
Pd-catalyzed Heck cyclization and in situ hydrocarboxylation or hydromethenylation via a hydrogen borrowing strategy†
Xianglei
Liu
and
Zhenhua
Gu
*
Department of Chemistry, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 20026, P. R. China. E-mail: zhgu@ustc.edu.cn
First published on 28th April 2015
Abstract
A Pd-catalyzed hydrocarboxylation of the in situ formed neopentylic alkenes by the use of “borrowed hydrogen” from Heck-type β-hydride elimination was reported. The advantage of this strategy is that no additional hydrogen sources or organometallic reagents were required. It was found that CHCl3 was not only used as a safe and convenient “CO” source but also proved superior in the linear/branched selectivity over carbon monoxide. Furthermore, the in situ formed neopentylic alkenes could be chemoselectively functionalized with other C–C unsaturated bonds being unchanged, even the less sterically hindered ones.
Introduction
The functionalization of unsaturated carbon–carbon bonds into carbonyl containing compounds are interesting and important transformations in organic synthesis,1 most notable representative achievements are including catalytic hydroformylation and hydroacylation of alkenes etc.2 Notwithstanding the great achievements of hydroformylation and hydroacylation, including their asymmetric versions, there are only limited methods for the preparation of carboxylic acids via direct hydrocarboxylation of alkenes. Alternatively, hydroformylation followed by oxidation suffered from the harsh oxidation conditions, which resulted in poor functional group tolerance (Scheme 1a). Ru-catalyzed intermolecular hydroesterification of alkenes was realized by Chang and co-workers, where the key point is the use of 2-pyridylmethyl formate as the reagent (Scheme 1b).3 Shi et al. developed a palladium-catalyzed hydrocarboxylation protocol of olefins in the presence of HCO2Ph and HCO2H.4 Recently, synthesis of carboxylic acids via catalytic fixation of CO2 has also been explored, which was applied to hydrocarboxylation of alkynes, diynes and enynes.5 In 2008 Rovis and co-workers described the first direct hydrocarboxylation of single alkenes with catalytic nickel and 1 atm of CO2 under mild conditions (Scheme 1c).6 Subsequently, Iwasawa, Hayashi and Thomas groups also developed efficient methods for hydrocarboxylation of dienes, allenes and alkenes via fixing CO2.7–9 However, it should be noted that in these reactions the addition