
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recycling of rare earths from NdFeB magnets using a combined leaching/extraction system based on the acidity and thermomorphism of the ionic liquid [Hbet][Tf2N]†
David
Dupont
and
Koen
Binnemans
*
KU Leuven, Department of Chemistry, Celestijnenlaan 200F – P.O. Box 2404, 3001 Leuven, Belgium. E-mail: Koen.Binnemans@chem.kuleuven.be
First published on 2nd March 2015
Abstract
The continuous miniaturization of electric motors, hard disks and wind turbines is causing an increasing demand for high-performance neodymium–iron–boron magnets (NdFeB). The supply risk for the rare-earth elements (REEs) used in these magnets is a growing concern and has sparked the development of recycling schemes for these end-of-life products. In this paper a new recycling process for (microwave) roasted NdFeB magnets is proposed, based on the carboxyl-functionalized ionic liquid: betainium bis(trifluoromethylsulfonyl)imide, [Hbet][Tf2N]. Using the thermomorphic properties of the [Hbet][Tf2N]–H2O system, a combined leaching/extraction step was designed. The change from a homogeneous system during leaching (80 °C) to a biphasic system at room temperature causes the dissolved metal ions to distribute themselves amongst the two phases. The valuable elements (Nd, Dy, Co) are thus separated from the iron with high separation factors. The stripping was done very efficiently using oxalic acid to precipitate the REE(III) and cobalt(II) ions while transferring the iron(III) from the ionic liquid to the water phase as a soluble oxalate complex. The cobalt (present in certain magnets) was removed by treating the mixed (REE/Co) oxalate precipitate with aqueous ammonia. The remaining REE oxalate was then calcined to form the REE oxides (99.9% pure). The ionic liquid is regenerated during the stripping step and contamination of the water phase was avoided by salting-out the ionic liquid with Na2SO4. This innovative recycling process features a combined leaching/extraction in mild conditions using a reusable acidic ionic liquid and an energy-efficient microwave roasting of the magnets. These aspects all contribute towards the green character of this process which can be considered as a sustainable and efficient alternative to mineral acid leaching and solvent extraction.
Introduction
The rare-earth elements (REEs) include the 15 lanthanides plus yttrium and scandium. They are used in many high-tech applications including wind turbines, electric vehicles, NiMH batteries, hard disk drives and fluorescent lamps.1–5 The demand for these elements is expected to grow by more than 8% per year, mainly driven by the increasing demand for the strong neodymium–iron–boron (NdFeB) magnets.2–6 NdFeB magnets contain two critical REEs, neodymium (Nd) and dysprosium (Dy), and the demand for these elements is expected to grow by 700% and 2600% respectively over the next 25 years as the NdFeB market expands rapidly.2 Currently, China dominates the REE supply from primary mining (>90% of the global production in 2013) and the worldwide recycling rate is still very low (<1%).4,6,7 Since the tightening of the Chinese export quota for rare earths in 2010, the rare-earth elements have been labeled as critical raw materials by the European Commission and the U.S. Department of Energy.1,3,4,6,8 Opening or reopening mines outside China requires time and large financial investments. Therefore recycling of REEs from end-user products like NdFeB magnets (38% of the market by value) could help to secure the supply of these critical elements.3 It is estimated that by 2020 over 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes of REEs will be held in NdFeB magnets worldwide in wind turbines, electric motors, hard disk drives, speakers, etc.3 Efficient recycling of these NdFeB magnets could therefore help to secure the supply of neodymium and dysprosium and to create a closed-loop system in order to solve the balance problem which is caused by the unwanted co-production of other rare-earth elements during primary mining.3,9 For example, the market for light rare earths is mainly driven by the demand for neodymium for NdFeB magnets, which means that enough ore has to be mined in order to meet the demand for neodymium, causing excess production of lanthanum and cerium which are far more abundant in most ores.9 This unwanted co-production has led to the accumulation of large stocks of certain elements (La, Ce) while facing shortages of others (Y, Eu, Tb, Nd, Dy).3,4,8–12 To remediate this problem and to secure the supply of certain critical REEs, new and increasingly efficient recycling schemes are being developed, opening up new possibilities for urban mining and rare-earth waste revalorization.1,3,10,11,13–18 The potential for direct recycling from end-user products instead of storing the materials in landfills is very high. Unfortunately, the collection of used NdFeB magnets is still not widely organized, but manufacturers are now putting in place initiatives to collect end-of-life consumer products to retrieve the valuable magnets. Examples include air conditioning manufacturer Hitachi, that has started collecting and recycling initiatives for the NdFeB magnets in their products.3 High quality cars are also being considered, since these can contain as much as hundred magnets per car. The number of recycling schemes for NdFeB magnets is growing rapidly as researchers try to develop more efficient processes to recover the rare earths. Many approaches have been proposed, not only hydrometallurgical routes, but also pyrometallurgical routes, glass slag methods, liquid metal extraction, gas-phase extraction methods, etc.3 Several hydrometallurgical processes involve a preliminary roasting step to transform the NdFeB magnets into the metal oxides.3,12 The dissolution of roasted magnets is partially selective since the dissolution of iron and cobalt oxides is kinetically slower than the dissolution of rare-earth oxides, but the iron and cobalt will always go into solution to a certain extent. It has been demonstrated by Vander Hoogerstraete et al. that by leaching with HCl while controlling the pH, the leached iron can be immediately reprecipitated as iron(III) hydroxide, effectively removing iron from the solution.12
000 tonnes of REEs will be held in NdFeB magnets worldwide in wind turbines, electric motors, hard disk drives, speakers, etc.3 Efficient recycling of these NdFeB magnets could therefore help to secure the supply of neodymium and dysprosium and to create a closed-loop system in order to solve the balance problem which is caused by the unwanted co-production of other rare-earth elements during primary mining.3,9 For example, the market for light rare earths is mainly driven by the demand for neodymium for NdFeB magnets, which means that enough ore has to be mined in order to meet the demand for neodymium, causing excess production of lanthanum and cerium which are far more abundant in most ores.9 This unwanted co-production has led to the accumulation of large stocks of certain elements (La, Ce) while facing shortages of others (Y, Eu, Tb, Nd, Dy).3,4,8–12 To remediate this problem and to secure the supply of certain critical REEs, new and increasingly efficient recycling schemes are being developed, opening up new possibilities for urban mining and rare-earth waste revalorization.1,3,10,11,13–18 The potential for direct recycling from end-user products instead of storing the materials in landfills is very high. Unfortunately, the collection of used NdFeB magnets is still not widely organized, but manufacturers are now putting in place initiatives to collect end-of-life consumer products to retrieve the valuable magnets. Examples include air conditioning manufacturer Hitachi, that has started collecting and recycling initiatives for the NdFeB magnets in their products.3 High quality cars are also being considered, since these can contain as much as hundred magnets per car. The number of recycling schemes for NdFeB magnets is growing rapidly as researchers try to develop more efficient processes to recover the rare earths. Many approaches have been proposed, not only hydrometallurgical routes, but also pyrometallurgical routes, glass slag methods, liquid metal extraction, gas-phase extraction methods, etc.3 Several hydrometallurgical processes involve a preliminary roasting step to transform the NdFeB magnets into the metal oxides.3,12 The dissolution of roasted magnets is partially selective since the dissolution of iron and cobalt oxides is kinetically slower than the dissolution of rare-earth oxides, but the iron and cobalt will always go into solution to a certain extent. It has been demonstrated by Vander Hoogerstraete et al. that by leaching with HCl while controlling the pH, the leached iron can be immediately reprecipitated as iron(III) hydroxide, effectively removing iron from the solution.12
The process described in this paper proposes an alternative that uses a fully reusable ionic liquid leaching agent instead of consuming mineral acids such as HCl, HNO3 or H2SO4.3 The carboxyl-functionalized ionic liquid betainium bis(trifluoromethylsulfonyl)imide, [Hbet][Tf2N], (Fig. 1) is a particularly interesting ionic liquid because of its ability to selectively dissolve metal oxides.19–22 It has also been used as organic phase in solvent extraction studies.23–29 Recently, [Hbet][Tf2N] was used for the design of a highly efficient and sustainable recycling process for rare earths from lamp phosphor waste powders.22 While that recycling process was based on a different concept, it does however show that this ionic liquid is very suitable for the green recycling and processing of metal oxide containing materials. Such task-specific ionic liquids and ionometallurgy in general, continue to show great potential for this type of specialized applications, opening up new possibilities compared to traditional hydrometallurgy.22,30–34 Many other domains such as extraction, electrochemistry, organic synthesis and catalysis, have also benefitted from the unique properties of ionic liquids. On many occasions, ionic liquids have shown to be very efficient green solvents and improve the safety of a process thanks to their negligible vapor pressure, low-flammability and relatively low toxicity.15,17,34–39
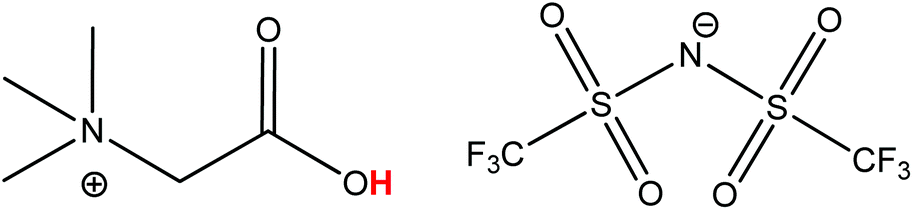 | ||
| Fig. 1 Structure of the ionic liquid betainium bis(trifluoromethylsulfonyl)imide [Hbet][Tf2N]. The acidic proton of the betaine group (pKa 1.83), responsible for the dissolution of metal oxides, is highlighted in red.40 | ||
Here, [Hbet][Tf2N] was used to selectively dissolve the valuable rare-earth oxides from the roasted NdFeB magnets, leaving most of the iron oxide matrix behind. Moreover, [Hbet][Tf2N]–H2O mixtures display thermomorphic behavior with an upper critical solution temperature (UCST) meaning that they will form one homogeneous phase above the cloud point temperature (55 °C for a 1:1 wt/wt system) and two phases below that temperature.21 The metals will then selectively distribute between both phases. Such a combined leaching/extraction system that automatically separates the metals when cooling down the leaching solution is innovative and applicable to NdFeB magnet recycling. The resulting process is a sustainable alternative to mineral acid leaching processes that often require solvent extraction and produce significant volumes of waste water. Since [Hbet][Tf2N] is a fluorinated ionic liquid, contamination of the water phase with ionic liquid should be avoided during the process. The loss of ionic liquid was circumvented by salting-out with Na2SO4.
Experimental
Chemicals
MgCl2 (98%), Mg(NO3)2·6H2O (99%), LiNO3 (99%), NaCl (98%), NaClO4·H2O (98%) and D2O (99.9 atom% D) were obtained from Sigma-Aldrich (Diegem, Belgium), CaCl2 (99.5%), NaNO3 (99%), KNO3 (99%), Ca(NO3)2·4H2O (99%) and Na2SO4 (99%) were obtained from Chem-Lab (Zedelgem, Belgium). KCl (99.5%) and NaI (99.5%) were purchased from AppliChem (Darmstadt, Germany) and LiCl (99%) from Fisher Chemical (UK). Nd(NO3)3·6H2O (99%) was obtained from Alfa Aeser (Karlsruhe Germany) and NH4NO3 (99%) from Chempur (Karlsruhe, Germany). NH4Cl (99.5%), Dy(NO3)3·6H2O (99%), Co(NO3)2·6H2O (99%), 1,4-dioxane (99.9 wt%) and betaine hydrochloride (HbetCl) (99%) were obtained from Acros Organics (Geel, Belgium). Lithium bis(trifluoromethylsulfonyl)imide (LiTf2N) (99%) was purchased from IoLiTec (Germany). Oxalic acid dihydrate (>99.5%) and Fe(NO3)3·9H2O (99%) were purchased from J.T. Baker. The silicone solution in isopropanol was obtained from SERVA Electrophoresis GmbH (Germany) and the praseodymium, holmium and erbium standard solutions (1000 mg L−1) were obtained from Merck (Overijse, Belgium). All chemicals were used as received without further purification.Equipment and characterization
1H NMR spectra were recorded on a Bruker Avance 300 spectrometer, operating at a frequency of 300 MHz. The samples were prepared by dissolving a small amount of product in heavy water (D2O). The viscosity of the ionic liquid was measured using an automatic Brookfield plate cone viscometer, Model LVDV-II CP (Brookfield Engineering Laboratories, USA). The morphology and size distribution was determined by scanning electron microscopy (SEM) using a Philips XL 30 FEG device equipped with EDX. Powder X-ray diffraction (XRD) was carried out on an Agilent SuperNova X-ray diffractometer, using Mo Kα radiation (λ = 0.71073 Å) and a CCD detector. A Retsch ZM100 centrifugal mill (Gemini BV), a Retsch RS100 vibratory disc mill and a Retsch PM400 planetary ball mill were used for grinding of the magnet and a muffle furnace for roasting. A Heraeus Megafuge 1.0 centrifuge was used to separate the magnet residue from the ionic liquid after the leaching experiments. Total reflection X-ray fluorescence spectroscopy (TXRF) was performed with a Bruker S2 Picofox TXRF spectrometer equipped with a molybdenum source. For the sample preparation, plastic microtubes were filled with a small amount of ionic liquid sample (50 mg), 1:1 vol/vol ethanol–H2O (700 μL) and a mix of standard solutions (1000 mg L−1): praseodymium (100 μL), holmium (50 μL) and erbium (25 μL). This mix is the result of an optimization procedure to find suitable standards with X-ray energies that closely match the energy of the probed elements (Nd, Fe, Dy, Co) in order to reduce the effects of secondary absorption of X-rays by the matrix (ESI, Fig. S3 and Table S1†). High-matrix ionic liquid containing samples require such a procedure, while low-matrix aqueous solutions can be accurately analyzed using just one standard. The microtubes were then vigorously shaken on a vibrating plate (IKA MS 3 basic). Finally, a 1 μL drop of this solution was put on a quartz plate, previously treated with a silicone–isopropanol solution (Serva®) to avoid spreading of the sample droplet on the quartz plate. The quartz plates were then dried for 30 min at 60 °C prior to analysis. Each sample was measured for 5 min.
