
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA noble metal-free proton-exchange membrane fuel cell based on bio-inspired molecular catalysts†
P. D.
Tran‡
a,
A.
Morozan‡
b,
S.
Archambault‡
c,
J.
Heidkamp‡
d,
P.
Chenevier
e,
H.
Dau
d,
M.
Fontecave
af,
A.
Martinent
*c,
B.
Jousselme
*b and
V.
Artero
*a
aLaboratoire de Chimie et Biologie des Métaux, Université Grenoble Alpes, CNRS, CEA, 17 rue des Martyrs, 38054 Grenoble Cedex 09, France. E-mail: vincent.artero@cea.fr
bCEA Saclay, IRAMIS, NIMBE/UMR 3685, Laboratory of Innovation in Surface Chemistry and Nanosciences (LICSEN), Gif sur Yvette F-91191, France
cInstitut LITEN CEA LITEN/DTNM/LCSN, Grenoble, France
dFB Physik, Free University Berlin, Berlin, Germany
eUniversité Grenoble Alpes, CNRS, CEA, INAC-SPRAM, F-38000 Grenoble, France
fCollège de France, 11 Place Marcelin Berthelot, 75005 Paris, France
First published on 6th January 2015
Abstract
Hydrogen is a promising energy vector for storing renewable energies: obtained from water-splitting, in electrolysers or photoelectrochemical cells, it can be turned back to electricity on demand in fuel cells (FCs). Proton exchange membrane (PEM) devices with low internal resistance, high compactness and stability are an attractive technology optimized over decades, affording fast start-up times and low operating temperatures. However, they rely on the powerful catalytic properties of noble metals such as platinum, while lower cost, more abundant materials would be needed for economic viability. Replacing these noble metals at both electrodes has long proven to be a difficult task, so far incompatible with PEM technologies. Here we take advantage of newly developed bio-inspired molecular H2 oxidation catalysts and noble metal-free O2-reducing materials, to fabricate a noble metal-free PEMFC, with an 0.74 V open circuit voltage and a 23 μW cm−2 output power under technologically relevant conditions. X-ray absorption spectroscopy measurements confirm that the catalysts are stable and retain their structure during turnover.
Proton exchange membrane (PEM) technology currently stands as the most promising fuel cell variety for both portable and automotive applications. PEMFCs have low internal resistance, high compactness and stability. Having been optimized over decades, they display high efficiency, start rapidly and operate at low temperature. However, they require platinum to catalyse both the H2 oxidation reaction (HOR) and the O2 reduction reaction (ORR) which hampers their economic viability and sustainability. Substituting earth-abundant catalysts for noble metals is thus critical. Significant progress has been achieved regarding the development of ORR catalysts1–7 but no PEMFC using earth-abundant catalysts at both electrodes has been reported so far.
We recently reported on noble metal-free catalysts for the HOR8,9 working under highly acidic conditions compatible with PEM technology. Hydrogenases10,11 are unique metalloproteins that catalyse HOR as efficiently as platinum nanoparticles do and in particular with remarkably high reaction rates (1500–9000 s−1 at pH 7 and 37 °C in water).12 Their active sites (Fig. 1) have inspired the design of new synthetic HOR catalysts, the most efficient ones being bisdiphosphine nickel complexes13 exquisitely combining a nickel centre in an electron-rich environment as found in NiFe hydrogenases with proton relays provided by a pendant base mimicking the aza-propanedithiolato cofactor of FeFe hydrogenases.14 In both classes of hydrogenases,15 the presence of basic residues at the vicinity of the catalytic metal centre indeed facilitates the activation and heterolytic cleavage of H2. Immobilization of such synthetic catalysts, either covalently or through π–π stacking interactions, on multiwall carbon nanotubes (CNTs) yielded Ni–CNT HOR catalytic nanomaterials.8,9 When interfaced with a Nafion membrane, these nanomaterials show catalytic activity for hydrogen evolution/uptake at the thermodynamic equilibrium, prolonged stability under turnover conditions and resistance to CO poisoning. It was thus tempting to implement these original materials as anode PEMFC catalysts. Here we report the first functional PEMFC based on earth-abundant materials at both anode and cathode.
