
Hydrogen-bond promoted nucleophilic fluorination: concept, mechanism and applications in positron emission tomography
Ji-Woong
Lee†
a,
Maria Teresa
Oliveira
a,
Hyeong Bin
Jang
a,
Sungyul
Lee
*b,
Dae Yoon
Chi
*c,
Dong Wook
Kim
*d and
Choong Eui
Song
*a
aDepartment of Chemistry, Sungkyunkwan University, 2066, Seobu-ro, Jangan-gu, Suwon-si, Gyeonggi-do 440-746, Korea. E-mail: s1673@skku.edu
bDepartment of Applied Chemistry, Kyung Hee University 1732, Duckyoung-daero 1732, Gihung-gu, Yongin-si, Gyeonggi-do 446-701, Korea. E-mail: sylee@khu.ac.kr
cDepartment of Chemistry, Sogang University, 35 Baekbeomro Mapogu, Seoul 121-742, Korea. E-mail: dychi@sogang.ac.kr
dDepartment of Chemistry, Inha University, 100 Inha-ro, Nam-gu, Incheon 402-751, Korea. E-mail: kimdw@inha.ac.kr
First published on 6th June 2016
Abstract
Due to the tremendous interest in carbon–fluorine bond-forming reactions, research efforts in this area have been dedicated to the development of facile processes to synthesize small fluorine-containing organic molecules. Among others, PET (Positron Emission Tomography) is one of the most important applications of fluorine chemistry. Recognizing the specific requirements of PET processes, some groups have focused on fluorination reactions using alkali metal fluorides, particularly through SN2-type reactions. However, a common “misconception” about the role of protic solvents and hydrogen bonding interactions in this class of reactions has hampered the employment of these excellent promoters. Herein, we would like to review recent discoveries in this context, showing straightforward nucleophilic fluorination reactions using alkali metal fluorides promoted by protic solvents. Simultaneous dual activation of reacting partners by intermolecular hydrogen bonding and the enhancement of the “effective fluoride nucleophilicity”, which is Nature's biocatalytic approach with the fluorinase enzyme, are the key to this unprecedentedly successful nucleophilic fluorination.
 Ji-Woong Lee and Maria Teresa Oliveira | Ji-Woong Lee received his MSc in chemistry from Sungkyunkwan University under the guidance of Prof. Choong Eui Song (2009). In 2013 he obtained his PhD under the supervision of Prof. Dr Benjamin List at the Max-Planck-Institute. After postdoctoral research at the UC-Berkeley, he is currently an assistant professor at the University of Copenhagen. Maria Teresa Oliveira received her MSc in chemistry from Instituto Superior Técnico, Lisbon (2008). In 2013 she obtained her PhD in organic chemistry under the supervision of Prof. Dr Nuno Maulide at the Max-Planck-Institute. Currently, she is a postdoctoral researcher at the University of Copenhagen. |
 Hyeong Bin Jang | Hyeong Bin Jang received her MSc in chemistry from Sungkyunkwan University under the guidance of Prof. Choong Eui Song (2011). She currently works as a research scientist at LG CHEM. |
 Sungyul Lee | Sungyul Lee received his PhD degree in physical chemistry from University of Chicago (1988). He has been appointed as an Assistant, Associate, and Professor at Kyung Hee University from 1989 to present. His research interests include the theoretical calculation of solvent catalysis and materials chemistry. |
 Dae Yoon Chi | Dae Yoon Chi received his PhD in organic chemistry from the University of Illinois at Urbana-Champaign (1986). Then he was appointed as a staff member of the Department of Nuclear Medicine, Samsung Medical Center in 1994. In 1995, he joined the Inha University and was promoted to a professor in 2000. In 2009, he moved to Sogang University. His main research spans medicinal chemistry and radiopharmaceutical chemistry. |
 Dong Wook Kim | Dong Wook Kim received his PhD in chemistry from Inha University (2004). He was a post-doctoral fellow at University of Illinois at Urbana-Champaign. In 2006 he became a professor at Chonbuk National University. In 2013, he moved to Inha University as a professor. His research interests focus on radiolabeling, medicinal chemistry and molecular imaging. |
 Choong Eui Song | Choong Eui Song (b. 1955) received his PhD from RWTH Aachen (1988). Since 1989, he has been working as a Principal Research Scientist at the Korea Institute of Science and Technology. In 2004, he moved to Sungkyunkwan University as a professor. His research interests focus on the development of new methodologies and chiral catalysts for asymmetric reactions. He has received several scientific awards, including the Scientist of the Month Award of Korea (2001), the distinguished contribution award for new drug development (Korea Drug Research Association, 2004) and the Scientific Excellence Award (Korean Chemical Society, 2013). |
Key learning points(1) Importance of hydrogen bonding interactions in enzymatic SN2 fluorination.(2) Alkali metal fluorides as efficient and safe nucleophilic sources in nucleophilic fluorination in protic solvents. (3) New mechanistic insights into the positive effects of protic solvents for SN2 reactions; simultaneous “electrophile–nucleophile” dual activation by intermolecular hydrogen bonding. (4) Synthesis of 18F-labeled PET radiotracers via protic solvent promoted nucleophilic fluorinations; high reaction rates and water tolerance. |
1. Introduction
Nucleophilic substitution is a fundamental class of reactions in chemistry. In this type of reaction, a nucleophile replaces a leaving group by attacking the positive or partially positive charge of an atom referred to as the electrophile. In the case of aliphatic bimolecular nucleophilic substitutions (SN2) at an sp3 carbon, a bond is broken and another bond is formed synchronously, i.e., in one step. SN2 reactions are usually severely affected by the solvent used.1 In most organic chemistry textbooks, polar aprotic solvents (e.g. dimethylformamide, dimethylsulfoxide, N-methylpyrrolidone, etc.) have been described as optimal for this type of chemical reaction. On the other hand, protic solvents such as water and alcohols have been deemed to be unsuitable due to the formation of “trapped” anionic nucleophiles by the hydrogen bonding donors of protic solvents, thus generating inert nucleophiles. For example, halides (F−, Cl−, Br− and I−) can be trapped in aqueous solutions and become unreactive, so their use in organic synthesis has been quite limited to polar aprotic solvent systems. However, Nature has utilized aqueous fluorides for nucleophilic substitution reactions,2 overcoming their low nucleophilicity in water, sometimes accelerating the reaction more than 108 fold over the non-catalyzed reaction. Unambiguous evidence of this operation has been obtained from the fluorinase enzyme isolated from Streptomyces cattleya.2 The active site for fluorination of SAM (S-adenosyl-L-methionine) is orchestrated to desolvate fluoride in a controlled manner by hydrogen bonding interactions, generating the fluoride nucleophile (Fig. 1).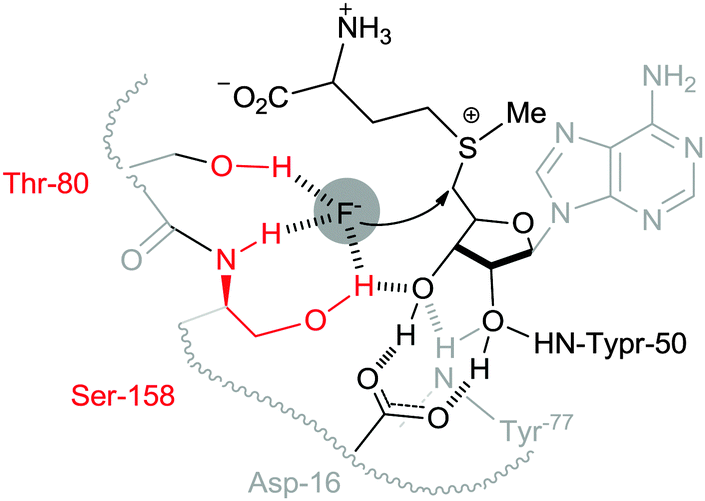 | ||
| Fig. 1 Schematic representation of the pre-reaction complex for the fluorination reaction of SAM at the reaction center of the fluorinase of S. cattleya. | ||
Despite their low natural occurrence in Nature, fluorinated organic molecules have received considerable attention in life sciences, particularly during the last decade.3,4 Fluorine is often incorporated to enhance the potency, lipophilicity, bioavailability, and metabolic stability of a molecule. Furthermore, radioactive fluorinated compounds (containing 18F) also serve as radiotracers for medical non-invasive imaging processes such as positron emission tomography (PET).5,6 In this regard, methods for introducing a single fluorine atom at a specific molecular site are considered to be of utmost importance.7,8
One possible fluorination approach involves utilizing highly reactive electrophilic “F+” equivalents to achieve the fluorination of nucleophilic substrates using transition metals, Lewis acids, or organocatalysts.9,10 However, these electrophilic fluorination methodologies have some inherent disadvantages such as expensive reagents, limited reactivity, and lower specific activity (GBq/μmol) in PET applications. When producing the 18F-labeled radiopharmaceuticals via18F− radiofluorination, their specific activities are quite important for determining PET imaging quality and the toxicity or side-effects of these radiotracers.7 In contrast, the use of more abundant and inexpensive nucleophilic fluorides such as metal fluorides (MFs) or HF-containing reagents is advantageous and provides complementary reactivity.11 Moreover, 18F-radiofluorination should ideally be performed using a nucleophilic [18F]fluoride source instead of [18F]F2 (or its derivatives) as an electrophilic fluorine source, because not only are most PET centres not equipped to handle this highly reactive and radioactive gaseous [18F]F2 reagent, but also it leads to radiopharmaceuticals possessing lower specific activities due to the inevitable use of [18F]F2 gas.11,12
The typical method for introducing a single fluorine atom at a specific aliphatic site by nucleophilic fluorination involves the displacement of a sulfonate or halide by a fluoride anion. The use of anhydrous HF (b.p = 19.6 °C) suffers from safety and handling problems. Alkali MFs such as KF and CsF are perhaps the most logically advantageous natural fluoride sources for nucleophilic fluorination because of their abundance, availability, and low cost. Moreover, the generation of 18F-labeled alkali MFs is convenient compared to other 18F-labeled fluorinating reagents, making them highly suitable for applications to medically relevant processes. However, the use of alkali MFs for fluorination is not straightforward, since the fluoride anion forms strong H-bonds with protic solvents, particularly water, significantly diminishing its nucleophilicity. Also, MFs' inherent high lattice energies make them virtually insoluble in most aprotic organic solvents. These facts indicate that SN2 chemistry with alkali MFs would suffer from issues dealing with unfavorable desolvation or a high kinetic barrier due to their low solubility.13 To solve this conundrum, much effort has been devoted to finding a suitable type of phase transfer catalyst and non-coordinating cationic species.14 However, despite being the state-of-the-art methodology since the 1970's, this approach generates a “naked” fluoride with high basicity, causing lower chemoselectivity (Scheme 1). This led to renewed interest in improving the nucleophilic fluorination processes using MFs, namely by generating a soluble but more “controllable” fluoride nucleophile from these sources.
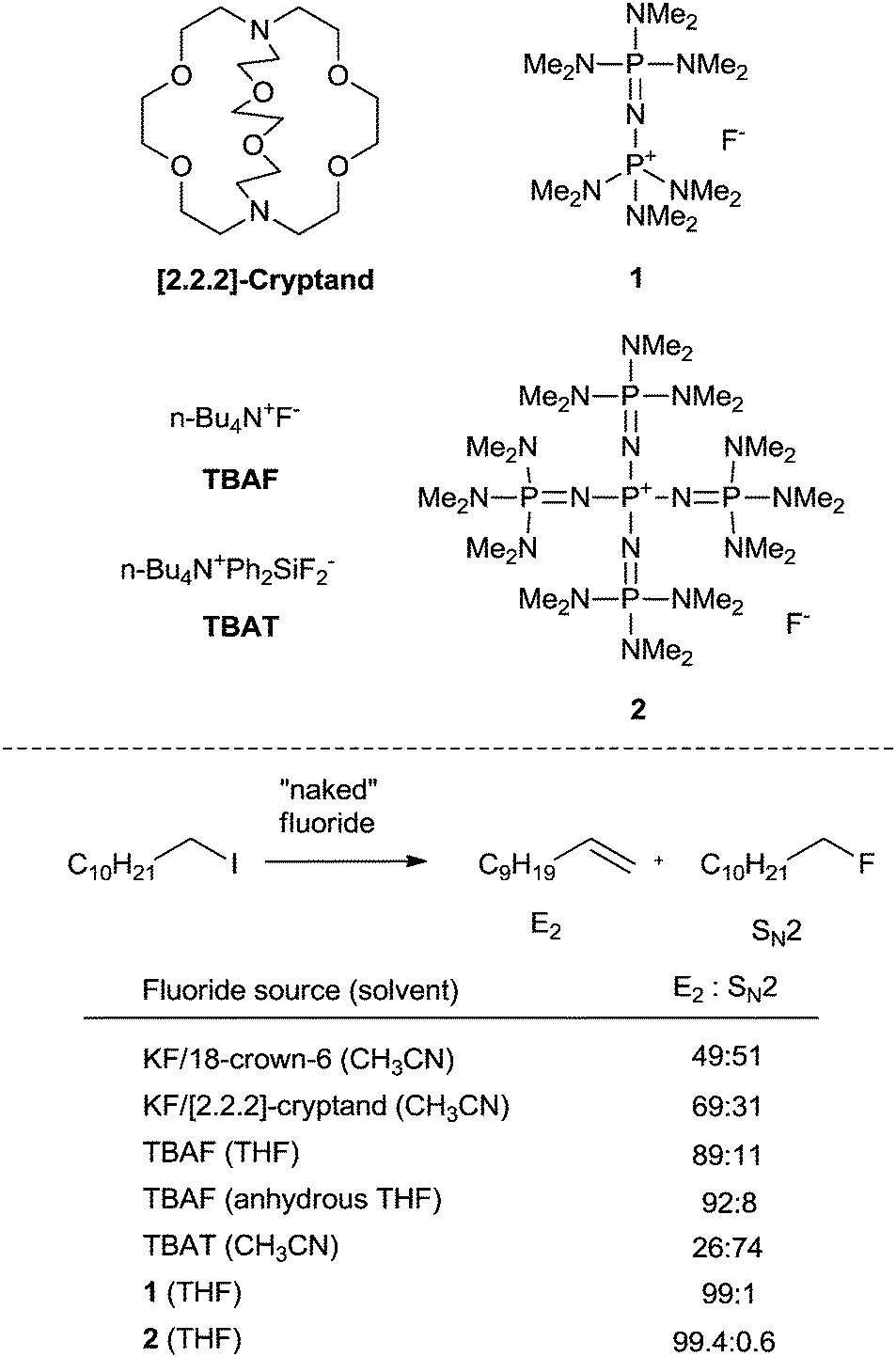 | ||
| Scheme 1 Selectivities in fluorination of 1-iodoundecane with naked fluorides. | ||
Over the last decade, we have developed several new methodologies employing protic solvents, which have been known to be inadequate for SN2 reactions, for facile fluorination reactions. Inspired by hydrogen-bond assisted/catalyzed enzymatic fluorination reactions, our solvent engineering approach has allowed us to generate more “controllable” fluoride nucleophiles, thus enabling practical and chemoselective nucleophilic fluorination reactions.