of stoichiometric or excess of organozinc, organoaluminum reagents or silane is required as the reductant. Although great progresses have been achieved, the development of efficient, mild and chemoselective protocols for the synthesis of carboxylic acids via hydrocarboxylation of alkenes is still urgently needed. In particular, the reaction should avoid the use of ZnEt2, AlEt3 and RMgBr, etc., as additional reducing reagents.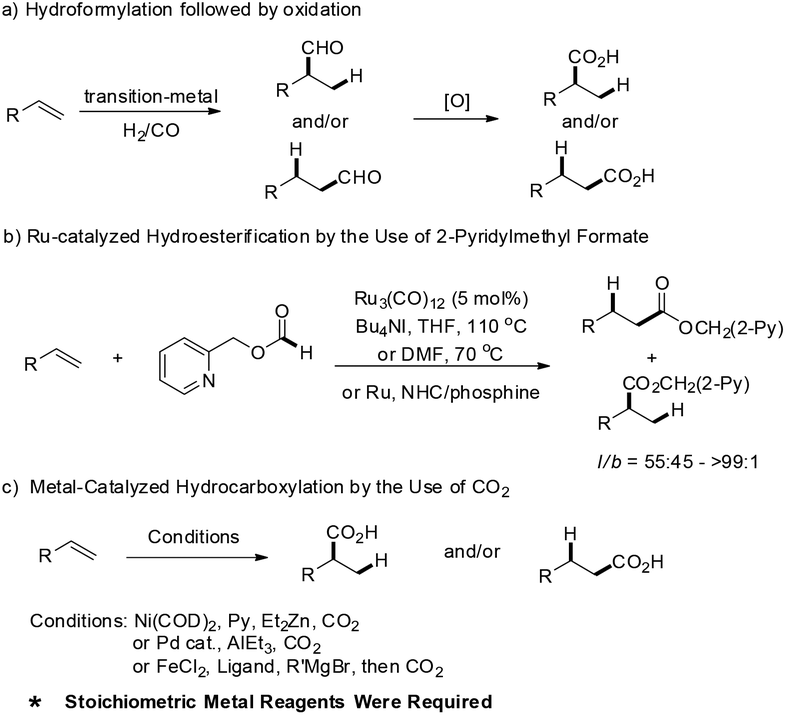 | ||
| Scheme 1 Representative synthesis of carboxylic acid via alkenes. | ||
Results and discussion
During the synthesis of the rhazinilam family of natural products we had difficulties with the functionalization of the newly formed neopentylic alkenes in the key intermediate A. Neither Ru-catalyzed alkene cross-metathesis nor hydrometallation (hydroboration or hydrozirconation) of the neopentylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond succeeded (Scheme 2), as a result an alternative strategy had to been chosen to complete the synthesis of rhazinal.10 The failure of hydrometallation of the neopentylic CC bond encouraged us to develop more efficient methods for hydrocarboxylation, especially for these sterically hindered alkenes. According to chemistry of Pd-catalyzed Heck reaction the H–Pd–X species were regarded as “waste”, which required bases to regenerate the catalytically active Pd(0) species by releasing the H–X byproduct. We reasoned that it might be possible to utilize the H–Pd–X species as hydrogen source via retro-hydropalladation of newly-formed CC double bonds, such as from III to IV (Scheme 3).11 Linear carboxylic acids would be formed after “CO” insertion in V, followed by reductive elimination. The advantages of this transformation are: (1) the bulky neopentylic alkenes could be chemoselectively hydrocarboxylated while other unsaturated C–C bonds, even less sterically hindered double bonds, could be tolerated; (2) the waste H–PdX could be utilized, and no additional organometallic reagents or hydrogen sources were needed, and as a result more functional groups could be tolerated. However, the challenges in this strategy should still be addressed: how to prevent the decomposition of palladium complex III releasing H–X under basic conditions;12 how to push intermediate IV to undergo “CO” insertion reaction while not β-hydride elimination.13 Herein, we report a palladium-catalyzed domino Heck cyclization, followed by an in situ hydrocarboxylation reaction via a hydrogen borrowing strategy for the synthesis of a variety of linear carboxylic acids.