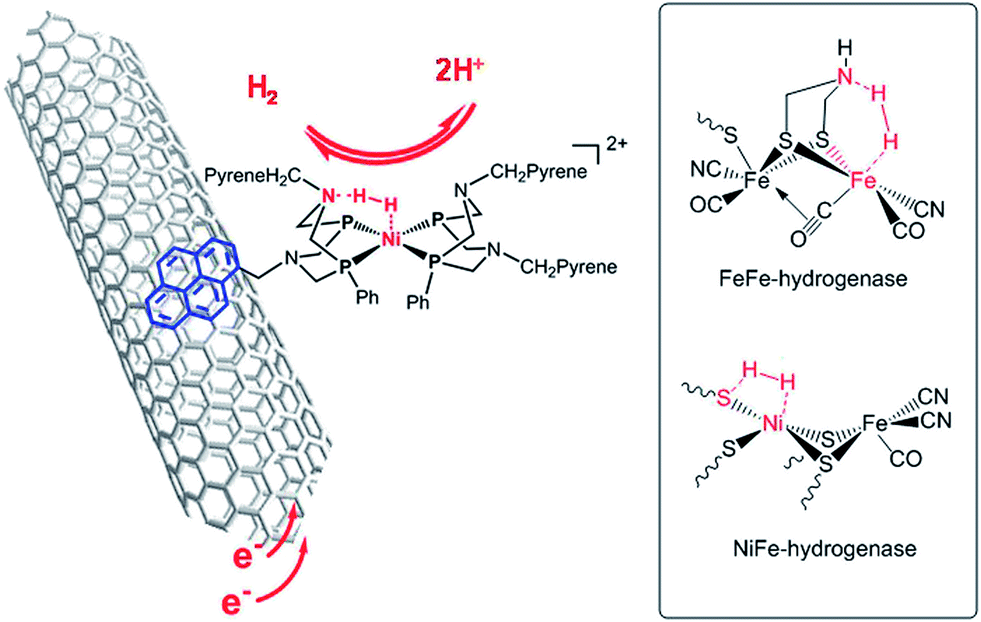 | ||
| Fig. 1 Structure of the Ni–CNT HOR catalyst based on a synthetic nickel bisdiphosphine complex inspired by the structures of the active sites of FeFe and NiFe hydrogenases shown in the inset. | ||
Deposition of one of these Ni–CNT HOR materials on a Nafion membrane was first achieved through spray coating.16 The preparation of a catalytic ink with suitable rheological properties required partial substitution of CNTs with carbon black nanoparticles with lower form factor. The anode Ni–CNT ink was then composed of a 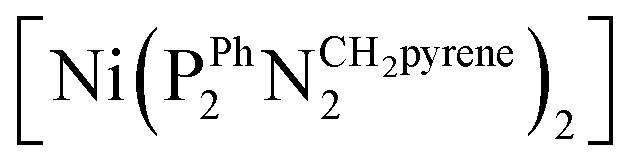 (BF4)2/CNTs (Ni–CNT, 30 wt% dry mass) material (Fig. 1),9 Vulcan XC-72 (40–50 wt%) and Nafion (20–30 wt%) in a water/isopropanol mixture (80/20 wt%). The use of a 5
(BF4)2/CNTs (Ni–CNT, 30 wt% dry mass) material (Fig. 1),9 Vulcan XC-72 (40–50 wt%) and Nafion (20–30 wt%) in a water/isopropanol mixture (80/20 wt%). The use of a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Vulcan XC-72:CNTs mass ratio led to particles in the 10–100 μm range as measured by dynamic light scattering.