Recently, the field of transition metal-catalyzed nucleophilic fluorinations9–12 to install fluorine substituents on a sp3-hybridized carbon15 has experienced rapid growth, offering a new and efficient synthetic strategy to prepare 18F-labeled radiopharmaceuticals.16 Moreover, a number of examples of nucleophilic deoxyfluorination reagents have shown their potential in facile nucleophilic fluorination reactions, although their applications are quite limited due to the instability of the reagents under ambient conditions.7,8
In this Tutorial Review, we focus on transition metal-free fluorination reaction protocols using alkali metal fluorides, mainly KF and CsF, as practical fluoride sources. Inspired by the operation of enzymatic fluorination reactions, we highlight the importance of weak forces such as hydrogen bonding interactions to promote selective fluorination reaction at sp3 carbon centers. The selected recent examples demonstrating the action of polar protic solvents in nucleophilic fluorination reactions are exemplified with ionic liquids, bulky alcohols and oligoethylene glycols, in which the C–F bond formation reaction is promoted mainly by H-bondings. Summarized experimental and mechanistic details shed light on the origin of positive effects of H-bondings in protic media for facile nucleophilic fluorination reactions. Recent applications of protic solvents in PET techniques with significant improvement compared to traditional 18F-fluorination protocols are also presented.
2. Fluorinase enzyme-catalyzed nucleophilic fluorination
In 2001, a seminal discovery regarding the unprecedented activity of two fluorinases for C–F bond formation was unveiled by the Withers group.17,18 They found that mutated enzymes such as glucosidase from Agrobacterium sp. can catalyse the β-glucosidic bond formation reaction of substrate A with R–OH in the presence of additional fluoride, KF (Scheme 2). In the absence of fluoride, the enzyme is totally inactive towards the β-glucosidic bond forming reaction. The intermediate, α-glucosyl fluoride C, was observed by 1H and 19F NMR spectroscopy. Subsequently, the resulting electrophilic fluoride C can participate in the substitution reaction with an incoming nucleophile (R–OH) to generate the desired β-glucosidic bond (D). The importance of Ser-358 is presumed to be that the hydrogen bonding interaction may play a role in the desolvation and stabilization of the fluoride, which acts not only as a nucleophile but also as a leaving group.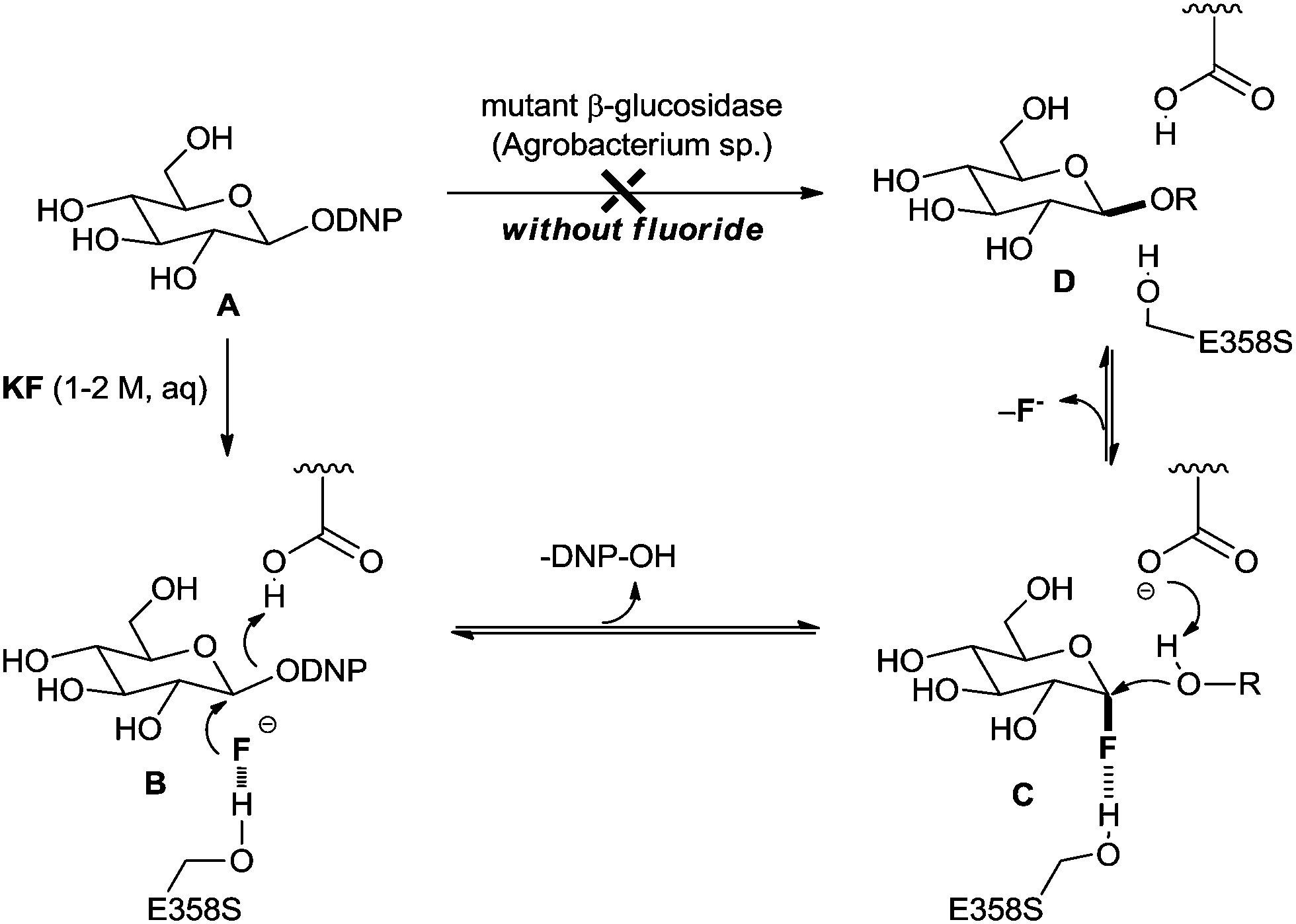 | ||
| Scheme 2 Enzymatic β-glucosidic bond formation reaction via nucleophilic fluorination and defluorination (DNP = 2,5-dinitrophenyl). | ||
Another example of an active fluorinase was reported by the O'Hagan group.19 The X-ray crystallographic structure of the fluorinase shows a snapshot of the fluorination reaction of SAM in the reaction pocket (Fig. 1).1 The hydroxy group of the Ser-158 and Thr-80 residues replaces water molecules, extracting the fluoride anion from aqueous medium and placing it at the active center of the enzyme, consequently increasing the nucleophilicity of the fluoride in the reaction site. These weak hydrogen bonding interactions of Ser-158 and neighboring Thr-80 are enough to compensate the high energy penalty of dehydration (activation barrier = ca. 400 kJ mol−1) to incorporate fluoride into the reaction center. Moreover, these hydrogen bonding interactions can reduce the basicity of fluoride, consequently enhancing “effective fluoride nucleophilicity”. These examples taken from Nature strongly suggest that the employment of hydrogen-bond interacting functional groups (e.g. serine and threonine) is critical for utilizing aqueous fluoride at extremely low concentrations. Therefore, it follows that fluorine chemistry in a flask may also benefit from hydrogen bonding interactions to employ inherently insoluble alkali fluoride in organic solvents.
3. SN2-type fluorination using alkali metal fluorides in aqueous ionic liquids
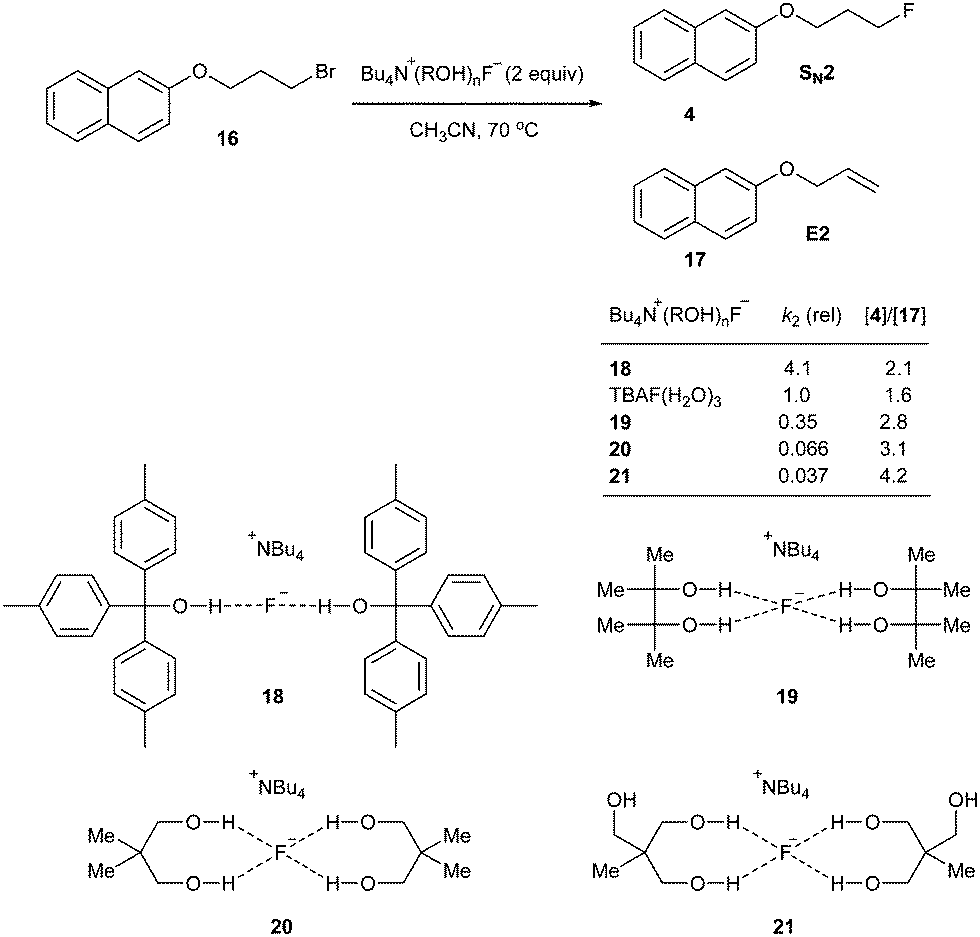
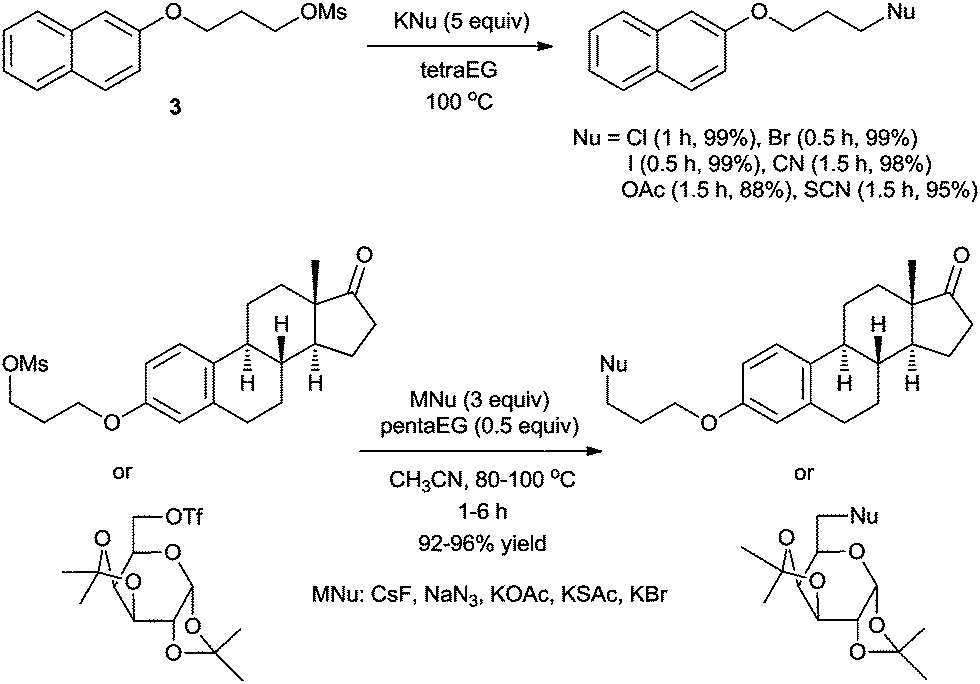
