
Au(I) π-bis(tert-butyldimethylsilyl)acetylene triphenylphosphine complex, an effective pre-catalyst for Au(I)-catalyzed reactions†
Dapeng
Zhu
,
Xin
Cao
and
Biao
Yu
*
State Key Laboratory of Bio-organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: byu@mail.sioc.ac.cn
First published on 11th February 2015
Abstract
A linear η2-alkyne π complex of [Ph3PAu]+, namely Au(I) π-bis(tert-butyldimethylsilyl)acetylene triphenylphosphine complex (1), was prepared and characterized. The new complex is found to be shelf-stable and effective for a range of the Au(I)-catalyzed alkyne π activation reactions, including a glycosylation reaction with glycosyl ortho-alkynylbenzoates as donors.
Introduction
The success of homogeneous gold catalysis with Au(I) complexes, such as [(L)Au][SbF6] (L = R3P, NHC, etc.), has been testified by numerous alkyne π activation towards nucleophilic attack in assembling structurally complex molecules (Fig. 1A).1 Among these, we have developed an effective glycosylation protocol employing glycosyl ortho-alkynylbenzoates as donors (Fig. 1C);2a–d the generality and versatility of this method has been demonstrated in the synthesis of complex oligosaccharides and glycoconjugates.2 Nevertheless, the loading of the cationic Au(I) complex in these Au(I)-catalyzed reactions is usually ≥10 mol%, due partly to loss of the active Au(I) species via disproportionation.3 In only a few gold-catalyzed reactions high turnover numbers have been achieved,4 but for the important class of glycosylation reactions such systems have so far not been developed. In this regard, new Au(I) complexes that own better a reactivity-stability balance profile are highly desirable for practical use in the Au(I)-catalyzed reactions.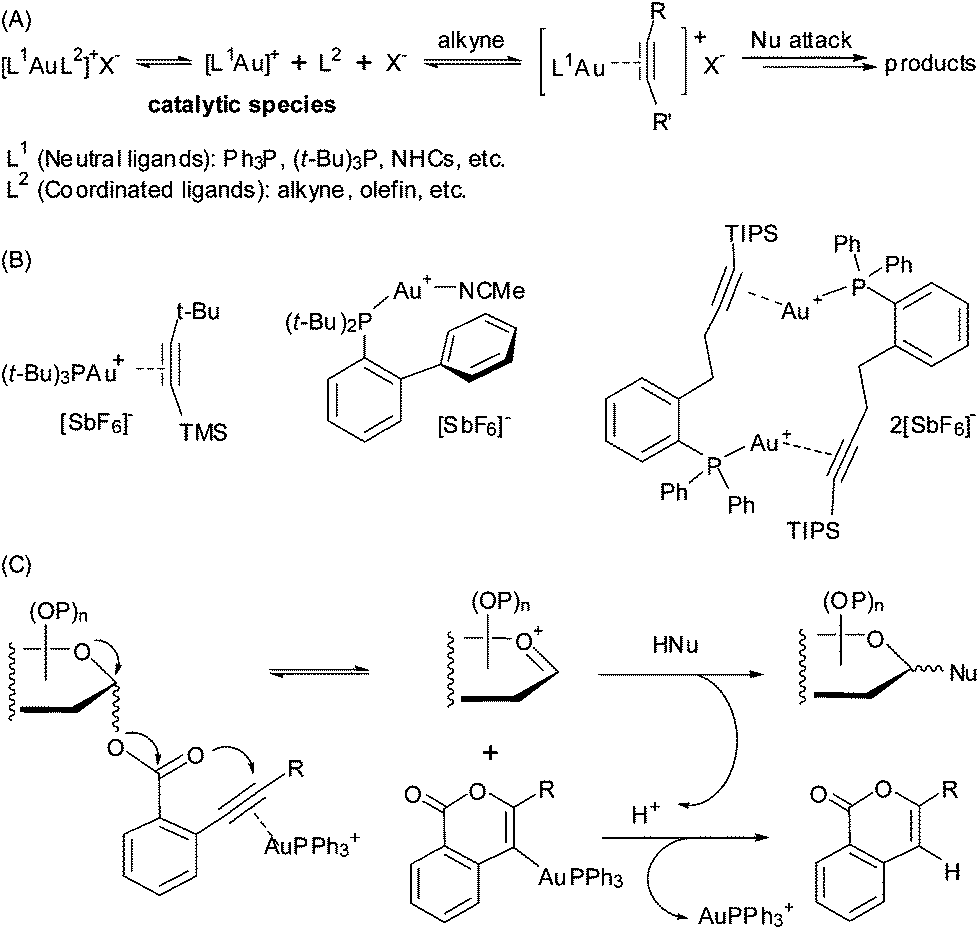 | ||
| Fig. 1 (A) Au(I)-catalyzed alkyne π activation transformations. (B) Representative [R3PAu(η2-alkyne)][SbF6] pattern complexes. (C) The gold(I)-catalyzed glycosidation of glycosyl ortho-alkynylbenzoates. | ||
Among the reported Au(I) coordination complexes,1 the [(R3P)Au(η2-alkyne)][SbF6] pattern (R = alkyl or aryl group) has been suggested as a compelling choice based on several notable merits.5 The formation of a Au(I) π-[η2-alkyne] coordination bond could give improved stability to the designed pre-catalysts, for example [(t-Bu3P)Au(η2-alkyne)][SbF6] (alkyne = MeC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-t-Bu or Me3SiCC-t-Bu) (Fig. 1B).5 In addition, this reversible coordination mode could regulate the release of the active catalytic Au(I)+ species, making the promoting activity of the catalyst in different reactions adjustable. While [Ph3PAu]+ is one of the most applied gold(I) species for alkyne π activation in Au(I)-catalyzed reactions,1a–c,5,6 to the best of our knowledge, a linear [Ph3PAu]+ catalyst in this coordination pattern has not yet been prepared and fully characterized with catalytic activity. Herein, we report a shelf-stable linear η2-alkyne π complex [Ph3PAu]+ species, namely Au(I) π-bis(tert-butyldimethylsilyl)acetylene triphenylphosphine, and its catalytic activity in the glycosylation reaction and a few other Au(I)-catalyzed reactions.
