
An alkyne hydrosilylation–Hiyama coupling approach to highly functionalised 1,3-dienes†
Catherine A.
McAdam
,
Mark G.
McLaughlin
and
Matthew J.
Cook
*
School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, Northern Ireland, UK. E-mail: m.cook@qub.ac.uk; Fax: +44 (0)28 90976524; Tel: +44 (0)28 90974682
First published on 17th March 2015
Abstract
A high yielding and robust protocol for the stereodefined synthesis of 1,3-dienes has been established through a hydrosilylation–Hiyama coupling strategy. In all cases the products were formed as single E,E isomers and conditions are tolerant of a wide range of functional groups not compatible with other methods.
1,3-Dienes are an important class of molecules which are prevalent in many natural products and display photophysical properties. They are also vital synthons in many synthetic campaigns as they can perform a number of key transformations such as the Diels–Alder reaction. Their synthesis can be performed using several strategies including olefinations, metathesis and cross-coupling reactions.
A major route to the formation of 1,3-dienes is via the use of palladium catalysed cross-coupling reactions (Scheme 1).1 While these reactions can be very effective for the formation of diene and polyene moieties, they can suffer from a variety of drawbacks including the use of toxic or sensitive reagents and a loss of geometric integrity during the coupling process. Both the traditional and oxidative Heck reactions have been reported with the latter operating under much milder conditions.2,3
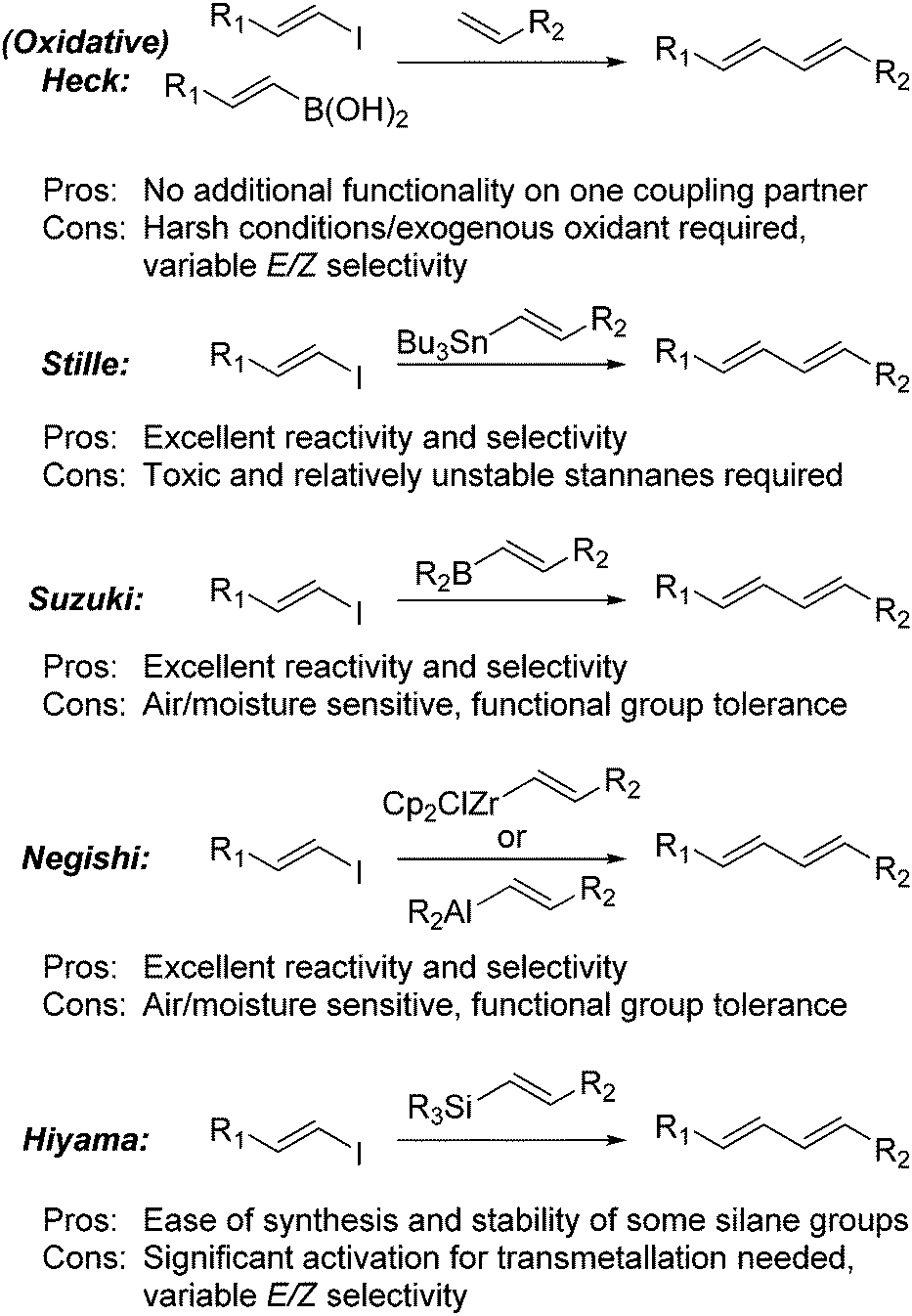 | ||
| Scheme 1 Cross-coupling methods for diene synthesis. | ||
The Stille reaction has been proven to be a valuable tool for the synthesis of dienes through the coupling of vinylstannanes with vinyl halides.4 The major issue to this approach is the use of toxic tin reagents, although this can be circumvented through the use of a “one-pot” hydrostannylation-Stille procedure5 which can even be rendered catalytic in tin.6
The Suzuki–Miyuara reaction has also proven itself to be a powerful tool for the construction of complex molecules.7 This approach generally requires the hydroboration of an alkyne using air and moisture sensitive boranes, such as 9-BBN. The corresponding vinylborane cannot be stored for prolonged periods of time and must be used immediately.8 Although the reaction provides high levels of reactivity and stereoselectivity, the boranes used are strong reducing agents which can cause issues with functional group compatibility. Alternatively, boronic acids or potassium trifluoroborates can be produced via a metallation reaction, either through metal-halogen exchange or deprotonation followed by trapping with borates.9,10 This approach is used widely, however functional group tolerance can be an issue, as can the hydroscopic