Synthesis of [Hbet][Tf2N]
The ionic liquid [Hbet][Tf2N] was synthesized according to a one-step literature method based on the reaction between HbetCl and LiTf2N.21,22 The level of chloride impurities was below 1 ppm (TXRF). Dry ionic liquid was obtained using a rotary evaporator. Increasing amounts of water were then added to obtain ionic liquids with different water contents. The water-saturated ionic liquid was prepared by keeping the ionic liquid in contact with an excess of water and then using the water-saturated ionic liquid phase (contains 14 wt% H2O).Magnet pre-processing: roasting and milling
The demagnetized NdFeB magnets were obtained from the University of Birmingham (UK) and Goudsmit Magnetics, and had the following composition (Table 1). The full ICP-MS trace analysis results are given in the ESI, Table S1.† The main difference between various NdFeB magnets lies in the cobalt and dysprosium content, which is added to high-end magnets in order to improve their magnetic and thermal properties. In reality, few magnets contain as much cobalt as magnet 1, but it is interesting to trace the movement of the valuable cobalt in any proposed recycling process. In this paper, the experiments were therefore mainly carried out with the magnet of type 1. At the end, the full recycling process was also tested for the magnet of type 2. Both magnets were demagnetized and then crushed and milled to obtain a magnet powder with a particle size < 400 μm.| Magnet 1 | Composition (wt%) | Magnet 2 | Composition (wt%) |
|---|---|---|---|
| a The total does not add up to 100% due to the presence of trace elements (Al, Nb, Pr, Cu, Mn, N, O, C, Si,…) and the analytical error associated with ICP-MS measurements. The full detailed composition is given in the ESI, Table S1. | |||
| Fe | 58.16 | Fe | 66.01 |
| Nd | 25.95 | Nd | 29.17 |
| Dy | 4.22 | Dy | 1.98 |
| Co | 4.21 | Co | 0.36 |
| B | 1.00 | B | 1.02 |
| Totala | 93.5 | Totala | 98.54 |
The powders were then roasted using a conventional oven at 950 °C for 15 h to assure full conversion to the metal oxides as described by Vander Hoogerstraete et al.12 An XRD diffractogram of the roasted magnet powder is given in the ESI (Fig. S2†). A proof-of-concept is also given for the roasting of magnets with microwaves. An analytical (monomodal) high-temperature microwave set-up was used to study the heating and roasting of NdFeB magnets with microwaves (Fig. 2).
 | ||
| Fig. 2 Analytical (monomodal) high-temperature microwave set-up, consisting of a microwave cavity (2) with a quartz table connected to a balance (1), an infra-red temperature probe (3) and a high-power microwave source (4). | ||
The oven-roasted particles were then crushed to obtain three different particle sizes. In the first case, the particles were crushed with a pestle and mortar to obtain large particles with an average diameter of (310 ± 140) μm. The other two particle sizes were obtained by using a ball mill and passing the powder through 90 μm, 60 μm and 40 μm sieves. Two fractions were retained: the fraction <90 μm that did not pass the 60 μm sieve, and the <40 μm fraction. These fractions had an average diameter of (73 ± 40) μm and (6 ± 3) μm, respectively. The particle size was determined by SEM and ImageJ software (Fig. S1†).
Leaching experiments
Small glass vials (4 mL) were filled with a fixed amount of [Hbet][Tf2N] (1 g) and 10 mg of roasted magnet particles which resulted in a solid/liquid ratio of 10 mg g−1. Water-containing ionic liquid (2.5–10 wt%) or a 1:1 wt/wt [Hbet][Tf2N]–H2O mixture was used depending on the experiment. A magnetic stirring bar was then added to each of the vials and they were sealed using a plastic screw cap. The dissolution experiments were carried out in an oil bath at a set temperature while stirring (600 rpm). The samples were then placed in a centrifuge (5300 rpm, 30 min) to precipitate the undissolved magnet residue. The metal content in the ionic liquid was determined using total reflection X-ray fluorescence spectroscopy (TXRF).
Quantitative 1H NMR
The dissolution of ionic liquid in the water phase was studied and optimized in order to assure that no ionic liquid would be lost during the process and no contamination of the water phase would occur. The concentration of ionic liquid in the aqueous phase was determined using quantitative 1H NMR with 1,4-dioxane as an internal standard. A sample of the water phase was taken (100 mg) and a known amount of 1,4-dioxane–D2O solution was added so that the concentration of 1,4-dioxane internal standard would be comparable to the amount of ionic liquid in the water phase. The 1H NMR spectrum of 1,4-dioxane shows one peak at δ = 3.6, corresponding to its 8 equivalent protons and does not overlap with the spectrum of water or [Hbet][Tf2N]. The relative concentration versus 1,4-dioxane and the absolute concentration of [Hbet][Tf2N] were calculated by integration of the peaks using Spinworks software.Optimized full recycling process
First the magnets were roasted. This was done in a conventional oven as described by Vander Hoogerstraete et al.,12 or in a microwave oven at 950 °C. The second method is preferred since it is more energy-efficient. The roasted magnets were then crushed/milled to reduce their particle size. This powder was leached in a 1:1 wt/wt [Hbet][Tf2N]–H2O mixture for 24–48 h (80 °C) to obtain full leaching of the REEs (Nd and Dy) and partial leaching of cobalt and iron. Thanks to the thermomorphic properties of the [Hbet][Tf2N]–H2O system, the solution is homogeneous during leaching (80 °C) but phase separates when cooling it to room temperature (25 °C). The dissolved metals distribute amongst the two phases with iron(III) going to the ionic liquid phase and the REEs and cobalt(II) going to the aqueous phase. NaNO3 is used as an additive to enhance the separation of the metals. The two phases were then separated from each other and the metal content was determined by TXRF. The water phase was stripped with stoichiometric amounts of pure (solid) oxalic acid (10 min, 25 °C, stirring) to precipitate the REEs and cobalt(II) as a mixed oxalate. The oxalate precipitate was removed by filtration and the cobalt(II) was fully removed by contacting the precipitate with aqueous ammonia and shaking it (2000 rpm) for 10 min at 25 °C. The resulting, highly pure, REE oxalate (>99.9 wt%) can be calcined to obtain the rare-earth oxides. The iron(III)-containing ionic liquid was stripped with an oxalic acid solution. This transfers the iron(III) to the water phase as a soluble oxalate complex, effectively cleaning the ionic liquid and regenerating it by reprotonation of the carboxylate groups. The waste water was cleaned using Na2SO4 to salt-out the remaining traces of ionic liquid, and Ca(OH)2 to precipitate the [Fe(C2O4)3]3− complex as Fe(OH)3(s) and CaC2O4(s).
Results and discussion
Roasting
Roasting or oxidative roasting of NdFeB magnets transforms all the elements in the magnet into their respective oxides. The resulting roasted magnet consisted mainly of Fe2O3, Nd2O3, Dy2O3 and CoO. The roasting of metallic cobalt at 950 °C leads to the formation of cobalt(II) oxide.41 The oxidative roasting of NdFeB magnets was done in an oven as described by Vander Hoogerstraete et al. (950 °C, 3–15 h).12 But here a proof-of-concept is also given for an alternative, more sustainable, roasting process based on microwave heating. Microwave energy has a lot of potential in mineral treatment operations such as heating, drying, leaching, roasting and smelting.42–44 Hua et al. reported for example a 4 to 17 times increase in roasting speed for a copper sulfide concentrate.44 Microwave heating is a non-contact and on/off heating technique that offers a number of advantages over conventional heating such as material-selective and rapid heating due to the fact that the material is heated from the inside (the material itself is the heating source). This significantly reduces the energy consumption and also improves the safety of the process.42 The strong coupling of microwaves with magnetic metals results in a very fast heating of the magnet (Fig. 3) and an energy-efficient roasting compared to traditional oven roasting where the air has to be heated first.45 The fact that the (demagnetized) magnet powder can be heated to 1000 °C in a few seconds is quite remarkable and shows the potential of microwave roasting. The very fast heating can also be used to quickly demagnetize NdFeB magnets by raising the temperature above the Curie temperature (312 °C).46 Slower heating speeds are of course also achievable by reducing the power input.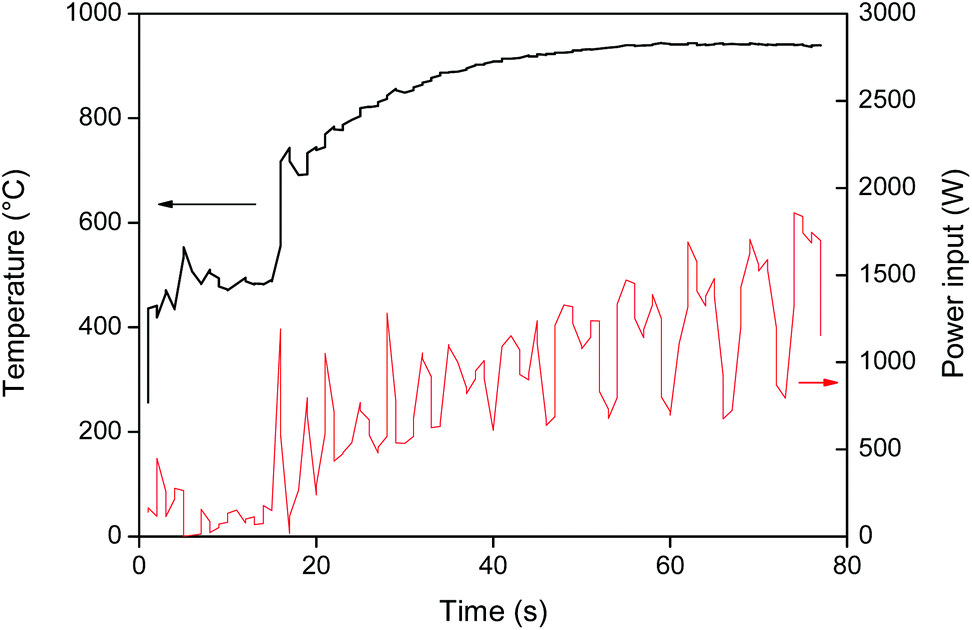 | ||
| Fig. 3 Heating of demagnetized NdFeB magnet particles (≈400 μm) with a monomodal high-temperature microwave set-up. The temperature was raised in two steps: first it was stabilized at 500 °C and then at 950 °C. The power input (W) and temperature (°C) are shown versus time (s). The temperature range under 250 °C was not accessible with the infra-red temperature probe. | ||
Leaching of NdFeB magnets in [Hbet][Tf2N]
The protonated carboxyl-functionalized ionic liquid betainium bis(trifluoromethylsulfonyl)imide [Hbet][Tf2N] (Fig. 1) has the ability to dissolve certain metal oxides, including rare-earth oxides (eqn (1)).20,21| REE2O3 + 6[Hbet][Tf2N] → 2[REE(bet)3][Tf2N]3 + 3H2O | (1) |
Other metal oxides like iron and cobalt oxides are poorly soluble in [Hbet][Tf2N].20,21 This difference in solubility can be exploited to obtain the (partially) selective dissolution of the valuable REE oxides from the roasted NdFeB magnets. Important parameters affecting the selectivity and kinetics are the roasted magnet particle size and the water content in the ionic liquid. Water accelerates the dissolution of metal oxides because it lowers the viscosity, facilitates the exchange of protons from the betaine groups and enhances the solvation of ions in the ionic liquid.20–22 The dissolution of metal oxides was studied by Nockemann et al. and is driven by the reactivity of the carboxylic acid group (pKa 1.83) located on the cation of the ionic liquid.20,21,40,47 The deprotonated zwitterionic betaine groups are also responsible for the complexation of the dissolved metal ions. The Tf2N− anions do not participate in the complex formation but they are essential for the formation of betainium-based hydrophobic ionic liquid, with a low viscosity and good thermal stability.22,47,48
First, water-containing ionic liquid was tested (2.5, 5, 7.5 and 10 wt% H2O) without exceeding the solubility limit of water in the ionic liquid (14 wt%). Roasted NdFeB magnet particles were then added to the ionic liquid (10 mg g−1) and stirred (600 rpm) at 90 °C for increasing amounts of time. The influence of the water content on the leaching kinetics of the roasted magnet is shown for three different particle sizes (310 ± 140) μm, (73 ± 40) μm and (6 ± 3) μm. The leaching of the largest particles (≈300 μm) is relatively slow but more selective towards the REE oxides (Fig. 4). The addition of water accelerates the leaching but it is clear that the leaching of the large particles is still too slow to fully dissolve the REE oxides from these roasted magnet particles within 100 h (Fig. 4). This is not due to the solubility of these oxides, since in the same conditions, commercial Nd2O3 and Dy2O3 could be fully dissolved in [Hbet][Tf2N] within 24 h. The reason is that the particles are large (≈300 μm) and consist mainly of iron oxides (≈60 wt%), which are very poorly soluble in [Hbet][Tf2N]. The leaching of the REE oxides embedded in this insoluble iron oxide matrix is therefore a slow process.