C-t-Bu or Me3SiCC-t-Bu) (Fig. 1B).5 In addition, this reversible coordination mode could regulate the release of the active catalytic Au(I)+ species, making the promoting activity of the catalyst in different reactions adjustable. While [Ph3PAu]+ is one of the most applied gold(I) species for alkyne π activation in Au(I)-catalyzed reactions,1a–c,5,6 to the best of our knowledge, a linear [Ph3PAu]+ catalyst in this coordination pattern has not yet been prepared and fully characterized with catalytic activity. Herein, we report a shelf-stable linear η2-alkyne π complex [Ph3PAu]+ species, namely Au(I) π-bis(tert-butyldimethylsilyl)acetylene triphenylphosphine, and its catalytic activity in the glycosylation reaction and a few other Au(I)-catalyzed reactions.
Results and discussion
Preparation of complex 1 and structural characterization
Given the alkyne ligand is activated once coordinated to Au(I) and could thus be readily attacked by nucleophiles, a sterically hindered alkyne is required in preparation of such a stable complex. We first tried to employ TMS-CC-TMS (2) as the ligand,7 however, the expected complex 3 made from PPh3AuCl/AgSbF6 (4) was found instable as indicated by the nonuniform 31P signals of the 31P NMR spectra at different temperatures. Thus, acetylene bearing bulky TBS groups, i.e., TBS-CC-TBS (5),8 was used as ligand. Upon addition of PPh3AuCl/AgSbF6 (4) into a solution of bis(tert-butyldimethylsilyl)acetylene (5) in dry CH2Cl2 at −70 °C and gradually warming the mixture up to rt, a sharp singlet at 39.0 ppm on 31P NMR showed up, which indicated clearly the formation of a single complex. After removal of the precipitated AgCl, concentration of the filtrate, and recrystallization of the resulting residue from CH2Cl2 and hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) at 0 °C, the desired Au(I) π-bis(tert-butyldimethylsilyl) acetylene triphenyl phosphine complex (1) was obtained in 87% yield as colourless crystals, which was stable on a shelf for at least half a year (Scheme 1).
:5) at 0 °C, the desired Au(I) π-bis(tert-butyldimethylsilyl) acetylene triphenyl phosphine complex (1) was obtained in 87% yield as colourless crystals, which was stable on a shelf for at least half a year (Scheme 1).
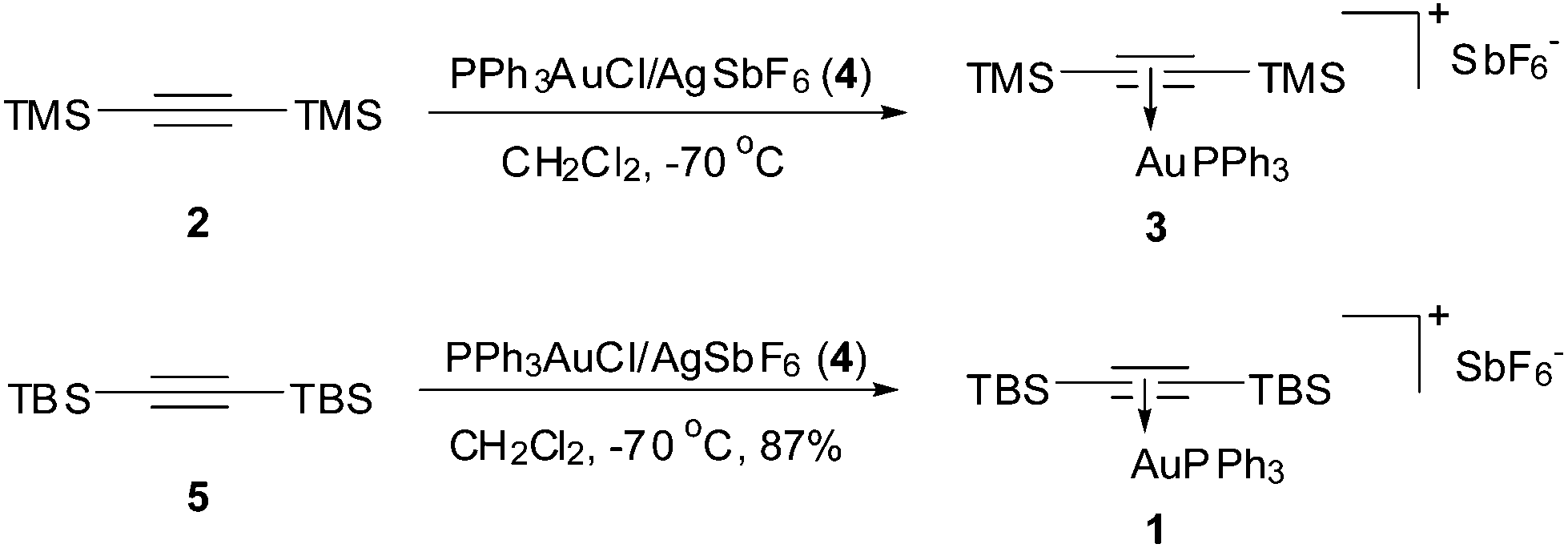 | ||
| Scheme 1 Preparation of Au(I) π-[η2-alkyne] complexes 3 and 1. | ||
The alkynyl carbon signal in complex 1 shifted downfield to 122.8 ppm and turned much wider than those of the free acetylene at 112.9 ppm on 13C NMR spectra. The overlapped signal of the two sp-hybridized carbon atoms demonstrated the existence of an symmetrical Au-[η2-acetylene] coordination, but not asymmetric bonding, i.e., η2→η1 slippage along the alkyne. The ligand's Raman spectra also showed big differences after coordination with an [AuPPh3]+ species, in which the νCC band signal shifted toward the red end at 1986 cm−1 relative to the free ligand peak at 2103 cm−1, which demonstrated the occurrence of the new π coordination.