C double bond succeeded (Scheme 2), as a result an alternative strategy had to been chosen to complete the synthesis of rhazinal.10 The failure of hydrometallation of the neopentylic CC bond encouraged us to develop more efficient methods for hydrocarboxylation, especially for these sterically hindered alkenes. According to chemistry of Pd-catalyzed Heck reaction the H–Pd–X species were regarded as “waste”, which required bases to regenerate the catalytically active Pd(0) species by releasing the H–X byproduct. We reasoned that it might be possible to utilize the H–Pd–X species as hydrogen source via retro-hydropalladation of newly-formed CC double bonds, such as from III to IV (Scheme 3).11 Linear carboxylic acids would be formed after “CO” insertion in V, followed by reductive elimination. The advantages of this transformation are: (1) the bulky neopentylic alkenes could be chemoselectively hydrocarboxylated while other unsaturated C–C bonds, even less sterically hindered double bonds, could be tolerated; (2) the waste H–PdX could be utilized, and no additional organometallic reagents or hydrogen sources were needed, and as a result more functional groups could be tolerated. However, the challenges in this strategy should still be addressed: how to prevent the decomposition of palladium complex III releasing H–X under basic conditions;12 how to push intermediate IV to undergo “CO” insertion reaction while not β-hydride elimination.13 Herein, we report a palladium-catalyzed domino Heck cyclization, followed by an in situ hydrocarboxylation reaction via a hydrogen borrowing strategy for the synthesis of a variety of linear carboxylic acids.
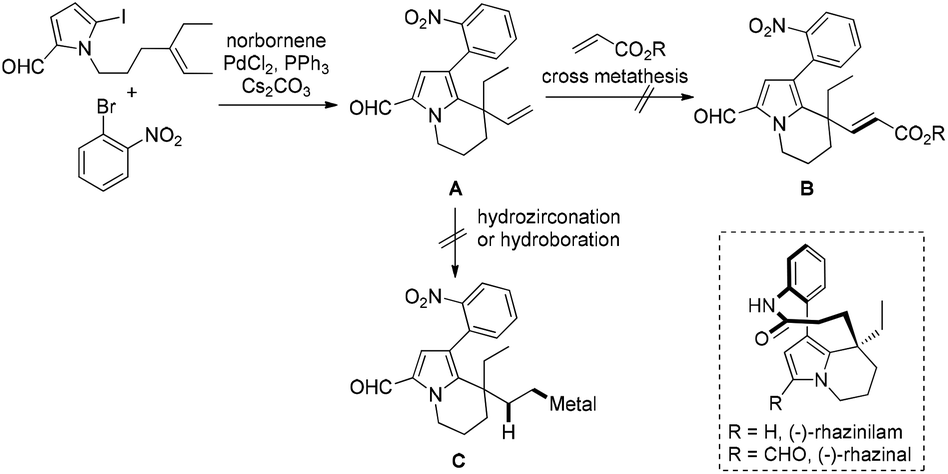 | ||
| Scheme 2 The failure for functionalization of neopentylic alkenes during the synthesis of rhazinilam family natural products. | ||
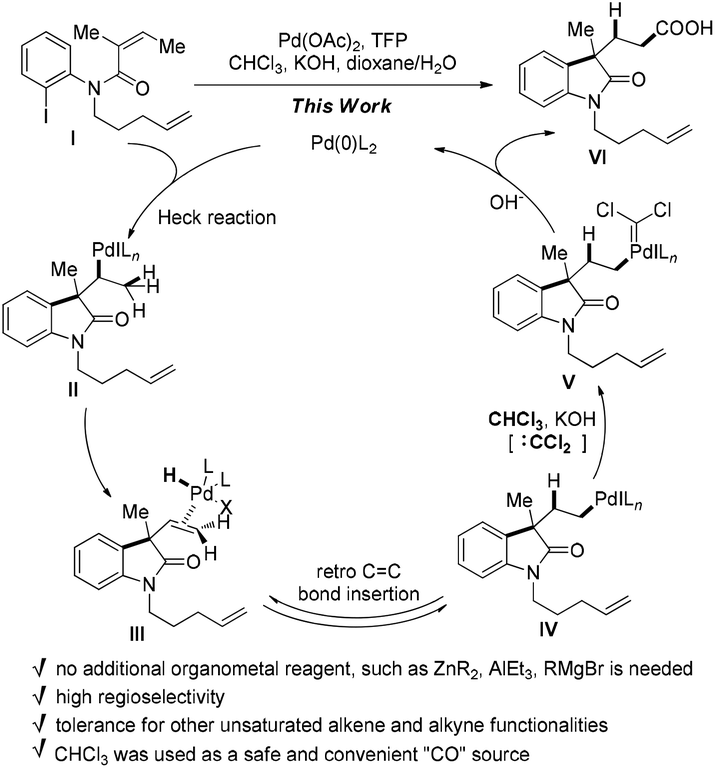 | ||
| Scheme 3 Hydrogen borrowing strategy for the hydrocarboxylation. | ||
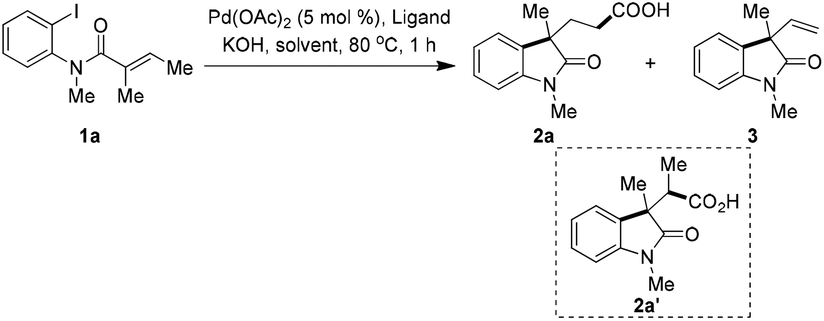
