
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeoxygenation of biobased molecules by decarboxylation and decarbonylation – a review on the role of heterogeneous, homogeneous and bio-catalysis
Gwen J S.
Dawes
,
Elinor L.
Scott
*,
Jérôme
Le Nôtre
,
Johan P. M.
Sanders
and
Johannes H.
Bitter
Biobased Chemistry and Technology, Wageningen University, Bornse Weilanden 9, 6708 WG Wageningen, The Netherlands. E-mail: Elinor.Scott@wur.nl
First published on 9th April 2015
Abstract
Use of biomass is crucial for a sustainable supply of chemicals and fuels for future generations. Compared to fossil feedstocks, biomass is more functionalized and requires defunctionalisation to make it suitable for use. Deoxygenation is an important method of defunctionalisation. While thermal deoxygenation is possible, high energy input and lower reaction selectivity makes it less suitable for producing the desired chemicals and fuels. Catalytic deoxygenation is more successful by lowering the activation energy of the reaction, and when designed correctly, is more selective. Catalytic deoxygenation can be performed in various ways. Here we focus on decarboxylation and decarbonylation. There are several classes of catalysts: heterogeneous, homogeneous, bio- and organocatalysts and all have limitations. Homogeneous catalysts generally have superior selectivity and specificity but separation from the reaction is cumbersome. Heterogeneous catalysts are more readily isolated and can be utilised at high temperatures, however they have lower selectivity in complex reaction mixtures. While bio-catalysts can operate at ambient temperatures, the volumetric productivity is lower. Therefore it is not always apparent in advance which catalyst is the most suitable in terms of conversion and selectivity under optimal process conditions. Here we compare classes of catalysts for the decarboxylation and decarbonylation of biobased molecules and discuss their limitations and advantages. We mainly focus on the activity of the catalysts and find there is a strong correlation between specific activity (turn over frequency) and temperature for metal based catalysts (homogeneous or heterogeneous). Thus one is not more active than the other at the same temperature. Alternatively, enzymes have a higher turnover frequency but drawbacks (low volumetric productivity) should be overcome.
 Gwen J S. Dawes | Gwen Dawes is a PhD student at Wageningen University, in the chair group Biobased Chemistry and Technology. She obtained her Bachelors and Masters degree at the University of Cambridge, UK. She has worked on the folding of spectrin at the University of Cambridge, and investigating drug delivery from implantable devices at TU Delft. |
 Elinor L. Scott | Elinor Scott is Assistant Professor at Wageningen University at the chair group of Biobased Chemistry and Technology. Since 1997 she has been concerned with the development of economically and ecologically attractive conversion routes and processes of biomass to industrial chemicals. She obtained her PhD in Chemistry at Heriot-Watt University. Following positions as Post-Doctoral researcher in the field of polymer chemistry at the University of Strathclyde (1994–1995) and Heriot-Watt University (1995–1996), she moved to the Netherlands where she worked as an industrial Post-Doctoral polymer scientist at DSM and later as a scientist at ATO looking into biomass conversions. |
 Jérôme Le Nôtre | Jérôme Le Nôtre is a Post-doctoral researcher in the chair group of Biobased Chemistry and Technology, Wageningen University working on catalytic conversions of biomass to bulk chemicals. He obtained his PhD in 2002 from the University of Rennes for research on ruthenium metathesis reactions. After performing post-doctoral work at Utrecht University and at the University of Bath he joined the group Biobased Chemistry and Technology. |
 Johan P. M. Sanders | Johan Sanders is Professor Emeritus of Valorisation of Plant Production Chains at Wageningen University. His work focuses on reducing the CO2 production in a cost effective way using chemistry, fermentation and biorefineries at a large, as well as at small scale, to enable optimal application of all plant components. From 1977 to 1993 he worked at Gist Brocades, working on various projects in the field of enzyme research; he became Associate Director of Food Research. From 1993 to 2001 he worked at AVEBE as R&D Director focusing on the enzymatic and genetic modification of starch. |
 Johannes H. Bitter | Harry Bitter is Professor of the group Biobased Chemistry and Technology at Wageningen University. His group focuses on the sustainable production of bulk chemicals and fuels from biomass by an integrated approach. The group motto is TIPTOP chemistry and technology: Turning variable InPut into Tailored OutPut, one of the great challenges in biomass conversion. He studied chemistry at the Radboud University and obtained his PhD at the University of Twente. He continued working in the field of heterogeneous catalysis at Utrecht University, focusing on the use of spectroscopy in catalysis. During the last years he has focused on the application of catalysis in biomass conversions and the use of carbon-based catalysts. |
1. Introduction
In order to ensure our planet remains habitable for future generations and to satisfy our needs for chemicals, fuels and energy, the use of renewable resources has become essential. This has motivated the European Commission to strive for the use of 20% by usage of renewable energy sources, 20% reduction in CO2 emission compared to 1990, and 20% increase in energy efficiency, by 2020.1 In order to achieve these ambitions alternative energy and fuel sources have been developed, such as solar,2 geothermal,3 hydropower4 and biofuel production from biomass.5 While a number of such developments are promising for energy supplies, only biomass feedstocks satisfy our need for the production of chemicals and materials.Biomass is extremely diverse in its composition, which necessitates the development of a biorefinery. A biorefinery is defined as “a facility that integrates biomass conversion processes and equipment to produce fuels, power, and chemicals from biomass. The biorefinery concept is analogous to today's petroleum refineries, which produce multiple fuels and products from petroleum”.6 One example of this in practice can be seen by looking into current-technology for bioethanol production, where a single plant7 produces not only ethanol, but also electricity and dried distillers grains and solubles (DDGS), which is sold as animal feed.
