
Gold-catalyzed bis(stannylation) of propiolates†
F.
Chahdoura
ab,
N.
Lassauque
ab,
D.
Bourissou
*ab and
A.
Amgoune
*ab
aUniversité de Toulouse, UPS, Laboratoire Hétérochimie Fondamentale Appliquée, 118 route de Narbonne, F-31062 Toulouse, France
bCNRS, LHFA UMR 5069, F-31062 Toulouse, France. E-mail: dbouriss@chimie.ups-tlse.fr; amgoune@chimie.ups-tlse.fr; Fax: +33 5618204; Tel: +33 561556803
First published on 10th May 2016
Abstract
The first example of gold-catalyzed addition of Sn–Sn bonds to alkynes is reported. A cationic gold(I) complex bearing a phosphite ligand is reported to efficiently catalyze the bis(stannylation) of propiolates with hexabutylditin under mild conditions.
 Abderrahmane Amgoune | Abderrahmane Amgoune received his PhD in 2006 at the University of Rennes (Profs. J.-F. Carpentier and C. Thomas), France. He then moved to the University of Konstanz as an Alexander-von-Humboldt postdoctoral research associate with Prof. S. Mecking. In 2008, he was appointed to a CNRS research position at the University of Toulouse. His current research interests are dedicated to coinage metal chemistry, gold chemistry in particular (coordination chemistry and reactivity), and his work involves rational design of gold complexes for the development of new catalytic reactions for applications in organic synthesis and polymer materials. |
Transition metal catalyzed addition of σ E–E and E–H bonds (E = B, Si, Sn…) to unsaturated substrates is a very efficient synthetic strategy for the direct functionalization of CC multiple bonds with reactive groups that can undergo a myriad of subsequent transformations.1 Group 10 metal catalysts occupy a forefront position in this area. In contrast, only sporadic examples of gold-catalyzed addition of σ E–E and E–H bonds (E = B, Si) to alkenes and alkynes have been described.2–9 The group of Stratakis has recently shown that gold nanoparticles supported on TiO2 are efficient catalysts for the disilylation,2 silaboration,3 and hydrosilylation4 of alkynes. Alkynes and alkenes have also been diborylated using nanoporous gold and binap-stabilized gold(0), as reported by Jin5 and Fernandez.6 Examples involving well-defined gold complexes are even rarer, with to the best of our knowledge only two reports by Marder7 and Corma8 dealing with the diboration of styrenes and the hydroboration of alkynes, respectively.
From a mechanistic viewpoint, while group 10 metals are known to readily achieve the main elementary steps involved in the catalytic cycle of E–E addition to alkynes and alkenes, i.e. the oxidative addition of the σ E–E bond, the migratory insertion of the CC multiple bond into the M–E bond and reductive elimination, these basic organometallic steps were unknown with gold until recently. However, over the last few years, gold complexes have been shown to display in fact versatile reactivity.10 For instance, we recently reported the oxidative addition of Si–Si,11,12 Sn–Sn,13 C–C14 and Csp2–X15,16 bonds to gold, as well as the insertion of alkynes17,18 and alkenes.19 Building on this knowledge, we envisioned to explore the reactivity of gold(I) complexes in catalytic bis(stannylation) of alkynes. Such a transformation has never been reported with gold, and it is actually limited to a few Pd,20–22 Mo23 and Cu24 complexes.
Herein, we report that cationic phosphine–gold(I) complexes are able to promote the bis(stannylation) of propiolates in catalytic conditions. The reaction is totally stereoselective and proceeds under mild conditions using hexabutyl- or hexamethyl-ditin reagents.