The X-ray structure of 1 (CCDC 1043329) (Fig. 2) showed an ion-separated complex with perfect symmetrical coordination of the TBS-CC-TBS unit to the gold(I) centre intuitively. The C1C2 bond distance of 1.204(7) Å is identical to a typical CC bond length of 1.202(5) Å within the limit of error.5b Because of the coordination effect of Au(I) as well as the steric hindrance from the PPh3 group, the two TBS groups bend significantly to the back of the acetylene, leading to the Si1–C1–C2 bond angle of 169.3(4)°, so that the CC bond is greatly sheltered by the t-butylsilyl groups from backside of the molecule. A search of the Cambridge Structural Database9 reveals that this titled complex is the first example of a linear [Ph3PAu]+ η2-alkyne π complex structurally identified by X-ray diffraction.5
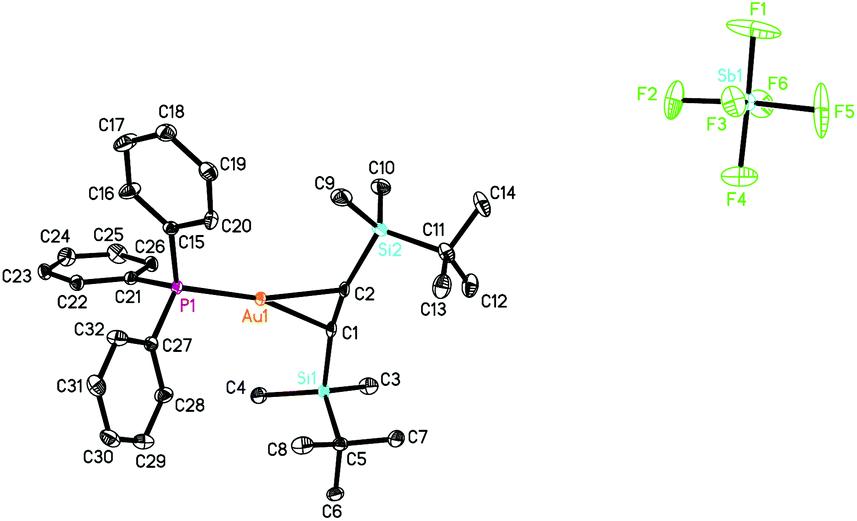 | ||
| Fig. 2 The X-ray structure of complex 1 (CCDC 1043329). Thermal ellipsoids are at 30% probability; all hydrogen atoms and the solvent of crystallization have been omitted for clarity. Key bond lengths (Å) and angles (°): Au(1)–C(1) 2.226(4), Au(1)–P(1) 2.260(1), C(1)–C(2) 1.204(7); C(15)–P(1)–Au(1) 112.9(1), P(1)–Au(1)–C(1) 164.7(1), Au(1)–C(1)–Si(1) 116.6(2), Si(1)–C(1)–C(2) 169.3(4). | ||
Glycosylation reactions catalyzed by complex 1
With the shelf-stable Au(I) complex 1 in hand, we investigated its catalytic activity in a few representative glycosylation reactions (Scheme 2 and Table 1). Using 2,3,4,6-tetra-O-benzoyl-D-glucopyranosyl ortho-cyclopropylethynylbenzoate (7)2b as donor and 1,2;3,4-di-O-isopropylidene-D-galactoside (10) as acceptor, the condensation in the presence of complex 1 (0.1 eq., CH2Cl2, 5 Å molecular sieves (MS), RT) proceeded smoothly, providing the coupled β-disaccharide 14 in nearly quantitative yield (97%; entry 1) within 10 h. In comparison, the completion of a similar reaction with Ph3PAuOTf (6) as the catalyst required 3 h (entry 2). This phenomenon could be rationalized assuming the reversible alkyne→[Ph3PAu]+ coordination was in action, resulting in a lower concentration of the catalytically active species [Ph3PAu]+ (than in Ph3PAuOTf) which reduces the rate.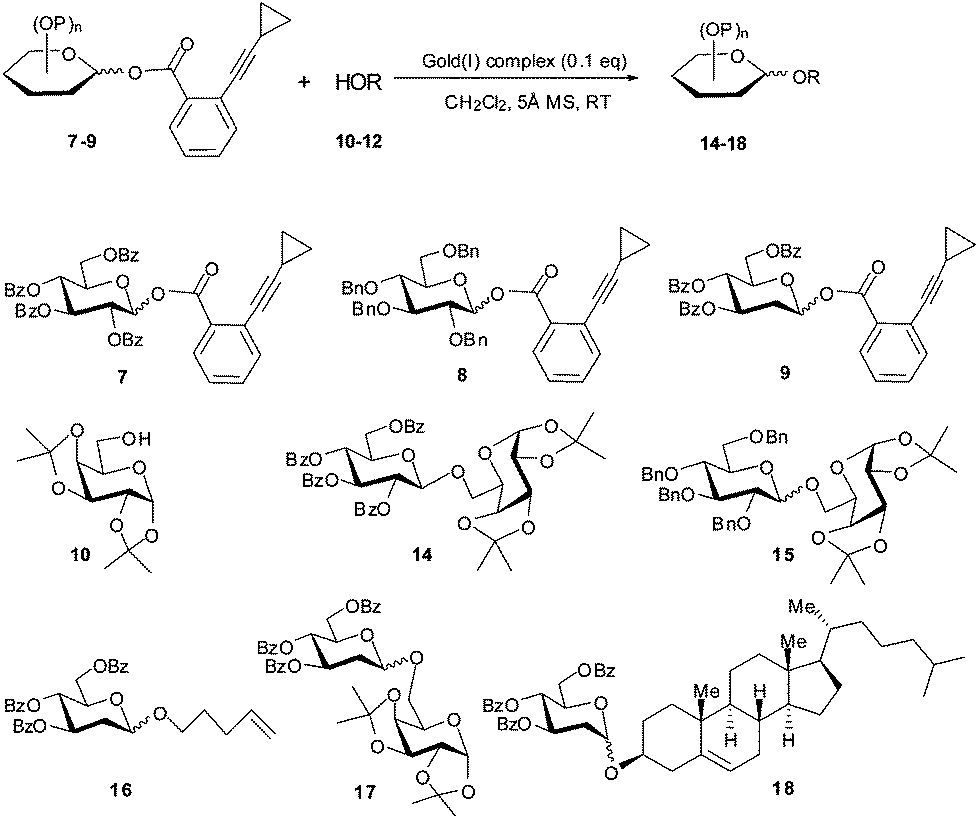 | ||
| Scheme 2 The Au(I)-catalyzed glycosylation reactions. | ||
| Entry | Au(I) | Donor | Acceptor | Product | Yielda (α/β ratio)b |
|---|---|---|---|---|---|
| a Isolated yields. b The α/β ratio was determined by 1H NMR spectroscopic measurements. | |||||
| 1 | 1 | 7 | 10 | 14 | 97% (β only) |
| 2 | 6 | 7 | 10 | 14 | 99% (β only) |
| 3 | 1 | 8 | 10 | 15 | 92% (α/β = 1.0:1) |
| 4 | 6 | 8 | 10 | 15 | 99% (α/β = 1.2:1) |
| 5 | 1 | 9 | 11 | 16 | 94% (α/β = 6.8:1) |
| 6 | 13 | 9 | 11 | 16 | 95% (α/β = 6.3:1) |
| 7 | 1 | 9 | 10 | 17 | 96% (α/β = 7.4:1) |
| 8 | 1 | 9 | 12 | 18 | 91% (α/β = 13.0:1) |