With this hypothesis in mind, compound 1a was chosen as the model substrate to optimize the reaction conditions. To our delight, under 1 atmosphere of CO gas, with Pd(PPh3)Cl2 as catalyst in DMF/H2O, 42% of desired product 2a was obtained,14 along with 14% of direct Heck-cyclization alkene 3, although the conversion was only about 60% (Table 1, entry 1). Increasing the pressure of CO gas (5 atm) significantly improved the conversion, however branched carboxylic acid 2a′ was formed in 32% yield as a single isomer and with no great improvement of the yield of 2a (entry 2). The reaction with new catalytic system Pd(OAc)2/tri(2-furyl)phosphine (TFP), 1 atm of CO in dioxane/H2O gave full conversion, however β-H elimination product 3 was formed predominantly and 2a was isolated in only 14% (entry 3). Chloroform, a useful reagent as a dichlorocarbene precursor to construct various dichlorocyclopropanes,15 was ever used as an alternative CO source for the synthesis of organic compounds16 or metalcarbonyl complexes.17 In our case in the presence of Pd(OAc)2/PPh3 and 4.0 equiv. of CHCl3, the reaction delivered 2a in 52% yield, and no branched product was detected (entry 4).18 Tri(2-furyl)phosphine was found to be superior over PPh3, and the corresponding reaction gave 79% yield of desired product (entry 5). Increasing the amount of CHCl3 to 8.0 equiv. did not further improve the isolated yield (entry 6). In the absence of H2O the reaction only gave a very low conversion in either THF or dioxane (entries 7 and 8). For comparison, by the use of HCO2Ph as “CO” source, the reaction only led to 90% of 3 in THF (entry 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | “CO” Source | Solvent | Yield/% | |
| 2a | 3 | ||||
| a The reaction was conducted in 0.20 mmol scale of 1a, 5 mol% of Pd(OAc)2, 15 mol% of phosphine at 80 °C for 1 h. b Pd(PPh3)2Cl2 was used, Et3N was used as base. c The conversion is 60%. d 32% of branched acid 2a′ was isolated. e No attempt for the isolation of 3. f 1.2 equiv. of HCO2Ph was used, Cs2CO3 was used as base. | |||||
| 1b,c | — | CO (1 atm) | DMF/H2O | 42 | 14 |
| 2b,d | — | CO (5 atm) | DMF/H2O | 47 | 16 |
| 3 | TFP | CO (1 atm) | Dioxane/H2O | 14 | 76 |
| 4 | PPh3 | CHCl3 (4 eq.) | Dioxane/H2O | 52 | |
| 5 | TFP | CHCl3 (4 eq.) | Dioxane/H2O | 79 | 17 |
| 6 | TFP | CHCl3 (8 eq.) | Dioxane/H2O | 80 | |
| 7 | TFP | CHCl3 (4 eq.) | THF | <5 | |
| 8 | TFP | CHCl3 (4 eq.) | Dioxane | 10 | 27 |
| 9 | TFP | HCO2Phf | THF | — | 90 |
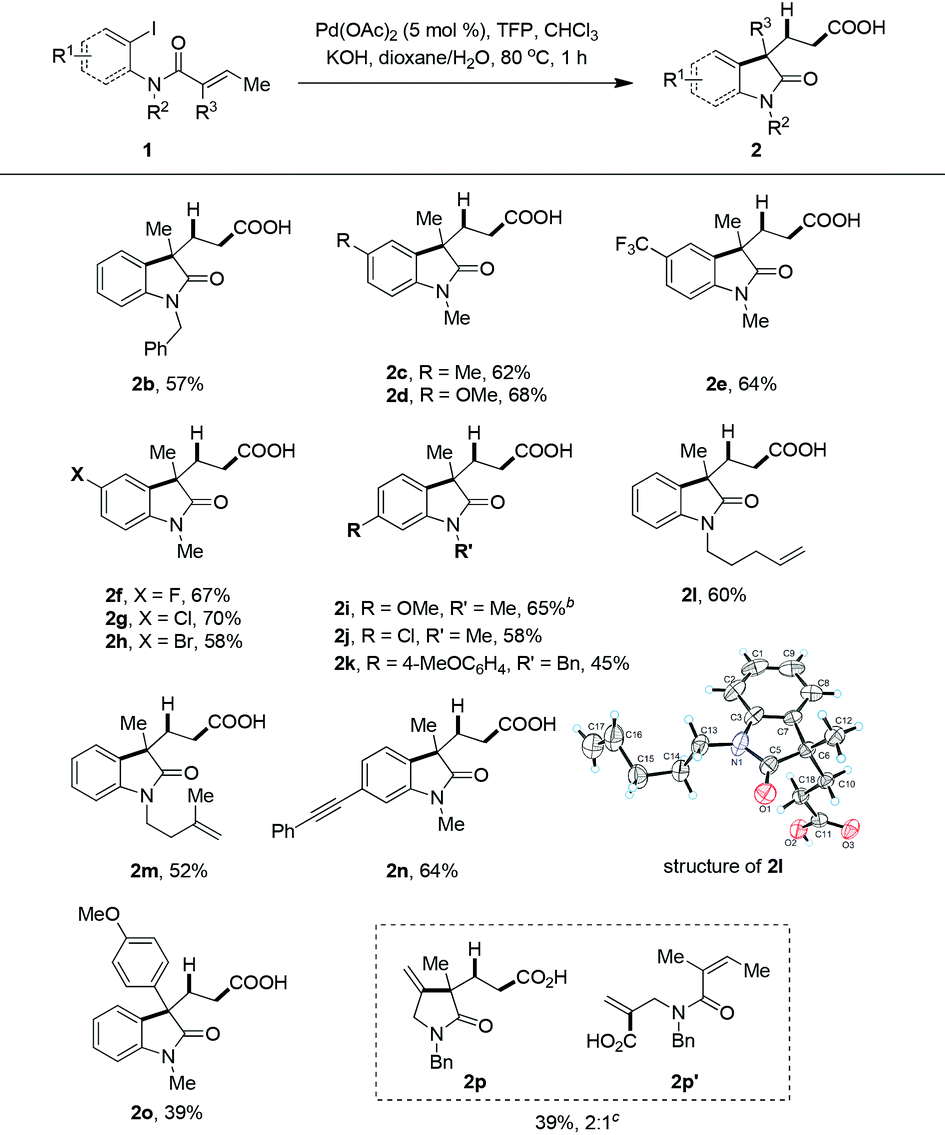