We previously reported a spontaneous intramolecular oxidative addition of a σ Sn–Sn bond to gold upon reaction of a diphosphine-distannane ligand with (Me2S)AuCl/NaBARF [BARF = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate] leading to a bis(stannyl) gold(III) complex (Scheme 1).13 A similar reaction was observed with copper, and the resulting bis(stannyl) copper(III) complex was found to undergo a clean insertion reaction with methyl propiolate to give a bis-stannylated alkene copper(I) complex.13
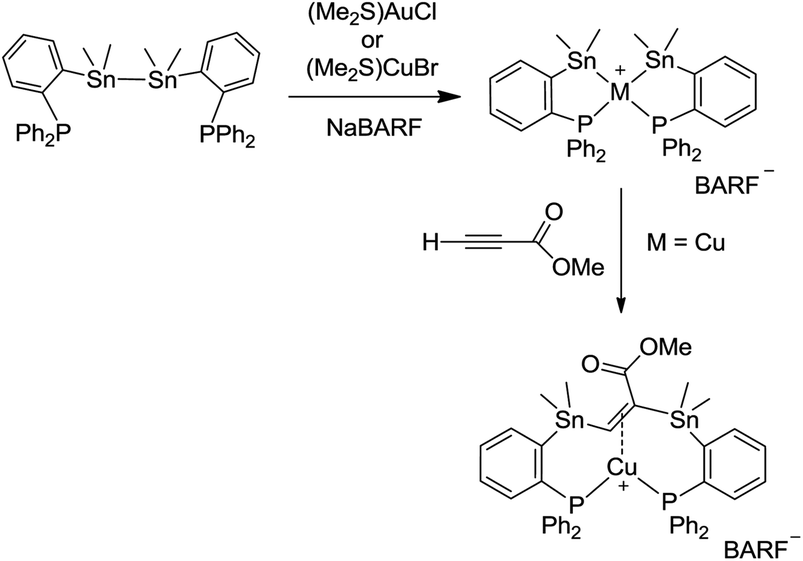 | ||
| Scheme 1 Chelation-assisted activation of a distannane at gold and copper, subsequent insertion of methyl propiolate. | ||
Based on these observations, we sought to test the reaction of hexabutylditin with methyl propiolate in the presence of catalytic amounts of gold. While no reaction was observed with (Ph3P)AuCl (20 mol%), 1H NMR monitoring of the reaction with (Ph3P)AuCl in the presence of NaBARF (20 mol%) indicated complete conversion of the starting materials after a period of 3 h in dichloromethane at room temperature (Scheme 2) and clean formation of the corresponding bis(stannylated) alkene.
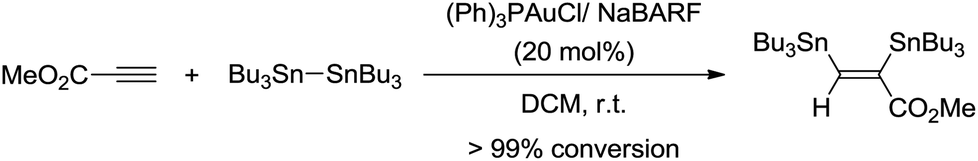 | ||
| Scheme 2 Bis(stannylation) of methyl propiolate catalyzed by gold. | ||
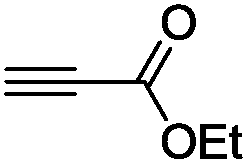
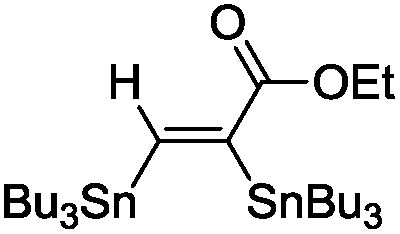
This first promising result prompted us to explore the activity of a series of LAuCl complexes (L = R3P, NHC). As indicated in Scheme 3, the best results in terms of activity, conversion and yields were obtained with P(OPh)3.25 Phosphines bearing highly electron-withdrawing substituents such as [P(C6F5)3] resulted in complete loss of activity, probably due to the low stability of the highly electrophilic cationic gold species. No reaction was observed with electron-rich ligands such as IPr [IPr = N,N’-(2,6-diisopropylphenyl)imidazol-2-ylidene] either. Several chloride abstracting agents were also screened (AgSbF6, AgOTf, AgBF4, see Table S1†),26 and the best results were obtained with NaBARF. As a result of these assays, the combination of [(PhO)3P]AuCl and NaBARF was selected for further studies. Reducing the catalyst down to 5 mol% and extending the reaction time to 4.5 h gave similar conversion and yield. Interestingly, very good isolated yields (>80%) were obtained after purification by column chromatography using deactivated silica gel.‡ The structure of the bis(stannylated) alkenes, including their cis configuration, was unambiguously established by 1H, 13C and 119Sn NMR characterization.26 The 119Sn NMR spectra systematically display two resonances signals for the two tin atoms at ca−46 and−61 ppm. The large 3JH–Sn coupling constant between the vinylic proton and the tin atom in trans position is particularly diagnostic (3JH–Sn ∼ 150–166 Hz).
