CrossRef CAS PubMed.
- In none of the reactions, full conversion was achieved. Yields are therefore based on the recovered starting materials.
- S. R. Chemler and W. R. Roush, J. Org. Chem., 1998, 63, 3800 CrossRef CAS.
- D. Grassi and A. Alexakis, Chem. Sci., 2014, 5, 3803 RSC.
- R. W. Hoffmann, M. Stahl, U. Schopfer and G. Frenking, Chem. – Eur. J., 1998, 4, 559 CrossRef CAS.
- S. Hanessian, Y. Yang, S. Giroux, V. Mascitti, J. Ma and F. Raeppel, J. Am. Chem. Soc., 2003, 125, 13784 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Barry Trost on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Optimization studies, full experimental procedures and 1H and 13C NMR spectra of new compounds. See DOI: 10.1039/c6qo00199h |
| § Present address: Center for Research in Biological Chemistry and Molecular Materials (CIQUS) and Department of Organic Chemistry, University of Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: E-mail: martin.fananas@usc.es |
| This journal is © the Partner Organisations 2016 |