The generality of this newly developed hydrocarboxylative method was applied to different substituted 2-iodoacrylamides (Table 2). The reaction of N-benzylated amide gave a slightly lower yield (2b). Both electron-donating or electron-withdrawing groups at the 4-position of the phenyl ring did not affect the reaction, and moderate yields could be achieved (2c–e). Some carbon–halogen bonds were tolerated (2f–h), and notably the C–Br bond, which could possibly be further functionalized by transition metal-catalyzed coupling reactions. Substrates with a substituent at the 5-position of the phenyl ring was also shown to be compatible (2i–k). The most striking point of this protocol is that other unsaturated C–C bonds were compatible, even those with less sterically hindered unsaturated C–C bonds, which was usually incompatible in previously reported hydroformylation and hydrocarboxylation methods. For example, as we expected compounds 2l and 2m were isolated in 60% and 52% yields, with mono- or 1,1-disubstituted CC bonds being unchanged. The structure of 2l was further established by single crystal X-ray analysis,19 and it clearly showed that the terminal mono-substituted alkene was unchanged. Carbon–carbon triple bond was also tolerated, and compound 2n were formed smoothly in 64% isolated yield. When R3 is an aryl substituent, such as a p-methoxyphenyl group, the yield dropped dramatically with the formation of a considerable amount of β-hydride elimination product (2o). The reaction afforded an inseparable mixture of cyclized product 2p and acyclic compound 2p′ in a total yield of 39% when vinyl iodide was used as starting material. These two compounds could be separated and characterized after being converted to the corresponding amides.
| a The reaction was conducted on a 0.20 mmol scale of 1, 4.0 equiv. of CHCl3, 8.0 equiv. of KOH, 5 mol% of Pd(OAc)2, 15 mol% of TFP in dioxane/H2O at 80 °C for 1 h. b The reaction time is 1.5 h. c It was isolated as an inseparable mixture, and 2p and 2p′ were separated and characterized by their corresponding amides with aniline. |
|---|
|
A control experiment was conducted, in which the reaction was performed under standard conditions in the presence of both 1g and 3 (Scheme 4). Besides the formation 57% of 2g, there was 9% of carboxylic acid 2a being detected, which was supposed to be the product from the hydrocarboxylation reaction of 3. These results clearly indicated that the palladium–hydride species partially released from the alkene before hydropalladation. Also intermediate IV in Scheme 3 might be stabilized by the coordination of oxygen atom of the amide group.
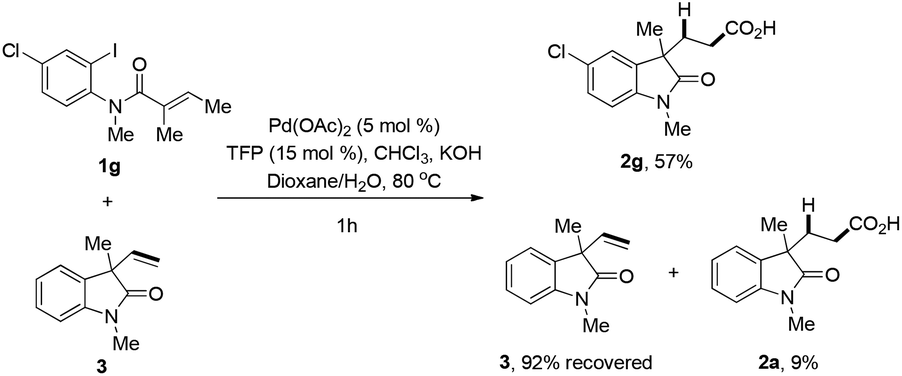 | ||
| Scheme 4 Cross reaction. | ||
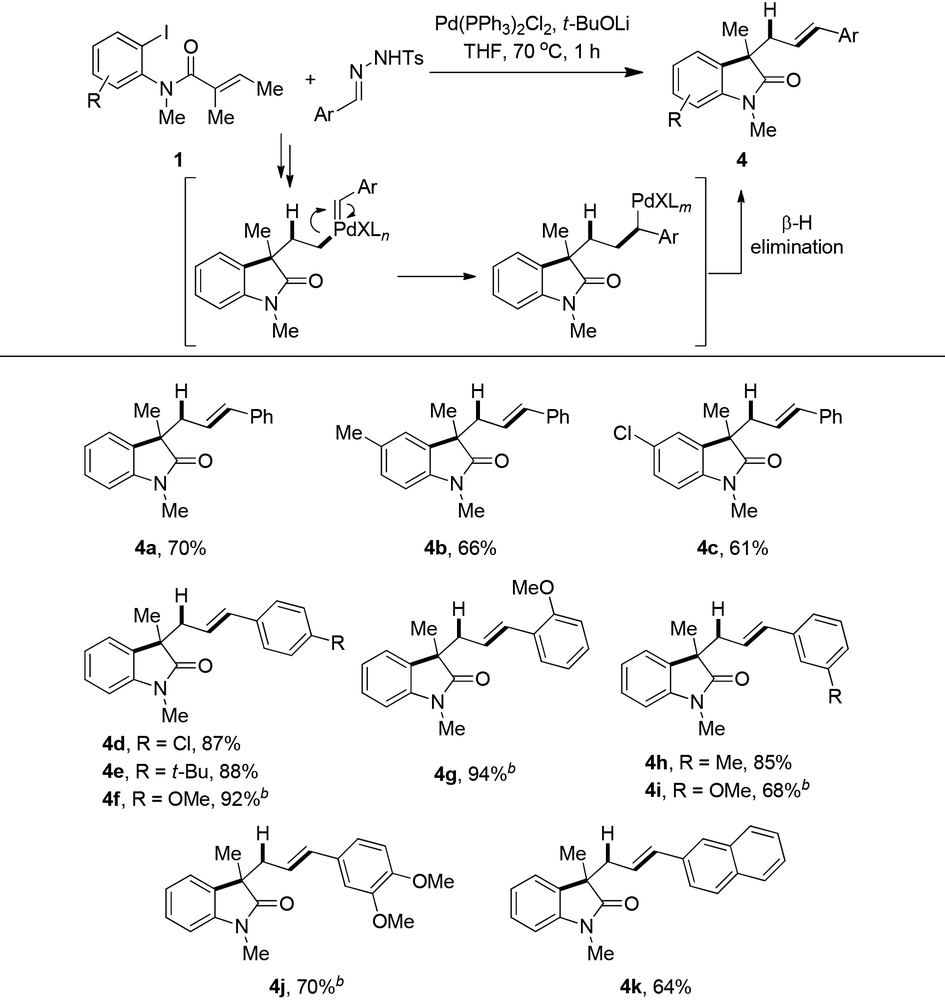