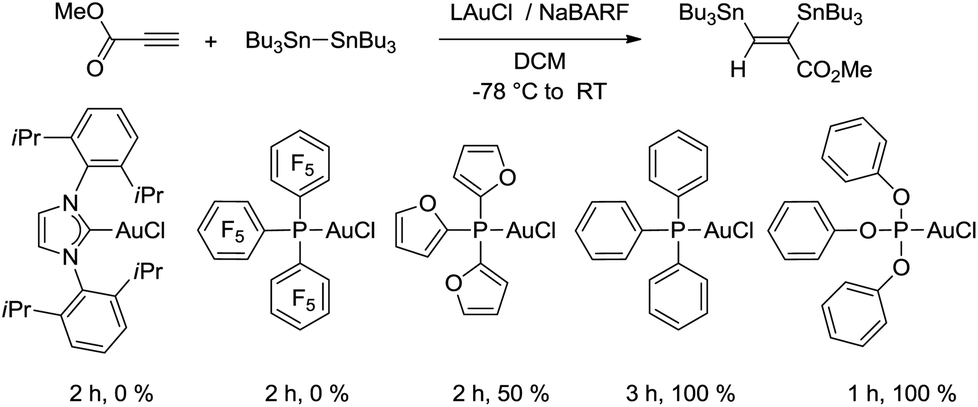 | ||
| Scheme 3 Gold(I) complexes screened in the bis(stannylation) of methyl propiolate. | ||
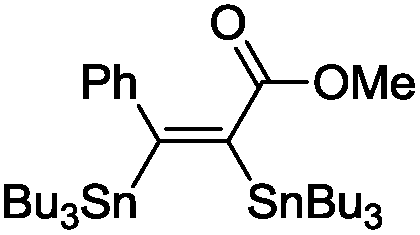
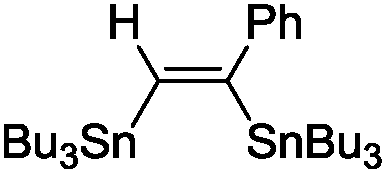
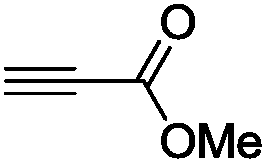
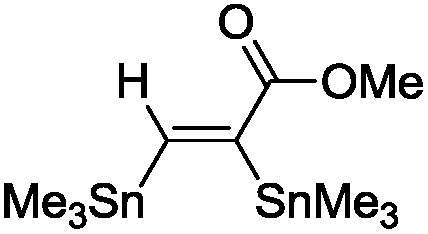
The scope of the transformation was then studied with a series of alkynes (Table 1). The reaction works well with a range of terminal propiolates, but not with internal ones (such as methyl phenylpropiolate, entry 2) and non-activated alkynes (such as phenyl acetylene, entry 3).27 The catalytic system shows good functional group compatibility, it tolerates thioether and vinylic groups (entries 8 and 9). Note also that the bis(stannylation) of the benzyl propiolate bearing an aryl iodide substituent (entry 10) proceeded in a selective manner. Oxidative addition of the Csp2–I bond, as would be typically observed with Pd catalysts, does not occur in that case.28 This observation highlights the orthogonal and complementary reactivity of gold and palladium complexes. Methyl propiolate can also be bis(stannylated) with hexamethyltin (entry 4). The reaction is clean and proceeds to completion, but the resulting product could only be isolated in 30% yield after chromatography due to its low stability.