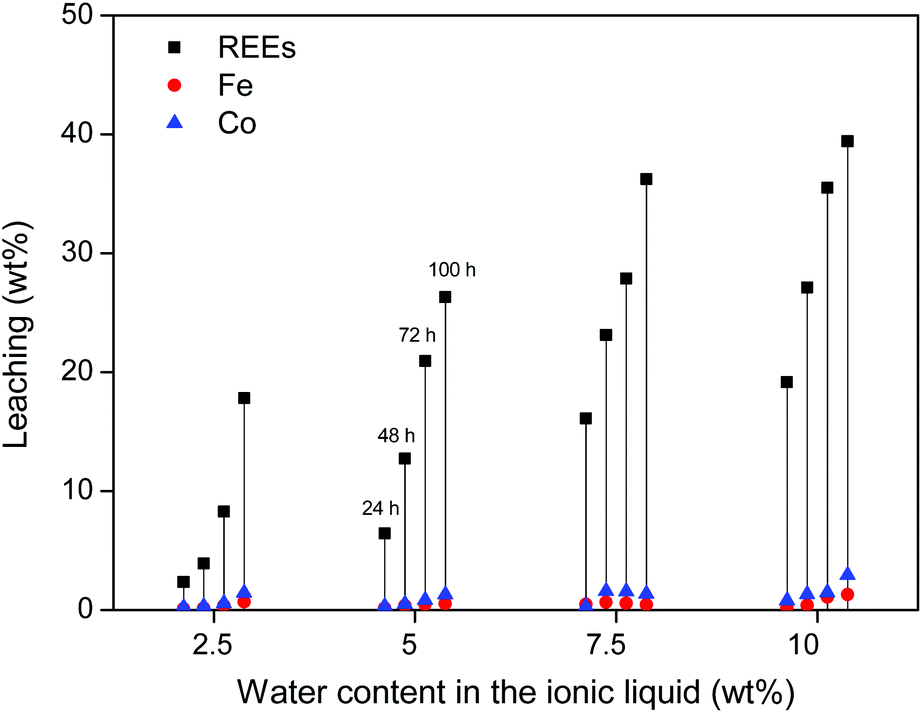 | ||
| Fig. 4 Leaching (%) of Co, Fe and REEs (Nd + Dy) from large roasted NdFeB particles (≈300 μm). The leaching is shown as function of time (24, 48, 72 and 100 h) and as function of the water content in the ionic liquid (2.5, 5, 7.5 and 10 wt%). The solutions were stirred at 600 rpm and 90 °C. | ||
The fact that the particles consist of a large insoluble Fe2O3 matrix hampers the efficient dissolution of the rare-earth oxides because it is very difficult for the ionic liquid to create deep channels in these iron oxide particles. SEM images and surface EDX analysis revealed that on the surface, all the Nd2O3 had been leached selectively without dissolving the Fe2O3 (Fig. 5). This confirms that it is possible to selectively leach the Nd2O3 from these particles. However, the penetration of the acidic ionic liquid in the particles was insufficient to dissolve the rare-earth oxides held in the core of the particles (Fig. 6). The particle size before and after leaching did not change significantly which shows that the large iron matrix is not dissolved and that the rare-earth oxides are leached, leaving behind the iron oxide matrix as a porous structure (Fig. 6). The visible denting of the surface is around 10–20 μm deep (Fig. 6) and EDX shows that the surface is free of Nd2O3 (Fig. 5). By reducing the particle size, the dissolution of rare-earth oxides should therefore be much faster, while retaining a certain selectivity towards the rare-earth oxides.
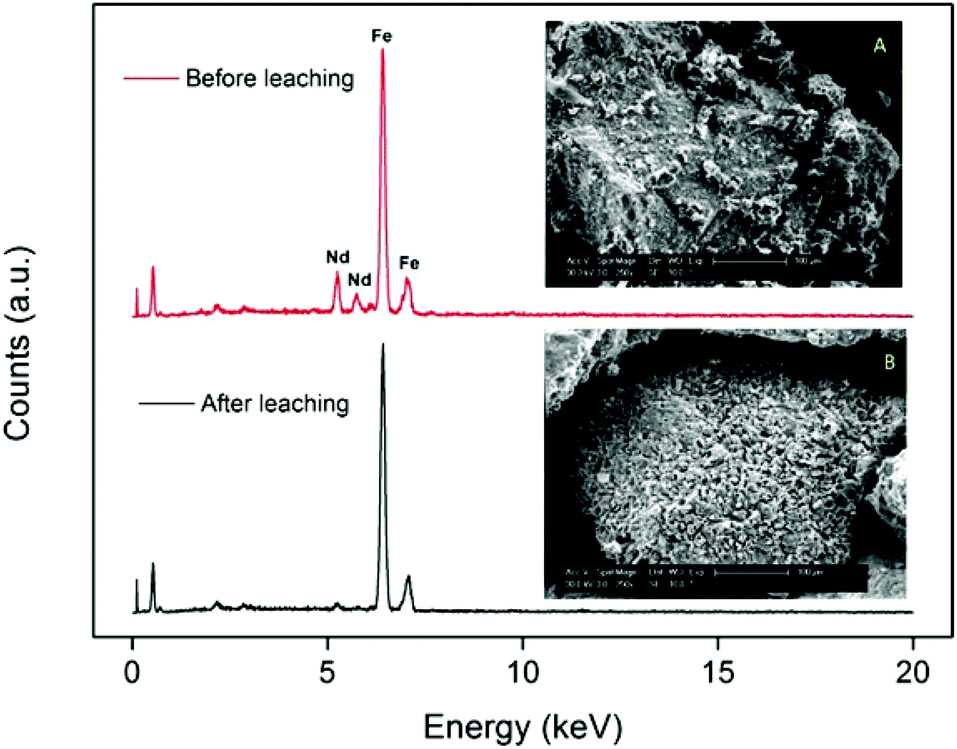 | ||
| Fig. 5 SEM images (magnified 250×) and EDX spectrum of the surface of the large NdFeB particles (≈300 μm) before (A) and after (B) leaching. | ||
 | ||
| Fig. 6 SEM images (magnified 3500×) of the surface of the particles before (A) and after (B) leaching. | ||
The leaching of these smaller particles (≈70 μm and 6 μm) was investigated in the same manner as the large particles, by measuring the leaching of rare earths, iron and cobalt in the ionic liquid with increasing water content. It is clear from Fig. 7 that the leaching of the milled powder is much faster and allows full leaching of the REE content within 48–72 h. Unfortunately, the leaching of iron and cobalt also increases significantly.
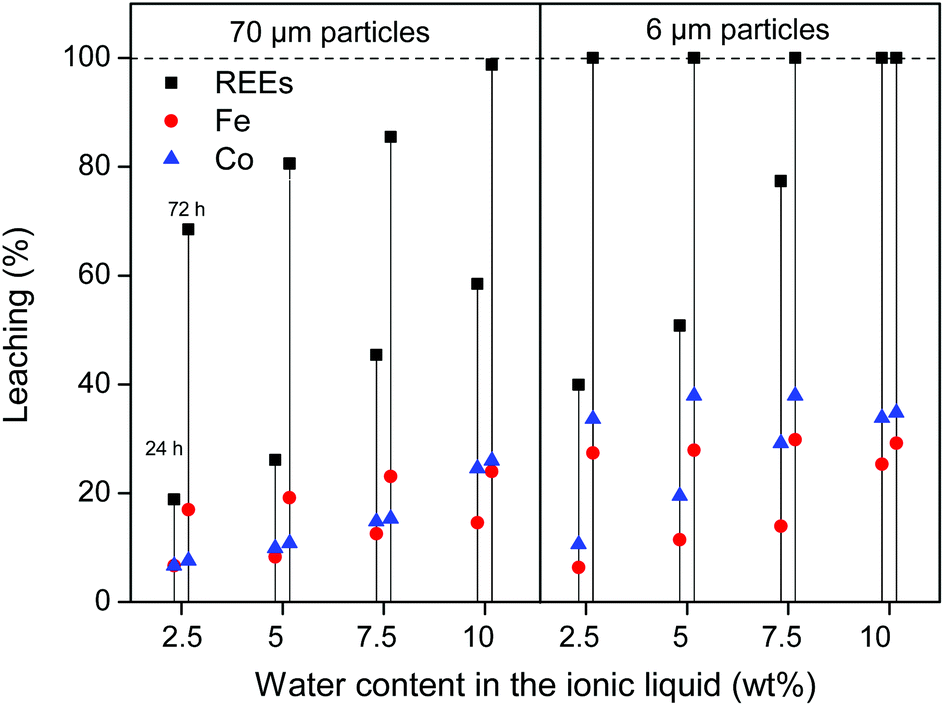 | ||
| Fig. 7 Leaching (%) of Co, Fe and REEs (Nd + Dy) from two different milled roasted NdFeB magnets (≈70 μm and 6 μm). The leaching is shown as function of time (24 and 72 h) and as function of the water content in the ionic liquid (2.5, 5, 7.5 and 10 wt%). The solutions were stirred 600 rpm at 90 °C. | ||
While these results are encouraging, the leaching in water-unsaturated ionic liquid is clearly slow and selective for large particles (Fig. 4) or fast and less selective for smaller particles (Fig. 7). To get full dissolution of the REEs, small particles must be used and too much iron and cobalt goes into solution. This would require additional solvent extraction steps to isolate the REEs and would complicate the recycling process. Instead, an alternative leaching system is proposed based on the same ionic liquid but with an excess of water (>13 wt%) so that it forms a biphasic system at room temperature. This system is very promising and innovative as it combines leaching with metal extraction (separation) in one convenient step thanks to the thermomorphic properties of this ionic liquid/water system.
Thermomorphic leaching/extraction in a [Hbet][Tf2N]–H2O system
The [Hbet][Tf2N]–H2O system shows thermomorphic behavior with an upper critical solution temperature (UCST).21,26,27 This means that above a certain temperature, called the cloud point temperature, it forms one homogeneous phase and under this temperature it is a biphasic system.26,27 In this biphasic system, the iron is mostly present in the ionic liquid phase, while the rare-earth ions and cobalt are mostly present in the water phase due to the higher affinity of Fe(III) ions for betaine compared to REE(III) and Co(II) ions. This observation led us to investigate the possibilities of a thermomorphic leaching system with its automatic metal separation once the system becomes biphasic at room temperature. Such a thermomorphic leaching/extraction system has to the best of our knowledge not been described in the literature yet. The combined leaching/extraction process starts by leaching the roasted NdFeB magnets in a 1:1 wt/wt blend of [Hbet][Tf2N]–H2O at 80 °C (above the cloud point temperature) so that it forms one homogeneous phase. Afterwards, the solution is cooled again to room temperature so the mixture can phase separate into a valuable REE/cobalt-enriched water phase and an iron-enriched ionic liquid phase (Fig. 8).
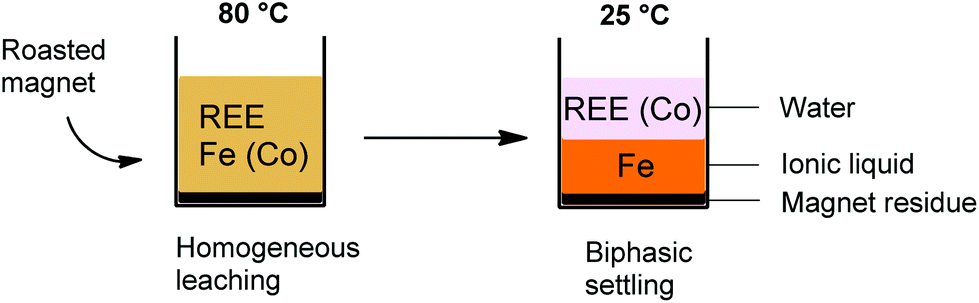 | ||
| Fig. 8 Schematic overview of the biphasic leaching-settling process using a thermomorphic ionic liquid. The [Hbet][Tf2N]–H2O mixture forms one homogeneous phase at 80 °C and forms two phases at room temperature. | ||
First, the leaching of the roasted NdFeB particles was tested using a 1:1 wt/wt [Hbet][Tf2N]–H2O mixture. The leaching of different particle sizes is again shown (300 μm, 70 μm and 6 μm) over time at 80 °C. At this temperature the system forms one homogeneous phase. The samples were then cooled to room temperature to phase separate and centrifuged to precipitate the remaining magnet residue. The total leaching percentage was determined by combining the measured metal content in the water phase and in the ionic liquid phase. The leaching is faster compared to experiments with lower water contents in the ionic liquid (Fig. 4 and 7). The effect of the particle size is similar in the sense that full leaching of the REEs is achieved faster for smaller particles but with less selectivity and slower for larger particles but with higher selectivity (Fig. 9).