As mentioned before, the high lattice energy of alkali MFs such as KF and CsF often hinders their use in organic synthesis because of their low solubility in organic solvents. It is well known that ionic liquids (ILs), being highly polarized ionic compounds, can provide stabilizing effects during the course of a reaction with a polar transition state and high solubility for metal salts.20In 2002, we first reported a facile nucleophilic fluorination of alkyl mesylates using KF and an IL, [bmim][BF4].21 For example, the fluorination of 3 with KF in a polar aprotic solvent such as CH3CN at 100 °C had hardly formed any product even after 24 h. The use of a crown ether also afforded only 40% yield even after 24 h. However, surprisingly, using [bmim][BF4] in the presence of water as the additive, the same reaction was completed within 1.5 h, affording the desired product 4 in up to 92% yield (Scheme 3). Our DFT calculations showed that the improved activity may be explained by the cooperative activity of the cation and the anion of ILs, i.e. the ionic liquid plays an ambiphilic “electrophile–nucleophile” dual activation role (Fig. 2).22 The counteranion of the ionic liquid acts as a Lewis base toward M+, drastically reducing its electrostatic effects and thereby “freeing” the F− nucleophile and the acidic C2-proton of the imidazolium cation interacts with the mesylate leaving group via hydrogen bonding, helping it to detach from the reactant. This nucleophilic substitution reaction in the presence of aqueous ILs can also successfully be extended to other nucleophiles such as other halogens, cyanide, acetate, azide and alkoxides to afford the corresponding substituted products with excellent yields.23
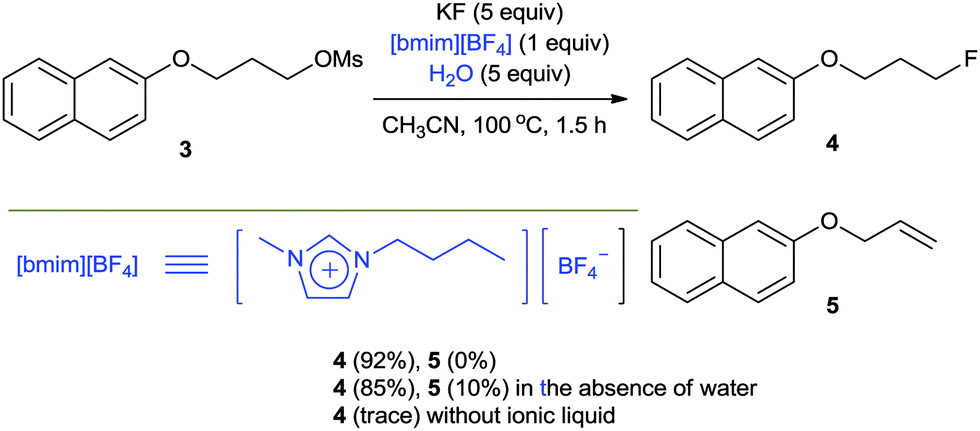 | ||
| Scheme 3 Ionic liquid promoted SN2-type fluorination. | ||
 | ||
| Fig. 2 Calculated transition state in SN2 fluorination using KF in ILs: cooperative activation by ILs. | ||
Very interestingly, the presence of water also turned out to be critical for the high selectivity of the reaction. The protic environment of water may reduce the basicity of the fluoride anion. In the absence of water, the fluorination of 3 afforded the desired product 4 (85%) together with the alkene byproduct 5 (10%). Very interestingly, however, the addition of water (5 equiv.) completely eliminated the formation of the undesired alkene product, consequently increasing the yield (92%) of the product 4 (Scheme 3). Moreover, even in the fluorination of a highly base-sensitive substrate, 5, which tends to undergo such an elimination reaction to produce styrene derivative 7, the use of water as a protic cosolvent suppressed the elimination reaction. On the other hand, in the absence of water, the same reaction produced a significant amount of elimination product 7 (39%) (Scheme 4).21
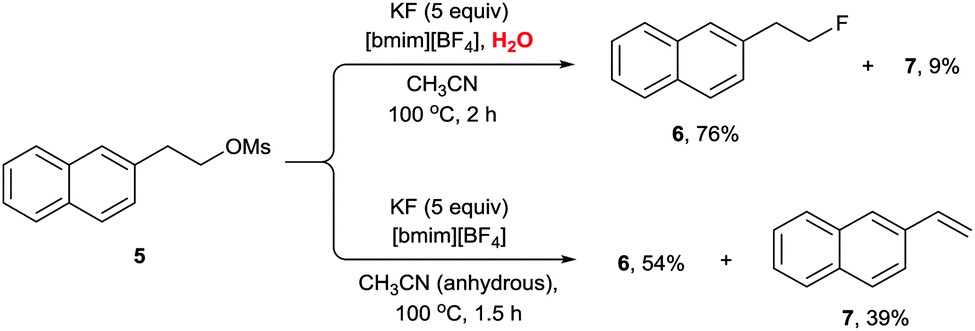 | ||
| Scheme 4 Nucleophilic fluorination of base-sensitive substrate 5 using KF. | ||
From this finding, we became aware of the important role of protic media in chemoselective fluorination by reducing the basicity of the naked fluoride anion. Moreover, the possibility of performing the reaction in the presence of water is noteworthy and highly beneficial for the applications to PET processes, since [18F]fluoride is generated from 18O-enriched water, and thus the obtained fluoride is in aqueous medium, which has to be completely dried before labeling of PET tracers. This is typically a time-consuming step required in conventional radiofluorination methods.
To enable facile purification of the final product, a polymer-supported IL 8 was reported (Scheme 5a).24 By using a substoichiometric amount of Merrifield resin-anchored polymeric IL 8, the fluorination of substrate 9 with CsF in acetonitrile/water for 2.5 h produced the desired product 10 in an excellent yield (98%). Surprisingly, the immobilized IL 8 showed superior activity to its non-immobilized version under identical reaction conditions. This could be ascribed to the synergistic effects between neighboring imidazolium salts, participating in efficient charge–charge interactions on the swellable polymer support. This bicipital effect of neighboring ion pairs has also been observed by Clark et al. using tetraarylphosphonium salt grafted silica gel as the catalyst for the iodination of alkyl bromides (Scheme 5b).25
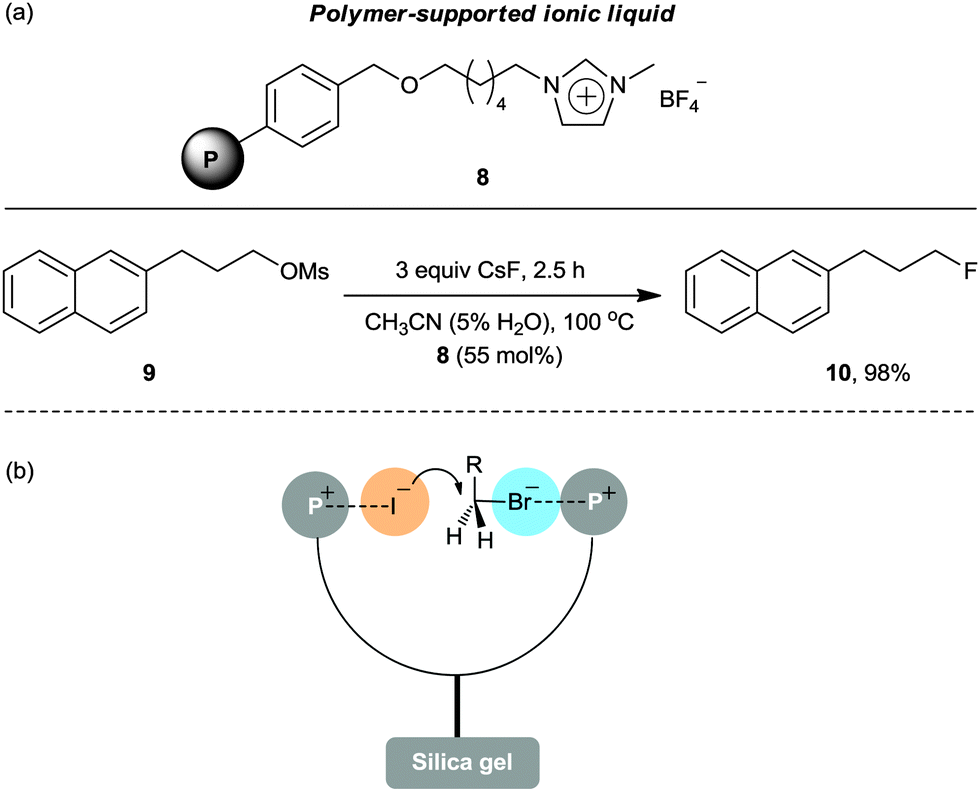 | ||
| Scheme 5 (a) Application of polymer-supported IL 8 in a fluorination reaction. (b) Bicipital effect of neighboring ion pairs using a polymer-supported phosphonium salt. | ||
As described above, polar media such as ILs offer a convenient reaction environment by generating highly reactive fluoride anions from MFs, while diminishing by-product formation. However, the utility of ILs for fluorination reactions is limited by gradual thermal degradation of ILs in the presence of basic fluoride anions.26 Therefore, we explored other more accessible, inexpensive, and robust reaction media for the activation of MFs towards fluorination reactions.