We then explored the catalytic activity of complex 1 in the glycosidation of armed donors, which are more active than those with a participating acyl group at 2-OH (such as 7). Not surprisingly, coupling of sugar alcohol 10 with 2,3,4,6-tetra-O-benzyl-D-glucopyranosyl cyclopropylethynylbenzoate (8) in the presence of complex 1 (0.1 eq., CH2Cl2, 5 Å MS, RT) completed within 10 h, providing the coupled glycoside 15 in 92% yield with no stereoselectivity (entry 3).10 These results are comparable to those obtained with Ph3PAuOTf (6) as the catalyst (entry 4). With 3,4,6-tri-O-benzoyl-2-deoxy-D-glucopyranosyl cyclopropylethynyl benzoate 9 as donor,11,12 the glycosylation of sugar alcohol 10 in the presence of complex 1 led to the coupled disaccharide 17 in excellent yield in favour of the thermodynamically more stable α anomer (α/β = 7.4/1; entry 7). Similar coupling of 9 with the simple alcohol pent-4-en-1-ol (11) provided the coupled glycoside 16 in high yield and α selectivity (entry 5). In comparison, similar results were obtained with PPh3AuNTf2 (13) as the catalyst (entry 6). Again, coupling of the 2-deoxy-sugar donor 9 with cholesterol (12) afforded the coupled glycoside 18 in high yield (91%) and excellent α selectivity (α/β = 13:1; entry 8).
Other reactions catalyzed by complex 1
To further demonstrate the catalytic profile of the Au(I) π-bis(tert-butyldimethylsilyl)acetylene triphenylphosphine complex (1), four well documented gold(I)-catalyzed alkyne π activation reactions were examined briefly under the catalysis of complex 1 to compare with the conventional gold(I) pre-catalyst PPh3AuCl/AgSbF6 (4), i.e. alkyne hydration,13 intramolecular rearrangement of alkynyl furan,14 1,6-enyne rearrangement,15 and a tandem 3,3-rearrangement–Nazarov reaction.16Thus, the alkyne substrate 19 in the presence of complex 1 (1 mol%) in wet 1,4-dioxane at RT, was activated and converted smoothly to the corresponding ketone 20 in a satisfactory yield of 93%, which is comparable to the reaction under catalysis of PPh3AuCl/AgSbF6 (4) (1 mol%) (Scheme 3). When the loading of the gold(I) pre-catalyst, either 1 or 4, was reduced to 0.1 mol%, the hydration of alkyne 19 proceeded sluggishly, leading to 93% recovery of the starting alkyne.
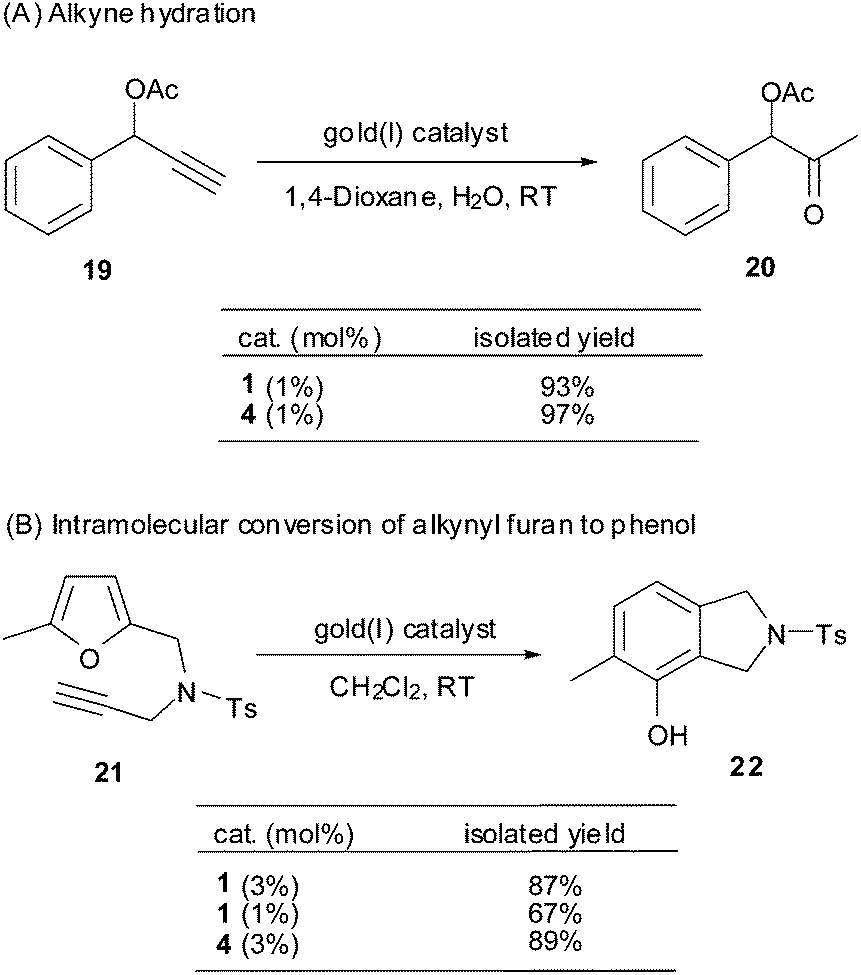 | ||
| Scheme 3 Complex 1 catalyzed reactions A and B. | ||
Complex 1 also proved to be effective in promoting the intramolecular conversion of alkynyl furan to phenol (Scheme 3).14a,b In the presence of 3 mol% or 1 mol% of the pre-catalyst 1 (CH2Cl2, RT), alkynyl furan 21 was converted to the corresponding isoindoline 22 in 87% and 67% yield, respectively. These results demonstrate that complex 1 is comparable or even more effective than the Schmidbaur–Bayler binuclear gold(I) complex ([(Mes3PAu)2Cl]BF4), 2.5 mol%, 65% yield) and PPh3AuCl/AgSbF6 (4) (3 mol%, 89%) in this phenol synthesis.