| Entry | Alkyne | (R′3Sn)2 | Product | Yield (%) |
|---|---|---|---|---|
| a Conditions: 0.4 mmol of ditin reagent, 0.4 mmol of alkyne, 5 mol% of [(PhO)3P]AuCl and NaBARF, rt, 4.5 h. All products isolated by column chromatography. | ||||
| 1 |

|
(Bu3Sn)2 |
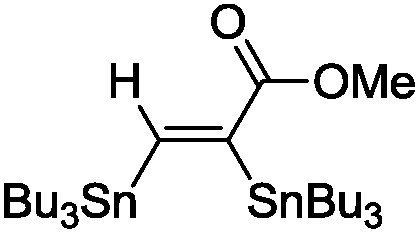
|
98 |
| 2 |
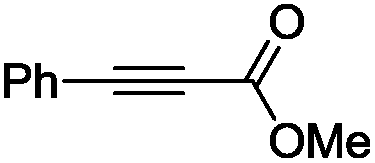
|
(Bu3Sn)2 |
|
0 |
| 3 |
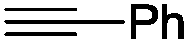
|
(Bu3Sn)2 |
|
0 |
| 4 |
|
(Me3Sn)2 |
|
30 |
| 5 |
|
(Bu3Sn)2 |
|
91 |
| 6 |
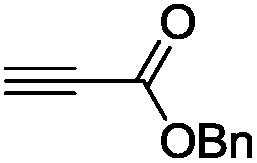
|
(Bu3Sn)2 |
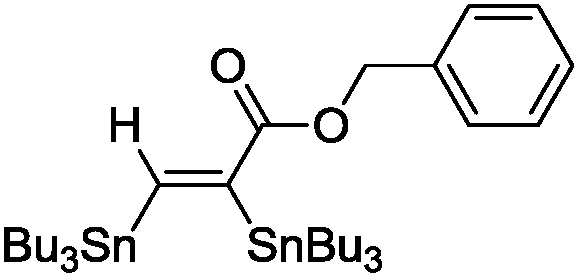
|
90 |
| 7 |
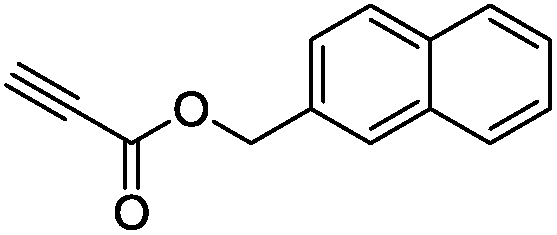
|
(Bu3Sn)2 |
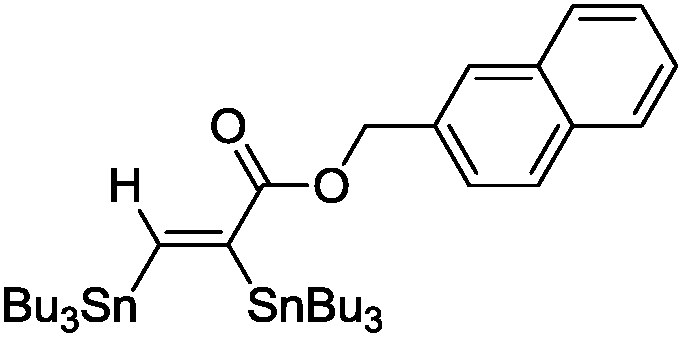
|
85 |