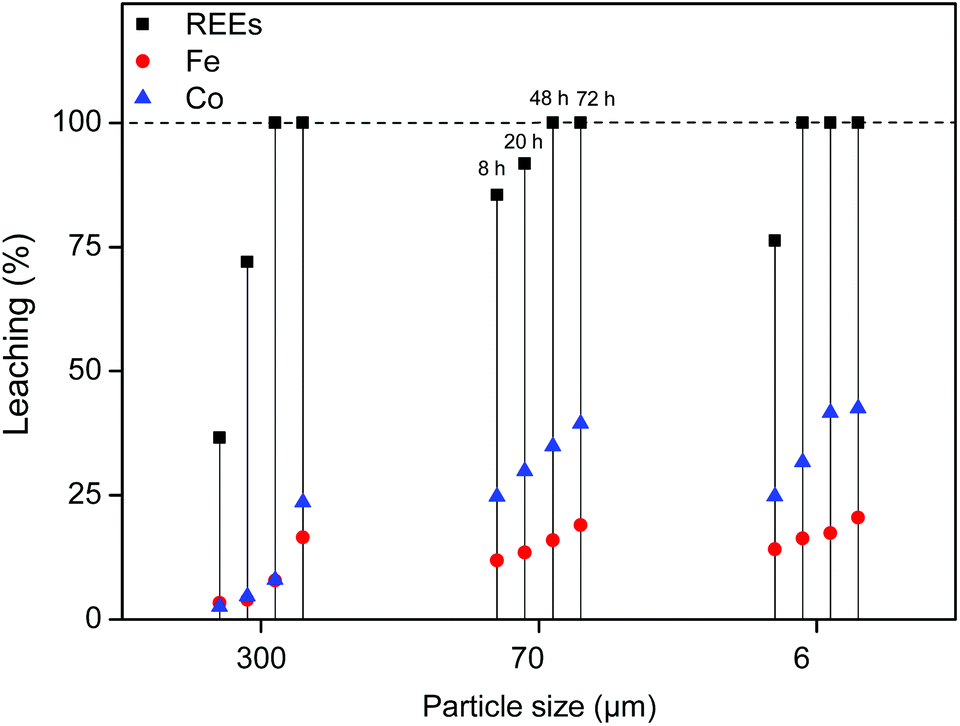 | ||
| Fig. 9 Leaching (%) of REEs, iron and cobalt from roasted NdFeB magnets (10 mg) using a 1:1 wt/wt [Hbet][Tf2N]–H2O system (2 g). The leaching was carried out at 80 °C for 8 h, 20 h and 48 h while stirring (600 rpm). | ||
The leaching temperature of the process was set at 80 °C, because the leaching is sufficiently fast at this temperature in this water-saturated system. At 80 °C the thermomorphic ionic liquid/water system is also certainly homogeneous (cloud point temperature is 55 °C for a 1:1 wt/wt [Hbet][Tf2N]–H2O system). Although closed vials were used, temperatures should not exceed 90 °C due to pressure buildup. At 80 °C, the viscosity of the ionic liquid/water system is also sufficiently low and almost equal to pure water (Fig. 10). The high temperature and low viscosity are important because the leaching kinetics of this material is slow, especially since the rare earths are held in a largely insoluble iron oxide matrix.
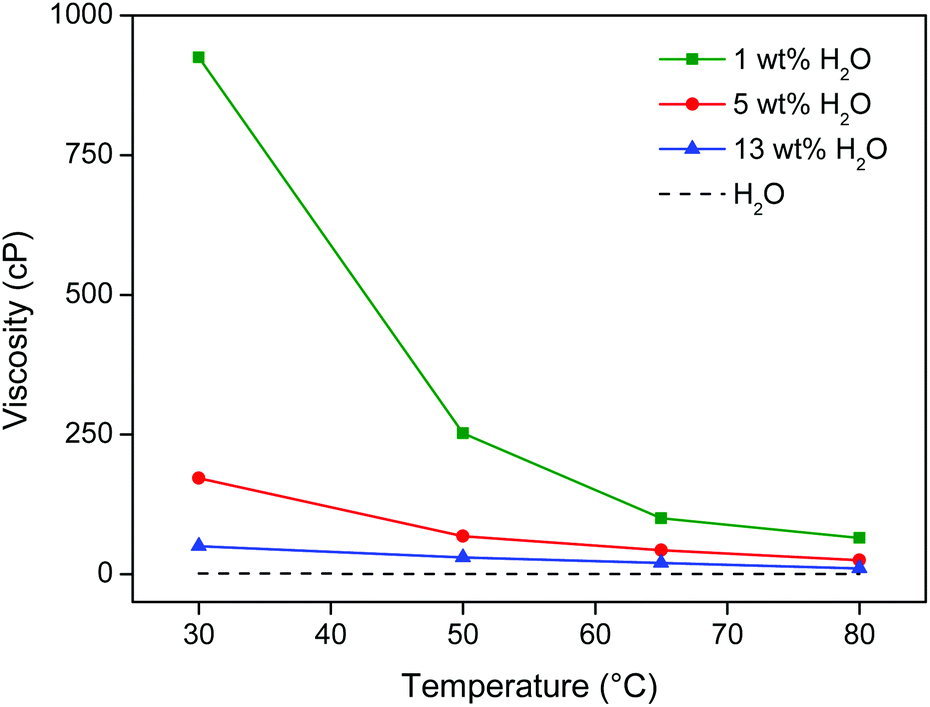 | ||
| Fig. 10 Viscosity (cP) of [Hbet][Tf2N] as function of its water content (wt%) and the temperature (°C). The viscosity of water is shown as a reference. | ||
Next, the metal separation over the two phases was optimized by studying the distribution of Nd(III), Dy(III), Fe(III) and Co(II) ions between the ionic liquid and the water phase. Onghena and Binnemans recently showed that Fe(III) can be extracted to the [Hbet][Tf2N] phase, while the REE(III) ions remain in the aqueous phase.29 We also tested the effect of inorganic salts in the water phase on the distribution of the metals. The advantage of the [Hbet][Tf2N] ionic liquid is that Tf2N− is one of the most difficult anions to displace from the ionic liquid so that no anion exchange reactions will occur for [Hbet][Tf2N]. A high salt concentration could therefore be used in the water phase without detecting any traces of the anions in the ionic liquid. A distinction must be made between the effect of salt anions and salt cations. Water-soluble salt anions can form complexes with the metal ions and therefore change the affinity of the metal ions for the water phase. Additionally, salt anions and cations can influence the solubility of the ionic liquid in the water phase.49 It was therefore interesting to investigate if salts could influence the betaine content in the water phase and thus influence the distribution of iron(III) as it forms iron-betaine complexes rather than aqua complexes. A synthetic mix (4 × 1000 ppm) of hydrated Nd(NO3)3, Dy(NO3)3, Fe(NO3)3 and Co(NO3)2 was mixed with a 1:1 wt/wt [Hbet][Tf2N]–H2O system. This ionic liquid/water mixture was heated for 15 min at 80 °C to obtain a homogeneous phase similar to the one formed during leaching. The mixture was then allowed to cool so that it would phase separate and equilibrate. The metal content was determined in each phase and expressed as the distribution ratio between the water phase and the ionic liquid phase (eqn (2)). A distribution ratio larger than 1 corresponds to a greater affinity for the water phase than for the ionic liquid phase.
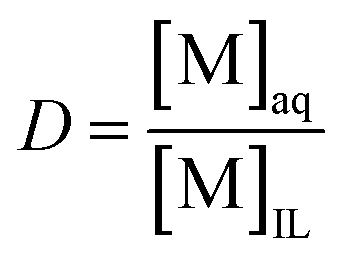 | (2) |
First the effect of salt anions is shown. It is clear from Fig. 11 that this biphasic leaching process effectively allows the separation of the valuable elements (Nd, Dy, Co) from iron. The separation of iron(III) from the REE(III) and Co(II) ions is quite efficient because the strong iron-betaine complexes cause the iron to stay in the ionic liquid, while the Nd, Dy and Co ions are present in the water phase as aqua complexes.
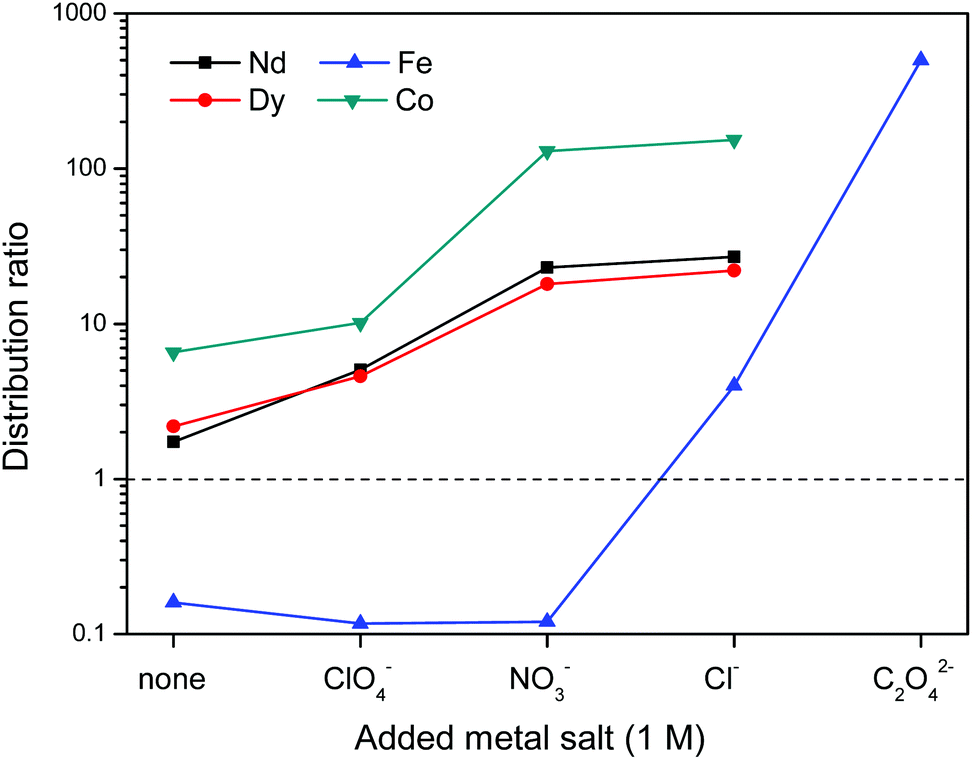 | ||
| Fig. 11 Influence of added metal salts (1 M) with varying anions on the distribution ratio of Nd(III), Dy(III), Fe(III) and Co(II) ions in a biphasic [Hbet][Tf2N]–H2O system. A high distribution ratio corresponds to a greater affinity for the water phase. Oxalate forms highly insoluble complexes with REE(III) and Co(II) ions. | ||
The increased distribution factors for Nd(III), Dy(III) and Co(II) when adding coordinating anions are due to a better coordination of metal ions in the water phase compared to the situation with non-coordinating ions such as Tf2N− (in the case of pure water) or ClO4−. The presence of coordinating anions such as nitrates or chlorides significantly improves the solubility of the REE(III) and Co(II) ions in the water phase, but too strongly coordinating anions such as sulfates or oxalates cause precipitation. However, the most striking feature in Fig. 11 is the transfer of iron(III) to the water phase in the presence of strongly coordinating anions such as chloride or oxalate anions (1 M). More weakly-coordinating anions such as nitrates and perchlorates do not cause transfer of iron(III) to the water phase because iron is preferentially present as the iron-betaine complex in that case. A photograph has been added to help visualize the reversal of the iron(III) distribution when adding coordinating anions such as chloride anions or oxalate anions (Fig. 12). To the best of our knowledge, this is the first time that this is described for the [Hbet][Tf2N]–H2O system. Switching between salt media effectively creates a tunable extraction system for iron(III). This phenomenon will also be used to strip iron from the water phase using oxalate anions which form the highly water-soluble [Fe(C2O4)3]3− complex, resulting in high distribution factors.