nature of these compounds and difficulties encountered during purification. An alternative approach was devised by Burke, who developed a series of MIDA boronate esters which are easily formed and purified and which do not suffer the same issues as the boronic acids and trifluoroborates.11 The approach developed by Negishi was to perform a hydroalumination, hydrozirconation or carboalumination reaction and the corresponding vinyl alane is used directly as a transmetallation partner.12 Although effective, these approaches can suffer from similar problems to the Suzuki protocols, do not allow for storage of the vinyl intermediate and require very air and moisture sensitive conditions.
Although vinylsilanes are readily accessible, Hiyama couplings are the least developed of the coupling reactions.13 This is mainly due to the activation required prior to the transmetallation event. The original variant required very strong activators such as DAST,14 however, solutions to this issue were found with the advent of new, more reactive silane species such as silanols15 and silacyclobutanes.16 Although effective for the coupling reactions, these groups are difficult to install and can provide mixture of geometric and regioisomers in the coupling reaction.
To circumvent these issues, we envisioned an approach to synthesize useful diene species, in which we could exploit the stability and accessibility of vinylsilanes. We began examining the formation of readily accessible vinylsilanes which could then be activated for Hiyama couplings. To achieve this we adopted our recently developed hydrosilylation methodology on propargylic alcohols to form the requisite vinylsilane.17 We utilised commercially available benzyldimethylsilane as our silane sources as the corresponding vinylsilane is very stable and can undergo simple activation with TBAF under Trost-type coupling conditions.18 Indeed, we have recently demonstrated this coupling in the synthesis of 1,3-diaryl allyl amines and stilnenes, utilising a sequential Suzuki–Hiyama approach.19 The synthetic utility of BnMe2Si-vinyl silanes, with both disubstituted vinylsilanes and trisubstituted variants have been previously demonstrated, including key steps in the synthesis of several polyene natural products.20
Under our standard PtCl2/XPhos hydrosilylation conditions we found excellent reactivity for the hydrosilylation conditions utilising just 0.5 mol% platinum (Table 1). In most cases the reactivity of these hydrosilylations was superior to phenyldimethylsilane which has been previously disclosed, demonstrating that these conditions are an excellent method for the installation of a benzyldimethylsilane group. The reaction was tolerant of a range of functional groups including heterocycles, ethers and aromatic groups with good to excellent yields being obtained in all cases.