The development of chemical production from biomass using fermentation is already becoming economically and industrially viable. Products such as 3-hydroxypropionic acid and lactic acid (for the production of acrylic acid) are currently emerging onto the market,8 and even hydrocarbon-based fuels are being produced fermentatively.9 The feedstock of choice for fermentation is glucose or glucose containing substrates such as starch or, more recently, cellulose.10 Recent research has focused on deriving these from sustainable secondary material streams e.g. second generation lignocellulosic fermentation,11 utilising cornstover or wood from otherwise unproductive land12–14 or chemical and thermal reclamation from waste such as slaughterhouse effluents or household garbage.15,16
So far, a significant focus has been on fermentative techniques for bio-based chemical production due to low temperature selective conversions and the ability for molecules to undergo various transformations in one processing step. However, fermentation processes can have low substrate/product concentrations in water which requires isolation. Another means of inducing chemical transformation is by using (bio)chemical transformation. Enzymes have also been successfully applied in industry for bulk chemical production. The use of nitrile hydratase17 for acrylamide production from acrylonitrile has been employed industrially since the 1990's and is preferred to the chemical conversion as it leads to lower side product formation. Amino acids can be produced enzymatically, e.g. alanine from fumaric acid using aspartase.18 Hydrolysis reactions in general are very important in industrial applications of enzymes, for example peptidases19 and lipases20 used in biological washing powders, and α-amylase21 and cellulase10 are being investigated for their application in the production of glucose for fermentation. The high selectivity and retention of chirality of enzymatic conversions is of particular interest for medicines, where single enantiomers are required.22,23 For example, ketoreductases can be used to selectively produce single enantiomeric products from racemic starting materials. However, the high selectivity of enzymes can be problematic when using racemic feedstocks, as up to 50% of the starting material may not be transformed.24 The use of enzymes is considered one of the most expensive components of biomass conversion.10,25
Chemo-catalytic conversion can be used to produce chemicals with high volumetric turnovers and are widely used in current chemical synthesis techniques. For the conversion of biomass current examples include high intensity techniques like gasification of biomass to form bio-syngas.26–28 This can be fed into Fischer–Tropsch reactors to produce waxes and paraffins for fuels.29–31 Pyrolysis of biomass such as woodchips allows for the production of bio-oil,32 lignin depolymerisation in supercritical carbon dioxide allows for the production of BTX-like aromatics for the chemicals industry.33 Lower temperature chemical conversions are also common, such as the production of isosorbide, a monomer and plasticising agent,34 from glucose35 or cellulose.36 Ethanol dehydration to green ethylene is a large and growing area.37 Furandicarboxylic acid, a novel plastic monomer38 can be produced from fructose via hydroxymethylfurfural.39 Fatty acid transesterification is also chemocatalysed40 and the byproduct, glycerol, is industrially converted into epichlorohydrin,41,42 a monomer used for resin production.43
An important trend in the conversion technologies used in combination with biomass is that they tend towards defunctionalisation, including deoxygenation. An example of this is the fermentation of ethanol from glucose, where the mass percentage of oxygen drops from 53% to 34%. This is due to the desire to supply ‘drop-in’ replacements (identical to current chemicals and products) to maintain the current market requirements, and crude oil-derived end products have comparatively low functionality compared to biomass. Decarboxylation offers opportunities to produce existing products from bio-based molecules to make products of a similar Gibbs free energy in a simple transformation. Decarboxylase enzymes comprise of 80 members, mostly amino acid decarboxylases. Examples of using these enzymes have been investigated44–46 in significant detail on the transformation of biomass,25,47 but reviews on chemocatalytic defunctionalisation are less prolific. Defunctionalisation by removing oxygen can be performed using hydrodeoxygenation, decarboxylation and decarbonylation. Hydrodeoxygenation has been reviewed previously for biobased feedstocks and will be left out of the current review.48 In our review we will focus on the decarboxylation and decarbonylation transformations of bio-based molecules such as a variety of aromatic and aliphatic molecules, including amino acids, into industrially relevant compounds using hetero-, homo- and bio- catalysis. It will attempt to draw parallels between the use of a specific catalytic systems on various substrates, and also between types of conversion, showing the resulting activity, selectivity's, and where appropriate, mechanistic details. Based on this review of the literature, we show the relation of activity for decarboxylation and decarbonylation as a function of the type of catalyst under various operating conditions.
2. Homogeneous catalysis
2.1. Silver/copper decarboxylation/decarbonylation
In the presence of catalytic quantities of silver(I) salts, it was originally shown that fatty acids are able to undergo decarboxylation to alkanes. Fatty acids are commonly found in nature as components of oils, such as those derived from plants (e.g. soybean oil, palm oil), algae, and animal sources (e.g. tallow, lard). They are typically pressed out of seeds as an oil, which usually consists of triacylglycerides, where three long chain fatty acids are attached to glycerol by ester linkages, and must be hydrolysed to form free fatty acids and glycerol. There are two broad classes of fatty acid: saturated fatty acids such as stearic acid (C18H36O2) and palmitic acid (C16H32O2) which typically come from animal fats, and unsaturated fatty acids such as oleic acid (C18H34O2) and linoleic acid (C18H32O2) which typically come from plant sources, and typically have lower melting points due to their more bulky structures. The distribution of these molecules from different sources is covered in other works.40 As these molecules are very rich in carbon and hydrogen and otherwise poor in functional groups, they are attractive for the formation of fuels and oils. Global production scale is around 6.5 billion litres, as per 2006.49 Current transformation methods for fatty acids involve the production of fatty acid methyl esters (FAMEs) by transesterification from glycerol, in order to produce bio-diesel, a fuel product that can supplement diesel fuels.50 Diesel typically comprises of aliphatic hydrocarbons of between 9 and 28 carbon constituents, with an aim to contain as little aromatic residues as possible in order to prevent carcinogenic effects.51 FAMEs from plant origin are between 9 and 21 carbons in content and do not contain aromatic moieties by default, therefore are an excellent replacement. As of 2011, the United States has a production capacity of 2.1 billion litres per year of biodiesel,52 and in Europe, the EU2020 directive promotes replacement of 10% transportation fuel by biofuel by the year 2020.1 However, FAMEs suffer a reduction in lower heating value from 43 MJ kg−1 to 37 MJ kg−1, and are considered to be only 91% as effective in a combustion engine when compared to conventional diesel.53 They also suffer from an increase of cloud point by 10 to 15 °C, decreasing the ability for this fuel to be effective in colder climates.54 Thus, current research has been focused on converting plant oils into a drop-in substitute for diesel fuel in order to maintain the desired properties. Thus, a decarboxylation reaction to remove oxygen is required to form alkanes or alkenes which can be used as fuel.Heavy metals have been known to allow the decarboxylation of fatty acids for many years, such as by the use of lead tetraacetate.55 However, as this reaction requires stoichiometric amounts of lead, it is not catalytic and will not be covered any further. The use of silver(I) salts and sodium peroxydisulphate to produce alkenes by oxidative decarboxylation has been observed previously.56,57 In these reactions the driving force comes from the peroxydisulphate ion, which produces silver(II) ions in solution. These ions are able to selectively decarboxylate fatty acids.58 The peroxydisulphate ion is a strong oxidising agent itself that can be used to oxidise several organic species such as formic acid and small alcohols in water, but is unselective.59 The mechanism for this reaction has been expanded upon in further work.60 Drawing together these references, we see that this reaction proceeds via the formation of a silver(II) ion in solution after reaction with the peroxydisulphate as shown in Fig. 1.