| 8 |
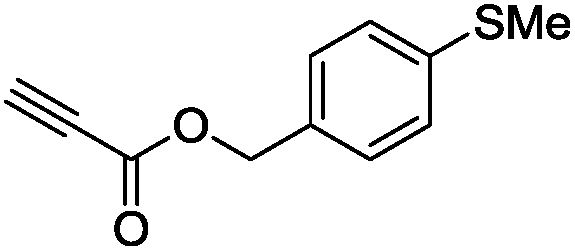
|
(Bu3Sn)2 |
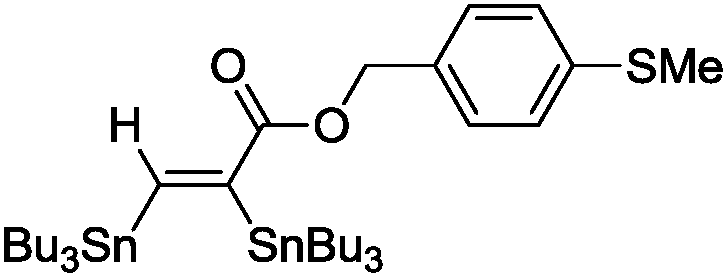
|
90 |
| 9 |
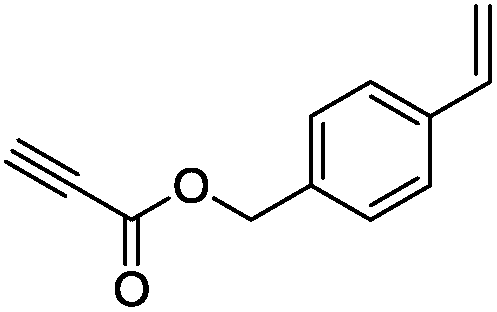
|
(Bu3Sn)2 |
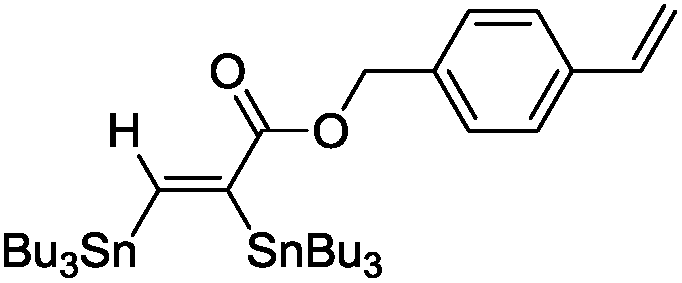
|
85 |
| 10 |
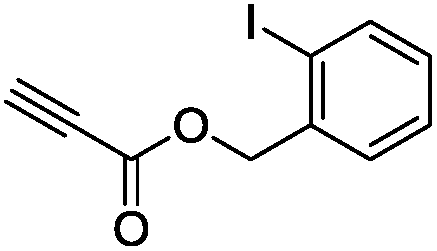
|
(Bu3Sn)2 |
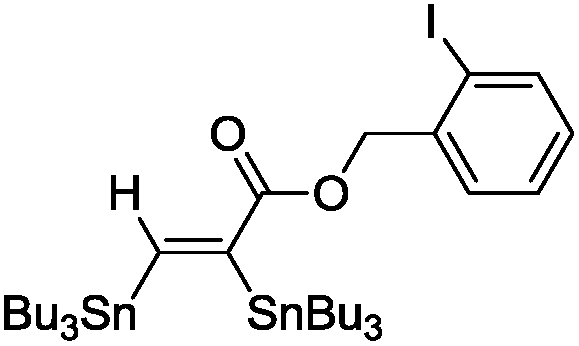
|
80 |
Interestingly, the gold catalytic system is also efficient for the stannylation of substrates featuring several CC triple bonds such as the tri-yne derivative 11. Varying the amount of hexabutylditin, it is possible to transform one or the three CC triple bonds. The bis and hexa-stannylated products 12a and 12b were thereby isolated in 89 and 70% yields, respectively (Scheme 4).