Considering a similar reactivity of palladium–carbeniod species, this hydrogen borrowing strategy was also applied to aryl carbenes by the use of N-tosyl hydrazones20 as precursors instead of chloroform (Table 3). With t-BuOLi as optimized base in THF the reaction could smoothly give various styrene-type products via a Pd–carbene migration–insertion process. Compound 4a was obtained in 70% isolated yield. A substitution at the para position of the 2-iodoaniline moiety slightly decreased the yields, such as 4b and 4c being isolated in 66% and 61% yields, respectively. Different functionalized aryl hydrazones were also suitable substrates. Para-, ortho- and meta-, as well as disubstituted phenyl N-tosyl hydrazones smoothly coupled with 1a to gave the corresponding products in good to excellent yields (4d–j). It should be noted that elevating the reaction temperature was necessary when electron-rich hydrazones were used. The reaction of β-naphthyl aldehyde derived hydrazone with 1a proceeded uneventfully to deliver 4k in 64% yield.
Conclusions
In conclusion, we report a domino palladium-catalyzed Heck cyclization and hydrocarboxylation reaction, where hydrogen was borrowed from a methyl group via β-hydride elimination. It should be noted that no additional reducing reagents or organometallic reagents were required. It is also striking that this hydrogen borrowing strategy could chemoselectively functionalize the in situ formed sterically hindered neopentylic CC bonds, while other unsaturated C–C bonds were unchanged, even the less sterically hindered ones. Through screening of the “CO” source, CHCl3 showed superiority not only in laboratory use but also in the linear/branched selectivity.
Acknowledgements
This work was financially supported by the ‘973’ project from the MOST of China (2015CB856600), NSFC (21272221, 21472179), the Recruitment Program of Global Experts, and the Fundamental Research Funds for the Central Universities (WK 2060190028, 2060190026 and 3430000001). Funding for the Author of National Excellent Doctoral Dissertation (201423) is also acknowledged.Notes and references
- For diesterification of styrenes, see: (a) M. Dai, C. Wang, G. Dong, J. Xiang, T. Luo, B. Liang, J. Chen and Z. Yang, Eur. J. Org. Chem., 2003, 4346–4348 CrossRef CAS PubMed; (b) B. Liang, J. Liu, Y.-X. Gao, K. Wongkhan, D.-X. Shu, Y. Lan, A. Li, A. S. Batsanov, J. A. H. Howard, T. B. Marder, J.-H. Chen and Z. Yang, Organometallics, 2007, 26, 4756–4762 CrossRef CAS.