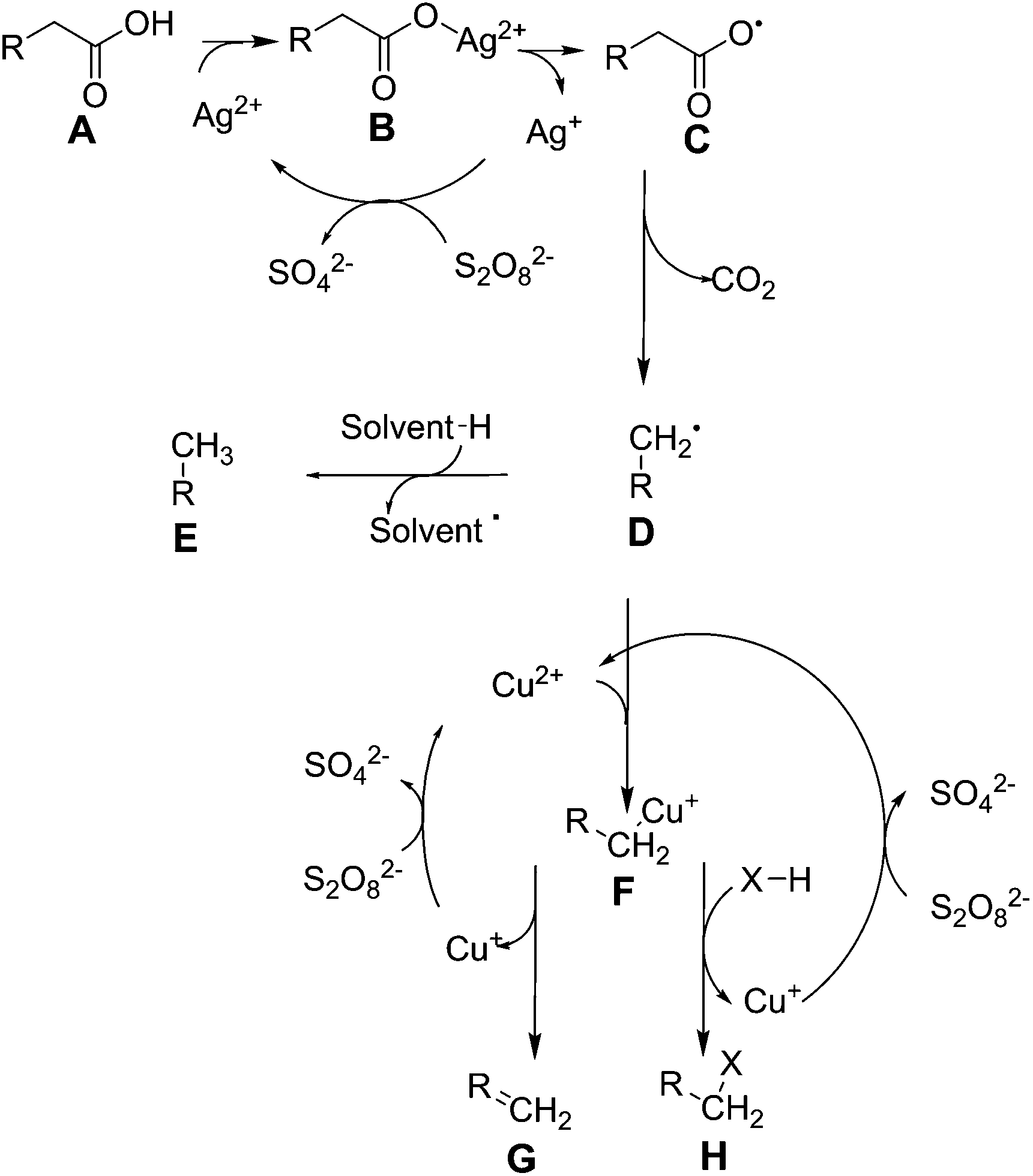 | ||
| Fig. 1 Reactions controlling silver(II) and copper(II) mediated fatty acid oxidative decarboxylation. | ||
This reaction proceeds via the formation of a silver–carboxylate complex B, then radical initiation to form an unstable carboxyl radical. This subsequently degrades into an alkyl group E. It was initially unclear whether the in situ formation of silver(II) ions proceeded via single electron oxidation to directly oxidise silver(I), or a two electron process to form silver(III) followed by disproportionation with silver(I) to form silver(II). Initially these were kinetically inseparable, but silver(III) ion disproportionation was confirmed as the correct mechanism in a later work.61 This is advantageous: silver(III) ions are capable of oxidizing water to oxygen at room temperature,58 and a low concentration of these should be maintained in order to keep efficiency high.62 As the reaction was shown to be proportional to pH, the reaction to form molecule B was stated to be the rate determining step. Normally, the formation of C, D and E proceed rapidly from there. With the addition of copper(II) salts to this reaction, additional coordination of the alkyl radical occurs to form F in Fig. 1, stabilising it and allowing oxidative elimination to occur. This enables the pathway to instead effectively decarbonylate, and produce alkenes G,63 or primary alcohols H, where X = OH. As the copper(II) ion is itself reduced by these actions, it must be regenerated, using more peroxydisulphate. In terms of a Green Chemistry approach, the use of further use of reagents is considered less attractive.