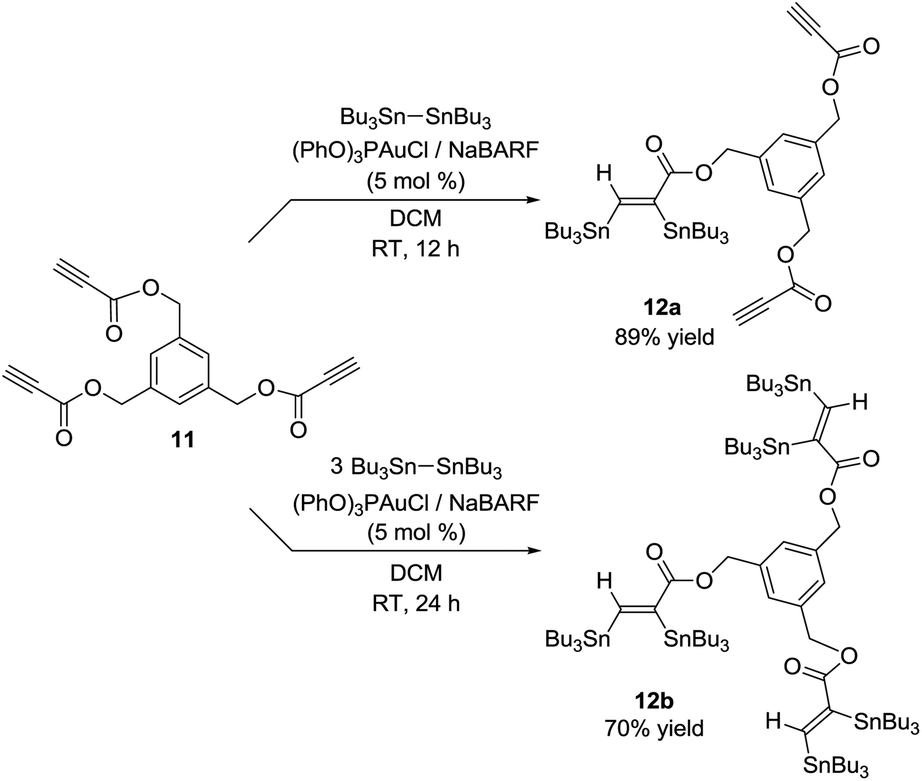 | ||
| Scheme 4 Bis and hexa-stannylation of the triyne substrate 9 catalyzed by gold. | ||
On the basis of the reaction stereoselectivity and of the analogous transformation with Pd complexes,20 the catalytic cycle shown in Scheme 5 is proposed.
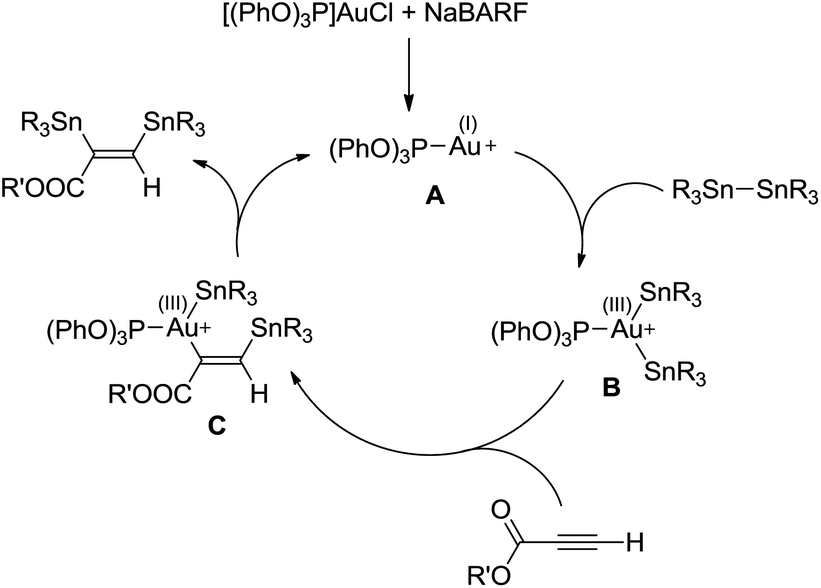 | ||
| Scheme 5 Catalytic cycle proposed to account for the bis(stannylation) of propriolates with gold complexes. | ||
Given our recent results on elementary reactions at gold,10–19 we postulate that the first step is the oxidative addition of the distannane to the cationic gold(I) species A to afford the gold(III) bis(stannyl) species B. Attempts to detect intermediate B by low temperature NMR spectroscopy, as we reported for the related bis(silyl) gold(III) complex,11 remained unsuccessful so far. It is worth mentioning that a phenyl/vinyl exchange between Au and Sn has been recently proposed to involve oxidative addition of a Sn–vinyl bond of Bu3Sn(vinyl) to (Me3P)AuPh.29 In the second step, insertion of the alkyne into the Au–Sn bond would occur in a regioselective manner, as observed with gold–silyl derivatives,17,18 to give the β-stannyl vinylgold(III) intermediate C. Finally, C–Sn reductive elimination from the transient tricoordinate gold(III) intermediate would release the bis(stannylated) alkene and regenerate the cationic gold(I) species A.30 Of note, this mechanistic picture differs from that generally operating for copper-catalyzed distannylation of C–C multiple bonds and related transformations. The latter reactions are typically performed under basic conditions, involve copper(I) alkoxide/stannyl species and proceed without change of the copper oxidation state.9,24
Conclusions
The first example of gold-catalyzed bis(stannylation) of CC multiple bonds is reported. Though limited to propiolate substrates, the reaction proceeds under mild conditions and is fully stereoselective. The cis-bis(stannylated) alkenes are obtained in high yields and as shown previously,31,32 they can be readily and broadly functionalized by cross-coupling reactions. From an organometallic viewpoint, these results show that gold complexes may readily achieve reaction sequences typically encountered with the group 10 metals, but not the coinage metals.33,34 Current work in our group aims at developing new gold-catalyzed transformations involving oxidative addition and/or syn addition processes.Notes and references