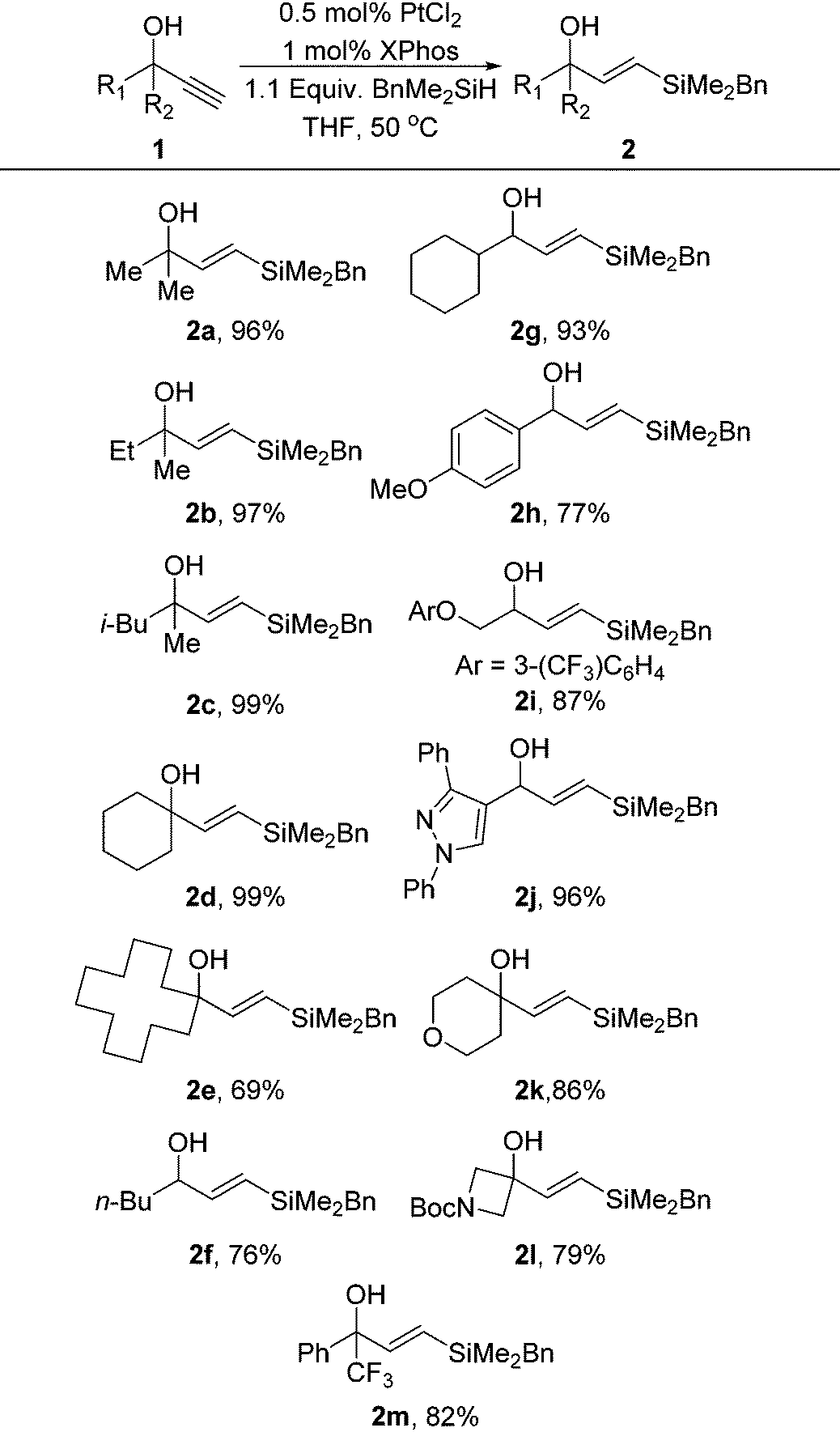
|
With the vinylsilanes 2 in hand, we began examining the Hiyama coupling to install the diene moiety (Table 2). We utilised β-iodostyrene, which can be readily prepared,21 to afford phenyl substituted diene 3a. Under standard Hiyama conditions that we and others have previously reported, the reaction proceeded smoothly forming the dienes in excellent yields. Once again the reaction was tolerant of many functional groups and provided a series of dienes in excellent yields. The reaction is not affected by the unprotected alcohol and this approach allows for functionality to be incorporated that would not be possible using the strongly reductive conditions required for hydroboration and hydroalumination strategies. The only class of substrate that was not amenable to this strategy was those containing a benzylic hydroxyl group. These substrates appear to undergo the Hiyama coupling readily, however, the resulting dienes were not stable under the reaction conditions leading to complex mixtures being obtained. This is presumably due to the extra stabilisation of the resulting carbocation following loss of OH.