4. t-Alcohol solvents for SN2 fluorinations with alkali metal fluorides
In most organic chemistry textbooks, polar aprotic solvents are described as optimal for SN2-type reactions whereas protic solvents such as water and alcohols are unsuitable. However, as described previously, a fluorinase enzyme catalyses a nucleophilic fluorination with MF in aqueous medium. In the enzymatic reaction, the H-bonding between the enzyme, fluoride, and substrate plays a crucial role.1,19 Moreover, in 1994, Yonezawa and co-workers observed that the t-BuOH–TBAF complexes showed the best reactivity in the SN2 fluorination reaction of benzyl bromide with a series of TBAF–alcohol complexes prepared from TBAF(H2O)3 and the corresponding alcohol solvents – such as t-BuOH, i-PrOH, n-BuOH, and n-PrOH.27 This indicates that fine engineering of the H-bonding network could enable the use of protic solvents in nucleophilic fluorination reactions. Indeed, a remarkable and unprecedented effect was observed by using bulky aliphatic alcohols (e.g., t-BuOH and t-amyl alcohol) as the solvents in the fluorinations with CsF.28Scheme 6 highlights the superior efficiency of sterically bulky t-alcohols compared with the conventional polar aprotic solvent, acetonitrile, in fluorination. The relatively nonpolar and bulky t-alcohols such as t-BuOH and t-amyl alcohol dramatically increased the reactivity of CsF, allowing much faster reactions compared with acetonitrile (Scheme 6a). Although a relatively nonpolar protic solvent, n-BuOH, can also promote the same reactions, a significant amount (30%) of ether side-product was produced due to its smaller size and weaker steric effects. Bulkier tert-alcohols, on the other hand, suppressed the ether-formation to much larger degrees. More interestingly, in the fluorination of base-sensitive substrates such as 13 even with a “naked” fluoride source, the use of steric bulky t-alcohol solvents significantly suppressed the formation of elimination byproducts.28 The protic environment of t-alcohols reduced the basicity of fluoride ions, thus effectively inhibiting the side reactions such as elimination or intramolecular cycloalkylation (Scheme 6b).28 Recently, these t-alcohol media have been used for the preparation of many fluorinated bio-active molecules such as 11 and 12 (Scheme 7).29,30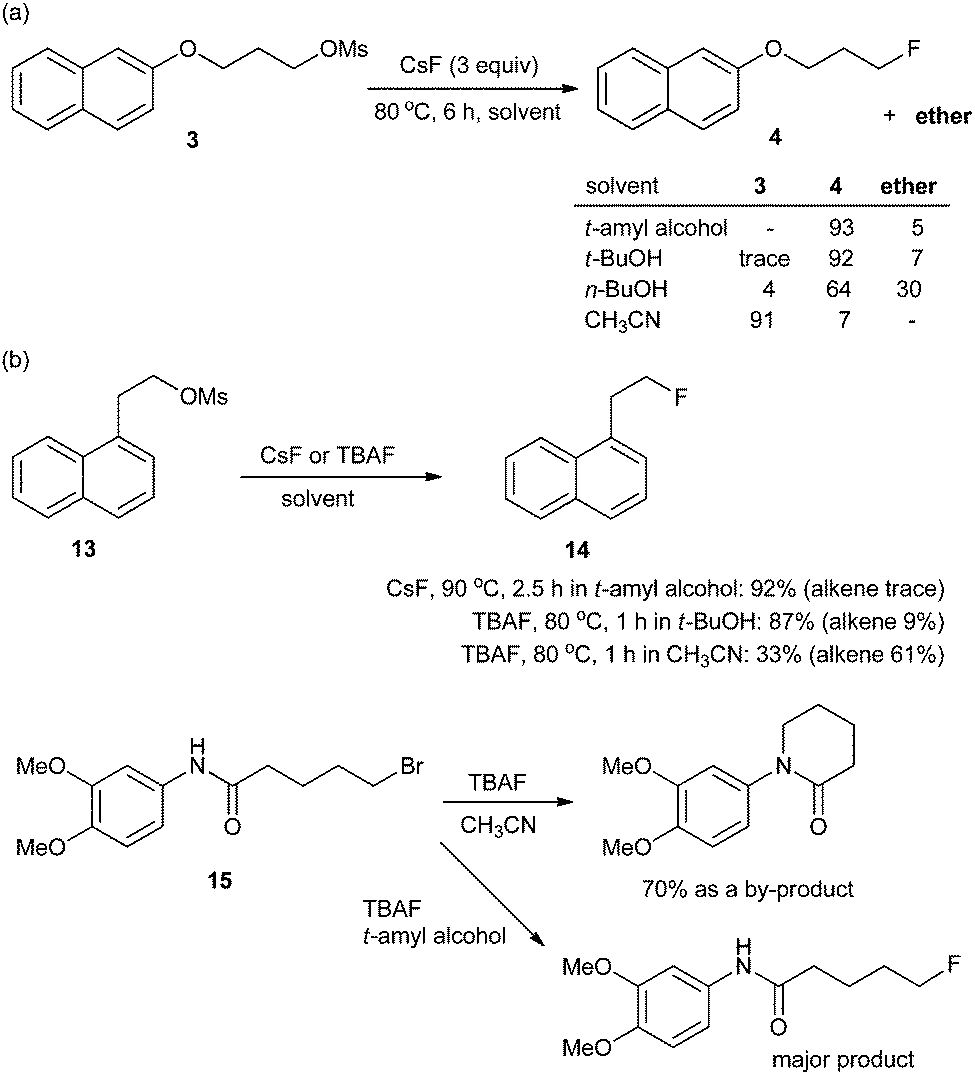 | ||
| Scheme 6 (a) Reactivity of CsF and (b) chemoselectivity of fluorination in t-alcohols and acetonitrile. | ||
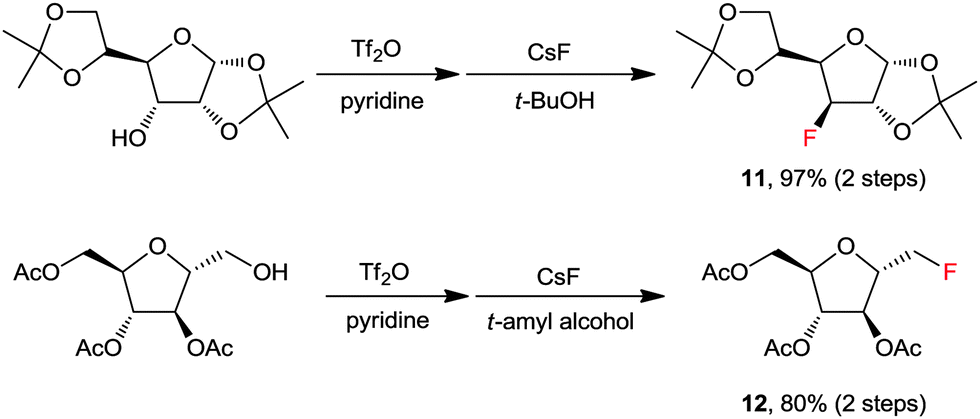 | ||
| Scheme 7 Selected examples for the preparation of fluorinated bio-active molecules by fluorination with CsF in a t-alcohol solvent. | ||
As shown in Fig. 3, t-alcohols are believed to affect the mechanism of this fluorination through the following H-bonding interactions: (1) the t-alcohol may form hydrogen bonds with the fluoride ion in the CsF lattice resulting in the selective solvation of the fluoride ion by t-alcohol molecules, thus reducing the strength of the CsF ionic bond. (2) The fluoride ion coordinated with bulky t-alcohols becomes a more “controllable” nucleophile, reducing the basicity of the fluoride ion and consequently suppressing side reactions such as eliminations. (3) The H-bonding of the t-alcohol solvents with the oxygen atoms of the sulfonate leaving group may improve its leaving group ability. Therefore, the use of bulky protic alcohols affords the possibility of generating a soluble, dissociated fluoride ion from CsF, while decreasing its Brønsted basicity.28,31
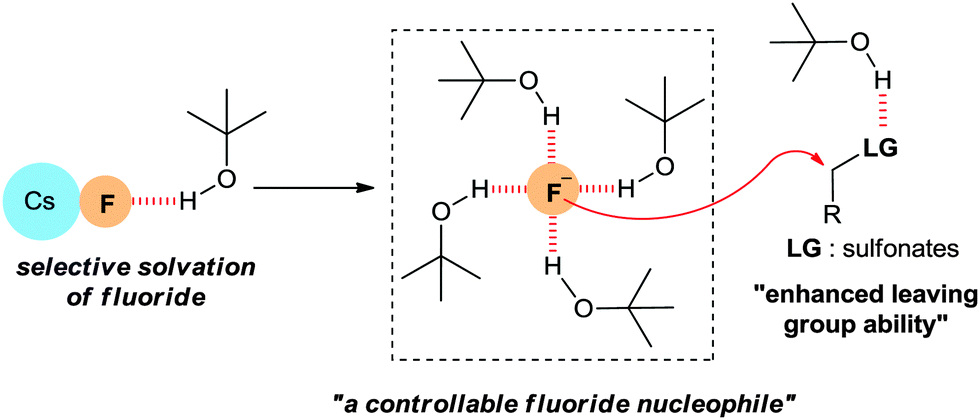 | ||
| Fig. 3 Positive effects of a t-alcohol medium on nucleophilic aliphatic fluorination reactions. | ||
More direct evidence that a “controllable” fluoride nucleophile can be generated during the t-alcohol media promoted fluorination process using an alkali metal fluoride was obtained by single crystal X-ray analysis of the TBAF(t-BuOH)4 complex which was simply formed from the mixture of commercially available TBAF and t-BuOH.32 In the X-ray structure of Fig. 4c, four t-BuOH molecules coordinate around a single fluoride by hydrogen bonds, consequently reducing the basicity of fluoride.
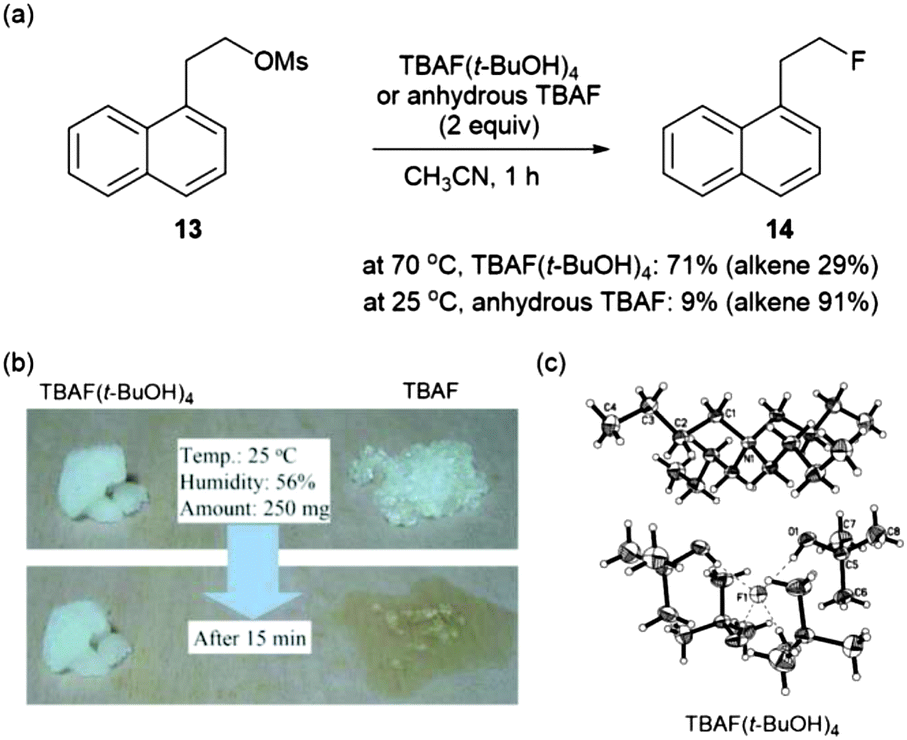 | ||
| Fig. 4 (a) Comparison of the reactivity and selectivity of TBAF(t-BuOH)4 and anhydrous TBAF in a nucleophilic fluorination reaction. (b) Reduced hygroscopicity of TBAF(t-BuOH)4. (c) X-ray crystal structure of TBAF(t-BuOH)4. | ||
The reduced basicity of TBAF(t-BuOH)4, due to the H-bonding interaction of fluoride, significantly lowered its reactivity compared with anhydrous TBAF, which generates a perfectly “naked” fluoride source in situ. Therefore, TBAF(t-BuOH)4 afforded higher yields of fluorinated product 14 while anhydrous TBAF led to the significant formation of elimination products (Fig. 4a). Additionally it possesses the inherent advantage of displaying lower hygroscopicity (Fig. 4b), as a consequence of its surrounding environment saturated with alcohol molecules, making it perfectly poised for strictly anhydrous reaction conditions.
Quite recently, Gouverneur and coworkers showed thorough examination of complexes of tetraalkyl ammonium fluoride with alcohol, 1,2-diols, 1,3-diols, triols and tetraols.33 According to the extensive X-ray crystallographic analysis, the coordination number of hydrogen bonding donors to fluoride is critical, affecting nucleophilicity and basicity of the fluoride (Scheme 8). It is believed that the reactivity of fluoride is governed by the dissociation from the alcohol ligands, indicating the importance of equilibrium of complexation and dissociation. Depending on the steric bulk and the branching degree of alcohols, the coordination environment varies and generates diverse character of fluoride, which can be applied in various catalysis and selective substitution reactions.
 | ||
| Scheme 8 Examples for the reactions of alcohol–fluoride complexes with 15. | ||
The characteristics of nucleophilic substitution reactions with a “controllable” fluoride nucleophile are in stark contrast to the two aspects generally associated with conventional SN2 reactions. First, as described previously, hindered protic solvents exhibited superior reactivity compared to aprotic dipolar solvents (CsF in t-BuOH vs. CsF in CH3CN, Fig. 5a). More interestingly, the relative reactivity of halide nucleophiles reversed in a bulky alcohol solvent such as t-BuOH (F− ≫ I− and Br−) from that typical for halide ions in conventional protic solvents such as MeOH (F− < Cl− < Br− < I−); the latter sequence was predicted by simply considering the differential strength of H-bonding between the halide and protic solvent (Fig. 5a).28,31 Moreover, Fig. 5b shows that the fluorination rate of a mesylate is significantly faster than those of the corresponding halides such as iodide and bromide. This result indicates that the reaction rate was determined not only by the nature of the leaving group, but also by other types of interactions such as those between the solvent (t-alcohol) and the leaving group. As proposed in Fig. 3, the H-bonding between the t-alcohol solvent and the oxygen atoms in the alkanesulfonate leaving group may improve its nucleofugic (leaving group) character. Although it is believed that organofluorines are hardly hydrogen bonding acceptors, it was recently discovered that even the strong benzylic C–F bonds can be activated by H-bonding donor molecules such as water (Scheme 9a).34 Similar to the mode of action of β-glycosidase (intermediate C in Scheme 2),18 hydrogen bonding interactions of water enabled the bimolecular nucleophilic substitution reactions of benzyl fluorides with diverse N-, O-, S-, and C-nucleophiles. The drastic change of leaving group ability of the reactant, benzyl fluoride, compared with benzyl bromide and chloride, was supported by performing an activation strain analysis in the gas phase. The most effective stabilization of the transition state was observed with benzyl fluoride in the presence of water. Soon after, the same group successfully applied this H-bonding strategy for the C–F bond activation of benzylic fluorides in the Friedel–Crafts benzylation reaction of arenes.35 From the comparison and competition experiments, only benzylic C–F bonds showed significant reactivity in the presence of other substrates (X = Cl, Br, OH, Scheme 9b), highlighting the importance of the hydrogen bonding interaction of H-bond acceptor Csp3–F with protic solvents.