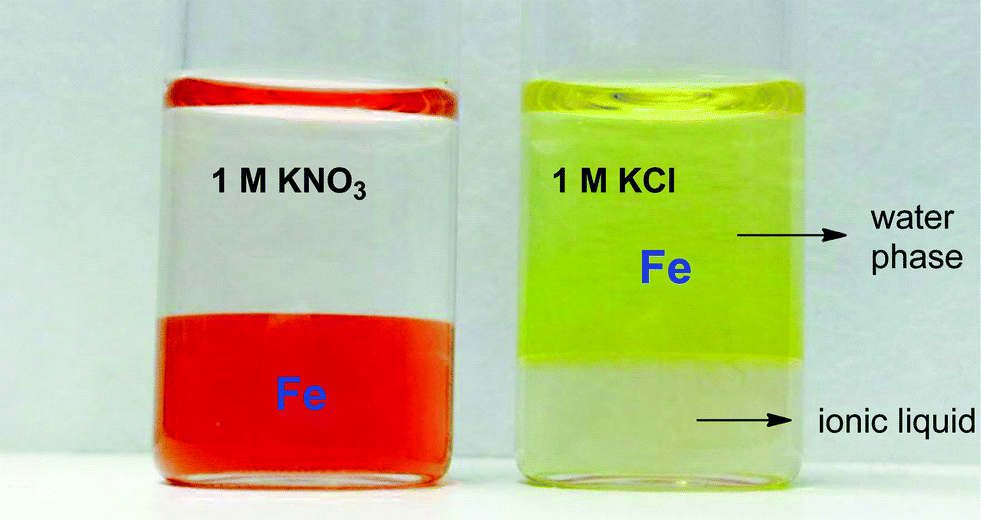 | ||
| Fig. 12 Reversal of iron(III) distribution when adding a non-coordinating salt (KNO3) compared to a salt with coordinating anions (KCl). The upper and lower phase consist of water and [Hbet][Tf2N], respectively. | ||
Salt cations on the other hand, cannot form complexes with the metal ions. They can however impact the solubility of ionic liquid in the water phase and therefore also the solubility of betaine, which would (slightly) affect the distribution factor for iron(III) which is present as an iron-betaine complex. In this context, the concept of salting-in and salting-out salts is important. The “Hofmeister series” ranks different anions and cations according to their salting-in or salting-out behavior, meaning their ability to influence the solubility of organic compounds or proteins in the water phase.50 Salting-out ions diminish the solubility of organic molecules in aqueous solutions and cause aggregation, while salting-in ions enhance the solubility of organic molecules (ionic liquid) in aqueous solutions.49–54 The effect of anions and cations on the ionic liquid [Hbet][Tf2N] was determined by measuring the [Hbet][Tf2N] content in the water phase when adding different salts. This was done using 1H NMR and 1,4-dioxane as an internal standard. The value obtained for the pure [Hbet][Tf2N]–H2O system was 14.0 wt% which is in good agreement with the values reported in the literature.29 The discussion of the salting-out effect is limited here to the effect of salt cations (Fig. 13) because salt anions form complexes with metal ions which affects the metal distribution much more than their salting-out effect. The salting-out effect of anions is however included in the ESI (Fig. S4†). The salting-out effect for cations follows the order NH4+ < K+ < Na+ < Li+ < Ca2+ < Mg2+ (Fig. 13) and the salting-out effect of anions follows the order ClO4− < I− < NO3− < Cl− < SO42− (Fig. S4†). This is in agreement with the effect of salts on other ionic liquid/water systems such as [C4mim][CF3SO3]–H2O and [C4mim][Tf2N]–H2O.51,53,54 Furthermore, a higher salt concentration increases the salting-in or salting-out effect of a salt, respectively.
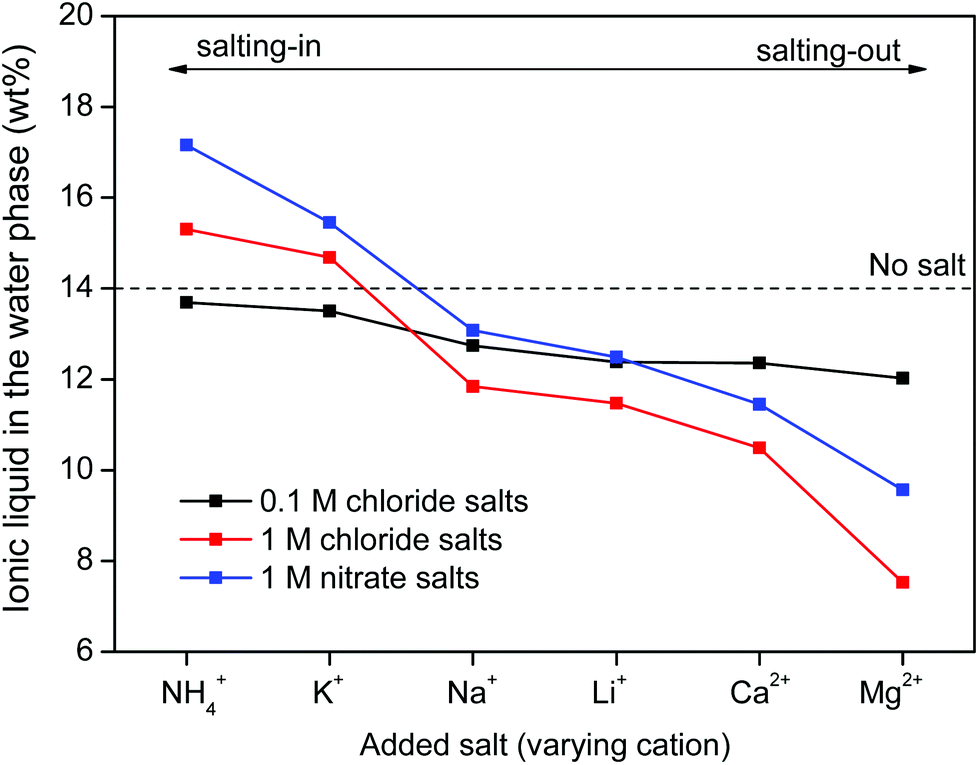 | ||
| Fig. 13 [Hbet][Tf2N] content (wt%) in the water phase when adding salts with varying cations and concentrations. The [Hbet][Tf2N] content was determined using 1H NMR and 1,4-dioxane as an internal standard. When no salt is added, the water phase contains 14 wt% of [Hbet][Tf2N].29 | ||
The distribution ratios for Co(II), Nd(III), Dy(III) and Fe(III) were then determined using these same salts (Fig. 14). When salts cause a salting-in effect compared to pure water (e.g. NH4NO3 or KNO3), the iron is more present in the water phase (D increases). For salts that cause a salting-out compared to pure water, the iron is less present in the water phase (D decreases). This is in agreement with the hypothesis that the solubility of betaine in the water phase is lowered by salting-out salts and therefore the affinity of iron(III) for the water phase is lowered since it has more affinity for betaine than water. Note however, that the salting-out effect of cations on the distribution factor of iron(III) (Fig. 14) is much smaller than the coordination effect of the anions (Fig. 11). The distribution of the other metal ions Nd(III), Dy(III) and Co(III) shows no dependence on salting-in or salting-out salts since they are present as aqua-complexes in the water phase as their affinity for betaine is much lower. They do however show a large difference between pure water and water + salt systems since the presence of counter ions such as nitrate ions significantly enhances the solubility of these aqua complexes by providing water-miscible counter ions, which are not formed during the dissolution of the metal oxides.
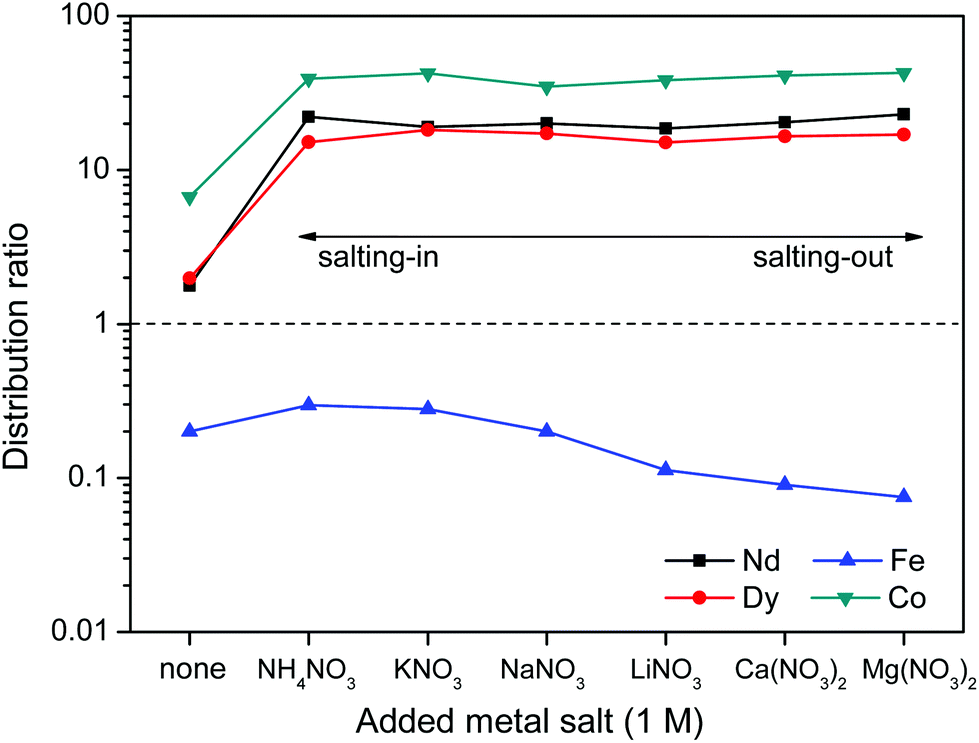 | ||
| Fig. 14 Influence of added metal salts (1 M) with varying cations on the distribution ratio of Nd(III), Dy(III), Fe(III) and Co(II) ions in a biphasic [Hbet][Tf2N]–H2O system. A high distribution ratio corresponds to a greater affinity for the water phase. | ||
To support the fact that salting-out behavior is directly responsible for the decrease of iron(III) in the water phase, several nitrate salts (1 M) were added and the resulting Nd/Fe separation factors α (eqn (3)) were plotted versus the ionic liquid content in the water phase (wt%) (Fig. 15). The separation factors are positive because DNd is larger than DFe, meaning that neodymium has a larger affinity for the water phase as defined by eqn (2).
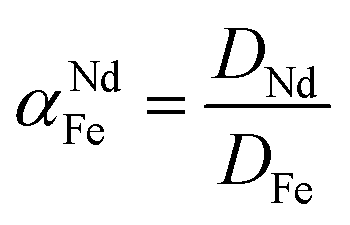 | (3) |
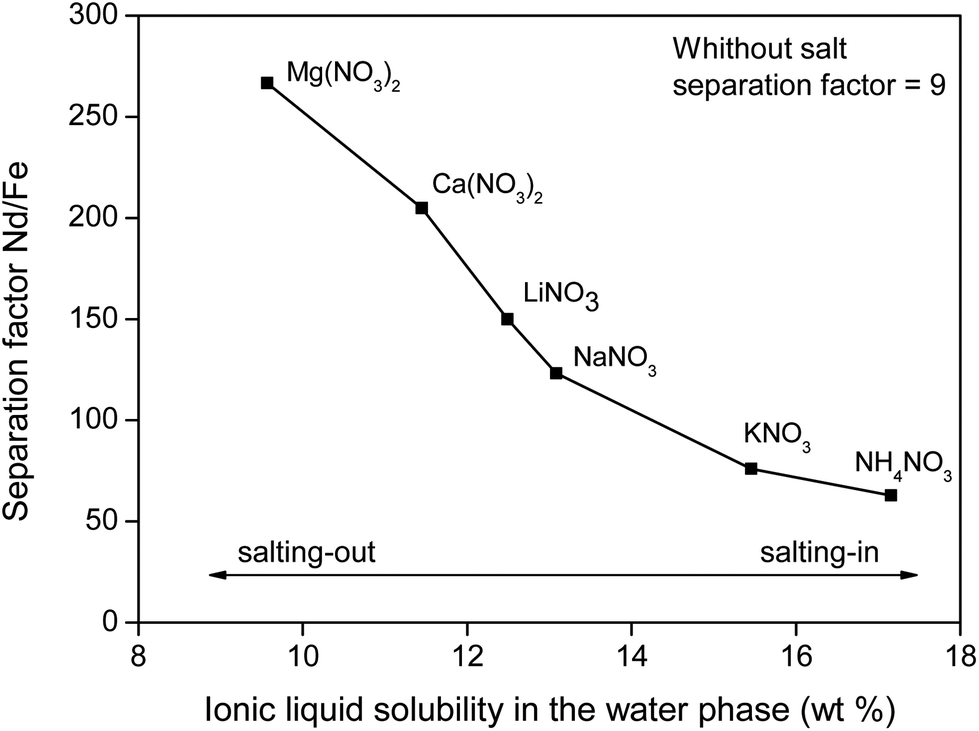 | ||
| Fig. 15 Nd/Fe separation factor as function of the [Hbet][Tf2N] content in the water phase (wt%). The corresponding salts (1 M) are shown as labels. | ||
Salt cations do not directly interact with the iron(III) cations, which is why the observed effects (Fig. 15) can be solely attributed to changes in the water-solubility of betaine due to salting-in or salting-out salts.