Echavarren et al. reported that N-propargyl-N-isoamylene toluene-4-sulfonylamine 23 underwent endo rearrangement to provide diene 24 in 96% yield under catalysis of PPh3AuCl/AgSbF6 (4) (2 mol%) in dry CH2Cl2 at RT (Scheme 4).15a,b With 2 mol% of the Au(I) complex 1, a similar reaction proceeded smoothly to afford diene 24 in 95% yield. When the catalyst loading was decreased to 1 mol% or even 0.1 mol%, the reaction still performed cleanly and afforded the desired product in 83% and 50% (44% starting material recovered) yield, respectively. In this case, the shelf-stable Au(I) complex 1 showed excellent catalytic efficiency comparable to the freshly prepared PPh3AuSbF6 (4).
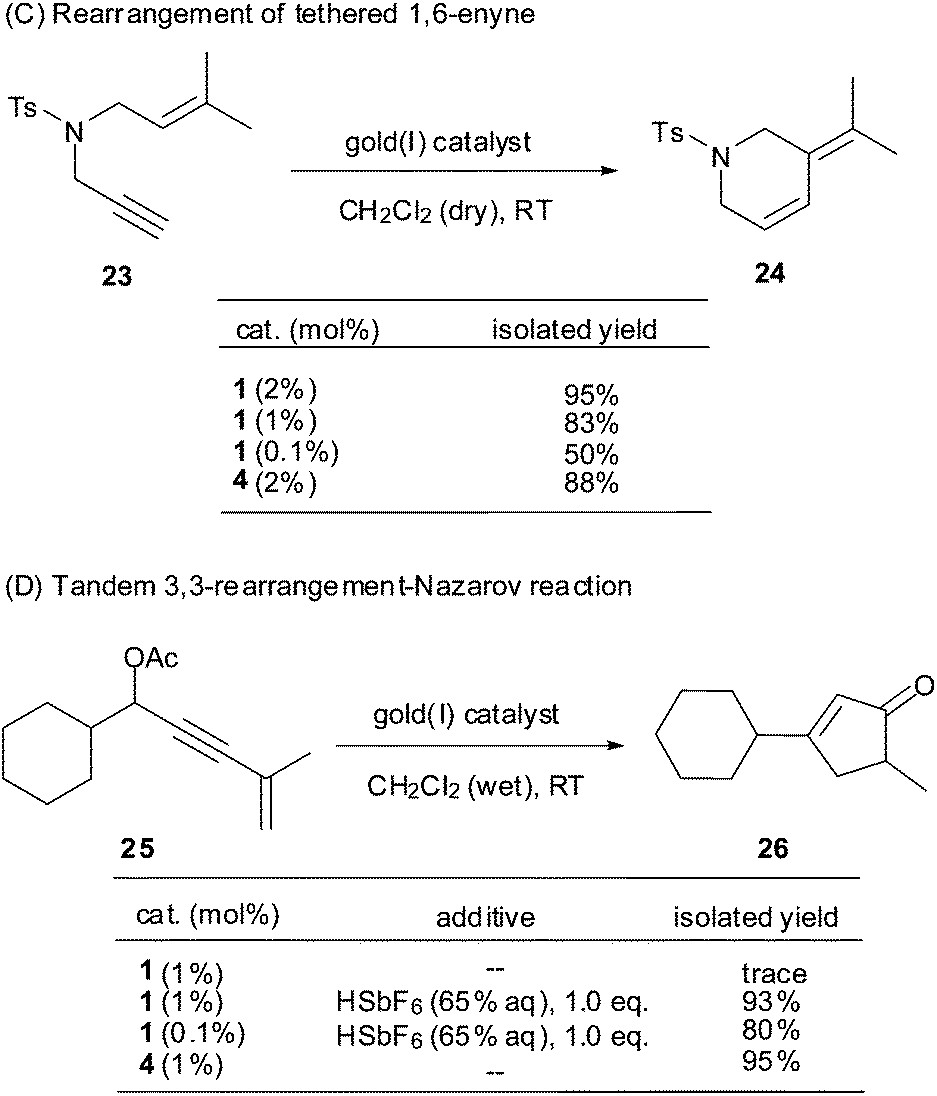 | ||
| Scheme 4 Complex 1 catalyzed reactions C and D. | ||
Zhang et al. reported the preparation of cyclopentenone 26 from enynyl acetate 25via a Au(I)-catalyzed tandem 3,3-rearrangement–Nazarov reaction (Scheme 4).16 In the presence of Au(I) complex 1 (1 mol%) in wet CH2Cl2 at RT, however, we were surprised to find that the conversion of 25 to 26 did not occur at all, whereas a similar transformation proceeded well in the presence of PPh3AuCl/AgSbF6 (1 mol%) to give cyclopentenone 26 in 95% yield. Our previous studies with a polystyrene-supported Au(I) catalyst showed that a Brönsted acid was crucial to facilitate this reaction.15c In fact, upon addition of 1.0 eq. HSbF6 (65% aq.) into the inert mixture of 25 and complex 1 (1 mol%) in CH2Cl2 at RT, 26 was obtained smoothly in 93% yield. In comparison, the reaction did not take place in the absence of complex 1. The present results imply that the release of catalytic [Ph3PAu]+ from the coordinated complex 1 is much milder than from PPh3AuCl/AgSbF6 (4).