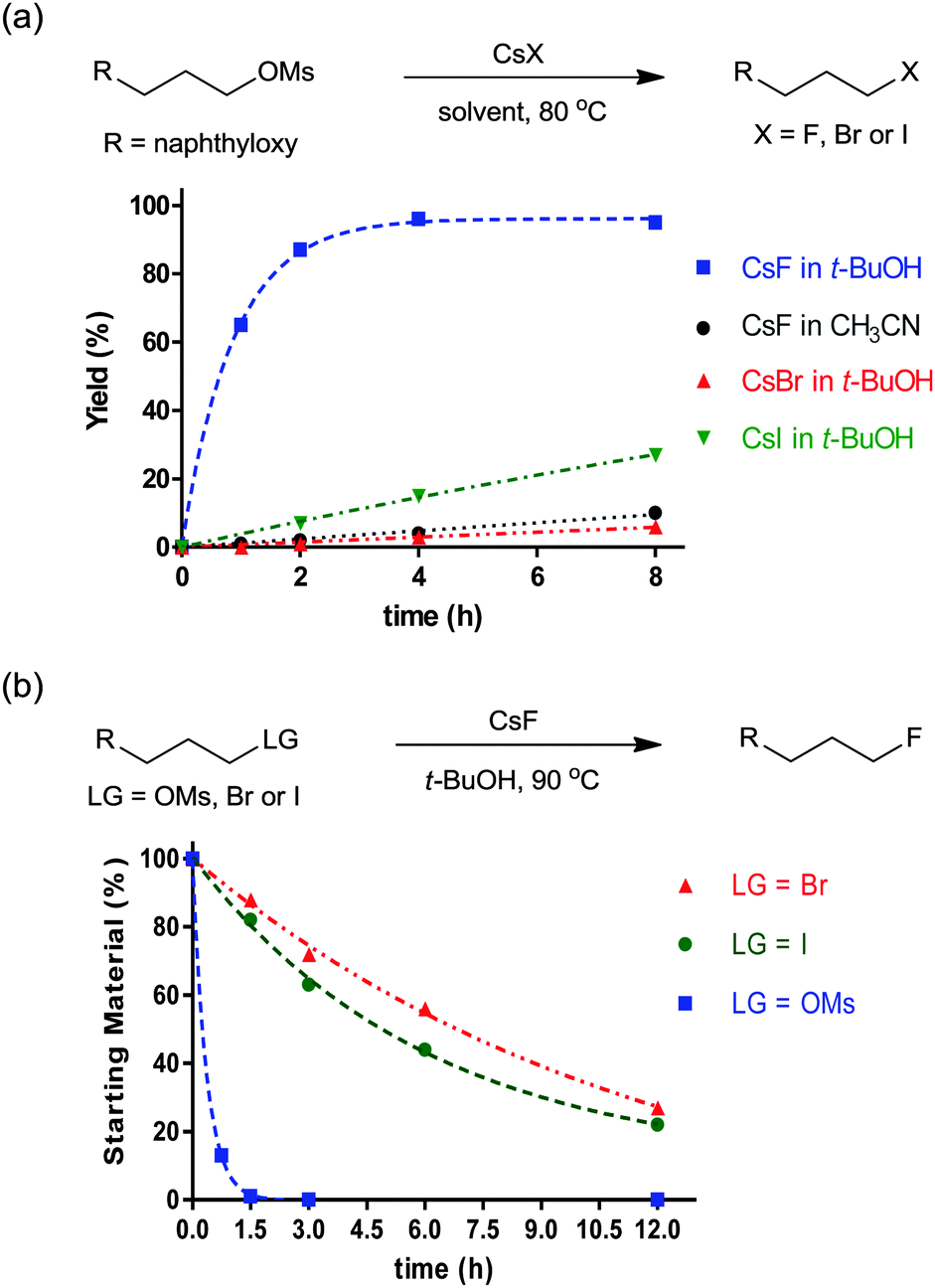 | ||
| Fig. 5 (a) Reactivity of CsF, CsBr, and CsI in t-BuOH or CH3CN. (b) Unusual leaving group ability in the t-BuOH reaction medium. | ||
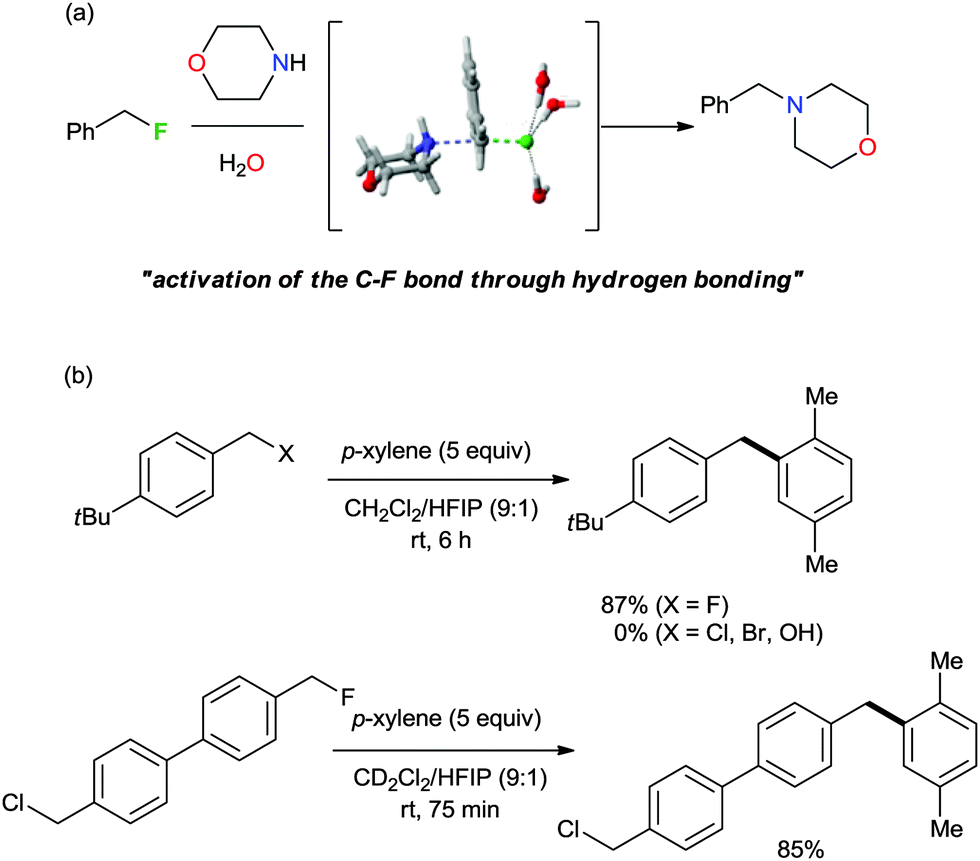 | ||
| Scheme 9 (a) Activation of the C–F bond through H-bonding. (b) Selective activation of a C–F bond in the presence of a similar C–X bond. HFIP: hexafluoroisopropanol. | ||
Synergistic effects of two promoters – ionic liquid and t-alcohol in one molecule
Recognizing that ILs and t-alcohols are beneficial for the activation of MFs via electrostatic and H-bonding interactions, synergistic effects of ILs and t-alcohols in one molecule could be expected to enhance selectivity and reactivity in fluorination reactions with MFs (Scheme 10a). Scheme 10b shows a comparison of different additives, including the combination of t-BuOH and nontethered [bmim][OMs]. In agreement with initial expectation, t-alcohol-tethered IL 22 proved to be the most successful promoter, inducing the conversion of substrate 3 to fluorinated product 4 in excellent yields.36 These intriguing cooperative effects of two promoters, i.e. ILs and t-BuOH, encouraged us to investigate a more efficient and easily accessible multifunctional promoter, which will be discussed in the next chapter. | ||
| Scheme 10 (a) Hybridization of ILs and t-alcohol in one molecule and (b) its application to fluorination with CsF. | ||
5. Oligoethylene glycols for nucleophilic fluorinations with metal fluorides
Traditional crown ethers have been extensively utilized in organic synthesis by generating naked anions from inorganic salts. However, the application of crown ethers in practical fluorination reactions using a metal salt is rare because of their low promoting effects. Moreover, the generation of a “naked” fluoride can induce various by-product formation, due to the high basicity.Oligoethylene glycols such as tetraethylene glycol (tetraEG) would be desirable candidates to overcome these drawbacks. By coordinating the potassium cation with polyethers, the solubility of potassium fluoride can be efficiently enhanced. On the other hand, one of the OH groups engages in controlled hydrogen bonding with the fluoride anion, thus decreasing the basicity, whereas the other OH group would interact with the leaving group, activating the electrophile towards nucleophilic attack (Fig. 6).37
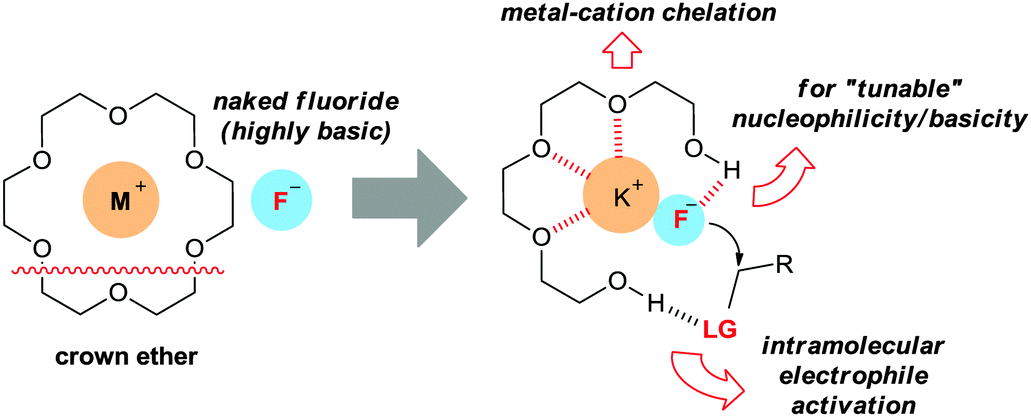 | ||
| Fig. 6 Bis-terminal hydroxyl polyethers as multifunctional promoters. | ||
To verify this hypothesis, we evaluated the potential of oligoethylene glycols as the promoters for nucleophilic fluorination with KF, which is the most inexpensive and convenient source of nucleophilic fluoride.37 Gratifyingly, in the presence of tetraEG or triethylene glycol (triEG), the reaction proceeded with unprecedented efficiency and completed within 1.5 h, giving the desired fluorinated product, 4, in almost quantitative yields (Scheme 11a). Notably, the thermal stability of oligoethylene glycols allowed the fluorination reaction to proceed under microwave conditions, affording the desired products in excellent yields in only 1 min (Scheme 11a). Considering the short half-life of the 18F radioisotope (t1/2, 109.8 min), this rapid fluorination may result in high radiochemical yields (RCYs) in the preparation of [18F]-radiotracers for PET imaging. As shown in Scheme 11b, our tetraEG medium protocol also allowed the fluorination of base-sensitive substrate 5 to produce the SN2 product 6 in 95% yield, along with 5% elimination product. The H-bonding between the terminal OH group and the fluoride anion probably reduced the basicity of the anion, consequently suppressing the elimination reaction. In contrast to these results, the reaction performed with KF in acetonitrile proceeded sluggishly, either in the presence or the absence of 18-crown-6-ether (Scheme 11a). Bulky protic solvents such as t-BuOH and t-amyl alcohol, which proved to be excellent media for fluorination with CsF, also showed almost no reactivity with KF probably because of its low solubility in these solvents.
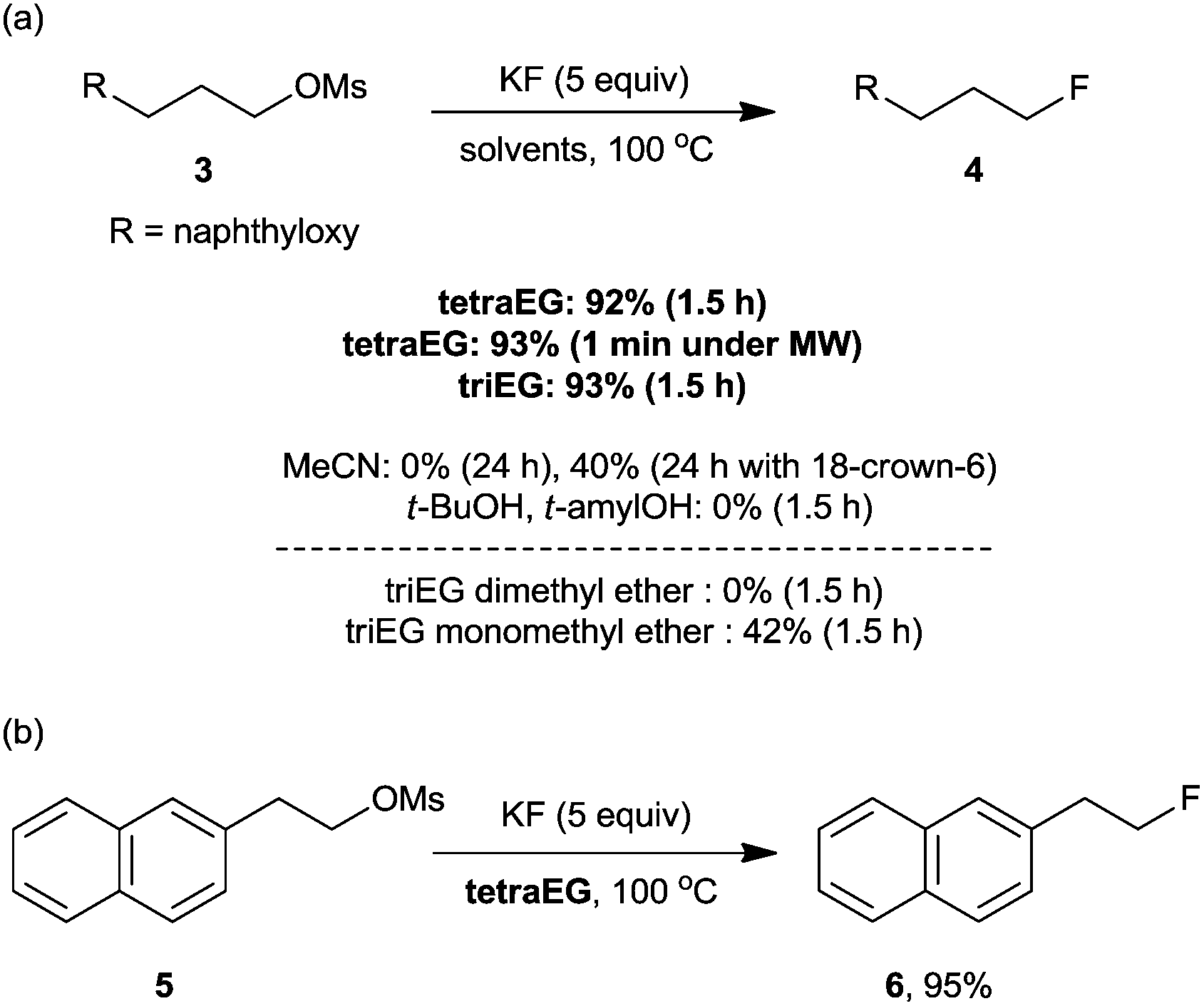 | ||
| Scheme 11 Nucleophilic fluorination with KF in various solvent systems. | ||
To further verify the importance of the cooperative multifunctionality of the promoter, fluorination with KF was carried out in various glycol derivatives. When triethylene glycol dimethyl ether (triglyme) was used as the solvent, the reaction did not proceed. On the other hand, triethylene glycol monoethyl ether showed approximately half of the reactivity of triEG under the same reaction conditions (Scheme 11a).37 Furthermore, the optimal reactivity with MFs can be easily achieved simply by varying the chain length of the oligoethylene glycols. As expected, in the case of CsF, the increase in chain length (e.g. penta- and hexaethylene glycol) resulted in an excellent yield of the product, while KF lost some reactivity in such solvents.38 All these experimental results strongly indicate that a cooperative activation mechanism is responsible for the observed rate accelerations in nucleophilic fluorinations.