The trend follows the cation salting-out series (Fig. 13) which is a strong indicator that the salting-out effect of the salts is indeed responsible for the trend in REE/iron separation. In general, the addition of nitrate salts also improves the separation compared to a system without nitrates, because of the availability of counter-ions (NO3−) for the dissolved REE(III) and Co(II) ions in the water phase. Based on Fig. 15, it is evident that the addition of the right inorganic salt is beneficial for this leaching/extraction system. It is however important to find a salt that does not interfere with the leaching or stripping process and that is not too expensive. NaNO3 was chosen because it significantly enhances the transfer of Nd(III), Dy(III) and Co(II) ions to the water phase while keeping iron(III) in the ionic liquid phase. NaNO3 is not the best salting-out agent but LiNO3 is too expensive and Ca2+ or Mg2+ salts cannot be used due to the fact that they would co-precipitate with oxalic acid, which was used for stripping the metal ions later on. NaCl cannot be used since chloride ions cause transfer of Fe(III) ions to the water phase. The NaNO3 present in [Hbet][Tf2N]–H2O enhances the properties of the extraction system but will not cause impurities in the ionic liquid (no extraction of NO3− and Na+ in the ionic liquid) and will not cause impurities in the final rare-earth product since it does not precipitate with oxalic acid. The salt can be added after leaching, just before cooling down (phase separation) or it can be added from the beginning of the process since it was tested that the presence of NaNO3 (1 M) had no effect on the leaching speed, efficiency or selectivity.
The result of this combined leaching/extraction process is that a REE-rich aqueous phase is obtained while keeping iron in the ionic liquid. This allows the use of less selective (faster) leaching conditions (e.g. 6 μm particles), since the co-leached iron is automatically separated from the leached REEs (Fig. 15) once the mixture cools down and phase separates. The resulting purity of the aqueous REE-rich phase after leaching for 48 h (full REE leaching) is shown in Table 2. Besides REEs, cobalt and traces of iron still remain in the water phase. These will be fully removed during the stripping step and a cobalt-removal step. In this way, a final REE oxide can be obtained with a purity >99.9 wt%, without the need for solvent extraction.15,17 The results are also shown for a magnet of type 2 that contains very little cobalt (Table 1).
| NdFeB Magnet | Type 1 | Type 1 | Type 1 | Type 2 |
|---|---|---|---|---|
| Particle size (μm) | 300 | 70 | 6 | 350 |
| REEs (wt%) | 98.5 | 93.8 | 92.1 | 98.1 |
| Fe (wt%) | 1.1 | 2.3 | 2.8 | 1.8 |
| Co (wt%) | 0.4 | 3.9 | 5.1 | <0.1 |
Metal stripping and recovery of ionic liquid
After separating the magnet residue, the metal ions are removed and the ionic liquid can be fully regenerated without losses. The most convenient technique is to separate the two phases and to add oxalic acid to the water phase, which forms polymeric precipitates with the REE(III) and Co(II) ions (eqn (4) and (5)). Any traces of iron in the water phase will not co-precipitate which results in a highly pure mixed REE/cobalt oxalate precipitate.55 The REE content in the oxalate varies between 95–100 wt% (Table 2) depending on the initial cobalt content (Table 1).| 2REE3+(aq.) + 3H2C2O4 → (REE)2(C2O4)3(s) + 6H+ | (4) |
| Co2+(aq.) + H2C2O4 → CoC2O4(s) + 2H+ | (5) |
The oxalate precipitation stripping method is very efficient, meaning that stoichiometric amounts of oxalic acid are sufficient to obtain 100% stripping (Fig. 16). The stripping is fast (complete within 10 min) even at room temperature.
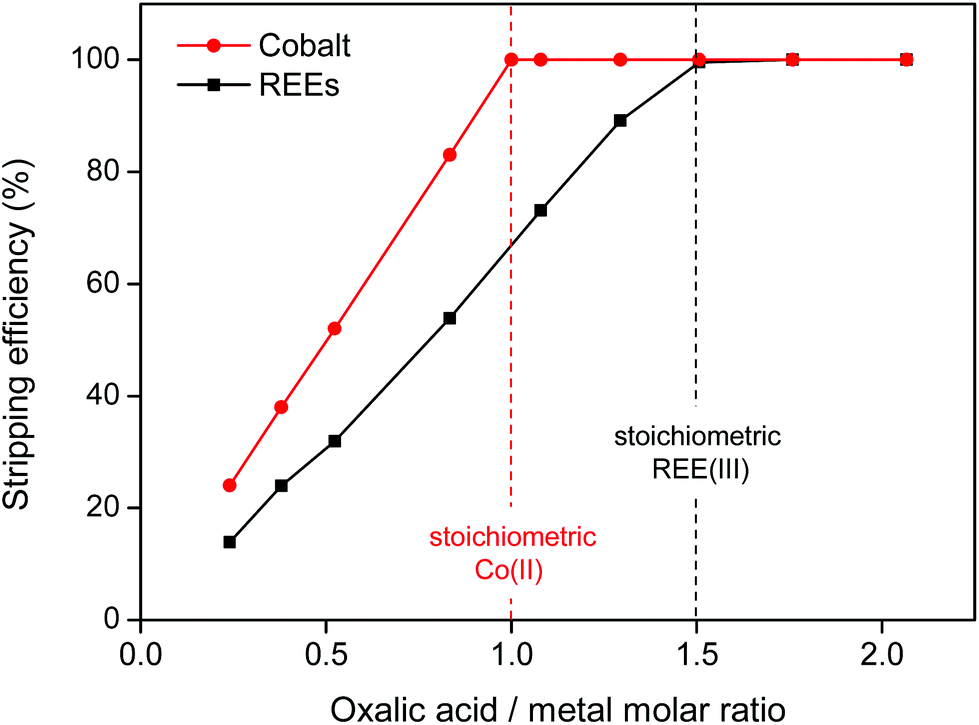 | ||
| Fig. 16 Precipitation stripping of REE(III) (Nd + Dy) and Co(II) ions from the water phase, using solid oxalic acid (stir 600 rpm, 10 min at 25 °C). A stoichiometric amount of oxalic acid (3:2 for REEs and 1:1 for Co) is sufficient to achieve full precipitation. | ||
This rare-earth oxalate product with traces of cobalt, can be further purified by treating it with aqueous ammonia which readily dissolves the cobalt(II) oxalate as an hexaamine complex (eqn (6)).56
| CoC2O4(s) + 6 NH3 → [Co(NH3)6]2+ + C2O42− | (6) |
This cobalt removal step is very efficient and fast (<10 min at 25 °C) and is capable of removing more than 99.9 wt% of the cobalt(II) (Fig. 17). An NH3/cobalt(II) molar ratio of 100 is required to obtain full removal of the cobalt (Fig. 17), but this is still a small amount since cobalt is only present in trace amounts in the oxalate product.
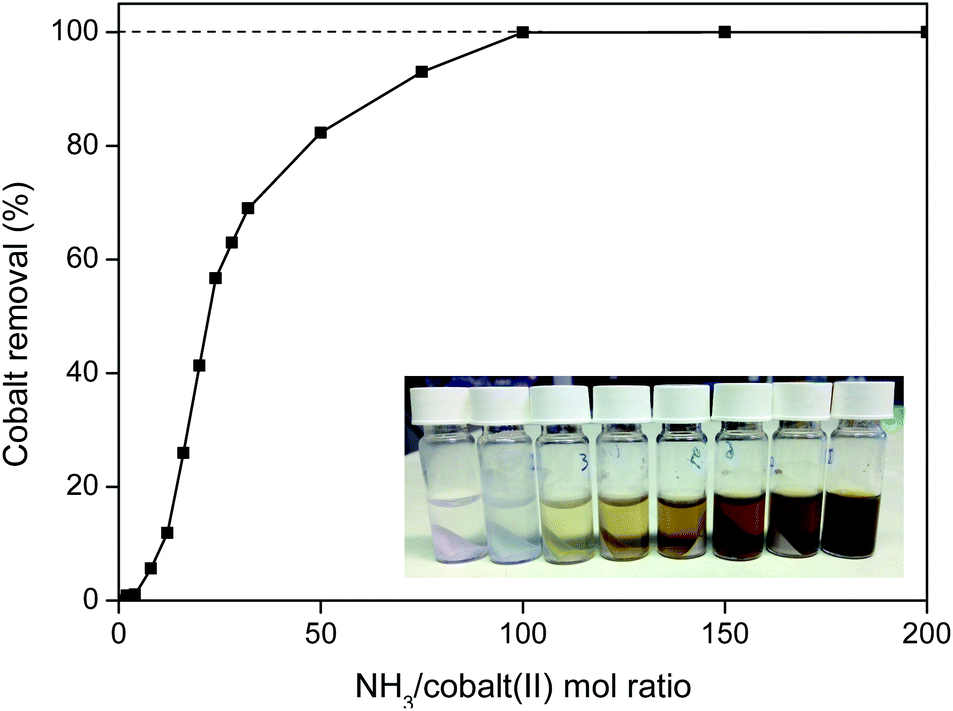 | ||
| Fig. 17 Cobalt(II) removal from the mixed REE/Co(II) oxalate precipitate, using increasing concentrations of aqueous ammonia. The ammonia/cobalt(II) ratio is plotted versus the cobalt removal efficiency (%). As inset, the color change is shown using some of the samples, with increasing amounts of ammonia from left to right. The samples were shaken (2000 rpm) for 10 min. | ||
The resulting pure REE oxalate after cobalt removal has a high purity (>99.9 wt%) and can be calcined in an oven to obtain the rare-earth oxides (Nd2O3 and Dy2O3) (eqn (7)) which can be used as precursor for the synthesis of NdFeB magnets.57,58
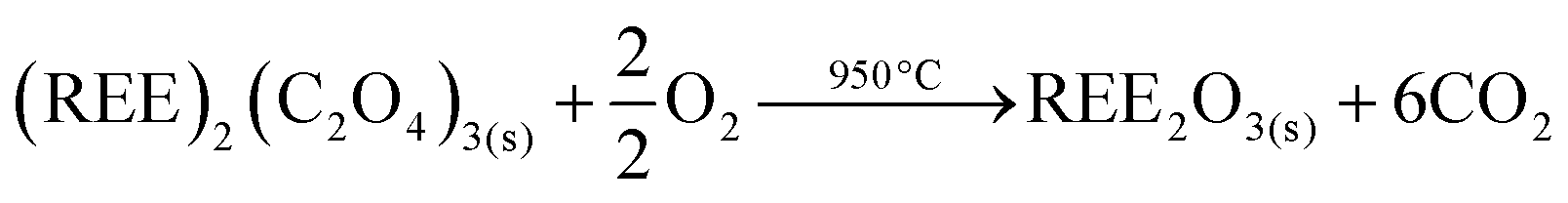 | (7) |
Next, the iron(III) is removed from the ionic liquid by contacting it with an oxalic acid (aqueous) solution or by reusing the (just cleaned) water phase and adding some solid oxalic acid. The samples are then shaken to form the iron(III) oxalate complex that is transferred to the water phase (eqn (8)).
| [Fe(bet)3][Tf2N]3 + 3H2C2O4 → [Fe(C2O4)3]3− + 3[Hbet][Tf2N] + 3H+ | (8) |
Iron(III) oxalate is very soluble in water as the complex [Fe(C2O4)3]3−, which means that the iron will be fully transferred to the water phase (Fig. 18) with a high distribution factor, as was shown previously (Fig. 11). A slight excess of oxalic acid is sufficient to obtain >99% stripping (back extraction) as shown in Fig. 18. The ionic liquid is also automatically regenerated by the reprotonation of the betaine groups (eqn (8)) and can be reused as such since it is now free of metal ions.
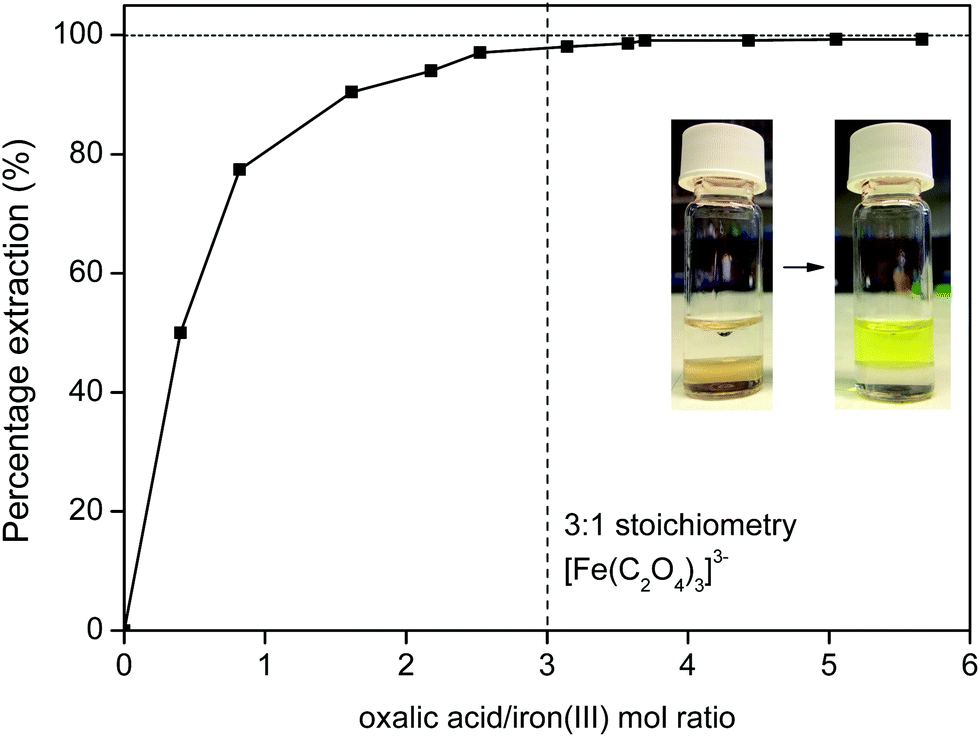 | ||
| Fig. 18 Stripping of iron(III) from the ionic liquid to the water phase using oxalic acid. The oxalic acid regenerates the ionic liquid and transfers the iron(III) ions to the water phase as the highly soluble Fe(C2O4)33− complex. The photograph shows the transfer of iron from the ionic liquid phase (left) to the water phase (right). | ||
The complete stripping process is summarized in Fig. 19. It is highly efficient since it only consumes small (stoichiometric) amounts of oxalic acid to achieve both the cleaning of the ionic liquid phase from iron(III) and the precipitation of REEs and cobalt(II) from the water phase. The REE oxalate product was calcined to obtain the corresponding oxides with high purity (>99.9 wt%). After stripping, the ionic liquid can be reused as such.