:1 Vulcan XC-72:CNTs mass ratio led to particles in the 10–100 μm range as measured by dynamic light scattering.
A novel Co–N–C ORR catalyst with suitable rheological properties was obtained through adaptation of a previous procedure developed by some of us.3,16 Pyrolysis of a mixture of a N-heterocyclic organic compound (triazolopyridine) and Vulcan XC-72 in the presence of Co(NO3)2 (ref. 3 and 17) at 700 °C gave a fine dispersion of 0.1–100 μm particles in Nafion (20–30 wt%) and water/isopropanol as a proper ink for spray coating. By analogy with the previously described material, the ORR activity is assigned to pyridinic-N functional groups located at the surface of the carbon matrix and binding cobalt ions as well as metallic cobalt nanoparticles.3 This material displays an overpotential for the ORR ∼300 mV higher than Pt/C in a half cell configuration and good stability for more than 15 h during chrono-amperometry measurements. A detailed structural and catalytic study is provided in the ESI.†
After spray coating of the catalytic inks (0.44 to 0.64 mg cm−2, corresponding to about 10 μm thickness) on both sides of a NRE-212 Nafion membrane (5.76 cm2) at 60–80 °C, the assembly was hot-pressed (50 °C, 2 × 105 Pa, 3 min) to improve catalyst–electrolyte contact and metal collectors were deposited on each side by evaporation of a gold layer, thin enough to allow for gas permeation. The resulting membrane–electrode assembly (MEA, Fig. 2) was assessed at 60 °C in air under natural convection and ambient humidity in a dedicated micro fuel cell test-rig. Partially humidified hydrogen (20 mL min−1 bubbling in water) was supplied to the anode, and air was passively supplied to the cathode.
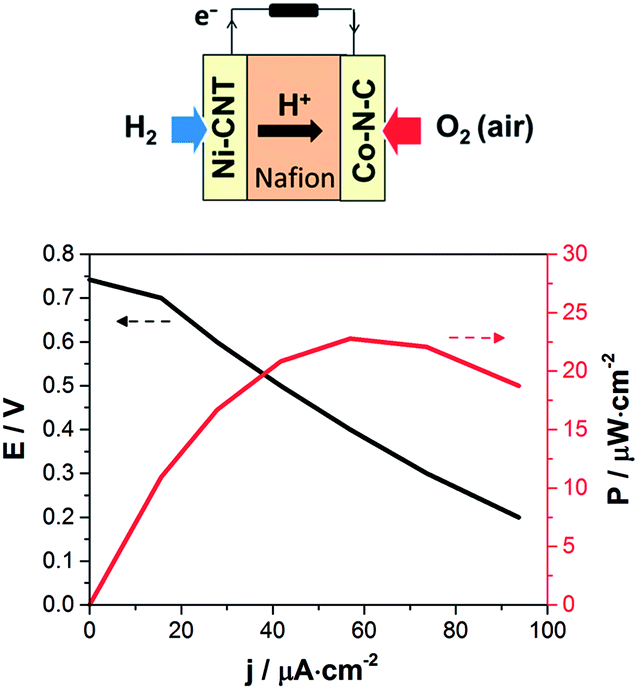 | ||
| Fig. 2 Left: schematic description of the PEMFC assembly; right: polarization and power density curves of the Ni–CNT/Co–N–C PEMFC recorded at 60 °C with a supply of partially humidified H2 (20 mL min−1) at the anode and passive air convection at the cathode. | ||
The polarization and power density curves for the resulting Ni–CNT/Co–N–C PEMFC are shown in Fig. 2. Table 1 compares the performances of this noble metal-free PEMFC with other cells in which one electrode has been replaced by a standard Pt-based electrode similarly prepared from 40% Pt/C catalytic material. The noble metal-free PEMFC proved functional with an open-circuit voltage (OCV) of 0.74 V, to be compared with the 1.0 V OCV value measured for a Pt/Pt PEMFC under the same conditions. The Ni–CNT/Pt and the Pt/Co–N–C control PEMFCs provided OCV values only 150 and 210 mV lower, respectively, than a standard Pt/Pt PEMFC, showing that each noble metal-free catalyst operates at reasonable overvoltages. The maximum power density of the noble metal-free PEMFC was 23 μW cm−2 