Conclusion
A linear η2-alkyne π complex of [Ph3PAu]+, namely Au(I) π-bis(tert-butyldimethylsilyl)acetylene triphenylphosphine complex (1), was readily prepared and characterized. This shelf-stable complex 1 is convenient to prepare, store, and handle, while it has been shown to be an effective pre-catalyst for a variety of the Au(I)-catalyzed alkyne π activation reactions, including the glycosidation of ortho-alkynylbenzoates, the hydration of alkynes, the intramolecular rearrangement of alkynyl furan, the 1,6-enyne rearrangement, and the tandem 3,3-rearrangement–Nazarov reaction. The mild release of catalytic [Ph3PAu]+ species in complex 1 might be beneficial in catalyzing certain transformations involving selectivities, and this is a topic of our current interest.Experimental section
The preparation of complex 1
To a 5 mL flask containing the commercially available TBS-CCH (0.5 mL) and dry THF (freshly distilled with Na, 3 mL) was added n-BuLi solution (2.5 M in hexane, 1.2 mL) under argon at −15 °C. After stirring for 1 h, a solution of TBSCl (484 mg in 3 mL dry THF) was introduced into the mixture. The mixture was slowly warmed to room temperature and stirred for another 1 h, and was then quenched by H2O (1 mL). The resulting mixture was extracted with hexane. The organic layer was dried over Na2SO4 and concentrated. The residue was recrystallized from MeOH to give the desired compound 5 as a white solid (444.7 mg, 65%): 1H NMR (400 MHz, CDCl3) δ 0.94 (s, 18H), 0.10 (s, 12H); 13C NMR (126 MHz, CDCl3) δ 112.9, 26.0, 16.5, −4.7; MS-EI+ 197 (100%), 155 (30%), 198 (22%), 199 (9%), 73 (8%), 254 (7%), 141 (6%), 156 (6%); HRMS-EI+ calcd for C14H30Si2 ([M]+) 254.1886, found 254.1885; Raman (532 nm, cm−1) νCC 2103.
A 5 mL flask containing TBS-CC-TBS (79.5 mg, 0.31 mmol), PPh3AuCl (76.5 mg, 0.15 mmol) and AgSbF6 (52.8 mg, 0.15 mmol) was cooled to −70 °C under argon. Then dry CH2Cl2 (4 mL, freshly distilled with CaH2) was introduced via a syringe. After stirring for 5 h, the mixture was warmed to room temperature slowly. The resulting AgCl was removed by cotton filtration and the filtrate was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (l mL) and then hexane (5 mL) was added. The white precipitate was collected to give the desired complex 1 as a white solid (127.9 mg, 87%). The sample for X-ray diffraction analysis was obtained by recrystallization from CH2Cl2–hexane via vapor diffusion at 0 °C. 1H NMR (400 MHz, CDCl3) δ 7.69–7.55 (m, 9H), 7.49 (dd, J = 13.8, 7.7 Hz, 6H), 1.01 (s, 18H), 0.32 (s, 12H); 13C NMR (101 MHz, CDCl3) δ 133.8 (d, J = 2.7 Hz), 133.3 (d, J = 2.7 Hz), 130.1 (d, J = 12.3 Hz), 125.9 (d, J = 64.0 Hz), 122.8 (s), 23.9 (s), 16.9 (s), −4.4 (s); 31P NMR (400 MHz, CDCl3) δ 39.0; HRMS (MALDI) calcd C32H45AuPSi2 ([M − SbF6]+) 713.2457, found 713.2449; Raman (532 nm, cm−1) νCC 1986.
General procedure for the glycosylation reactions (14–18)
To a 25 mL flask containing the donor (0.10 mmol, 1.0 eq.), the acceptor (0.12 mmol, 1.2 eq.) and activated 5 Å molecular sieves (weight equal to the combined weight of the donor and the acceptor), was added dry CH2Cl2 to maintain a concentration of 0.05 M under Ar. The gold(I) catalyst 1 (0.0102 mmol, 0.1 eq.) was added 30 minutes later. The mixture was stirred until TLC indicated disappearance of the donor. The molecular sieves were filtered off over Celite and washed with CH2Cl2 twice. The filtrate was concentrated under reduced pressure and chromatographed with a gradient of petroleum ether–EtOAc to afford the coupled glycosides (14–18).The hydration of alkyne 19
To a 5 mL Schlenk tube was added a solution of gold(I) catalyst 1 (2.1 mg in 0.5 mL CH2Cl2, 0.45 mL), and the solvent was removed under reduced pressure. Then compound 19 (35.5 mg, 2.0 mmol) was added, and the reaction system was protected with Ar. Wet 1,4-dioxane (36 μL H2O in 1 mL dry 1,4-dioxane, 0.3 mL) was added to the mixture. After stirring for 12 h, the mixture was concentrated in vacuo. The residue was chromatographed (petroleum ether–EtOAc, 8:1) to yield compound 20 as a colorless oil (36.6 mg, 93%): 1H NMR (500 MHz, CDCl3) δ 7.42–7.39 (m, 5H), 5.98 (s, 1H), 2.19 (s, 3H), 2.11 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 201.59, 170.16, 133.10, 129.30, 129.01, 127.99, 80.87, 26.04, 20.63; MS-EI+ 149 (100%), 107 (96%), 43 (32%), 79 (21%), 77 (14%), 150 (11%), 105 (11%), 108 (10%); HRMS-EI+ calcd for C11H12O3 ([M]+) 192.0786, found 192.0788.