Evidence for the cooperative mechanism has also been obtained by quantum chemical calculations.37Fig. 7 shows the mechanism of the reaction of C3H7–OMs with KF in tetraEG. The five oxygen atoms in tetraEG act as the Lewis base toward K+, thus drastically lowering its electrostatic effects on the fluoride, thereby “freeing” the fluoride. The two terminal-OH groups of the tetraEG also play a profound role in solvent catalysis: one of them interacts with the fluoride ion by H-bonding, thus reducing the basicity of fluoride. The other OH interacts with the mesylate leaving group, helping it to detach from the reactant. This compact and highly organized arrangement of the substrate, counter cation, nucleophile, and solvent provides an ideal environment with a very low activation barrier (E‡ = 18.6 kcal mol−1 and G‡ (100 °C) = 19.9 kcal mol−1), even lower than those calculated using similar methods for the same reaction with CsF (in t-BuOH, E‡ = 23.5 kcal mol−1 and G‡ (80 °C) = 23.1 kcal mol−1) and TBAF (in t-BuOH, E‡ = 25.3 kcal mol−1 and G‡ (80 °C) = 22.3 kcal mol−1).
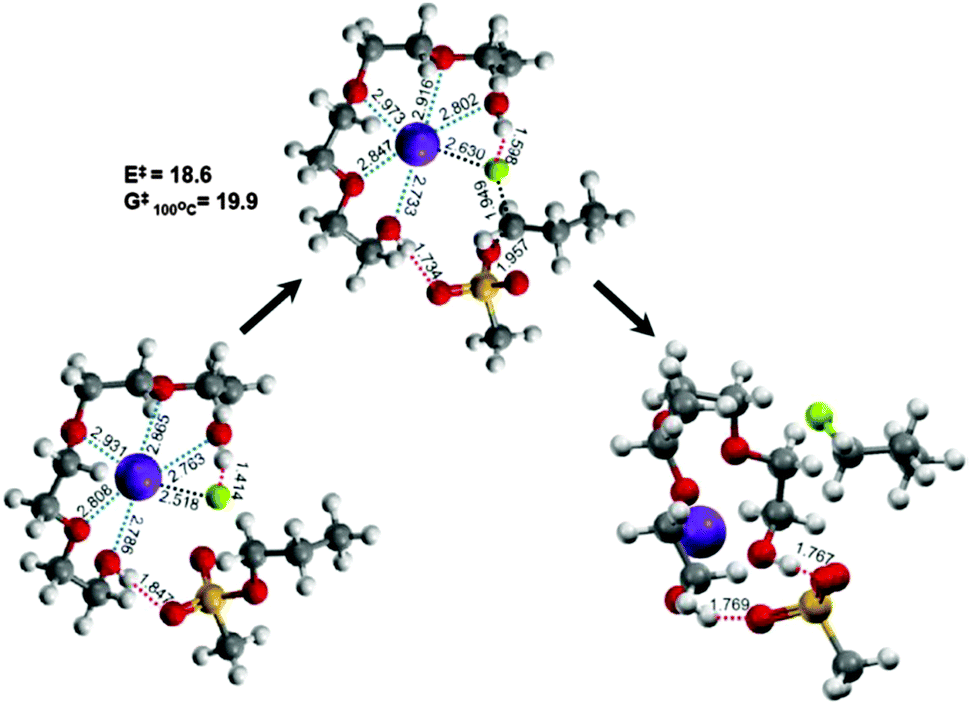 | ||
| Fig. 7 Calculated structures of pre-reaction complexes, transition states and post-reaction complexes in the mechanism for the reaction [K+F− + C3H7–OMs → K+OMs− + C3H7–F] in tetraEG. Energy and Gibbs free energy in kcal mol−1 and bond lengths in Å (MPW1K/6-31++G**). | ||
Very recently, Pliego reported a similar theoretical assumption to accelerate SN2 fluorination reactions by generating a transition state with double hydrogen bonding donors.39 The authors claimed that a protic catalyst can stabilize the transition state of SN2 reactions, which is in contrast to conventional wisdom for SN2 reaction conditions. According to the calculated results, 1,4-benzenedimethanol was able to render two H-bonds towards the fluoride anion and the leaving group, promoting substitution reaction simultaneously. In the presence of 1,4-benzenedimethanol as a catalyst, the SN2 reactions can selectively be accelerated over the E2 pathway compared to the non-catalysed one (ΔE between catalyzed SN2 and non-catalyzed SN2 = 5 kcal mol−1) (Fig. 8).
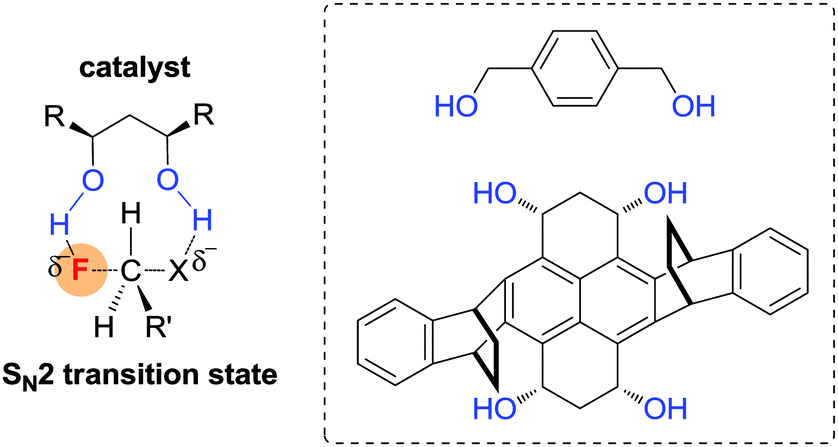 | ||
| Fig. 8 Stabilization of the transition state of SN2 reaction by using protic catalysts. | ||
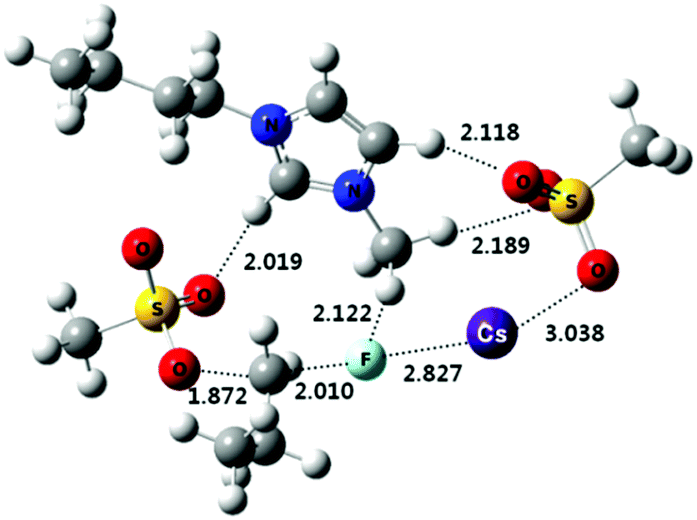
More direct experimental evidence for the activation of KF with oligoethylene glycols was also obtained by the single crystal X-ray structure analysis of the complex of KF and a 1,1′-bi-2-naphthol(BINOL)-based chiral analog of oligoethylene glycol (Fig. 9).40 As shown in the crystal structure (Fig. 9), the primary interaction (O–H–F) of terminal phenolic protons with fluoride is clear (O–F distance: 2.9–3.0 Å). Due to the dimer formation during the single crystal preparation, another hydrogen bonding interaction is observed with the other extremity phenolic protons of the other unit of the glycol-derivative (O–F distance: 3.2–3.3 Å), exhibiting weaker interactions. It is believed that the unique chelating environment using BINOL-based chiral oligoethylene glycol with KF can generate fluoride anions with a low coordination number, inducing higher reactivity for nucleophilic substitution reactions and other catalytic applications.40–42
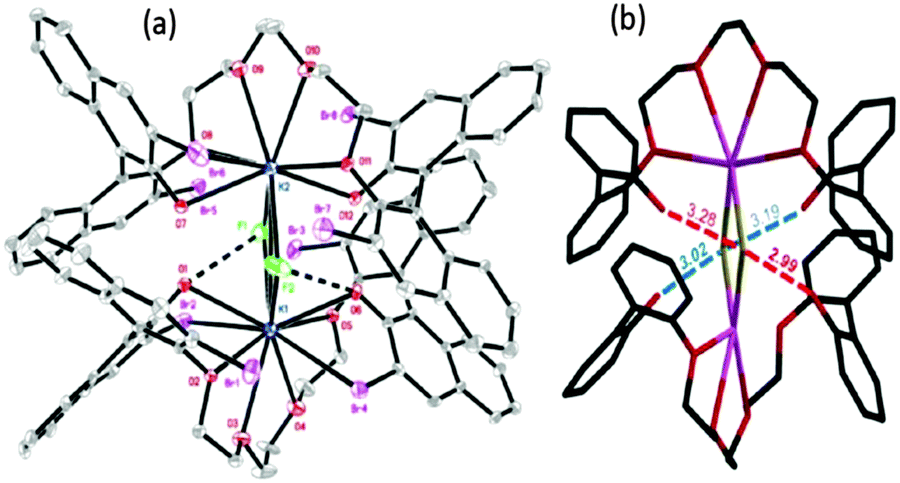 | ||
| Fig. 9 Experimental evidence for the activation of KF with a protic solvent such as tetraethylene glycol. (a) ORTEP structure as a dimeric form of the complex of KF and a binol-based chiral oligoethylene glycol. (b) BINOL backbone and bromine on 3,3′-positions are omitted for clarity; H-bond interactions between OH–F are highlighted in red and blue with distance (Å). | ||
The simultaneous activation of both the nucleophile and the electrophile by oligoethylene glycols seems to be a more general phenomenon, promoting SN2 reactions with different sources of nucleophiles (Scheme 12).37,40 Thus, the nucleophilic substitution reactions proceeded smoothly with a diverse set of carbon, nitrogen, oxygen, sulfur and halogen nucleophiles to give the corresponding products in almost quantitative yields. This reaction profile highlights the advantage of using oligoethylene glycols as the promoters for general nucleophilic substitution reactions. Furthermore, the concept for the ambiphilic activation using oligoEGs has been successfully extended to its asymmetric version.40–42 The employment of 1,1′-bi-2-naphthol-based chiral analogs of oligoethylene glycols showed unprecedented catalytic performance in desilylative kinetic resolution,40 asymmetric Strecker synthesis41 and asymmetric silylation reactions.42 Owing to its robustness and simplicity, the fluorination protocol with oligoethylene glycol promoters can be applied in various fields. In fact, it was recently applied to a selective electrochemical fluorination reaction with KF and tertiary carbon cation reactants by the Fuchigami research group (Scheme 13).43
 | ||
| Scheme 12 Nucleophilic substitutions in oligoEGs using various salts. | ||
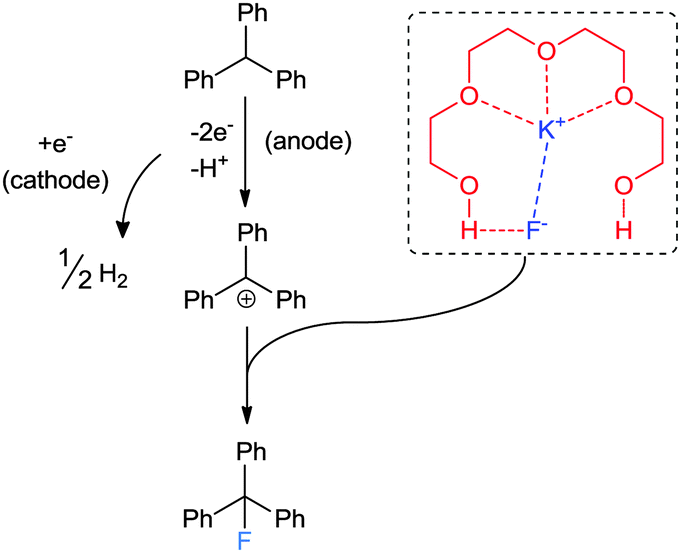 | ||
| Scheme 13 Schematic representation of electrochemical fluorination using tetraEG. | ||
Bis-tert-alcohol-functionalized crown-6-calix[4]arene (BACCA)
Most recently, a bis-tert-alcohol-functionalized crown-6-calix[4]arene (BACCA) as another type of bis-terminal hydroxyl polyether was reported by Kim and his coworkers.44 As shown in Scheme 14a, the binding of Cs+ to the crown-6-calix[4]arene subunit of BACCA separates CsF to a large distance (>8 Å) with the release of an essentially free fluoride, as established in a quantum chemical study. Moreover, H-bonding between the fluoride and the bis-terminal t-alcohol subunit of BACCA can reduce the basicity of fluoride, thus enhancing the effective nucleophilicity. As shown in Scheme 14b, BACCA showed a high performance in the nucleophilic fluorination reaction using CsF. On the other hand, the methylated BACCA, in which the two terminal tert-alcohol groups were methylated to prevent H-bonding with the fluoride, showed inferior activity in the same reaction. This result concludes that two H-bonds of OH groups towards fluoride and the leaving group seem to be important for promoting nucleophilic substitution reaction. Moreover, the use of BACCA allowed fluorination of base-sensitive substrates to proceed chemoselectively even in polar aprotic solvent such as CH3CN (Scheme 14c).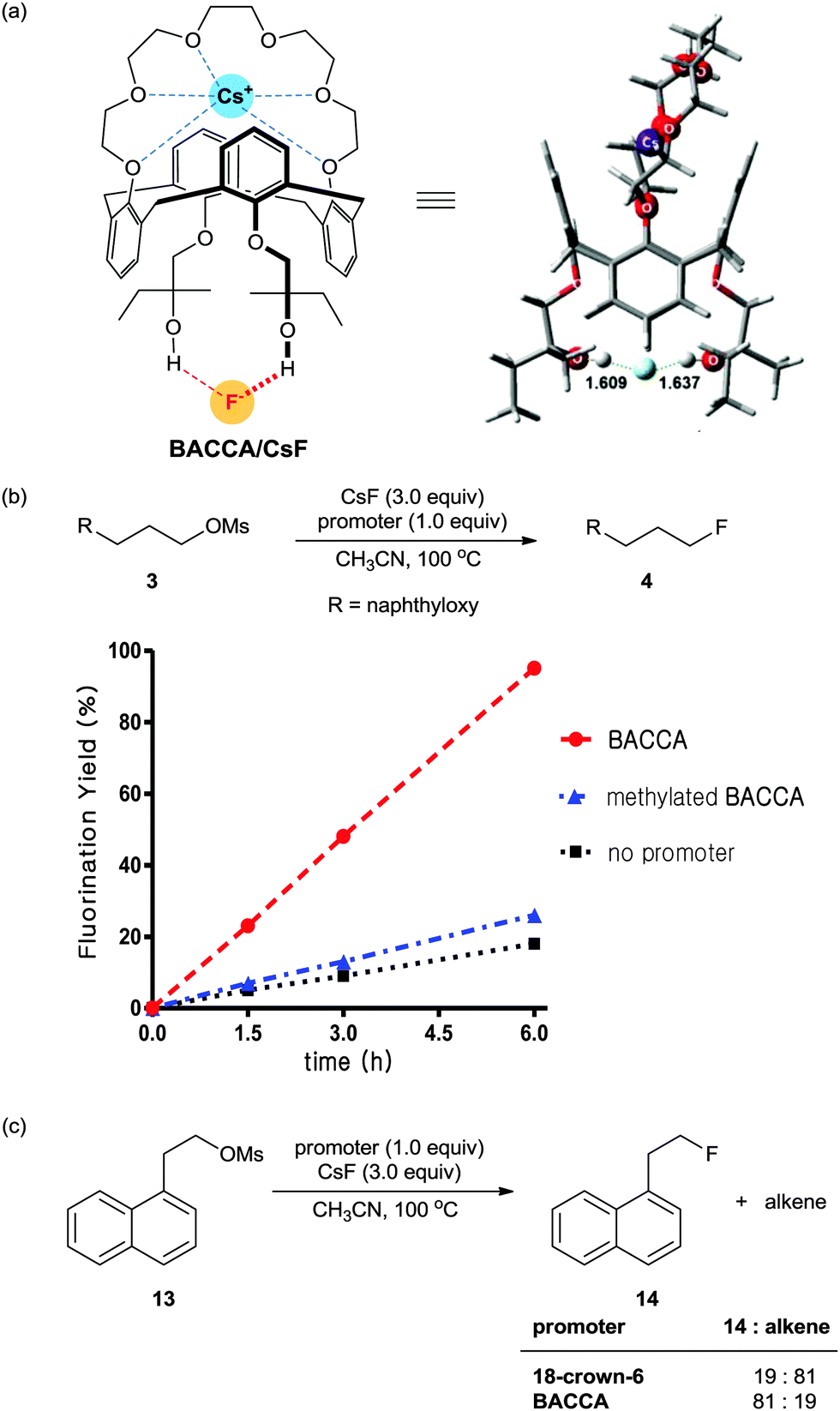 | ||