under dynamic conditions and it stabilized at 20 μW cm−2 under stationary conditions. From Table 1, it can be concluded that the ORR catalyst, tested against Pt in a full PEMFC device, shows a higher current density (13 mA cm−2) than in half-cell configuration (1–3 mA cm−2, Fig. S4 and S5†) but the reverse is observed for the HOR catalyst (0.224 mA cm−2 in a PEMFC versus 1 mA cm−2 in half-cell configuration, Fig. S6†). The limitation in terms of current density and output power thus comes here from the bio-inspired anode material and its interface with the membrane rather than from the cathode catalyst, indicating a direction for further improvement of performances. Finally, the operation of Pt/Co–N–C and Ni–CNT/Pt PEMFCs at temperatures increasing from 25 °C to 60 °C showed a low increase in current density by a factor of ≤1.5, corresponding to an activation energy below 10 kJ mol−1 and 20 kJ mol−1, respectively. These low activation energy values show that the current is likely limited by the gas transport processes in the bulk of the electrode materials.
| Anode | Cathode | OCV/V | I@0.2 V/μA cm−2 | P max/μW cm−2 |
|---|---|---|---|---|
| Pt | Co–N–C | 0.79 | 1.3 × 104 | 2600 |
| Ni–CNT | Pt | 0.85 | 224 | 70 |
| Ni–CNT | Co–N–C | 0.74 | 94 | 23 |
| Pt | Pt | 1.00 | 1.75 × 105 | 1.05 × 103 |
We then performed X-ray absorption spectroscopy (XAS) measurements to identify the active species at work in the device. X-ray absorption spectra were measured directly on the MEA, before and after 1 hour of continuous operation.
X-ray absorption near edge structure (XANES) data recorded at the Co K edge on Co–N–C cathodes before and after O2-reduction catalysis are presented in Fig. 3. The spectra exhibit two distinct contributions from metallic Co and a non-metallic ionic cobalt compound. Metallic cobalt is the major species in the as-prepared material but this contribution is lowered to 28 ± 15% of the total Co concentration after fuel cell operation. This oxidative transformation is not correlated to any loss of activity. Rather, it is consistent with the relatively positive potential of the cathode during turnover in the contact with air. Comparison with the Co K edge positions of [Co(H2O)6](NO3)2 and LiCoO2, containing CoII and CoIII ions respectively, indicates an oxidation state between +II and +III for the cobalt ions in the catalytic material during operation, while extended X-ray absorption fine structure (EXAFS) analysis indicates coordination of cobalt ions to O or N atoms (Fig. S9†). The Co–N–C material thus behaves similarly to the cobalt–polypyrrole composite material previously reported by Bashyam and Zelenay.2
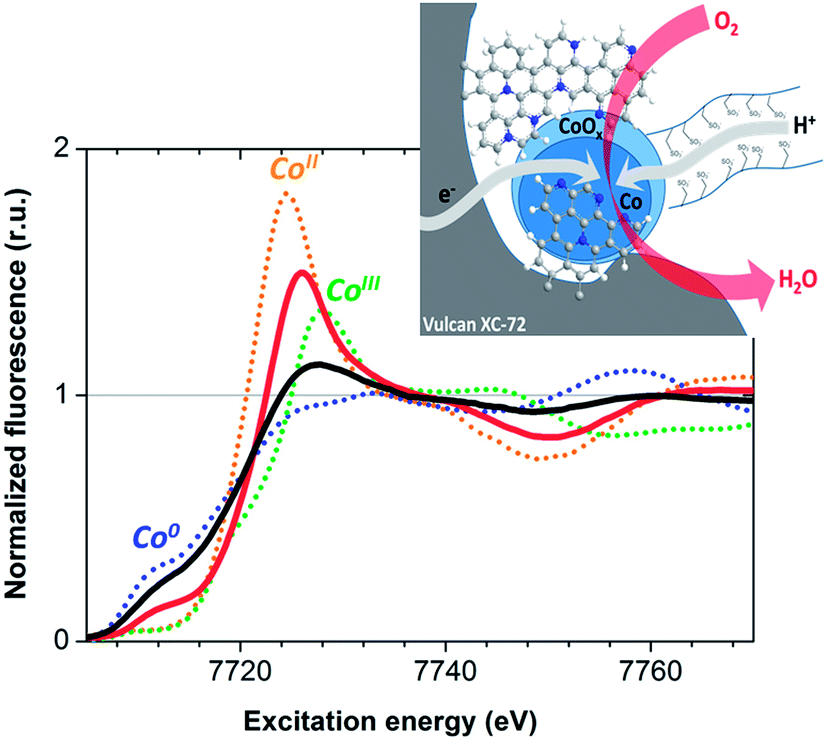 | ||