Recently, silver nitrate was used as a catalyst for decarboxylation of several saturated (stearic and palmitic acid) and unsaturated (oleic, ricinoleic, eliadic, linoleic, linolenic) fatty acids.64 In the case of saturated fatty acids, it was shown that a selectivity for alkanes of max. 50 mol% at complete conversion when using palmitic acid as a substrate. This reaction was performed at 78 °C for 20 min, in water–acetonitrile 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 in the presence of silver nitrate and sodium peroxydisulphate. The details and yields can be seen in Table 1. The other side products for this reaction are not characterised, but a small fraction of 1-pentadecenol and pentadecene were observed in the reaction mixture. By using thermogravimetric analysis, as much as 35 mol% of the material is noticed at 200–500 °C, and up to 10 mol% from 500–800 °C, implying that a significant amount of polymerisation is occurring in the radical-based reaction. With the addition of copper(II) sulphate, 37 mol% 1-alkene and 22 mol% 1-alkanol was produced from palmitic acid. The authors note that the terminal alkene is not the only product of this reaction. As the copper(II) salt can coordinate with alkenes, at the reaction temperature of 78 °C it allows for an isomerisation to occur, moving the double bond along the molecule, and thus affecting the purity of the final product, however the overall selectivity for monomeric products was increased. When using unsaturated fatty acids the problem of incomplete mass balance becomes more significant due to the presence of reactive double bonds in the molecule, increasing the probability of polymerisation, however 10–35% of the desired alkane or alkene was obtained. Other authors have further investigated and reviewed the mechanistic pathways in the deoxygenation of vegetable oils.65
:9 in the presence of silver nitrate and sodium peroxydisulphate. The details and yields can be seen in Table 1. The other side products for this reaction are not characterised, but a small fraction of 1-pentadecenol and pentadecene were observed in the reaction mixture. By using thermogravimetric analysis, as much as 35 mol% of the material is noticed at 200–500 °C, and up to 10 mol% from 500–800 °C, implying that a significant amount of polymerisation is occurring in the radical-based reaction. With the addition of copper(II) sulphate, 37 mol% 1-alkene and 22 mol% 1-alkanol was produced from palmitic acid. The authors note that the terminal alkene is not the only product of this reaction. As the copper(II) salt can coordinate with alkenes, at the reaction temperature of 78 °C it allows for an isomerisation to occur, moving the double bond along the molecule, and thus affecting the purity of the final product, however the overall selectivity for monomeric products was increased. When using unsaturated fatty acids the problem of incomplete mass balance becomes more significant due to the presence of reactive double bonds in the molecule, increasing the probability of polymerisation, however 10–35% of the desired alkane or alkene was obtained. Other authors have further investigated and reviewed the mechanistic pathways in the deoxygenation of vegetable oils.65
| AgNO3 | CuSO4 | Na2S2O8 | Conversion % | A | B | C |
|---|---|---|---|---|---|---|
|
a Reaction conditions: Acetonitrile:H2O, 9:5, 20 min, 78 °C A: pentadecane, B: 1-pentadecene, C: 1-pentadecanol.
|
||||||
| 1 | — | 0.5 | 80 | <1 | 19.6 | <1 |
| 1 | — | 2.5 | 97 | <1 | 49.9 | <1 |
| 1 | 1 | 0.5 | 93 | 12.3 | <1 | 7.4 |
| 1 | 1 | 2.5 | 99 | 35.4 | 2.2 | 16.5 |
Aromatic molecules are produced in plants and animals on a regular basis (such as products of the shikimic acid pathway66), and some aromatic chemicals can be made from the resultant biomass components. For example, plant oils derived from terpenes can be rich in benzoic acid, for example benzoin resin. The latter can be derived from the bark of the Styrax species of tree where up to 40% is benzoic acid, and 40% cinnamic acid can be present.67 Another source could be citrus peel oil, where p-cymene can be found, which can be subsequently converted into other compounds.68 However, no method of producing aromatic compounds from biomass is yet adopted by the market, largely due to plant oils being either in very low concentrations, or the plants very slow growing and thus have a low productivity. Alternatively, a substantial source of biomass-derived aryl rings could be lignin. Lignin is described in more detail later in this review, but briefly, depolymerised lignin can contain oxygenated aromatic structures such as vanillic acid and syringic acid and phenol.33,69 Silver(I) has been shown to act as an efficient decarboxylating agent of aryl carboxylates such as ortho-anisic acid, producing 98 mol% anisole in the presence of 10 mol% silver(I) acetate and 15 mol% potassium carbonate in N-methylpyrrolidone (NMP) at 120 °C for 16 hours, as shown in Table 2. They also showed that silver(I) can decrease the activation energy for the decarboxylation of 2-fluorobenzoic acid to as little as 29 kcal mol−1 when NMP is used as a solvent.70,71
| Catalyst | Amount catalyst (mol%) | Ligandb | Additivec | Solvent | Temp (°C) | Yield (mol%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 16 h. b 10 mol%. c 15 mol%. d 1,10 phenanthroline. e Quinolone. | ||||||
| Ag2O | 5 | Phend | — | NMP/quine | 170 | 22 |
| Ag2O | 5 | Phend | — | NMP | 170 | 85 |
| Ag2O | 5 | Phend | — | NMP | 120 | 60 |
| AgOTf | 10 | Phend | — | NMP | 120 | 0 |
| AgOAc | 10 | Phend | — | NMP | 120 | 70 |
| AgOAc | 10 | PPh3 | — | NMP | 120 | 0 |
| AgOAc | 10 | — | — | NMP | 120 | 24 |
| AgOAc | 10 | — | K2CO3 | NMP | 120 | 98 |
| AgOAc | 10 | — | K2CO3 | NMP | 80 | 60 |
Besides silver salts, copper(I) has also been shown to act as a catalyst for the decarboxylation of compounds such as cinnamic acid.72 Here, 20 mol% of copper(I) bromide was used, in addition to 240 °C and 2 equivalents of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) for a 30 minute period to produce styrene at 75% yield.
Silver(II) and peroxydisulphate mixtures can also be used to decarboxylate amino acids.73 Amino acids could be an interesting feedstock for the formation of chemicals, as several are structurally similar to current industrial chemicals.74 With the use of decarboxylation reactions a number of relevant products could be obtained. At catalytic concentrations of silver(II) in water they successfully showed that a radical mechanism occurs, shown in Fig. 2. Interestingly, they show that at higher concentrations a cyclic structure is formed, where the silver atom bridges between the amine and carboxyl groups, potentially directing towards alpha decarboxylation. The main product of this reaction is an imine, which is not a commonly found intermediate in industrial applications. If this reaction were to be performed in a more selective approach it would produce an amine.