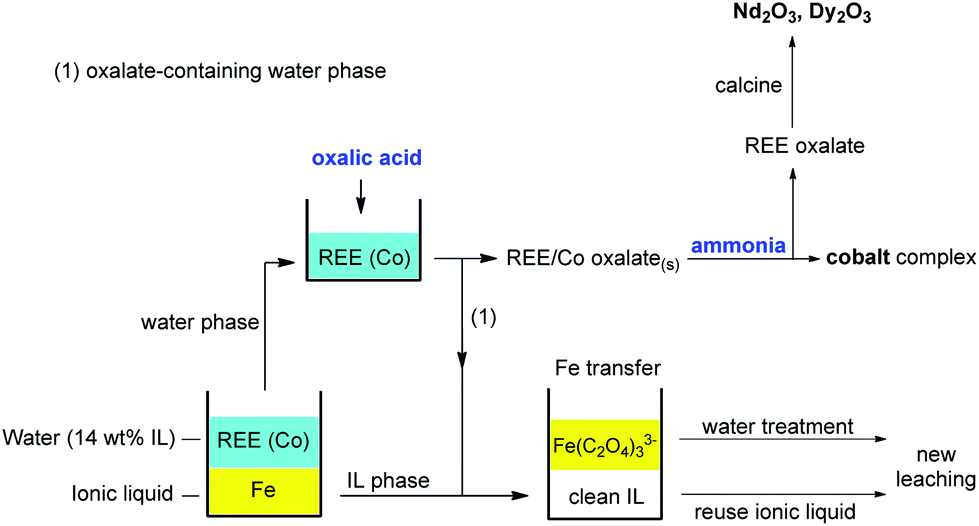 | ||
| Fig. 19 Overview of the proposed stripping process based on oxalic acid precipitation of REE(III) and Co(II) ions and phase transfer of iron(III) as a water-soluble oxalate complex. This process allows full recovery of the ionic liquid. The cobalt can be removed by treating the mixed REE/Co oxalate precipitate with aqueous ammonia. | ||
The water phase can be further treated to remove the iron(III) oxalate complex and fully retrieve the ionic liquid that is still dissolved in the water phase. An efficient water treatment method was designed, which is discussed in detail in the ESI (Fig. S5†). Basically, the iron is removed by adding Ca(OH)2 to precipitate the soluble [Fe(C2O4)3]3− complex as Fe(OH)3(s) and CaC2O4(s). The recovery of ionic liquid from the water phase can be done with various techniques such as adsorbents, specialized membranes, electrodialysis or nanofiltration.59–63 However, a very straightforward approach is the salting-out of the ionic liquid using strong (cheap) salting-out agents such as Na2SO4 (Fig. 20). This strong salting-out agent reduces the loss of ionic liquid in the water phase to less than 0.15 wt% (compared to 14 wt% without salt). This guarantees an almost full recovery of the ionic liquid which is required to avoid contamination of the water phase. In order to avoid consumption of Na2SO4, this salting-out step can be performed in an evaporation pond in which the rate of incoming water is equal to the evaporation rate, effectively keeping the Na2SO4 concentration constant while continuously salting-out the ionic liquid from the incoming waste water.
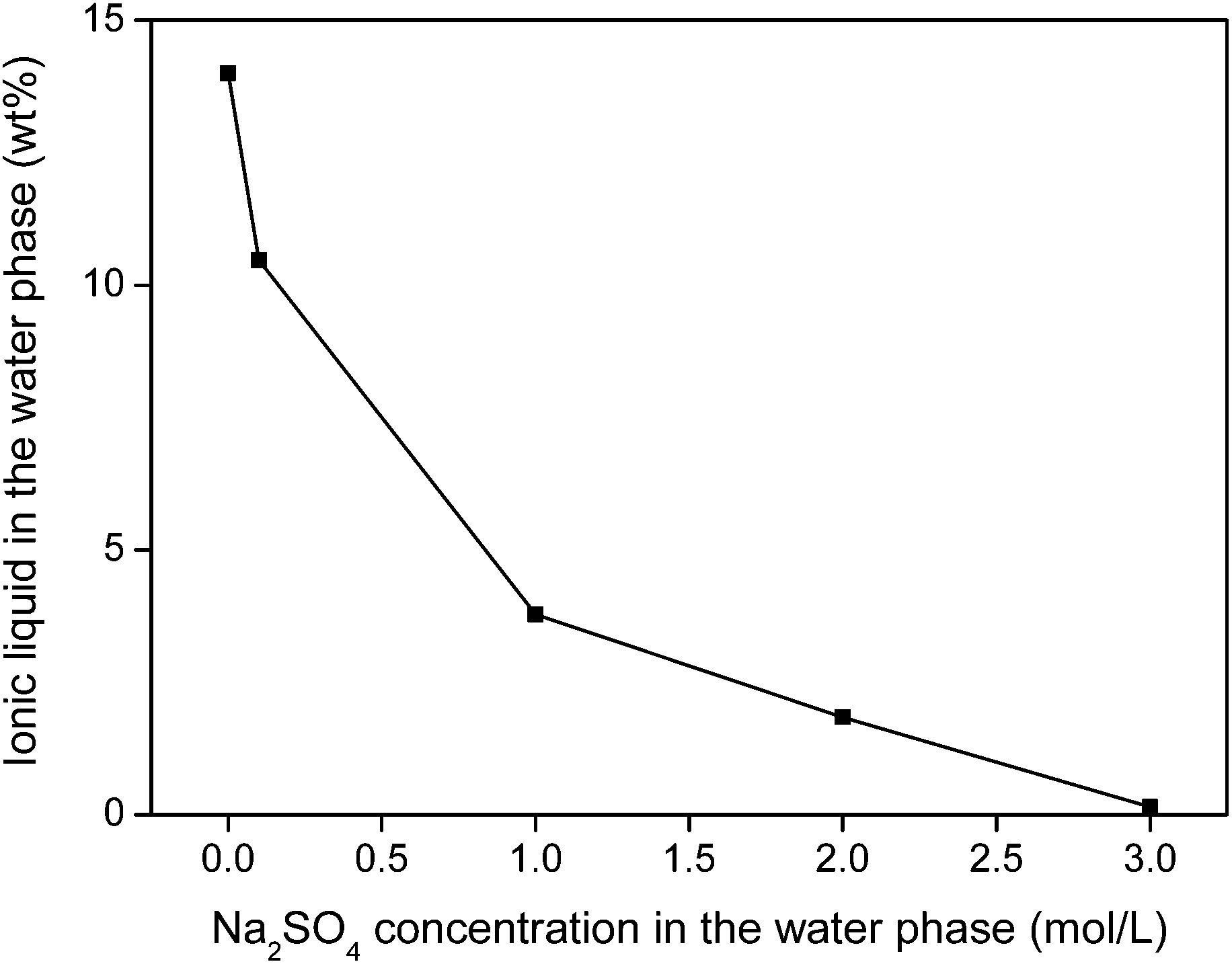 | ||
| Fig. 20 Salting-out of ionic liquid from water using Na2SO4. This way, loss of ionic liquid can be avoided. | ||
No regeneration or cleaning of the ionic liquid is necessary because [Hbet][Tf2N] does not undergo anion exchange. According to the Hofmeister series, Tf2N− cannot be displaced by salt anions such as NO3−, Cl− or SO42− which lie at the opposite side of the Hofmeister series. High concentrations of Na2SO4 will therefore not affect the purity of the ionic liquid. Even at 3 M Na2SO4, the sulfate content in the ionic liquid was below detection limit (TXRF). It was shown in previous work that the ionic liquid also had an excellent thermal stability up to 200 °C.22 This guarantees a good reusability and explains why no loss of efficiency was observed after 3 leaching/stripping cycles. An overview of the proposed recycling process is shown in Fig. 21. This process recovers over 99% of the rare earths in the NdFeB magnet with a purity >99.9 wt%, without the need for solvent extraction. The ionic liquid was also fully recovered to avoid contamination of the water phase. This process is therefore a sustainable alternative compared to mineral acid leaching and solvent extraction.
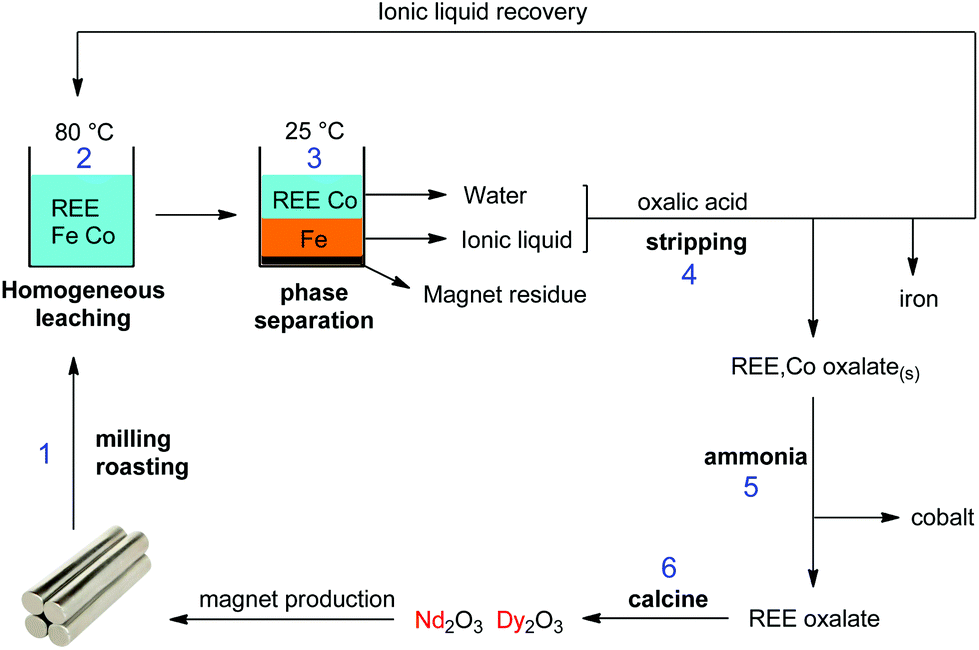 | ||
| Fig. 21 Overview of the proposed recycling process for roasted NdFeB magnets, based on the acidity and thermomorphism of the functionalized ionic liquid [Hbet][Tf2N]. Step 1: crushing and (microwave) roasting of the magnets. Step 2: Leaching of the roasted magnets in [Hbet][Tf2N]–H2O at 80 °C (homogeneous phase). Step 3: Biphasic settling at room temperature and automatic distribution of the metals across the two phases. Step 4: stripping of both separate phases with oxalic acid, which regenerates the ionic liquid, removes the iron and produces a mixed REE/Co oxalate. Step 5: removal of the cobalt by treatment with aqueous ammonia. Step 6: Calcination of the REE oxalate to form the oxides (purity > 99.9 wt%), which can be used as a precursor for the production of new NdFeB magnets.58 | ||
As a final remark it is interesting to discuss the possibility of microwave heating of ionic liquids. In most of our experiments, the ionic liquid was heated by a conventional heating source. However, since the leaching of the REE oxides in the ionic liquid requires heating (80–90 °C) over prolonged periods of time, it is interesting to note that ionic liquids can be heated in a very energy-efficient way by microwave irradiation. Ionic liquids consist entirely of ions as opposed to water or organic solvents which explains the very high adsorption of microwave radiation.22,64 Microwaves do not interfere with the chemical dissolution process but provide a more economical way to heat an ionic liquid in an industrial context.42,43,64,65 Several reactor designs exist where the radiation of an external microwave source is guided through a nonmagnetic hollow metal tube (waveguide) into the reaction vessel.42 The immediate on/off behavior of microwave irradiation and the fact that the liquid is heated from the inside as opposed to conventional heating, is also useful for process control and safety.42,64,66
Conclusion
The combined ionic liquid leaching/extraction system described in this work is an innovative approach to NdFeB magnet recycling. The process is based on the leaching of (microwave) roasted NdFeB magnets in the carboxyl-functionalized ionic liquid [Hbet][Tf2N]. The special thermomorphic properties of the [Hbet][Tf2N]–H2O system cause the mixture to be homogeneous during leaching at 80 °C (temperature above the cloud point temperature) and biphasic when cooling back down to room temperature. The formation of a biphasic system induces metal separation where Fe(III) goes to the ionic liquid phase and the REE(III) and Co(II) ions to the water phase with high separation factors. Such a thermomorphic leaching system offers many advantages and was studied in detail, including the effect of inorganic salts on the metal distribution. Oxalic acid was used to precipitate the REE(III) and cobalt(II) ions, while stripping iron(III) by back extraction as a soluble iron oxalate complex. The cobalt was then removed from the oxalate product using aqueous ammonia. The final REE oxalate had a purity above 99.9 wt% and could be calcined to obtain the pure oxides which can be used as precursors for the production of new NdFeB magnets.58 The stripping step also automatically regenerated the ionic liquid and a salting-out method was designed (using Na2SO4) to exclude any loss of ionic liquid or contaminate on of the water phase. Therefore, this closed-loop system only generates little waste and offers selectivity, mild conditions and reusability, which make this a promising green technology for the targeted recovery of REEs from NdFeB magnets. Considering the supply risk for these elements (mainly Nd and Dy) and the high demand for NdFeB magnets this technology could help to secure the demand for these critical elements by providing a sustainable and efficient alternative for the recycling of spent NdFeB magnets.Acknowledgements
The authors wish to thank the KU Leuven (projects GOA/13/008 and IOF-KP RARE3), the FWO Flanders (PhD fellowship to DD and research project G.0900.13) for financial support. The University of Birmingham (UK) and Goudsmit Magnetics are acknowledged for providing the demagnetized NdFeB magnets, Jeroen Sniekers for helping with the SEM and EDX measurements, Neil Brooks for powder XRD characterization, Tom Vander Hoogerstraete for the help with milling and roasting of the magnets, Clio Deferm for milling of the magnets, Jef Vleugels for the useful discussions and Carlo Groffils, Sam Buls and Olivier Van Roey for their help with the microwave set-up and the microwave experiments.Notes and references
- D. Schüler, M. Buchert, R. Liu, G. S. Dittrich and C. Merz, Study on rare earths and their recycling, Öko-Institut, Darmstadt, 2011 Search PubMed.