| Fig. 3 X-ray absorption near edge structure spectra (Co K edge) of the Co–N–C material measured for the as-prepared material (black solid line) and after O2-reduction catalytic operation (red solid line). The spectra of metallic Co (blue dotted line), [CoII(H2O)6](NO3)2 (orange dotted line) and LiCoIIIO2 (green dotted line) standards are also shown for comparison. Inset: representation of the Co–N–C material with carbon and nitrogen atoms depicted in grey and blue respectively. | ||
The as-prepared Ni–CNT anode nanomaterial was compared with the molecular Ni catalyst (Fig. 4 and S10†) to gain insight into the molecular state of the metal catalytic centers. Detailed analysis of the Ni K edge XAS signal measured for the complete cell proved impossible, so we turned to previously described half-cell experiments with the Ni–CNT material deposited on a gas diffusion layer.8,9 As previously reported,9 the Ni-edge EXAFS spectrum of the Ni–CNT material before operation could be decomposed into the weighted addition of the spectra for pure molecular 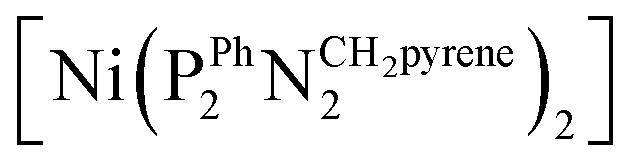 catalyst and for NiII ions coordinated to lighter atoms as in [Ni(H2O)6]2+, indicating that about 2/3rd of the Ni complex remains in the NiP4 state (Table S2†). After 1 hour of operation for H2 oxidation at 0.25 V vs. RHE and in direct contact with 0.5 M aqueous H2SO4 electrolyte, the signal assigned to decomposed Ni complex disappears, leaving a pure NiP4 signature (Fig. 4, Fig. S10 and Table S2†).18 As the current density remains constant during the whole measurement (Fig. S4†), corresponding to 4000 turnovers achieved for each NiP4 species, we conclude that the decomposed Ni compound was washed off from the material during the electrochemical measurement. Additionally this experiment tells us that the bio-inspired NiP4 complex is the only species responsible for catalysis and that it does withstand extensive PEM fuel cell operation with complete retention of its molecular structure.
catalyst and for NiII ions coordinated to lighter atoms as in [Ni(H2O)6]2+, indicating that about 2/3rd of the Ni complex remains in the NiP4 state (Table S2†). After 1 hour of operation for H2 oxidation at 0.25 V vs. RHE and in direct contact with 0.5 M aqueous H2SO4 electrolyte, the signal assigned to decomposed Ni complex disappears, leaving a pure NiP4 signature (Fig. 4, Fig. S10 and Table S2†).18 As the current density remains constant during the whole measurement (Fig. S4†), corresponding to 4000 turnovers achieved for each NiP4 species, we conclude that the decomposed Ni compound was washed off from the material during the electrochemical measurement. Additionally this experiment tells us that the bio-inspired NiP4 complex is the only species responsible for catalysis and that it does withstand extensive PEM fuel cell operation with complete retention of its molecular structure.