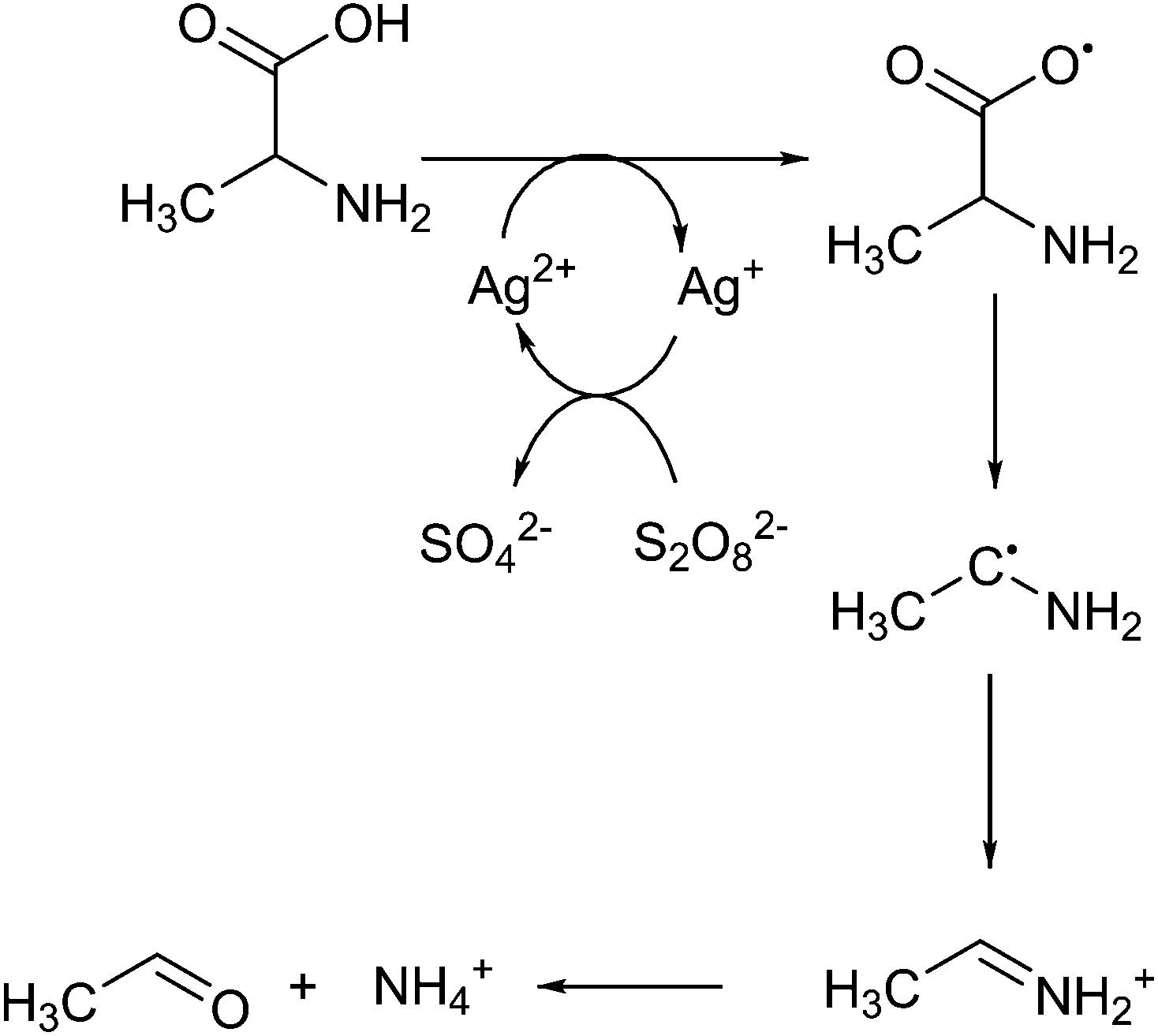 | ||
| Fig. 2 Amino acid decarboxylation by silver(II). | ||
Butanone, which can be produced from levulinic acid by decarboxylation, is used as an industrial solvent, and has a market scale of about 0.3 million tons per year.75 Levulinic acid is a compound formed by the hydration of hydroxymethylfurfural (HMF), which itself is formed by the dehydration of glucose (or fructose) in the presence of (Lewis or Brønsted) acid catalysts.76 These steps are shown in Fig. 3. HMF is a platform chemical in its own right,39 being capable of being transformed into several other industrially relevant compounds including furandicarboxylic acid (FDCA) or caprolactam.77 HMF has also been shown to undergo selective decarbonylation to furfural alcohol using Ir based catalysts.78 Fructose is either found in nature as a monosaccharide and is significantly more active than glucose for the formation of HMF and levulinic acid.79 The reaction from glucose is considered to proceed via the transformation into fructose, but is shown in the diagram as glucose is sourced from both starch and cane sugar sources and thus has a wider application. Levulinic acid has the potential to be a platform molecule for the production of chemicals such as 2-methyl tetrahydrofuran (a novel solvent or fuel oxygenate),80 diphenolic acid (a novel monomer),81 or used in the form of levulinic esters as fuel.82 However, originally the impact of this molecule may be somewhat diminished as the estimated cost of production has risen in recent years from a low $0.10/kg−1 up to a substantially higher $10 per kg.76 In the decarboxylation of levulinic acid with silver(I) phosphate and sodium peroxydisulphate in an aqueous buffer solution of citric acid and sodium hydrogen phosphate, the rate of reaction relies upon the availability of carboxylate groups for the silver(I) ions to coordinate to.83 Thus, a sharp peak of activity occurs at pH 4 and above, resulting in an increase from almost 0 to 30% yield of butanone, with the conversion of levulinic acid changing from <5% to nearly 80%. At a pH lower than 4, they report no butanone production and that the main product is acetone. This is reported to be a side product of their buffer, citric acid, degrading in the reaction conditions instead of levulinic acid. The best yield of butanone was 43% after 30 minutes at 140 °C in an autoclave, at a selectivity of just over 60%. They also report production of methyl vinyl ketone when copper(I) was used in place of silver(I), but no clear yield analysis was undertaken.