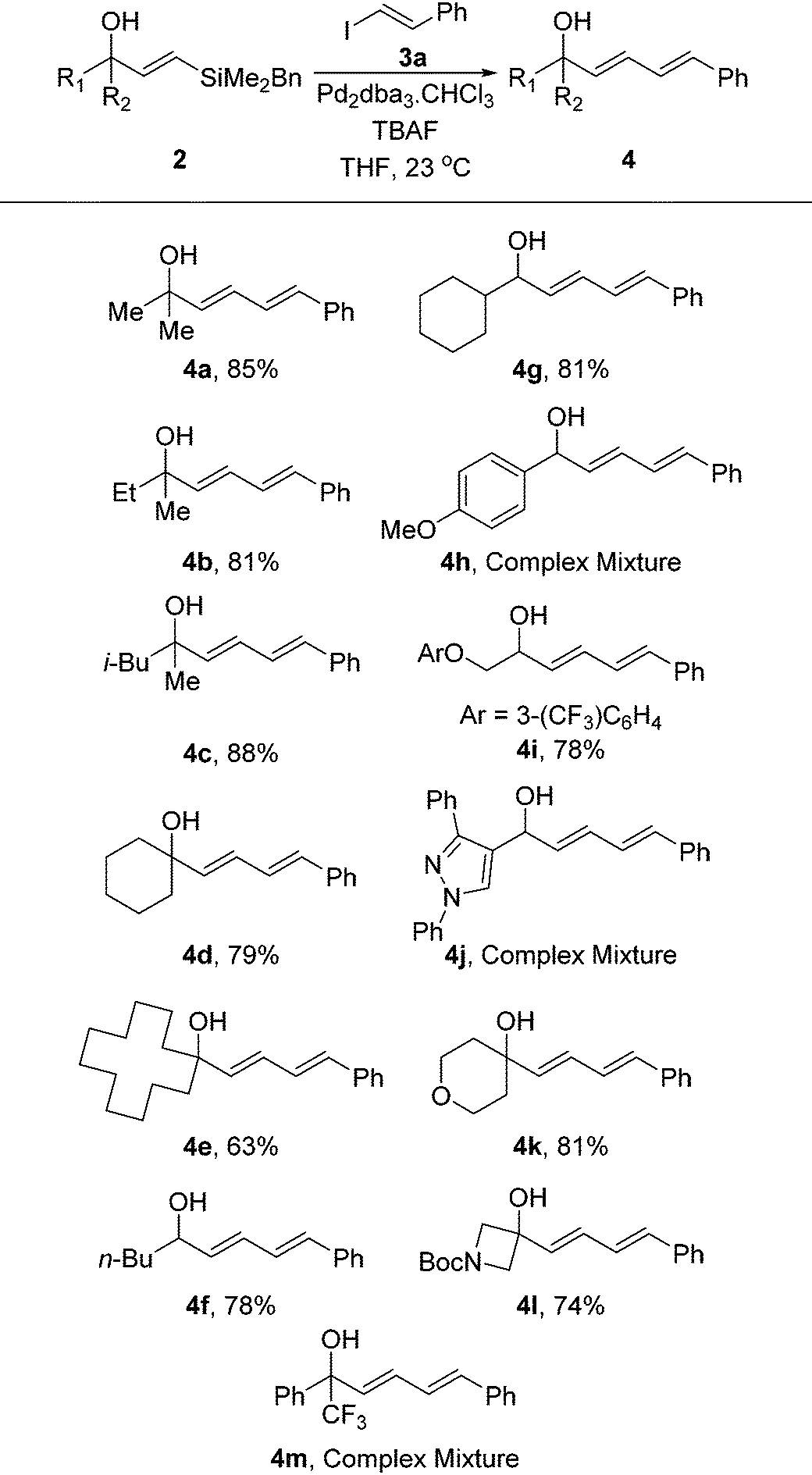
|
We also examined the use of other vinyliodides (Table 3). Alongside the parent iodostyrene coupling partner, both electron rich and deficient aromatic vinyliodides (3b–c) perform well in the coupling. Non-aromatic substrates such as 1-iodo-hex-1-ene 3d also coupled to with vinylsilane 2d to provide the corresponding diene 7 in good yields.
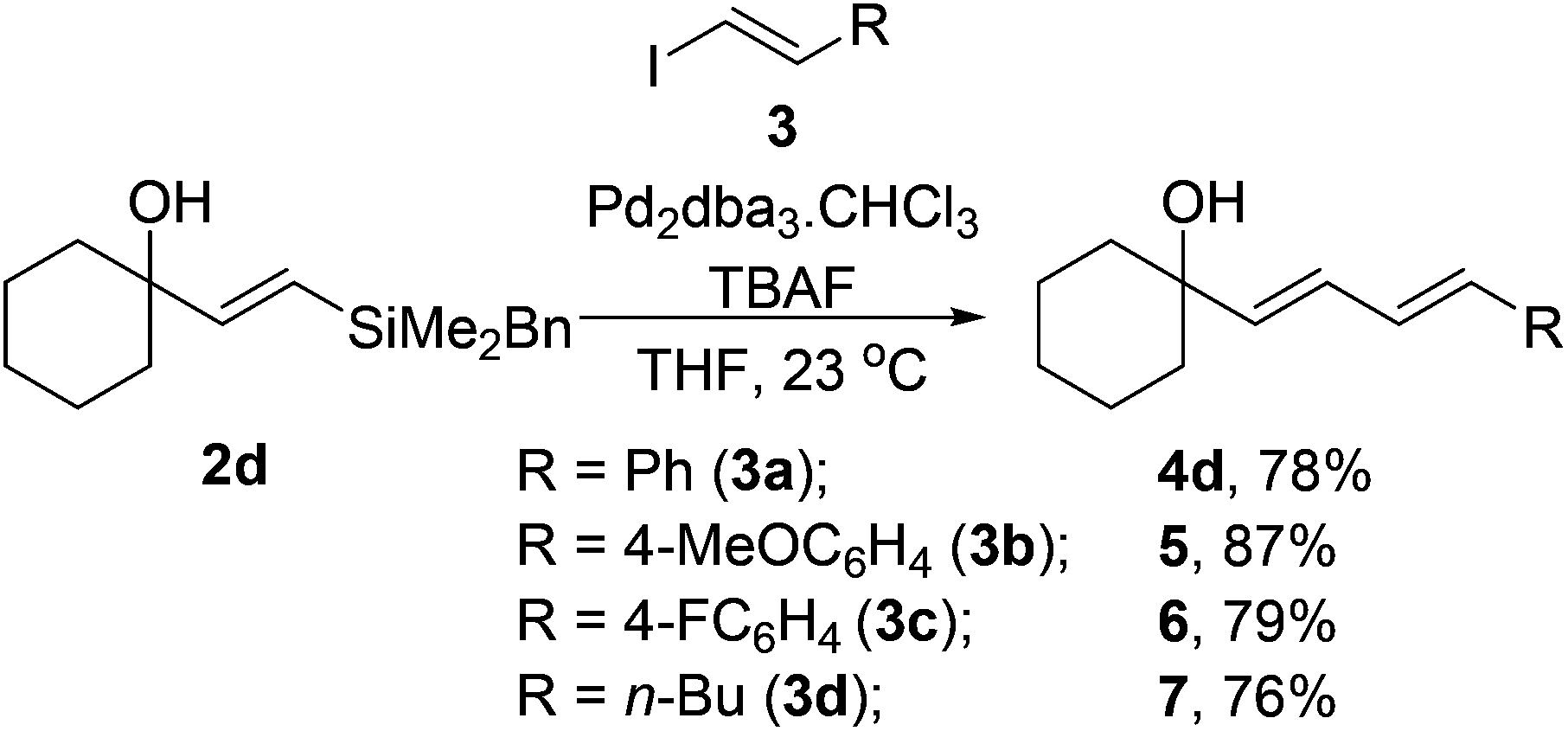
|
With both components of the protocol optimised, we examined the possibility of performing the entire procedure in a one-pot manner. The combination of hydrosilylations and Hiyama couplings have also been demonstrated in the synthesis of retinoid natural products.22 In these reports, the use and isolation of the benzyldimethylsilane was necessary prior to the Hiyama coupling. The use of 1,1,3,3-tetramethylsiloxane [(HMe2Si)2O] as the hydrosilylating agent was effective using Denmark's protocol,23 however this can lead to a diminished E/Z ratio compared to the use of BnMe2SiH. As with the previous report by López,22 all attempts to perform