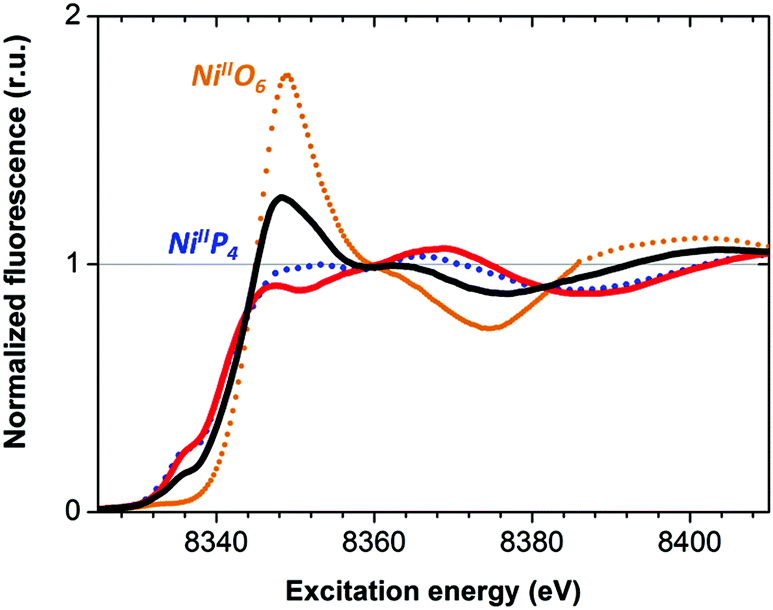 | ||
Fig. 4 X-ray absorption near edge structure spectra (Ni K edge) of the Ni–CNT material as prepared (black solid line), and after 1 h H2 oxidation (red solid line). Spectra of pristine 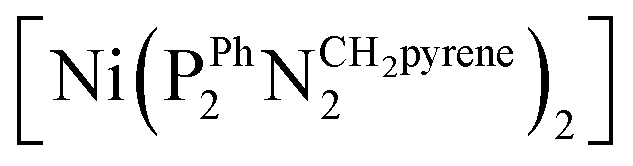 (blue dotted line) and of [Ni(OH2)6]2+ (orange dotted line) are shown for comparison. See Fig. S10† for a magnification of the K edge region. (blue dotted line) and of [Ni(OH2)6]2+ (orange dotted line) are shown for comparison. See Fig. S10† for a magnification of the K edge region. | ||
We have thus successfully demonstrated the potential of the bio-inspired chemistry approach for the development of novel fuel cells based exclusively on non-noble metals. Previously described earth-abundant catalytic HOR and ORR materials have been assessed either in half-cell measurements or in combination with a platinum-based counter electrode material. Here, we report the first example of an operational PEMFC using a bio-inspired Ni-based material at the anode and a Co-based material at the cathode. Importantly, the operation and feeding conditions used in our study correspond to the expected daily use of market-dedicated micro devices and the performances reported here compare well with those of the first generation of H2/O2 enzymatic fuel cells.19–21 Further improvements of the performances of the devices can be expected from optimization of the ink formulation and control of the tridimensional structure of the electrodes as previously demonstrated for H2/O222–25 or glucose/O226 biofuel cells.