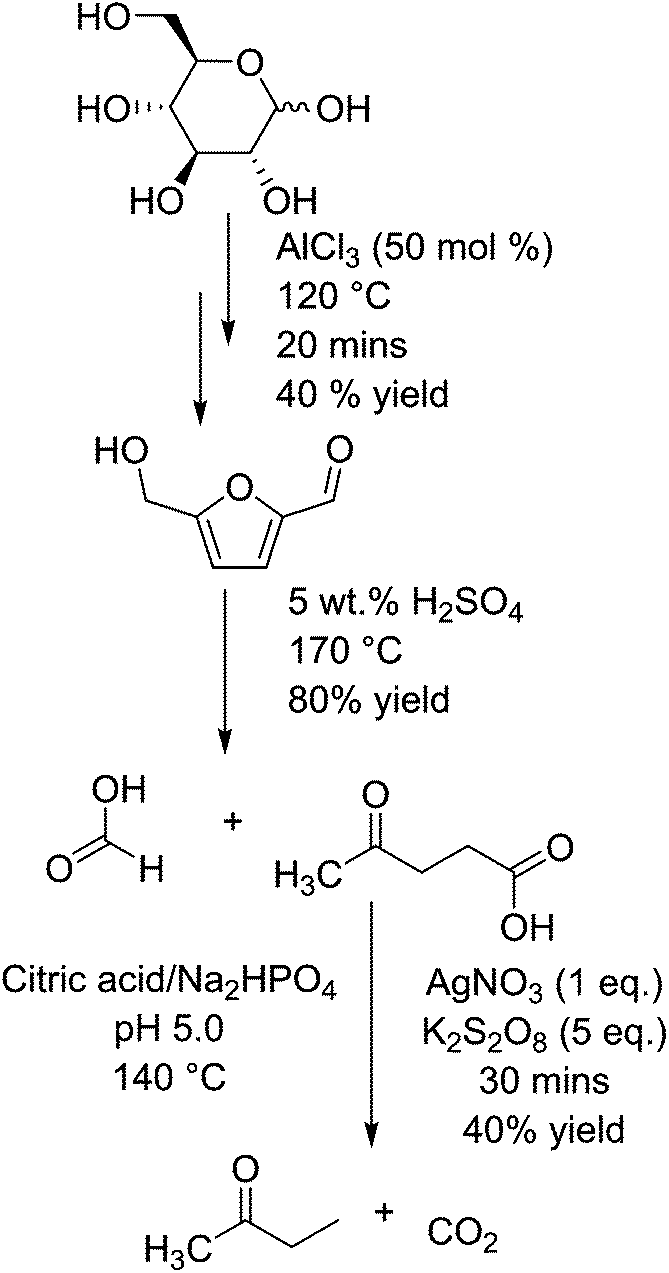 | ||
| Fig. 3 Formation of butanone from glucose via hydroxymethylfurfural and levulinic acid. | ||
2.2. Palladium decarbonylation
Carbonylation reactions are readily used in industrial scales, such as in the petrochemical synthesis of acetic acid amongst others.84 This process relies upon the presence of an Ir(CO)2I2 catalyst in solution. Before this was the Monsanto process based on Rh(CO)2I2 catalysis.85 Palladium(II) also shows carbonylation activity. However, its activity is less attractive than rhodium or iridium.86 However, when it comes to decarbonylation, at least of furfural, palladium-based catalysts show markedly higher activity compared to other transition metals.87Furfural is a major product from the dehydration of C5 sugars, mainly xylose and arabinose, originating from the hemicellulose component of lignocellulose.88 The amount of hemicellulose is between 5% for softwoods and up to 25% of the dry weight for hardwoods.89 Bagasse from sugarcane also contains between 26 and 30% pentosan sugars by weight,90 and some foodstuff by-products such as corn husks contain up to 28 wt% xylose.91 Given that these materials would otherwise be used for their caloric value, these feedstocks are preferable to wood in an industrial capacity. Due to the low overall yield involved with the production of furfural at large scale, the estimated market price of furfural is around 1700 $ per ton.88 The decarbonylation of furfural can be used to produce furan, an intermediate in the production of tetrahydrofuran (THF), a widely used solvent (Fig. 4). Furfural is also a promising intermediate for the synthesis of chemicals or fuel applications. Examples include methyl tetrahydrofuran and ethylfurfuryl ether as fuel oxygenates, as well as complex aldol condensation routes to C10 and C15 molecules which can be hydrogenated to form branched alkanes.82
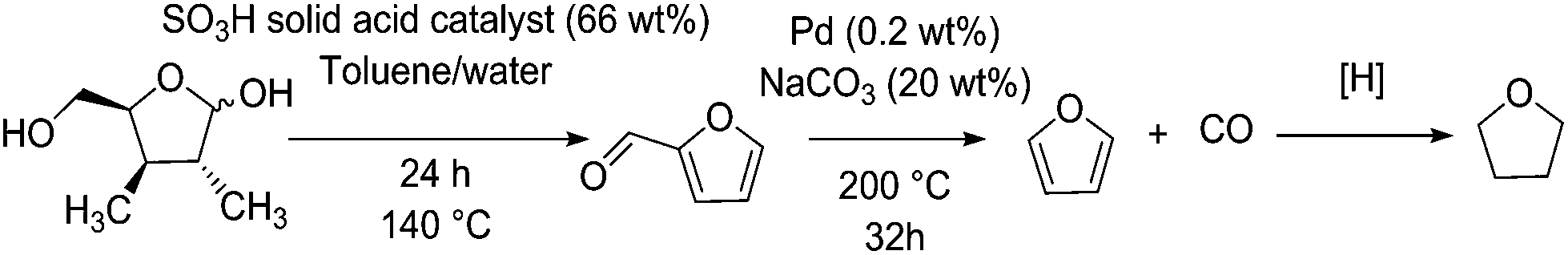 | ||
| Fig. 4 Formation of tetrahydrofuran (THF) via furfural from xylose. | ||
Shown in Fig. 5 is the mechanism elucidated for the decarbonylation of fatty acids by palladium(II)-based homogeneous catalysts, as proposed by Gooßen et al.92 and extended by others. This mechanism is reinforced by the detail that Gooßen et al. showed that the reaction takes place in an analogous fashion when an enol ester is substituted for the anhydride.93,94 It is thought that decarbonylation proceeds via the formation of an anhydride, which then allows the coordination of the palladium(II) salt. The anhydride is cleaved to produce an acyl anion, which then coordinated to the palladium atom. As the product of this reaction is an alkene, isomerisation of the double bond is an issue, but it can be eliminated by the use of correct conditions and ligands.