this in a true one-pot sequence led to very poor yields in the Hiyama step. The reaction could, however be streamlined through the development of a “telescoped” procedure (Scheme 2). On completion of the hydrosilylation reaction, the mixture was filtered and concentrated. The crude residue was subjected to the Hiyama coupling conditions to provide diene 4d with no isolation or purification of vinylsilane 2d required.
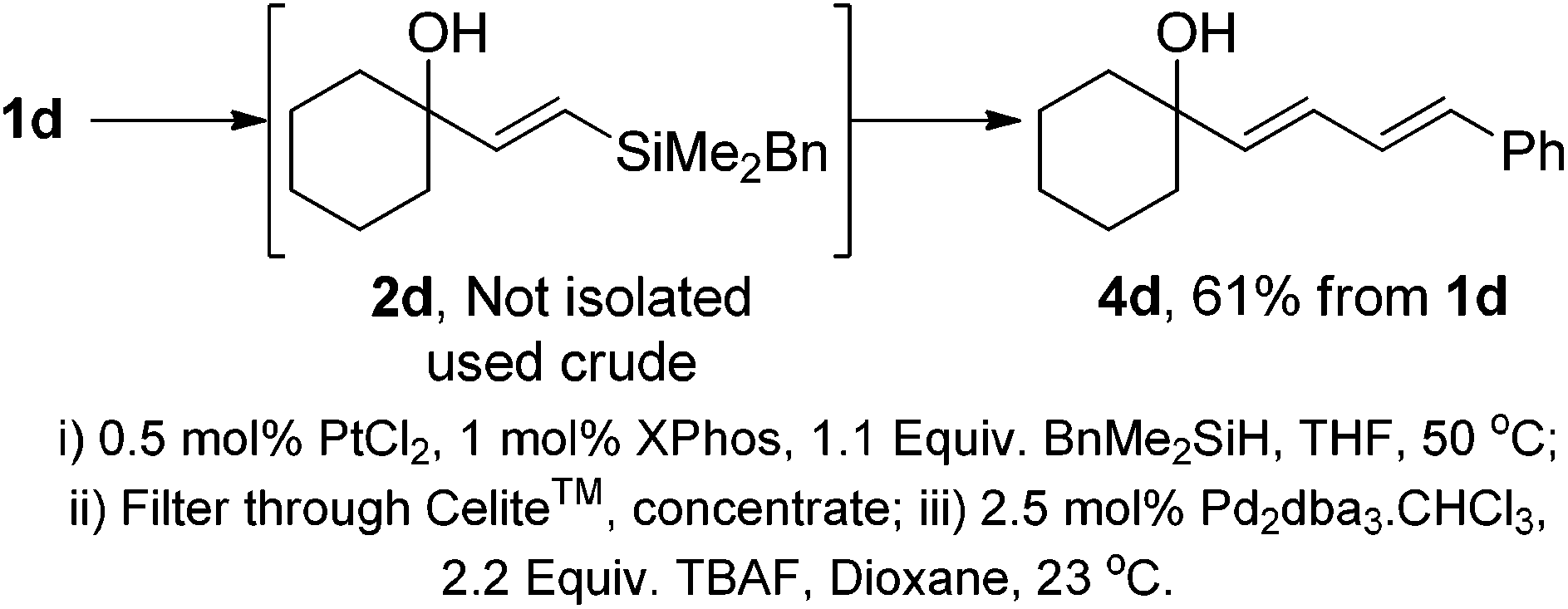 | ||
| Scheme 2 Telescoped hydrosilylation–Hiyama coupling. | ||
With the Hiyama coupling strategy in hand we examined the uses of the diene products as synthetic handles. We were intrigued to discover whether the diene could undergo a intramolecular Diels–Alder reaction with a tethered dieneophile. We took pyran substituted diene 4k and were able to allylate the allylic alcohol position to form our Diels–Alder precursor 8. This could undergo a thermally promoted intramolecular Diels–Alder which provided tricyclic product 9 in excellent yield and as a 1.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of exo:endo diastereoisomers (Scheme 3).