Acknowledgements
This work was supported by ANR (EnzHyd project, ANR-08-PANH-008 and Labex program ARCANE, ANR-11-LABX-0003-01) and the FCH Joint Undertaking (Nano-Cat project, grant no. 325239). The XAS measurements were carried out at beamline KMC-1 of the BESSY synchrotron operated by the Helmholtz Zentrum Berlin (HZB); we thank M. Mertin and Dr F. Schäfers (both HZB) for their technical support and the Berlin cluster of excellence on Unifying Concepts in Catalysis (UniCat) for the financial support.References
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS PubMed.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- A. Morozan, P. Jegou, B. Jousselme and S. Palacin, Phys. Chem. Chem. Phys., 2011, 13, 21600–21607 RSC.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 CAS.
- S. Pylypenko, S. Mukherjee, T. S. Olson and P. Atanassov, Electrochim. Acta, 2008, 53, 7875–7883 CrossRef CAS PubMed.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Metaye, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371–1374 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- P. Chenevier, L. Mugherli, S. Darbe, L. Darchy, S. DiManno, P. D. Tran, F. Valentino, M. Iannello, A. Volbeda, C. Cavazza and V. Artero, C. R. Chim., 2013, 16, 491–516 CrossRef CAS PubMed.
- A. K. Jones, E. Sillery, S. P. J. Albracht and F. A. Armstrong, Chem. Commun., 2002, 866–867 RSC.
- T. Ueno, M. Ohashi, M. Kono, K. Kondo, A. Suzuki, T. Yamane and Y. Watanabe, Inorg. Chem., 2004, 43, 2852–2858 CrossRef CAS PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS PubMed.
- Y. Matsubara, E. Fujita, M. D. Doherty, J. T. Muckerman and C. Creutz, J. Am. Chem. Soc., 2012, 134, 15743–15757 CrossRef CAS PubMed.
- A. Morozan, P. Jegou, M. Pinault, S. Campidelli, B. Jousselme and S. Palacin, Chemsuschem, 2012, 5, 647–651 CrossRef CAS PubMed.
- The same behavior is observed if the material is simply equilibrated with the aqueous electrolyte during the measure of a few cyclic voltammograms (Fig. S10†) or if the experiment is carried out during 1 h under H2 evolution conditions (Fig. S11 and Table S2†). If the same measurement is carried out on the Ni–CNT material coated with a Nafion membrane, the XANES signal is unmodified even after 1 hour of operation for H2 evolution at –0.3 V vs. RHE (Fig. S11 and Table S2†).
- K. A. Vincent, J. A. Cracknell, O. Lenz, I. Zebger, B. Friedrich and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16951–16954 CrossRef CAS PubMed.
- K. A. Vincent, J. A. Cracknell, J. R. Clark, M. Ludwig, O. Lenz, B. Friedrich and F. A. Armstrong, Chem. Commun., 2006, 5033–5035 RSC.
- A. F. Wait, A. Parkin, G. M. Morley, L. dos Santos and F. A. Armstrong, J. Phys. Chem. C, 2010, 114, 12003–12009 CAS.
- L. Xu and F. A. Armstrong, Energy Environ. Sci., 2013, 6, 2166–2171 CAS.
- S. Krishnan and F. A. Armstrong, Chem. Sci., 2012, 3, 1015–1023 RSC.
- A. Ciaccafava, A. De Poulpiquet, V. Techer, M. T. Giudici-Orticoni, S. Tingry, C. Innocent and E. Lojou, Electrochem. Commun., 2012, 23, 25–28 CrossRef CAS PubMed.
- A. de Poulpiquet, H. Marques-Knopf, V. Wernert, M. T. Giudici-Orticoni, R. Gadiou and E. Lojou, Phys. Chem. Chem. Phys., 2014, 16, 1366–1378 RSC.
- A. Zebda, C. Gondran, A. Le Goff, M. Holzinger, P. Cinquin and S. Cosnier, Nat. Commun., 2011, 2, 370 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc03774j |
| ‡ These authors equally contributed to the work. |
| This journal is © The Royal Society of Chemistry 2015 |