- E. Alonso, A. M. Sherman, T. J. Wallington, M. P. Everson, F. R. Field, R. Roth and R. E. Kirchain, Environ. Sci. Technol., 2012, 46, 3406–3414 CrossRef CAS PubMed.
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Cleaner Prod., 2013, 51, 1–22 CrossRef CAS.
- M. Humphries, Rare Earth Elements: The Global Supply Chain, Congressional Research Service, USA, 2013 Search PubMed.
- S. Massari and M. Ruberti, Resour. Policy, 2013, 38, 36–43 CrossRef.
- Report on Critical raw materials for the EU, European Commission, DG Enterprise & Industry, Brussels, 2014.
- A. Golev, M. Scott, P. D. Erskine, S. H. Ali and G. R. Ballantyne, Resour. Policy, 2014, 41, 52–59 CrossRef.
- U.S. Department of Energy, Critical Materials Strategy, USA, 2011 Search PubMed.
- K. Binnemans, P. T. Jones, K. Acker, B. Blanpain, B. Mishra and D. Apelian, JOM, 2013, 65, 846–848 CrossRef.
- M. Tanaka, T. Oki, K. Koyama, H. Narita and T. Oishi, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, Amsterdam, 2013, ch. 255, vol. 43, pp. 159–212 Search PubMed.
- Y. Wu, X. Yin, Q. Zhang, W. Wang and X. Mu, Resour., Conserv. Recycl., 2014, 88, 21–31 CrossRef.
- T. Vander Hoogerstraete, B. Blanpain, T. Van Gerven and K. Binnemans, RSC Adv., 2014, 4, 64099–64111 RSC.
- Solvay, Press release, http://www.solvay.com, 2012.
- K. Binnemans and P. T. Jones, J. Rare Earths, 2014, 32, 195–200 CrossRef CAS.
- T. Vander Hoogerstraete, S. Wellens, K. Verachtert and K. Binnemans, Green Chem., 2013, 15, 919–927 RSC.
- S. Wellens, R. Goovaerts, C. Moller, J. Luyten, B. Thijs and K. Binnemans, Green Chem., 2013, 15, 3160–3164 RSC.
- T. Vander Hoogerstraete and K. Binnemans, Green Chem., 2014, 16, 1594–1606 RSC.
- K. Larsson, C. Ekberg and A. Ødegaard-Jensen, Waste Manage., 2013, 33, 689–698 CrossRef CAS PubMed.
- C. Jagadeeswara Rao, K. A. Venkatesan, K. Nagarajan and T. G. Srinivasan, Radiochim. Acta, 2008, 96, 403–409 Search PubMed.
- P. Nockemann, B. Thijs, T. N. Parac-Vogt, K. Van Hecke, L. Van Meervelt, B. Tinant, I. Hartenbach, T. Schleid, V. T. Ngan, M. T. Nguyen and K. Binnemans, Inorg. Chem., 2008, 47, 9987–9999 CrossRef CAS PubMed.
- P. Nockemann, B. Thijs, S. Pittois, J. Thoen, C. Glorieux, K. Van Hecke, L. Van Meervelt, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2006, 110, 20978–20992 CrossRef CAS PubMed.
- D. Dupont and K. Binnemans, Green Chem., 2015, 17, 856–868 RSC.
- K. Sasaki, K. Takao, T. Suzuki, T. Mori, T. Arai and Y. Ikeda, Dalton Trans., 2014, 43, 5648–5651 RSC.
- K. Sasaki, T. Suzuki, T. Mori, T. Arai, K. Takao and Y. Ikeda, Chem. Lett., 2014, 43, 775–777 CrossRef CAS.
- D. P. Fagnant, G. S. Goff, B. L. Scott, W. Runde and J. F. Brennecke, Inorg. Chem., 2013, 52, 549–551 CrossRef CAS PubMed.
- T. Vander Hoogerstraete, B. Onghena and K. Binnemans, Int. J. Mol. Sci., 2013, 14, 21353–21377 CrossRef PubMed.
- T. Vander Hoogerstraete, B. Onghena and K. Binnemans, J. Phys. Chem. Lett., 2013, 4, 1659–1663 CrossRef PubMed.
- B. Onghena, J. Jacobs, L. Van Meervelt and K. Binnemans, Dalton Trans., 2014, 43, 11566–11578 RSC.
- B. Onghena and K. Binnemans, Ind. Eng. Chem. Res., 2015, 54, 1887–1898 CrossRef CAS.
- A. P. Abbott, G. Frisch, S. J. Gurman, A. R. Hillman, J. Hartley, F. Holyoak and K. S. Ryder, Chem. Commun., 2011, 47, 10031–10033 RSC.
- S. Wellens, T. Vander Hoogerstraete, C. Möller, B. Thijs, J. Luyten and K. Binnemans, Hydrometallurgy, 2014, 144–145, 27–33 CrossRef CAS.
- A. Rout, S. Wellens and K. Binnemans, RSC Adv., 2014, 4, 5753–5758 RSC.
- S. Wellens, B. Thijs, C. Moller and K. Binnemans, Phys. Chem. Chem. Phys., 2013, 15, 9663–9669 RSC.
- A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 13, 471–481 RSC.
- S. Wellens, B. Thijs and K. Binnemans, Green Chem., 2012, 14, 1657–1665 RSC.
- Y. Gu, Green Chem., 2012, 14, 2091–2128 RSC.
- Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- T. Welton, Green Chem., 2011, 13, 225–225 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- R. M. C. Dawson, in Data for Biochemical Research, Oxford University press, New York, 3rd edn, 1986, pp. 8–9 Search PubMed.
- G. Brauer, in Handbook of Preparative Inorganic Chemistry, Academic Press, New York, 2nd edn, 1963, ch. 28, pp. 1513–1559 Search PubMed.
- K. E. Haque, Int. J. Miner. Process., 1999, 57, 1–24 CrossRef CAS.
- M. Al-Harahsheh and S. W. Kingman, Hydrometallurgy, 2004, 73, 189–203 CrossRef CAS.
- Y. Hua, C. Cai and Y. Cui, Sep. Purif. Technol., 2006, 50, 22–29 CrossRef CAS.
- K. Miura, M. Masuda, M. Itoh, T. Horikawa and K.-i. Machida, J. Alloys Compd., 2006, 408–412, 1391–1395 CrossRef CAS.
- S. Pan, Rare Earth Permanent-Magnet Alloys’ High Temperature Phase Transformation: In Situ and Dynamic Observation and Its Application in Material Design, Springer, Berlin, Heidelberg, 2014 Search PubMed.
- P. Nockemann, B. Thijs, K. Lunstroot, T. N. Parac-Vogt, C. Görller-Walrand, K. Binnemans, K. Van Hecke, L. Van Meervelt, S. Nikitenko, J. Daniels, C. Hennig and R. Van Deun, Chem. – Eur. J., 2009, 15, 1449–1461 CrossRef CAS PubMed.
- R. Reddy, J. Phase Equilib. Diffus., 2006, 27, 210–211 CrossRef CAS.
- M. G. Freire, A. R. R. Teles, J. N. Canongia Lopes, L. P. N. Rebelo, I. M. Marrucho and J. A. P. Coutinho, Sep. Sci. Technol., 2011, 47, 284–291 CrossRef.
- Y. Zhang and P. S. Cremer, Curr. Opin. Chem. Biol., 2006, 10, 658–663 CrossRef CAS PubMed.
- M. G. Freire, P. J. Carvalho, A. M. S. Silva, L. M. N. B. F. Santos, L. P. N. Rebelo, I. M. Marrucho and J. A. P. Coutinho, J. Phys. Chem. B, 2009, 113, 202–211 CrossRef CAS PubMed.
- M. G. Freire, C. M. S. S. Neves, P. J. Carvalho, R. L. Gardas, A. M. Fernandes, I. M. Marrucho, L. M. N. B. F. Santos and J. A. P. Coutinho, J. Phys. Chem. B, 2007, 111, 13082–13089 CrossRef CAS PubMed.
- S. Shahriari, C. M. S. S. Neves, M. G. Freire and J. A. P. Coutinho, J. Phys. Chem. B, 2012, 116, 7252–7258 CrossRef CAS PubMed.
- L. I. N. Tomé, F. R. Varanda, M. G. Freire, I. M. Marrucho and J. A. P. Coutinho, J. Phys. Chem. B, 2009, 113, 2815–2825 CrossRef PubMed.
- J.-S. Sohn, D.-H. Yang, S.-M. Shin and J.-G. Kang, Geosyst. Eng., 2006, 9, 81–86 CrossRef.
- D. Nicholls, in The Chemistry of Iron, Cobalt and Nickel, Pergamon, Exeter, 1st edn, 1973, pp. 1053–1107 Search PubMed.
- A. K. Galwey and E. Brown, Thermal Decomposition of Ionic Solids: Chemical Properties and Reactivities of Ionic Crystalline Phases, Elsevier Science, 1999 Search PubMed.
- G. Qi, M. Hino and A. Yazawa, Mater. Trans. JIM, 1990, 31, 463–470 CrossRef CAS.
- L. Bai, X. Wang, Y. Nie, H. Dong, X. Zhang and S. Zhang, Sci. China: Chem., 2013, 56, 1811–1816 CrossRef CAS.
- C. M. S. S. Neves, M. G. Freire and J. A. P. Coutinho, RSC Adv., 2012, 2, 10882–10890 RSC.
- C. Abels, C. Redepenning, A. Moll, T. Melin and M. Wessling, J. Membr. Sci., 2012, 405–406, 1–10 CrossRef CAS.
- J. F. Fernández, D. Waterkamp and J. Thöming, Desalination, 2008, 224, 52–56 CrossRef.
- J. Lemus, J. Palomar, F. Heras, M. A. Gilarranz and J. J. Rodriguez, Sep. Purif. Technol., 2012, 97, 11–19 CrossRef CAS.
- J. Hoffmann, M. Nuchter, B. Ondruschka and P. Wasserscheid, Green Chem., 2003, 5, 296–299 RSC.
- A. Arfan and J. P. Bazureau, Org. Process Res. Dev., 2005, 9, 743–748 CrossRef CAS.
- C. O. Kappe and D. Dallinger, Nat. Rev. Drug Discovery, 2006, 5, 51–63 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of the NdFeB magnet particles with full ICP-MS analysis, XRD diffractograms and SEM images. The TXRF analysis procedure and water treatment process are also discussed. Finally, the salting-out effect of salt anions on the water-solubility of [Hbet][Tf2N] is shown. See DOI: 10.1039/c5gc00155b |
| This journal is © The Royal Society of Chemistry 2015 |