- For reviews, see: (a) F. Agbossou, J.-F. Carpentier and A. Mortreux, Chem. Rev., 1995, 95, 2485–2506 CrossRef CAS; (b) P. Eilbracht, L. Bärfacker, C. Buss, C. Hollmann, B. E. Kitsos-Rzychon, C. L. Kranemann, T. Rische, R. Roggenbuck and A. Schmidt, Chem. Rev., 1999, 99, 3329–3366 CrossRef CAS PubMed; (c) B. Breit, Acc. Chem. Res., 2003, 36, 264–275 CrossRef CAS PubMed; (d) J. Klosin and C. R. Landis, Acc. Chem. Res., 2007, 40, 1251–1259 CrossRef CAS PubMed; (e) M. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS PubMed; (f) X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041–1051 CrossRef CAS PubMed.
- (a) S. Ko, Y. Na and S. Chang, J. Am. Chem. Soc., 2002, 124, 750–751 CrossRef CAS PubMed; (b) E. J. Park, J. M. Lee, H. Han and S. Chang, Org. Lett., 2006, 8, 4355–4358 CrossRef CAS PubMed; (c) B. Li, S. Lee, K. Shin and S. Chang, Org. Lett., 2014, 16, 2010–2013 CrossRef CAS PubMed.
- Y. Wang, W. Ren, J. Li, H. Wang and Y. Shi, Org. Lett., 2014, 16, 5960–5963 CrossRef CAS PubMed.
- (a) Y. Zhang and S. N. Riduan, Angew. Chem., Int. Ed., 2011, 50, 6210–6212 CrossRef CAS PubMed; (b) B. Miao and S. Ma, Org. Chem. Front., 2015, 2, 65–68 RSC.
- M. C. Williams, J. B. Johnson and T. Rovis, J. Am. Chem. Soc., 2008, 130, 14936–14937 CrossRef PubMed.
- J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2008, 130, 15254–15255 CrossRef CAS PubMed.
- (a) T. Fujihara, T. Xu, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2011, 50, 523 CrossRef CAS PubMed; (b) S. Li, W. Yuan and S. Ma, Angew. Chem., Int. Ed., 2011, 50, 2578–2582 CrossRef CAS PubMed.
- (a) E. Shirakawa, D. Ikeda, S. Masui, M. Yoshida and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 272–279 CrossRef CAS PubMed; (b) M. D. Greenhalgh and S. P. Thomas, J. Am. Chem. Soc., 2012, 134, 11900–11903 CrossRef CAS PubMed.
- (a) X. Sui, R. Zhu, G. Li, X. Ma and Z. Gu, J. Am. Chem. Soc., 2013, 135, 9318–9321 CrossRef CAS PubMed; (b) X. Sui, R. Zhu and Z. Gu, Synlett, 2013, 2023–2031 CAS.
- For some typical reductive couplings reactions, see: (a) H. Iida and M. J. Krische, Top. Curr. Chem., 2007, 279, 77–104 CrossRef CAS; (b) H.-Y. Jang, R. R. Huddleston and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 15156–15157 CrossRef CAS PubMed; (c) R. R. Huddleston, H.-Y. Jang and M. J. Krische, J. Am. Chem. Soc., 2003, 125, 11488–11489 CrossRef CAS PubMed; (d) S. B. Han, H. Han and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 1760–1761 CrossRef CAS PubMed; (e) J. R. Zbieg, E. Yamahuchi, E. L. Mclnturff and M. J. Krische, Science, 2012, 336, 324–327 CrossRef CAS PubMed.
- For some examples of retro-insertion of C–C double bonds to HPdX, see: (a) S. Ma and Z. Yu, Angew. Chem., Int. Ed., 2003, 42, 1955–1957 CrossRef CAS PubMed; (b) E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455–1458 CrossRef CAS PubMed; (c) T. Kochi, T. Hamasaki, Y. Aoyama, J. Kawasaki and F. Kakiuchi, J. Am. Chem. Soc., 2012, 134, 16544–16547 CrossRef CAS PubMed; (d) T. S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830–6833 CrossRef CAS PubMed; (e) T. S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340–344 CrossRef CAS PubMed.
- For Pd-catalyzed cross-coupling of alkyl halides bearing β-hydride, see: (a) M. R. Netherton, C. Dai, K. Neuschütz and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 10099–10100 CrossRef CAS; (b) J. H. Kirchhoff, M. R. Netherton, I. D. Hills and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 13662–14663 CrossRef CAS PubMed; (c) K. Menzel and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 3718–3719 CrossRef CAS PubMed; (d) J.-Y. Lee and G. C. Fu, J. Am. Chem. Soc., 2002, 125, 5616–5617 CrossRef PubMed; (e) J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 12527–12530 CrossRef CAS PubMed; (f) M. Eckhardt and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 13642–13643 CrossRef CAS PubMed.