- I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320 CrossRef CAS PubMed.
- (a) C. Gryparis, M. Kidonakis and M. Stratakis, Org. Lett., 2013, 15, 6038 CrossRef CAS PubMed; (b) I. Titilas, M. Kidonakis, C. Gryparis and M. Stratakis, Organometallics, 2015, 34, 1597 CrossRef CAS.
- C. Gryparis and M. Stratakis, Org. Lett., 2014, 16, 1430 CrossRef CAS PubMed.
- (a) M. Kidonakis and M. Stratakis, Org. Lett., 2015, 17, 4538 CrossRef CAS PubMed; (b) A. Psyllaki, I. N. Lykakis and M. Stratakis, Tetrahedron, 2012, 68, 8724 CrossRef CAS.
- Q. Chen, J. Zhao, Y. Ishikawa, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2013, 15, 5766 CrossRef CAS PubMed.
- J. Ramirez, M. Sanau and E. Fernandez, Angew. Chem., Int. Ed., 2008, 47, 5194 CrossRef CAS PubMed.
- R. T. Baker, P. Nguyen, T. B. Marder and S. A. Westcott, Angew. Chem., Int. Ed., 1995, 34, 1336 CrossRef CAS.
- A. Leyva, X. Zhang and A. Corma, Chem. Commun., 2009, 4947 RSC.
- For selected references on copper-catalyzed addition reactions of E–E bonds to alkenes/alynes, see: (a) H. Yoshida, ACS Catal., 2016, 6, 1799 CrossRef CAS; (b) H. Yoshida, Y. Hayashi, Y. Ito and K. Takaki, Chem. Commun., 2015, 51, 9440 RSC; (c) H. Yoshida, Y. Takemoto and K. Takaki, Chem. Commun., 2015, 51, 6297 RSC.
- M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022 CrossRef CAS PubMed.
- M. Joost, P. Gualco, Y. Coppel, K. Miqueu, C. E. Kefalidis, L. Maron, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2014, 53, 747 CrossRef CAS PubMed.
- P. Gualco, S. Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2011, 50, 8320 CrossRef CAS PubMed.
- N. Lassauque, P. Gualco, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2013, 135, 13827 CrossRef CAS PubMed.
- M. Joost, L. Estévez, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5236 CrossRef CAS PubMed.
- J. Guenther, S. Mallet-Ladeira, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 1778 CrossRef CAS PubMed.
- M. Joost, A. Zeineddine, L. Estévez, S. Mallet Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654 CrossRef CAS PubMed.