:1 mixture of exo:endo diastereoisomers (Scheme 3).
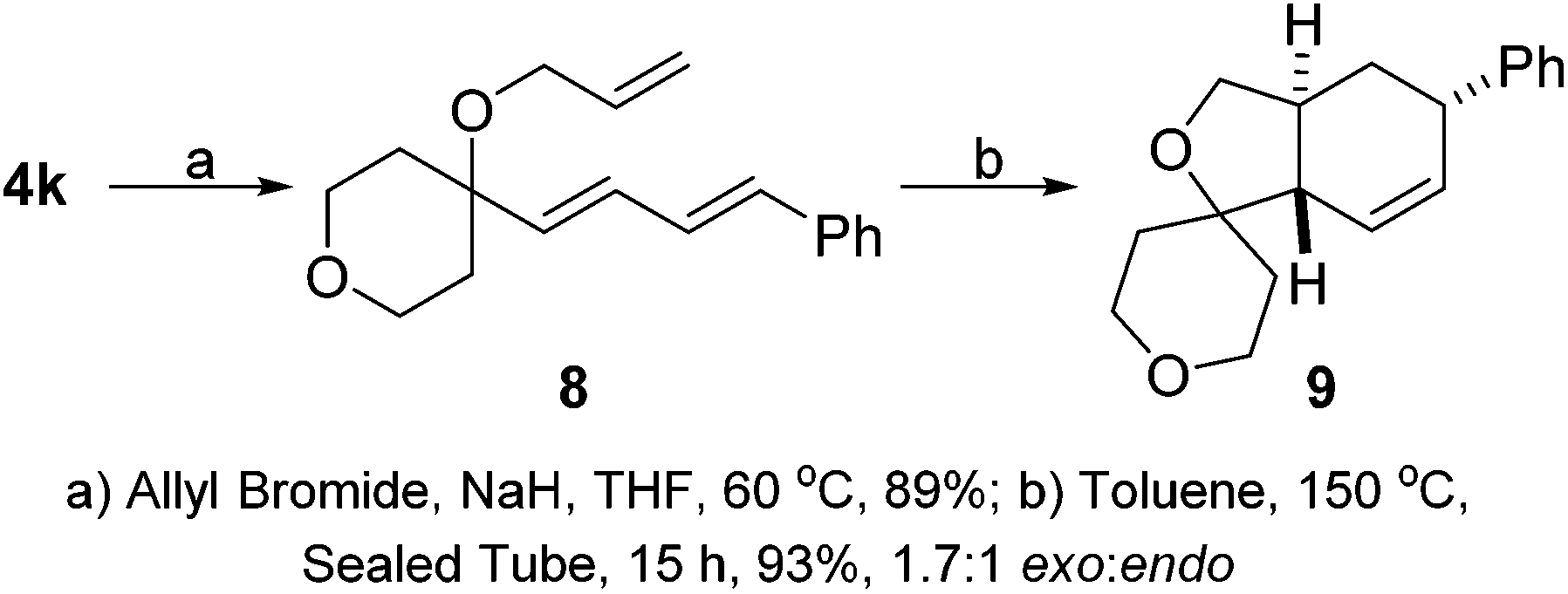 | ||
| Scheme 3 Diels–Alder reaction of diene. | ||
Conclusions
To conclude, we have developed a robust and reliable method for the formation of highly functionalised 1,3-dienes through a hydrosilylation–Hiyama protocol. The substrate scope is good although benzylic products are not stable to the reaction conditions and form complex mixtures. The reaction is tolerant of both aromatic and aliphatic substituted alkenyl iodides with little difference in yields or reactivity. We have demonstrated the application of these products in a intramolecular Diels–Alder reaction to form a novel sprio-tricycle in excellent yields.Experimental24
General procedure for the hydrosilylation reaction
To an oven dried 5 mL round bottom flask equipped with a reflux condenser and magnetic stirrer was added PtCl2 (0.5 mol%) and 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (1 mol%) (XPhos). The flask was then flushed with argon and dry THF was added. The mixture was then stirred at 50 °C for 30 minutes until a yellow homogenous mixture was obtained. The corresponding propargyl alcohol (1.0 equiv.) was added followed by benzyldimethylsilane (1.2 equiv.) via syringe (CAUTION: rapid evolution of hydrogen gas) and the solution was stirred at 50 °C overnight. The solvent was evaporated and the crude mixture was applied to the top of a column and chromatographed to afford the requisite (E)-vinyl silane.General procedure for the Hiyama coupling
Desired substrate (1.0 equiv.) was added to an oven dried reaction vessel and placed under argon. Dioxane (1 mL) was added, followed by tetra-n-butylammonium fluoride (1 M solution in THF) (2.2 equiv.). The orange solution was stirred at room temperature for 30 minutes. A solution of vinyl iodide (1.1 equiv.) in dioxane (1 mL) was then added. Pd2dba3·CHCl3 (5 mol%) was immediately added as a single portion.Reaction was stirred for 4–6 hours (as determined by thin layer chromatography), at which time it was filtered through a pad of silica and concentrated under reduced pressure. Column chromatography (silica gel) afforded the requisite diene.