| Scheme 14 (a) Bis-tert-alcohol-functionalized crown-6-calix[4]arene (BACCA) and (b) SN2 fluorination reaction using CsF in the presence of BACCA, methylated BACCA, or without any promoter. (c) Chemoselective fluorination of base-sensitive substrate 13 in the presence of BACCA. | ||
6. 18F-radiofluorination using [18F]fluoride for PET applications
Fluorine-18 is regarded as one of the best positron emitters because of its many desirable characteristics such as a small steric size, stable bonding to carbon, a suitable decay rate, low positron energy, and ease of production. As a result of our research program with protic solvents for nucleophilic fluorination reactions, clinically relevant [18F]radiopharmaceuticals now can be accessed for wide use in clinical fields and PET molecular imaging studies.Synthesis of PET radiopharmaceuticals in ionic liquid media
The practicability of the reaction implied by the fact that IL-based fluorinations do not require anhydrous conditions21 compelled us to apply them to the 18F-labeling of various molecules, a process in which it is difficult to insure the complete absence of water. In these experiments, we used no-carrier-added [18F]fluoride in water as obtained from the production target (18O(p,n)18F). The 18F-radiofluorination of an alkyl mesylate precursor was attempted using [18F]fluoride in water with Cs2CO3 in IL, [bmim][OTf]. Gratifyingly, even in the presence of water, the reaction gave the corresponding 18F-fluorinated product ([18F]-4) in 93 ± 1.2% RCY (Scheme 15a).45 This method not only provides the product rapidly in a good yield and with high effective specific activity, but is also particularly simple to operate. Practically, [18F]fluoride is obtained from the production target and it can be directly added to the reaction media without the dehydrating step (Scheme 15b).45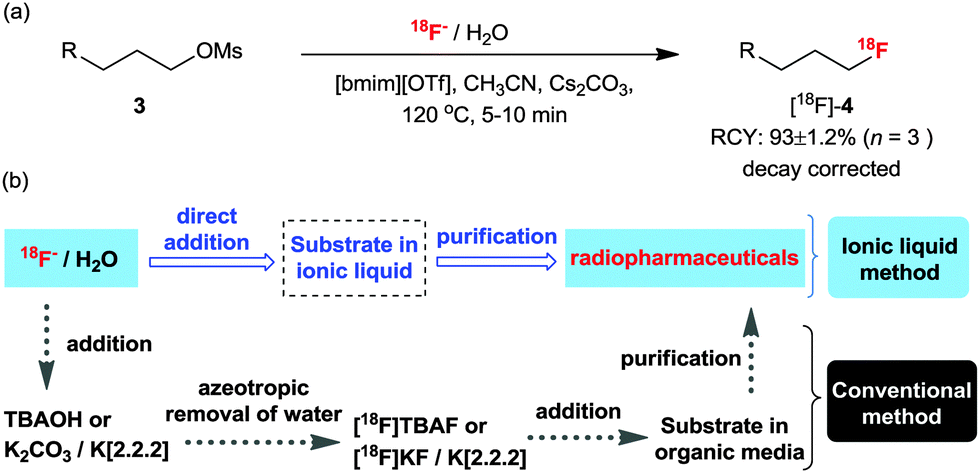 | ||
| Scheme 15 (a) 18F-radiofluorination in IL [bmim][OTf]. (b) A schematic comparison of the conventional 18F labeling process and the IL-based process. | ||
Taking advantage of the merits (high selectivity, high reaction rate, and convenient introduction of fluorine-18) of the 18F-radiofluorination method in aqueous ILs, we successfully applied this protocol to an improved preparation of 3′-deoxy-3′-[18F]fluorothymidine ([18F]FLT) (23)46 and 2-[18F]fluoro-2-deoxyglucose ([18F]FDG) (24)47 which are the most popular radiopharmaceuticals for PET imaging (Scheme 16).
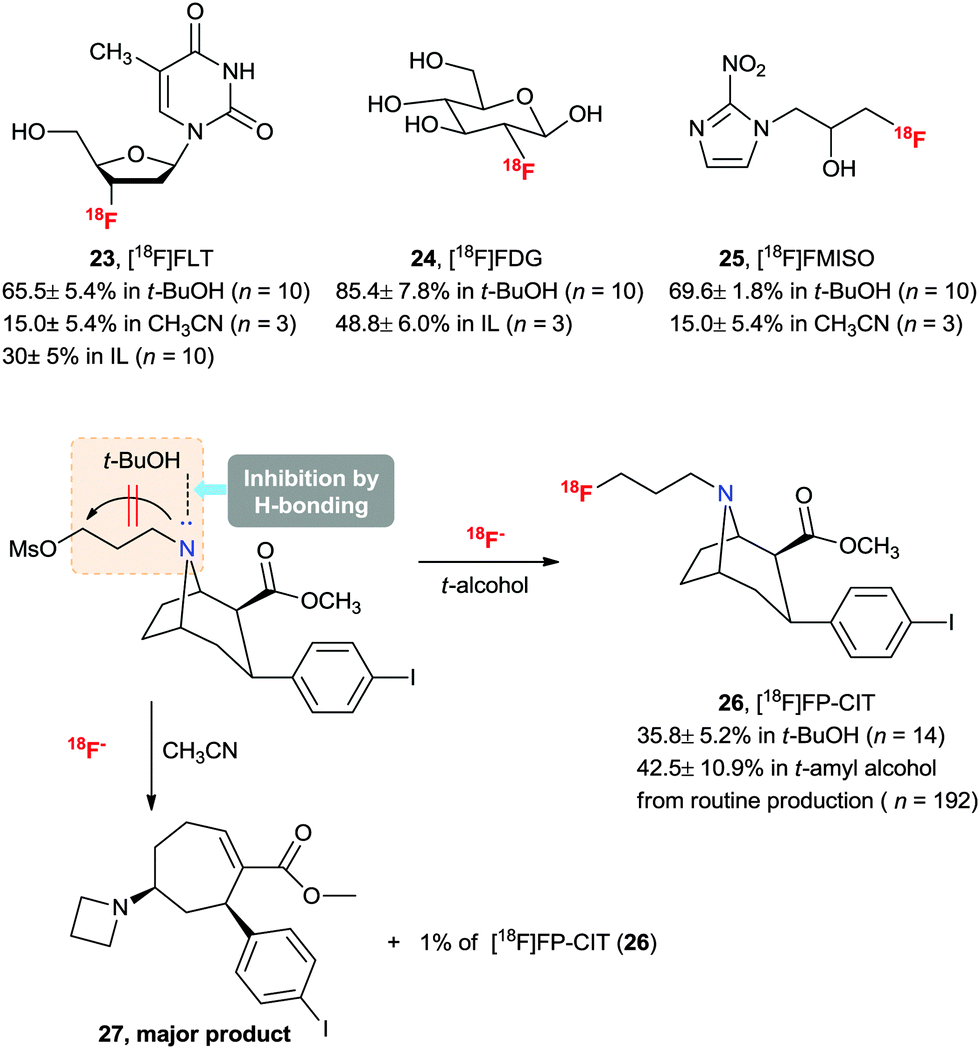 | ||
| Scheme 16 Synthesis of key 18F-labeled PET radiopharmaceuticals using t-alcohol solvent and their RCYs. | ||
Synthesis of PET radiopharmaceuticals in t-alcohol media
The practical fluorination protocol in t-alcohol media renders it perfectly suited for application in the 18F radiolabeling of important PET radiopharmaceuticals for automated mass production. Generally, when using no-carrier-added [18F]fluoride, 18F radiolabeling for the production of PET radiopharmaceuticals requires basic reaction conditions. Therefore, the protic atmosphere emerging from t-alcohol media could help to reduce the basicity of the labeling reaction condition and allow the radiofluorination to proceed chemo-selectively, consequently increasing the RCY. In addition, due to the lower boiling points of t-alcohols (e.g. bp of t-BuOH, 81 °C) than those of typical polar aprotic solvents such as DMF and DMSO, t-alcohols can easily be removed using nitrogen gas stream after the 18F fluorination reaction. We aimed at radiopharmaceuticals that are challenging to label such as [18F]FLT (23), [18F]FDG (24), 1-[18F]fluoro-3-(2-nitroimidazol-1-yl)propan-2-ol ([18F]FMISO) (25) and N-[18F]fluoropropyl-2β-carbomethoxy-3β-(4-iodophenyl)nortropane ([18F]FP-CIT) (26).28 As shown from the results in Scheme 16, all the examples showcase the utility of this t-alcohol medium radiofluorination compared to a conventional polar aprotic solvent, e.g. acetonitrile. Significantly, higher RCYs of 18F-fluorinated compounds 23–26 were obtained by automatic synthesis using the t-alcohol solvent system compared to other reported methods.28,47 In particular, [18F]FP-CIT (26) is an important radiopharmaceutical for the PET imaging of dopamine transporters. However, the extremely low RCY yield (1%) of 26 in CH3CN hampered its application. A dramatic improvement was observed by using t-BuOH, affording 36% RCY. Furthermore, [18F]FP-CIT (26) can now be obtained in 42.5 ± 10.9% RCY from recent routine 192 productions for 1.5 years using an auto-synthesizer module in t-amyl alcohol.48 As shown in Scheme 16, the protic atmosphere of t-alcohol as well as H-bonding between t-alcohol and the N atom of the FP-CIT precursor can suppress side reactions such as ring opening by two sequential steps of bimolecular elimination and the intramolecular N-alkylation reaction, thereby increasing the RCY. Bayer Healthcare® has also tried to extend this methodology to automatic mass production of other PET radiopharmaceuticals.It would also be instructive to note that the t-alcohol fluorination protocol was recently applied by us and other research groups to synthesize interesting PET radiotracers, such as [18F]fluorine-substituted tanaproget (28) as a progesterone receptor imaging agent49 and N4′-[18F]fluoropropyl ciprofloxacin (29) as a bacterial infection imaging agent (Scheme 17).50 It is noteworthy here that mild radiofluorination reaction conditions allowed us to access biologically relevant molecules with diverse functional groups including nitriles, pyrroles, carbamates, esters, enone and cyclopropyl groups. Further applications of the 18F-radiofluorination reaction protocol using the protic engineering solvents will give facile access to different types of radiotracers of interest.