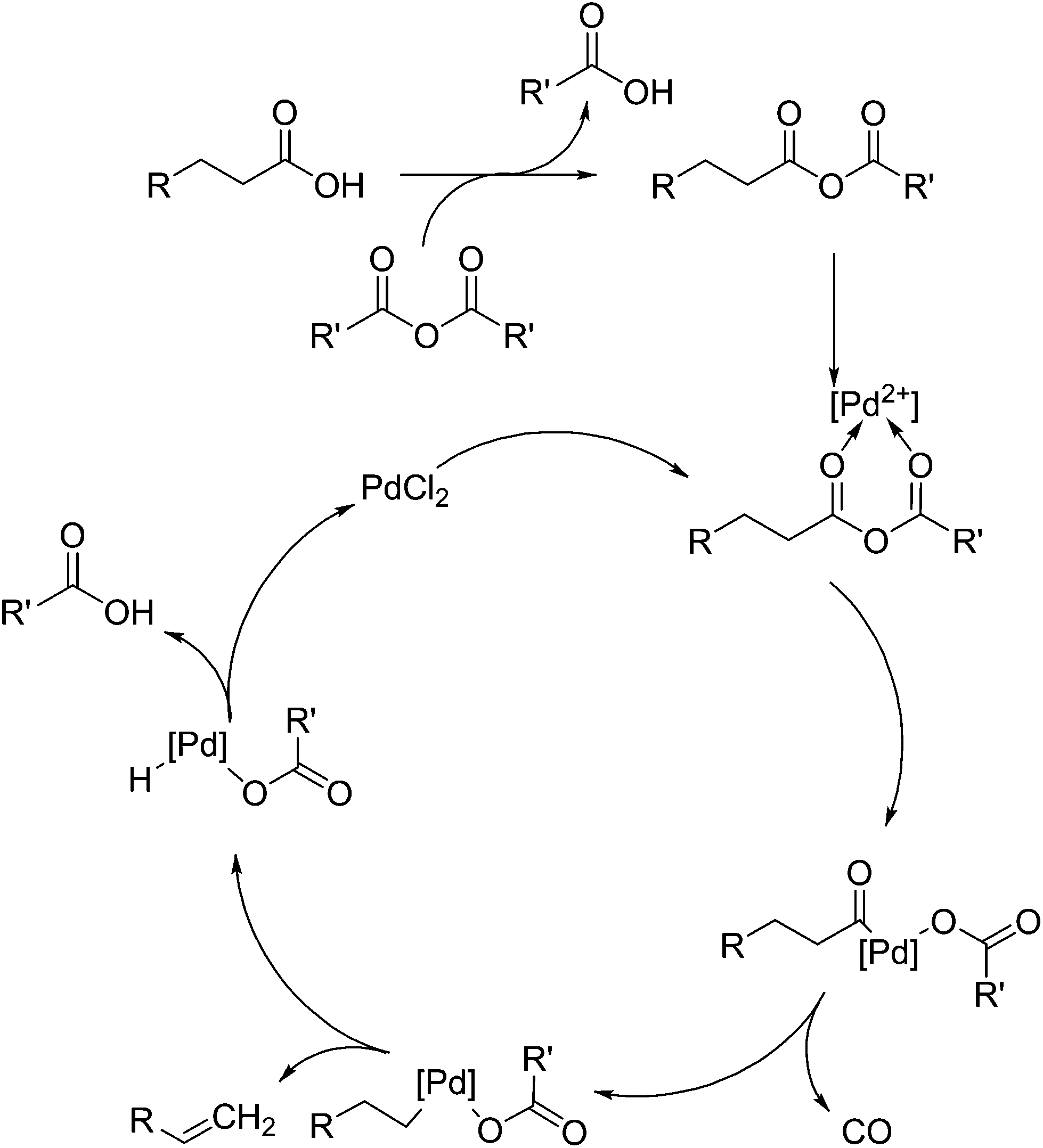 | ||
| Fig. 5 Palladium-catalysed decarbonylation of fatty acids via anhydride formation. | ||
Originally, the use of bis(2-diphenylphosphinophenyl)ether (DPEPhos) ligands and 125 °C was selected as a means of limiting the products to only 1-alkenes. It was shown that the use of pivalic anhydride can produce the required anhydride to promote decarbonylation, but further research shows the use of acetic anhydride produced comparable results.95 The use of stearic acid anhydride as a substrate also proceeded to 94% yield with a production of 93% stearic acid, indicating the addition of an auxiliary anhydride may not be necessary. Maetani et al. also showed that palladium(II) is not necessary either, showing a successful decarbonylation occurring with iron(II) chloride as a catalyst.96
One other conclusion from Gooßens work is that DPEPhos is needed as a ligand for PdCl2 to improve activity and selectivity in the transformation of phenylbutanoic acid, as the use of other phosphorus-based ligands allows for too-rapid isomerisation and loss of selectivity. By this same transformation method it was shown that palmitic acid could be transformed into 1-pentadecene with 64% yield after 16 h reaction time with 3 mol% catalyst in 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU), see Table 3. DPEPhos is not always required when isomerisation is not an issue, such as the decarbonylation of hydrocinnamic acid (described in 3.5) to hydroxystyrene, or of succinic acid monoester to acrylic acid ester.97 The conclusion drawn is that the much cheaper PPh3 ligand is almost as effective as DPEPhos in these reactions, indicating the role of this ligand as a moderator for by-product formation. In a similar reaction with IrCl(CO)(PPh3)2, the reaction conditions required to achieve good yields are much more stringent (250 °C rather than 110 °C).98 Under most circumstances, this catalyst shows a high selectivity for isomerisation at such high temperatures and in some cases produces less than 1% terminal alkenes. The follow-up work of this paper shows that the complex Fe(CO)(Cl)2 can be substituted for palladium in these reactions, although they show that the problem of isomerisation of the double bond can account for up to 9% of the final product, such as in the use of phenylbutanoic acid.