The Hashmi transformation of alkynyl furan 21 to phenol 5-methyl-2-tosylisoindolin-4-ol (22)
To a 25 mL flask containing compound 21 (60.7 mg, 0.2 mmol) and gold(I) catalyst 1 (5.5 mg, 0.006 mmol) was added CH2Cl2 (2 mL) at room temperature. After stirring for 1 h, the mixture was concentrated in vacuo. The residue was purified by silica gel chromatography (petroleum ether–EtOAc–CH2Cl2, 5:1:1) to give compound 22 as a white solid (52.8 mg, 87%): 1H NMR (400 MHz, d6-DMSO) δ 8.97 (s, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 6.96 (d, J = 7.6 Hz, 1H), 6.59 (d, J = 7.5 Hz, 1H), 4.49 (s, 2H), 4.46 (s, 2H), 2.36 (s, 3H), 2.08 (s, 3H); 13C NMR (101 MHz, d6-DMSO) δ 149.8, 143.7, 134.8, 132.9, 130.5, 123.0, 127.4, 123.4, 122.1, 113.3, 53.7, 51.9, 20.9, 15.7; HRMS-ESI+ calcd for C16H17NO3SNa ([M + Na]+) 326.0821, found 326.0827.
Synthesis of 3-(propan-2-ylidene)-1-tosyl-1,2,3,6-tetrahydro-pyridine (24) via the 1,6-enyne rearrangement of 23
To a 25 mL flask containing compound 23 (54.7 mg, 0.2 mmol) and gold(I) catalyst 1 (3.79 mg, 0.004 mmol) was added dry CH2Cl2 (2 mL) at room temperature under Ar. After stirring for 30 minutes, the solution was concentrated in vacuo. The mixture was purified by silica gel chromatography (petroleum ether–EtOAc–CH2Cl2, 10:1:1) to give compound 24 as a white solid (52.2 mg, 95%): 1H NMR (300 MHz, CDCl3) δ 7.65 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 6.7 Hz, 2H), 6.34 (d, J = 10.2 Hz, 1H), 5.53 (d, J = 10.0 Hz, 1H), 3.90 (s, 2H), 3.77 (s, 2H), 2.42 (s, 3H), 1.77 (s, 3H), 1.68 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 143.3, 134.1, 129.9, 129.3, 127.6, 124.5, 122.3, 120.9, 45.03, 45.00, 21.5, 20.3, 19.6; HRMS-ESI+ calcd for C15H19NO2SNa ([M + Na]+) 300.1029, found 300.1034.
The synthesis of 3-cyclohexyl-5-methylcyclopent-2-enone (26) via a tandem 3,3-rearrangement–Nazarov reaction of 25
To a 25 mL flask containing compound 25 (46.3 mg, 0.2 mmol), HSbF6 (∼65% aq., 76.5 mg), and gold(I) catalyst 1 (1.86 mg, 0.002 mmol), was added wet CH2Cl2 (2 mL) at room temperature. After being stirred for 2 h, the mixture was concentrated in vacuo. The residue was purified by silica gel chromatography (petroleum ether–EtOAc, 8:1) to give compound 26 as a colorless oil (35.0 mg, 93%): 1H NMR (400 MHz, CDCl3) δ 5.88 (s, 1H), 2.85 (dd, J = 18.3, 6.7 Hz, 1H), 2.40 (pd, J = 7.3, 2.4 Hz, 1H), 2.28 (t, J = 11.1 Hz, 1H), 2.19 (d, J = 18.4 Hz, 1H), 1.89 (d, J = 12.6 Hz, 2H), 1.81 (d, J = 12.5 Hz, 2H), 1.73 (d, J = 12.7 Hz, 1H), 1.43–1.19 (m, 5H), 1.17 (d, J = 7.5 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 212.74, 185.52, 126.47, 41.75, 40.35, 38.41, 31.17, 31.14, 25.96, 25.95, 25.90, 16.42. MS-EI+: 178 (100%), 122 (97%), 136 (38%), 107 (37%), 163 (26%), 79 (24%), 95 (20%), 93 (19%); HRMS-EI+ calcd for C12H18O ([M]+) 178.1358, found 178.1356.
Acknowledgements
Financial support from the Ministry of Science and Technology of China (2012ZX09502-002) and the National Natural Science Foundation of China (21432012 and 21102169) is gratefully acknowledged.Notes and references
- For selected reviews, see: (a) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed; (b) A. Arcadi, Chem. Rev., 2008, 108, 3266–3325 CrossRef CAS PubMed; (c) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed; (d) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (e) Y. Yamamoto, I. D. Gridnev, N. T. Patil and T. Jin, Chem. Commun., 2009, 5075–5087 RSC; (f) S. P. Nolan, Acc. Chem. Res., 2010, 44, 91–100 CrossRef PubMed; (g) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed; (h) A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864–876 CrossRef CAS PubMed; (i) H. Schmidbaur and A. Schier, Organometallics, 2009, 29, 2–23 CrossRef; (j) R. E. M. Brooner and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2013, 52, 11714–11724 CrossRef CAS PubMed.
- (a) Y. Li, Y. Yang and B. Yu, Tetrahedron Lett., 2008, 49, 3604–3608 CrossRef CAS PubMed; (b) Y. Li, X. Yang, Y. Liu, C. Zhu, Y. Yang and B. Yu, Chem. – Eur. J., 2010, 16, 1871–1882 CrossRef CAS PubMed; (c) Y. Zhu and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 8329–8332 CrossRef CAS PubMed; (d) Y. Tang, J. Li, Y. Zhu, Y. Li and B. Yu, J. Am. Chem. Soc., 2013, 135, 18396–18405 CrossRef CAS PubMed; (e) Y. Yang, Y. Li and B. Yu, J. Am. Chem. Soc., 2009, 131, 12076–12077 CrossRef CAS PubMed; (f) Y. Li, J. Sun and B. Yu, Org. Lett., 2011, 13, 5508–5511 CrossRef CAS PubMed; (g) J. Zhang, H. Shi, Y. Ma and B. Yu, Chem. Commun., 2012, 48, 8679–8681 RSC; (h) G. Xiao and B. Yu, Chem. – Eur. J., 2013, 19, 7708–7712 CrossRef CAS PubMed; (i) J. Yu, J. Sun, Y. Niu, R. Li, J. Liao, F. Zhang and B. Yu, Chem. Sci., 2013, 4, 3899–3905 RSC; (j) S. Nie, W. Li and B. Yu, J. Am. Chem. Soc., 2014, 136, 4157–4160 CrossRef CAS PubMed; (k) P. Xu, W. Xu, Y. Dai, Y. Yang and B. Yu, Org. Chem. Front., 2014, 1, 405–414 RSC.