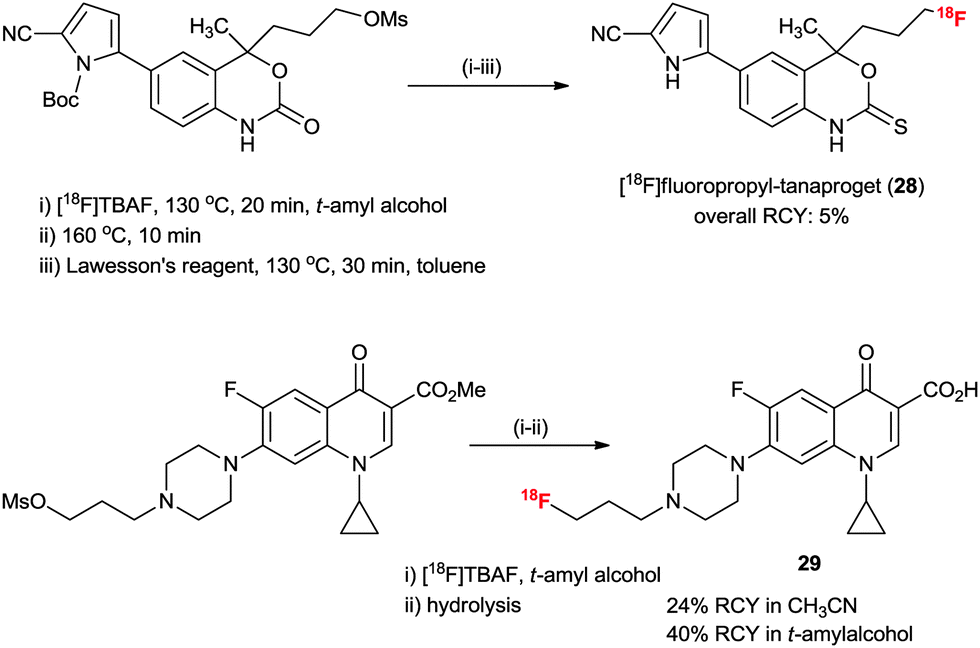 | ||
| Scheme 17 Synthesis of N4′-[18F]fluoropropyl-tanaproget and N4′-[18F]fluoropropyl ciprofloxacin using t-alcohol solvent. | ||
7. Conclusions
This Tutorial Review summarizes the recent breakthroughs in bimolecular nucleophilic fluorination reactions at an sp3-hybridized carbon, through the activation of alkali MFs in protic reaction media. Although conventional wisdom deems protic solvents as unsuitable for nucleophilic displacement reactions, we found that t-alcohols, oligoEGs, and aqueous ILs led to remarkable and unprecedented efficiency in nucleophilic fluorinations. Thus, by evolving from ILs to t-alcohols, and recently to oligoethylene glycols as the promoters, MFs have become superior fluoride reagents and thus now applicable to diverse base-sensitive substrates, affording excellent yields of the fluorinated products with high reaction rates. The simultaneous dual activation of reacting partners by intermolecular hydrogen bonding and the enhancement of the “effective fluoride nucleophilicity” by hydrogen bond interactions, mimicking Nature's biocatalytic approach in the fluorinase enzyme, are the key to this unprecedentedly straightforward nucleophilic fluorination. The advantages of these protocols become particularly practical in the context of 18F labeling for PET imaging, where the short half-life of the radionuclide leads to an enormous dependency on reaction speed and efficiency.Acknowledgements
This work was supported by grants NRF-2014R1A2A1A01005794, NRF-2014R1A2A2A03007401, NRF-2012-0008397, NRF-2013-030262, NRF-2012R1A2A2A02013289, NRF-2011-0021836, and NRF-2015M2A2A6A01045378, the KISTI supercomputer Center (2015), and the Converging Research Center Program (2014M3C1A8066306).Notes and references
- J. Xie and W. L. Hase, Science, 2016, 352, 32–33 CrossRef CAS PubMed.
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- A. Harsanyi and G. Sandford, Green Chem., 2015, 17, 2081–2086 RSC.
- L. Zhu, K. Ploessl and H. F. Kung, Science, 2013, 342, 429–430 CrossRef CAS PubMed.
- S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef CAS PubMed.
- F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378 CrossRef CAS PubMed.
- P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073 CrossRef CAS PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef CAS PubMed.
- C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC.
- E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639–642 CrossRef CAS PubMed.
- C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216–3221 CrossRef CAS PubMed.
- R. Schwesinger, R. Link, P. Wenzl and S. Kossek, Chem. – Eur. J., 2006, 12, 438–445 CrossRef PubMed.
- J. Wu, Tetrahedron Lett., 2014, 55, 4289–4294 CrossRef CAS.
- S. H. Liang and N. Vasdev, Angew. Chem., Int. Ed., 2014, 53, 11416–11418 CrossRef CAS PubMed.
- O. Nashiru, D. L. Zechel, D. Stoll, T. Mohammadzadeh, R. A. J. Warren and S. G. Withers, Angew. Chem., Int. Ed., 2001, 40, 417–420 CrossRef CAS.
- D. L. Zechel, S. P. Reid, O. Nashiru, C. Mayer, D. Stoll, D. L. Jakeman, P. A. J. Warren and S. G. Withers, J. Am. Chem. Soc., 2001, 123, 4350–4351 CrossRef CAS PubMed.
- D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef PubMed.
- J.-W. Lee, J. Y. Shin, Y. S. Chun, H. B. Jang, C. E. Song and S. G. Lee, Acc. Chem. Res., 2010, 43, 985–994 CrossRef CAS PubMed.
- D. W. Kim, C. E. Song and D. Y. Chi, J. Am. Chem. Soc., 2002, 124, 10278–10279 CrossRef CAS PubMed.
- Y.-H. Oh, H. B. Jang, S. Im, M. J. Song, S.-Y. Kim, S.-W. Park, D. Y. Chi, C. E. Song and S. Lee, Org. Biomol. Chem., 2011, 9, 418–422 CAS.
- D. W. Kim, C. E. Song and D. Y. Chi, J. Org. Chem., 2003, 68, 4281–4285 CrossRef CAS PubMed.
- D. W. Kim and D. Y. Chi, Angew. Chem., Int. Ed., 2004, 43, 483–485 CrossRef CAS PubMed.
- J. H. Clark, S. J. Tavener and S. J. Barlow, Chem. Commun., 1996, 2429–2430 RSC.
- C. B. Murray, G. Sandford and S. R. Korn, J. Fluorine Chem., 2003, 123, 81–84 CrossRef CAS.
- T. Yonezawa, Y. Sakamoto and K. Nogawa, Jpn. Kokai Tokkyo Koho, 1994, JP 06316551 A Search PubMed.
- D. W. Kim, D.-S. Ahn, Y.-H. Oh, S. Lee, H. S. Kil, S. J. Oh, S. J. Lee, J. S. Kim, J. S. Ryu, D. H. Moon and D. Y. Chi, J. Am. Chem. Soc., 2006, 128, 16394–16397 CrossRef CAS PubMed.
- M. Egli, P. S. Pallan, C. R. Allerson, T. P. Prakash, A. Berdeja, J. Yu, S. Lee, A. Watt, H. Gaus, B. Bhat, E. E. Swayze and P. P. Seth, J. Am. Chem. Soc., 2011, 133, 16642–16649 CrossRef CAS PubMed.
- O.-M. Soueidan, B. J. Trayner, T. N. Grant, J. R. Henderson, F. Wuest, F. G. West and C. I. Cheeseman, Org. Biomol. Chem., 2015, 13, 6511–6521 CAS.
- D. W. Kim, H.-J. Jeong, S. T. Lim, M.-H. Sohn, J. A. Katzenellenbogen and D. Y. Chi, J. Org. Chem., 2008, 73, 957–962 CrossRef CAS PubMed.
- D. W. Kim, H.-J. Jeong, S. T. Lim and M.-H. Sohn, Angew. Chem., Int. Ed., 2008, 47, 8404–8406 CrossRef CAS PubMed.
- K. M. Engle, L. Pfeifer, G. W. Pidgeon, G. T. Giuffredi, A. L. Thompson, R. S. Paton, J. M. Brown and V. Gouverneur, Chem. Sci., 2015, 6, 5293–5302 RSC.
- P. A. Champagne, J. Pomarole, M. E. Therien, Y. Benhassine, S. Beaulieu, C. Y. Legault and J. F. Paquin, Org. Lett., 2013, 15, 2210–2213 CrossRef CAS PubMed.
- P. A. Champagne, Y. Benhassine, S. Beaulieu, J. Desroches and J. F. Paquin, Angew. Chem., Int. Ed., 2014, 53, 13835–13839 CrossRef CAS PubMed.
- S. S. Shinde, Y. S. Lee and D. Y. Chi, Org. Lett., 2008, 10, 733–735 CrossRef CAS PubMed.
- J.-W. Lee, H. Yan, H. B. Jang, H. K. Kim, S. W. Park, S. Lee, D. Y. Chi and C. E. Song, Angew. Chem., Int. Ed., 2009, 48, 7683–7686 CrossRef CAS PubMed.
- V. H. Jadhav, S. H. Jang, H.-J. Jeong, S. T. Lim, M.-H. Sohn, J. Y. Kim, S. Lee, J. W. Lee, C. E. Song and D. W. Kim, Chem. – Eur. J., 2012, 18, 3918–3924 CrossRef CAS PubMed.
- J. R. Pliego Jr., Phys. Chem. Chem. Phys., 2011, 13, 779–782 RSC.
- H. Yan, H. B. Jang, J. W. Lee, H. K. Kim, S. W. Lee, J. W. Yang and C. E. Song, Angew. Chem., Int. Ed., 2010, 49, 8915–8917 CrossRef CAS PubMed.
- H. Yan, J. S. Oh, J. W. Lee and C. E. Song, Nat. Commun., 2012, 3, 1212 CrossRef PubMed.
- S. Y. Park, J.-W. Lee and C. E. Song, Nat. Commun., 2015, 6, 7512 CrossRef PubMed.
- T. Sawamura, K. Takahashi, S. Inagi and T. Fuchigami, Angew. Chem., Int. Ed., 2012, 51, 4413–4416 CrossRef CAS PubMed.
- V. H. Jadhav, W. Choi, S.-S. Lee, S. Lee and D. W. Kim, Chem. – Eur. J., 2016, 22, 4515–4520 CrossRef CAS PubMed.
- D. W. Kim, Y. S. Choe and D. Y. Chi, Nucl. Med. Biol., 2003, 30, 345–350 CrossRef CAS PubMed.
- B. S. Moon, K. C. Lee, G. I. An, D. Y. Chi, S. D. Yang, C. W. Choi, S. M. Lim and K. S. Chun, J. Labelled Compd. Radiopharm., 2006, 49, 287–293 CrossRef CAS.
- H. W. Kim, J. M. Jeong, Y.-S. Lee, D. Y. Chi, K.-H. Chung, D. S. Lee, J.-K. Chung and M. C. Lee, Appl. Radiat. Isot., 2004, 61, 1241–1246 CrossRef CAS PubMed.
- S. J. Lee, S. J. Oh, W. Y. Moon, M. S. Choi, J. S. Kim, D. Y. Chi, D. H. Moon and J. S. Ryu, Nucl. Med. Biol., 2011, 38, 593–597 CrossRef CAS PubMed.
- J. H. Lee, H.-B. Zhou, C. S. Dence, K. E. Carlson, M. J. Welch and J. A. Katzenellenbogen, Bioconjugate Chem., 2010, 21, 1096–1104 CrossRef CAS PubMed.
- K. Sachin, E.-M. Kim, S.-J. Cheong, H.-J. Jeong, S. T. Lim, M.-H. Sohn and D. W. Kim, Bioconjugate Chem., 2010, 21, 2282–2288 CrossRef CAS PubMed.
Footnote |
| † Current address: Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark |
| This journal is © The Royal Society of Chemistry 2016 |