- H. J. Lim and T. V. RajanBabu, Org. Lett., 2011, 13, 6596–6599 CrossRef CAS PubMed.
- (a) G. L. Ciamician and M. Dennsted, Chem. Ber., 1881, 14, 1153–1163 CrossRef PubMed; (b) W. O. Kermack, W. H. Perkin and R. Robinson, J. Chem. Soc., 1922, 121, 1872–1896 RSC.
- (a) V. V. Grushin and H. Alper, Organometallics, 1993, 12, 3846–3850 CrossRef CAS; (b) I. A. de Carcer Garcia, A. M. Sevim, A. de la Escosura and T. Torres, Org. Biomol. Chem., 2013, 11, 2237–2240 RSC. For an early study on the mechanism of generation CO from chloroform, see: J. Hine, J. Am. Chem. Soc., 1950, 72, 2438–2445 CrossRef CAS.
- (a) G. R. Clark, K. Mareden, W. R. Roper and L. J. Wright, J. Am. Chem. Soc., 1980, 102, 1206–1207 CrossRef CAS; (b) W. R. Roper and A. H. Wright, J. Organomet. Chem., 1982, 233, C59–C63 CrossRef CAS; (c) G. R. Clark, W. R. Roper and A. H. Wright, J. Organomet. Chem., 1982, 236, C7–C10 CrossRef CAS; (d) E. B. Boyar and S. D. Robinson, J. Chem. Soc., Dalton Trans., 1985, 2113–2119 RSC; (e) W. R. Roper, J. Organomet. Chem., 1986, 300, 167–190 CrossRef CAS; (f) J. H. Park, J. H. Koh and J. Park, Organometallics, 2001, 20, 1892–1894 CrossRef CAS; (g) C. S. Chin, M. Kim and H. Lee, Organometallics, 2002, 21, 1679–1683 CrossRef CAS; (h) D. Solé, L. Vallverdú, X. Solans, M. Font-Bardia and J. Bonjoch, Organometallics, 2004, 23, 1438–1447 CrossRef.
- For other reagents worked as carbon monoxide source, see a review: T. Morimoto and K. Kakiuchi, Angew. Chem., Int. Ed., 2004, 43, 5580–5588 CrossRef CAS PubMed . For other typical results, see: (a) S. Cacchi, G. Fabrizi and A. Goggiamani, Org. Lett., 2003, 5, 4269–4272 CrossRef CAS PubMed; (b) K. Hosoi, K. Nozaki and T. Hiyama, Org. Lett., 2002, 4, 2849–2851 CrossRef CAS PubMed; (c) Y. Wan, M. Alterman, M. Larhed and A. Hallberg, J. Org. Chem., 2002, 67, 6232–6235 CrossRef CAS PubMed; (d) T. Schareina, A. Zapf, A. Cotté, M. Gotta and M. Beller, Adv. Synth. Catal., 2010, 352, 1205–1209 CrossRef CAS PubMed; (e) D. N. Sawant, Y. S. Wagh, K. D. Bhatte and B. M. Bhanage, J. Org. Chem., 2011, 76, 5489–5494 CrossRef CAS PubMed; (f) P. Hermange, A. T. Lindhardt, R. H. Taaning, K. Bjerglund, D. Lupp and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 6061–6071 CrossRef CAS PubMed; (g) P. Hermange, T. M. Gøgsig, A. T. Lindhardt, R. H. Taaning and T. Skrydstrup, Org. Lett., 2011, 13, 2444–2447 CrossRef CAS PubMed; (h) M. L. N. Rao, V. Venkatesh and P. Dasgupta, Tetrahedron Lett., 2010, 51, 4975–4980 CrossRef CAS PubMed; (i) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed; (j) Z. Lian, S. D. Friis and T. Skrydstrup, Chem. Commun., 2015, 51, 3600–3603 RSC.
- CCDC 1047496. For details see the ESI.†.
- For some reviews, see: (a) Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577–6605 CrossRef CAS PubMed; (b) N. M. G. Franssen, A. J. C. Walters, J. N. H. Reek and B. de Bruin, Catal. Sci. Technol., 2011, 1, 153–165 RSC; (c) J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 7486–7500 CrossRef CAS PubMed; (d) Z. Zhang, Y. Zhang and J. Wang, ACS Catal., 2011, 1, 1621–1630 CrossRef CAS; (e) Y. Zhang and J. Wang, Top. Curr. Chem., 2012, 327, 239–269 CrossRef CAS; (f) Y. Zhang and J. Wang, Eur. J. Org. Chem., 2011, 1015–1026 CrossRef PubMed; (g) Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560–572 RSC; (h) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236–247 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1047496. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00091b |
| This journal is © the Partner Organisations 2015 |