- (a) A. S. K. Hashmi, L. Schwarz, J.-H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285–2288 CrossRef CAS; (b) M. Kumar, J. Jasinski, G. B. Hammond and B. Xu, Chem. – Eur. J., 2014, 20, 3113–3119 CrossRef CAS PubMed.
- (a) A. S. K. Hashmi, Science, 2012, 338, 1434–1434 CrossRef CAS PubMed; (b) M. C. B. Jaimes, C. R. N. Böhling, J. M. Serrano-Becerra and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 7963–7966 CrossRef PubMed; (c) M. C. B. Jaimes, F. Rominger, M. M. Pereira, R. M. B. Carrilho, S. A. C. Carabineiroc and A. S. K. Hashmi, Chem. Commun., 2014, 50, 4937–4940 RSC.
- (a) H. V. R. Dias, J. A. Flores, J. Wu and P. Kroll, J. Am. Chem. Soc., 2009, 131, 11249–11255 CrossRef CAS PubMed; (b) T. N. Hooper, M. Green, J. E. McGrady, J. R. Patel and C. A. Russell, Chem. Commun., 2009, 3877–3879 RSC; (c) T. N. Hooper, M. Green and C. A. Russell, Chem. Commun., 2010, 46, 2313–2315 RSC; (d) P. P. Sharp, M. G. Banwell, J. Renner, K. Lohmann and A. C. Willis, Org. Lett., 2013, 15, 2616–2619 CrossRef CAS PubMed; (e) N. D. Shapiro and F. D. Toste, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2779–2782 CrossRef CAS; (f) D. Zuccaccia, L. Belpassi, L. Rocchigiani, F. Tarantelli and A. Macchioni, Inorg. Chem., 2010, 49, 3080–3082 CrossRef CAS PubMed; (g) T. J. Brown and R. A. Widenhoefer, J. Organomet. Chem., 2011, 696, 1216–1220 CrossRef CAS PubMed; (h) M. A. Celik, C. Dash, V. A. K. Adiraju, A. Das, M. Yousufuddin, G. Frenking and H. V. R. Dias, Inorg. Chem., 2012, 52, 729–742 CrossRef PubMed; (i) A. S. K. Hashmi, T. Lauterbach, P. Nösel, M. H. Vilhelmsen, M. Rudolph and F. Rominger, Chem. – Eur. J., 2013, 19, 1058–1065 CrossRef CAS PubMed.
- (a) C. N. Oberhuber, S. Lopez and A. M. Echavarren, J. Am. Chem. Soc., 2005, 127, 6178–6179 CrossRef PubMed; (b) M. Munoz, J. Adrio, J. C. Carretero and A. M. Echavarren, Organometallics, 2005, 24, 1293–1300 CrossRef CAS; (c) N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133–4136 CrossRef PubMed.
- D. Zhao, A. D. Allen and T. T. Tidwell, J. Am. Chem. Soc., 1993, 115, 10097–10103 CrossRef CAS.
- R. West and L. C. Quass, J. Organomet. Chem., 1969, 18, 55–67 CrossRef CAS.
- The website of Cambridge Structural Database: http://www.ccdc.cam.ac.uk/pages/Home.aspx.
- (a) A. Demchenko, T. Stauch and G. J. Boons, Synlett, 1997, 818–819 CrossRef CAS PubMed; (b) M. Adinolfi, A. Iadonisi, A. Ravida and M. Schiattarella, Tetrahedron Lett., 2004, 45, 4485–4488 CrossRef CAS PubMed; (c) M. A. Oberli, P. Bindschadler, D. B. Werz and P. H. Seeberger, Org. Lett., 2008, 10, 905–908 CrossRef CAS PubMed.
- (a) C. H. Marzabadi and R. W. Frank, Tetrahedron, 2000, 56, 8385–8417 CrossRef CAS; (b) D. Hou and T. L. Lowary, Carbohydr. Res., 2009, 344, 1911–1940 CrossRef CAS PubMed; (c) J. Park, T. J. Boltje and G. J. Boons, Org. Lett., 2008, 10, 4367–4370 CrossRef CAS PubMed; (d) Y. S. Lu, Q. Li, L. H. Zhang and X. S. Ye, Org. Lett., 2008, 10, 3445–3448 CrossRef CAS PubMed.
- Y. Ma, Z. Li, H. Shi, J. Zhang and B. Yu, J. Org. Chem., 2011, 76, 9748–9756 CrossRef CAS PubMed.
- (a) N. Ghosh, S. Nayak and A. K. Sahoo, J. Org. Chem., 2011, 76, 500–511 CrossRef CAS PubMed; (b) D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012–9019 CrossRef CAS PubMed; (c) M. Kumar, G. B. Hammond and B. Xu, Org. Lett., 2014, 16, 3452–3455 CrossRef CAS PubMed.
- (a) A. S. K. Hashmi and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef CAS; (b) A. S. K. Hashmi, M. C. Blanco, E. Kurpejović, W. Frey and J. W. Bats, Adv. Synth. Catal., 2006, 348, 709–713 CrossRef CAS; (c) A. S. K. Hashmi, M. Rudolph, H.-U. Siehl, M. Tanaka, J. W. Bats and W. Frey, Chem. – Eur. J., 2008, 14, 3703–3708 CrossRef CAS PubMed; (d) Y. Chen, W. Yan, N. G. Akhmedov and X. Shi, Org. Lett., 2010, 12, 344–347 CrossRef CAS PubMed.
- (a) C. N. Oberhuber, M. P. Munoz, E. Bunuel, C. Nevada, D. J. Cardenas and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 2402–2406 CrossRef PubMed; (b) C. N. Oberhuber, M. P. Muñoz, S. López, E. Jiménez-Núñez, C. Nevado, E. Herrero-Gómez, M. Raducan and A. M. Echavarren, Chem. – Eur. J., 2006, 12, 1677–1693 CrossRef PubMed; (c) W. Cao and B. Yu, Adv. Synth. Catal., 2011, 353, 1903–1907 CrossRef CAS.
- L. Zhang and S. Wang, J. Am. Chem. Soc., 2006, 128, 1442–1443 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1043329. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00023h |
| This journal is © the Partner Organisations 2015 |