Notes and references
- For review of palladium catalysed cross-couplings in total synthesis see: K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed.
- (a) A. L. Hansen, J.-P. Ebran, M. Ahlquist, P.-O. Norrby and T. Skydstrup, Angew. Chem., Int. Ed., 2006, 45, 3349 CrossRef CAS PubMed; (b) M. Lemhadri, A. Battace, F. Berthiol, T. Zair, H. Doucet and M. Santelli, Synthesis, 2008, 1142 CAS.
- For oxidative Heck reaction see: (a) K. S. Yoo, C. H. Yoon and J. W. Jung, J. Am. Chem. Soc., 2006, 128, 16348 Search PubMed; (b) C. Zheng, D. Wang and S. S. Stahl, J. Am. Chem. Soc., 2012, 134, 16496 CrossRef CAS PubMed; (c) J.-J. Dai, C. Fang, B. Xiao, J. Yi, J. Xu, Z.-J. Liu, X. Lu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 8436 CrossRef CAS PubMed.
- Reviews: (a) Chemistry of Tin, ed. P. J. Smith, Balckie Academic & Professional, New York, 1997 Search PubMed; (b) A. G. Davies, in Organotin Chemistry, VCH, New York, 1998 Search PubMed.
- R. E. Maleczka, Jr., J. M. Lavis, D. H. Clark and W. P. Gallagher, Org. Lett., 2000, 2, 3655 CrossRef.
- (a) R. E. Maleczka, Jr., W. P. Gallagher and I. Tersiege, J. Am. Chem. Soc., 2000, 122, 384 CrossRef; (b) W. P. Gallagher, I. Terstiege and R. E. Maleczka, Jr., J. Am. Chem. Soc., 2001, 123, 3194 CrossRef CAS PubMed; (c) R. E. Maleczka, Jr. and W. P. Gallagher, Org. Lett., 2001, 3, 4173 CrossRef.
- For review of Suzuki–Miyaura coupling reaction see: N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- Selected examples: (a) D. A. Singleton and J. P. Martinez, Tetrahedron Lett., 1991, 32, 7365 CrossRef CAS; (b) S. Pereira and M. Srebnik, Organometallics, 1995, 14, 3127 CrossRef CAS; (c) A. V. Kalinin, S. Scherer and V. Snieckus, Angew. Chem., Int. Ed., 2003, 42, 3399 CrossRef CAS PubMed; (d) N. PraveenGanesh, S. D'Hondt and P. Y. Chavant, J. Org. Chem., 2007, 72, 4510 CrossRef CAS PubMed.
- (a) L. Ricardo and W. R. Roush, Org. Lett., 2007, 9, 4315 CrossRef PubMed; (b) M. Binanzer, G. Y. Fang and V. K. Aggarwal, Angew. Chem., Int. Ed., 2010, 49, 4264 CrossRef CAS PubMed; (c) A. Robinson and V. K. Aggarwal, Org. Biomol. Chem., 2012, 10, 1795 RSC.
- (a) G. A. Molander and L. A. Felix, J. Org. Chem., 2005, 70, 3950 CrossRef CAS PubMed; (b) G. A. Molander and Y. Yokoyama, J. Org. Chem., 2006, 71, 2493 CrossRef CAS PubMed; (c) For mechanism of RBF3K reactions see: A. J. J. Lennox and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2012, 134, 7431 CrossRef CAS PubMed.
- (a) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716 CrossRef CAS PubMed; (b) S. J. Lee, K. C. Gray, J. S. Paek and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 466 CrossRef CAS PubMed; (c) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 14084 CrossRef CAS PubMed; (d) B. E. Uno, E. P. Gillis and M. D. Burke, Tetrahedron, 2009, 65, 3130 CrossRef CAS; (e) D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961 CrossRef CAS PubMed; (f) G. R. Dick, D. Knapp, E. P. Gillis and M. D. Burke, Org. Lett., 2010, 12, 2314 CrossRef CAS PubMed; (g) J. R. Struble, S. J. Lee and M. D. Burke, Tetrahedron, 2010, 66, 4710 CrossRef CAS; (h) E. M. Woerly, A. H. Cherney, E. K. Davies and M. D. Burke, J. Am. Chem. Soc., 2010, 132, 6941 CrossRef CAS PubMed; (i) S. J. Lee, T. M. Anderson and M. D. Burke, Angew. Chem., Int. Ed., 2010, 49, 8860 CrossRef CAS PubMed; (j) S. Fujii, S. Y. Chang and M. D. Burke, Angew. Chem., Int. Ed., 2011, 50, 7862 CrossRef CAS PubMed; (k) G. R. Dick, E. M. Woerly and M. D. Burke, Angew. Chem., Int. Ed., 2012, 51, 2667 CrossRef CAS PubMed; (l) E. M. Woerly, J. Roy and M. D. Burke, Nat. Chem., 2014, 6, 484 CrossRef CAS PubMed.