- M. Joost, P. Gualco, S. Mallet-Ladeira, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2013, 52, 7160 CrossRef CAS PubMed.
- M. Joost, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 10373 CrossRef CAS PubMed.
- (a) F. Rekhroukh, R. Brousses, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 1266 CrossRef CAS PubMed; (b) F. Rekhroukh, L. Estévez, C. Bijani, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2016, 55, 3414 CrossRef CAS PubMed; (c) F. Rekhroukh, L. Estévez, C. Bijani, K. Miqueu, A. Amgoune and D. Bourissou, Organometallics, 2016, 35, 995 CrossRef CAS.
- J. Mancuso and M. Lautens, Org. Lett., 2003, 5, 1653 CrossRef CAS PubMed.
- H. Yoshida, K. Tanino, J. Ohshita and A. Kunai, Angew. Chem., Int. Ed., 2004, 43, 5052 CrossRef CAS PubMed.
- T. N. Mitchell, A. Amamria, H. Killing and D. Rutschow, J. Organomet. Chem., 1986, 304, 257 CrossRef CAS.
- S. Braune, M. Pohlman and U. Kazmaier, J. Org. Chem., 2004, 69, 468 CrossRef CAS PubMed.
- H. Yoshida, A. Shinke and K. Takaki, Chem. Commun., 2013, 49, 11671 RSC.
- For examples of gold-catalyzed reactions for which phosphites give the best results, see: (a) I. Alonso, H. Faustino, F. López and J. L. Mascareñas, Angew. Chem., Int. Ed., 2011, 50, 11496 CrossRef CAS PubMed; (b) M. C. B. Jaimes, F. Rominger, M. M. Pereira, R. M. B. Carrilho, S. A. C. Carabineiro and A. S. K. Hashmi, Chem. Commun., 2014, 50, 4937 RSC.
- See ESI.†.
- A competitive tin redistribution reaction readily occurs after oxidative addition of hexabutylditin to gold, giving SnBu4 as main compound along with unidentified decomposition products. This side reaction may explain the absence of reactivity with internal and less activated alkynes.
- Oxidative addition of Csp2–X bonds to gold is extremely scarce and requires specific environments around gold, see ref. 16.
- D. Carrasco, M. García-Melchor, J. A. Casares and P. Espinet, Chem. Commun., 2016, 52, 4305 RSC.
- Nevertheless, it is noteworthy that the reaction can be carried out in a stepwise manner by adding the hexabutylditin to the cationic gold species at −80 °C followed by addition of the alkyne.
- F. R. Wust, A. Hohne and P. Metz, Org. Biomol. Chem., 2005, 3, 503 Search PubMed.
- R. Mabon, A. M. E. Richecoeur and J. B. Sweeney, J. Org. Chem., 1999, 64, 328 CrossRef CAS.
- For selected examples of gold-catalyzed 2e redox coupling involving external oxidants, see: (a) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS PubMed; (b) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, J. Am. Chem. Soc., 2014, 136, 254 CrossRef CAS PubMed; (c) X. C. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636 CrossRef CAS PubMed; (d) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2011, 17, 8248 CrossRef CAS PubMed.
- For rare examples of redox gold catalysis without external oxidant, see: (a) M. D. Levin and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 6211 CrossRef CAS PubMed; (b) R. Cai, M. Lu, E. Y. Aguilera, Y. Xi, N. G. Akhmedov, J. L. Petersen, H. Chen and X. Shi, Angew. Chem., Int. Ed., 2015, 54, 8772 CrossRef CAS PubMed; (c) J. Serra, C. J. Whiteoak, F. Acuña-Parés, M. Font, J. M. Luis, J. Lloret-Fillol and X. Ribas, J. Am. Chem. Soc., 2015, 137, 13389 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data. See DOI: 10.1039/c6qo00128a |
| ‡ Purification of bis(stannylated) alkenes is difficult due to protodestannylation side-reaction during column chromatography. |
| This journal is © the Partner Organisations 2016 |