- (a) S. Baba and E.-i. Negishi, J. Am. Chem. Soc., 1976, 98, 6729 CrossRef CAS; (b) F. Feng and E.-i. Negish, Org. Lett., 2001, 3, 719 CrossRef; (c) Z. Huang and E.-i. Negishi, Org. Lett., 2006, 8, 3675 CrossRef CAS PubMed.
- For a comprehensive review of organosilicon couplings see: W. T. Chang, R. C. Smith, C. S. Regens, A. D. Bailey, N. S. Werner and S. E. Denmark, Org. React., 2011, 75, 213 CrossRef CAS.
- Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918 CrossRef CAS.
- S. E. Denmark and M. H. Ober, Org. Lett., 2003, 5, 1357 CrossRef CAS PubMed.
- (a) S. E. Denmark and J. Y. Choi, J. Am. Chem. Soc., 1999, 121, 5821 CrossRef CAS; (b) S. E. Denmark, D. Werhli and J. Y. Choi, Org. Lett., 2000, 2, 2491 CrossRef CAS PubMed; (c) For sequential cross-coupling of 1,4-bissilylbutadienes see: S. E. Denmark and S. A. Tymonko, J. Am. Chem. Soc., 2005, 127, 8004 CrossRef CAS PubMed.
- (a) M. G. McLaughlin and M. J. Cook, Chem. Commun., 2011, 47, 11104 RSC; (b) M. G. McLaughlin and M. J. Cook, J. Org. Chem., 2012, 77, 2058 CrossRef CAS PubMed; (c) C. A. McAdam, M. G. McLaughlin, A. J. S. Johnston, J. Chen, M. W. Walter and M. J. Cook, Org. Biomol. Chem., 2013, 11, 4488 RSC; (d) A. J. S. Johnston, M. G. McLaughlin, J. P. Reid and M. J. Cook, Org. Biomol. Chem., 2013, 11, 7662 RSC; (e) M. G. McLaughlin and M. J. Cook, Chem. Commun., 2014, 50, 3501 RSC; (f) J. P. Reid, C. A. McAdam, A. J. S. Johnston, M. N. Grayson, J. M. Goodman and M. J. Cook, J. Org. Chem., 2015, 80, 1472 CrossRef CAS PubMed.
- B. M. Trost, M. R. Machacek and Z. T. Ball, Org. Lett., 2003, 5, 1895 CrossRef CAS PubMed.
- (a) S. W. Chavhan and M. J. Cook, Chem. – Eur. J., 2014, 20, 4891 CrossRef CAS PubMed; (b) M. G. McLaughlin, C. A. McAdam and M. J. Cook, Org. Lett., 2015, 17, 10 CrossRef CAS PubMed.
- (a) B. M. Trost, M. U. Frederiksen, J. P. N. Papillon, P. E. Harrington, S. Shin and B. T. Shireman, J. Am. Chem. Soc., 2005, 127, 3666 CrossRef CAS PubMed; (b) S. E. Denmark and S. Fujimori, J. Am. Chem. Soc., 2005, 127, 8971 CrossRef CAS PubMed; (c) Y. Zhang and J. S. Panek, Org. Lett., 2007, 9, 3141 CrossRef CAS PubMed; (d) S. E. Denmark, J. H.-C. Liu and J. M. Muhuhi, J. Org. Chem., 2010, 76, 201 CrossRef PubMed; (e) T. Takeda, H. Wasa and A. Tsubouchi, Tetrahedron Lett., 2011, 52, 4575 CrossRef CAS.
- Y. Zhao and V. Snieckus, Org. Lett., 2013, 16, 390 CrossRef PubMed.
- (a) J. Montenegro, J. Bergueiro, C. Saá and S. López, Org. Lett., 2009, 11, 141 CrossRef CAS PubMed; (b) J. Bergueiro, J. Montenegro, F. Cambeiro, C. Saá and S. López, Chem. – Eur. J., 2012, 18, 4401 CrossRef CAS PubMed; (c) J. Montenegro, J. Bergueiro, C. Saá and S. López, Chem. – Eur. J., 2012, 18, 14100 CrossRef PubMed.
- (a) S. E. Denmark and Z. Wang, Org. Lett., 2001, 3, 1073 CrossRef CAS PubMed; (b) S. E. Denmark and Z. Wang, Org. Synth., 2005, 81, 54 CrossRef CAS , Coll. Vol., 2009, 11, 367.
- For full experimental details and compound characterisation see ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, compound characterisation and spectra of the compounds reported (1H and 13C NMR). See DOI: 10.1039/c5qo00002e |
| This journal is © the Partner Organisations 2015 |