| Fatty acid R = | Anhydride R′ = | Catalyst | Ligand | Solvent | Yield 1-alkene (mol%) | Yield other alkene (mol%) |
|---|---|---|---|---|---|---|
| a 2 eq. Anhydride, 3 mol% cat. 9 mol% ligand, 110 °C, 16 h. b 10 mol% cat. 20 mol% ligand, 1 eq. KI, 1 eq. anhydride, 240 °C, 3 h, CO (20 atm). c 1,5-Bis(diphenylphosphino)pentane. | ||||||
| PhCH2CH2 | t Bua | PdCl2 | DPE-Phos | NMP | 70 | <2 |
| n-C13H27 | t Bua | PdCl2 | DPE-Phos | DMPU | 64 | — |
| PhCH2CH2 | Meb | FeCl2 | DPPPentc | — | 56 | 2 |
| n-C13H27 | Meb | FeCl2 | DPPPentc | — | 65 | 6 |
| n-C15H31 | n-C17H35b | FeCl2 | DPPPentc | — | 94 | — |
Significant work has been performed by the group of Gooßen et al. into the use of palladium(II) based catalysts as centres for aryl carboxylate decarboxylation, particularly for coupling reactions (Fig. 6).99,100 For Heck-type coupling reactions, the mechanism proceeds via a reductive elimination in order to remove carbon dioxide, so an oxidant is required to drive the reaction. The original work on this field uses silver nitrate as an oxidant,101 but this can be replaced with oxygen as the electron acceptor,102 without significant impact on yields or selectivity. For decarboxylative cross coupling, the primary mechanism is by transmetalation of aryl ligands and subsequent reductive elimination, shown below in Fig. 6. Because palladium(II) is not a very active decarboxylation catalyst, copper(I) or silver(I) is used to promote decarboxylation, and subsequently transfer the resulting aryl groups over to palladium.103 There they can be coupled with groups from aryl halides,104 other aryl carboxylates,105 or aryl groups produced by C–H activation.106 This reaction is useful for the utilisation of benzoic acid derivatives and producing compounds for azo dyes or other coloured compounds. In addition, the ability for this to activate C–H aryl bonds gives it added purpose in the utilisation of lignin-derived compounds.
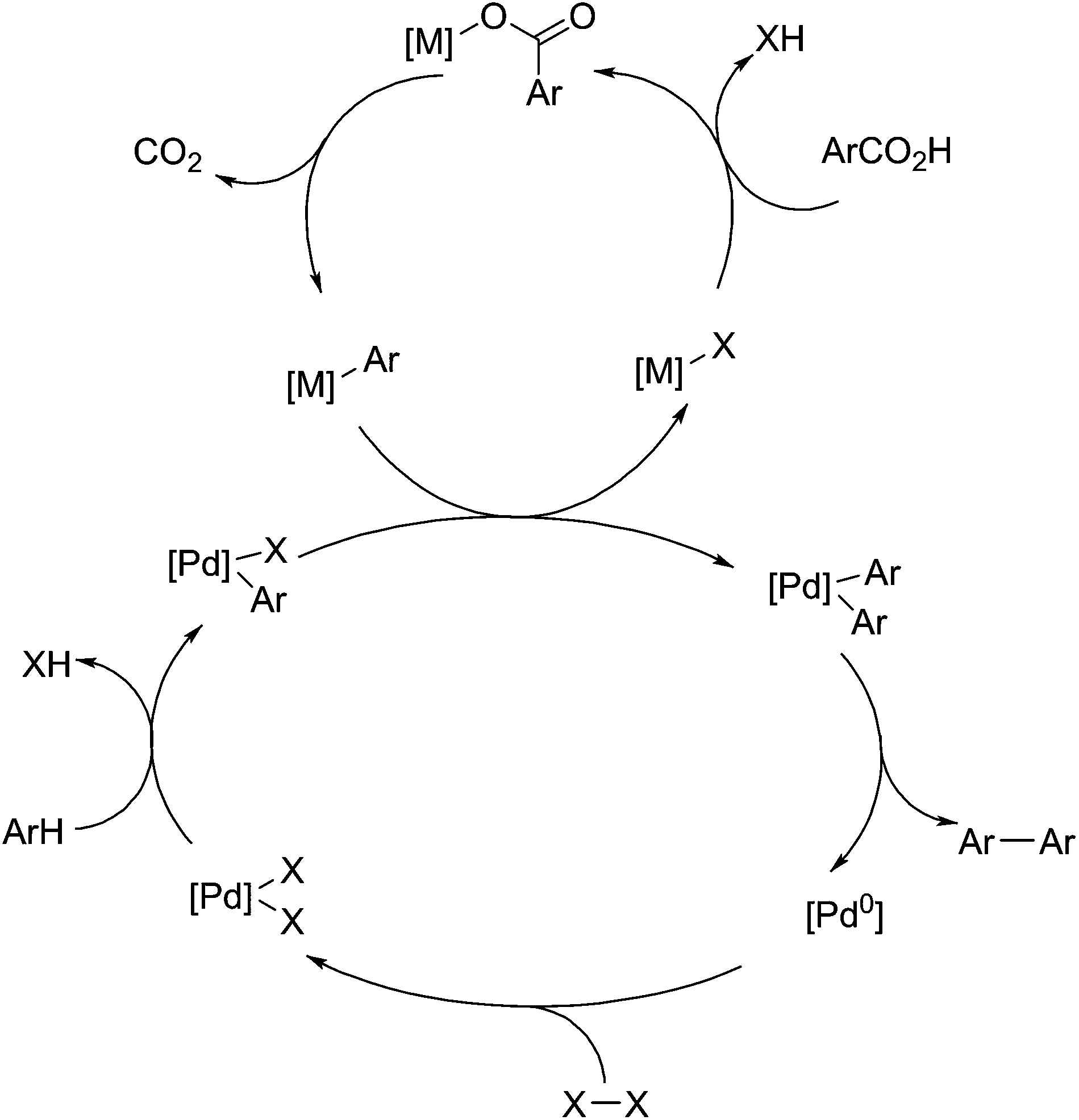 | ||
| Fig. 6 Decarboxylative coupling with Pd(II) and a second metal (M = Cu(I), Ag(I)). | ||
2.3. Henkel reaction
Henkel decarboxylation can be used to disproportionate the presence of carboxyl groups on aromatic molecules. Research on this reaction was originally performed for the production of terephthalate from benzoate, discovered in 1952 by Henkel et Cie.107 to work over cadmium salts at 500 °C. Terephthalic acid is used in the production of poly(ethylene terephthalate) (PET), a plastic used for its controllable crystallinity, transparency and low permeability of gases, and has a global production of above 40 million tons per year.108 Unlike the mechanism for silver-catalysed decarboxylation, this reaction proceeds via two-electron abstraction of CO2, and the use of cadmium(II) allows for proton abstraction, as shown diagrammatically in Fig. 7. It would appear that the catalyst is required for the selectivity for thermodynamic products, as Henkel-like activity has been observed in the pyrolysis of coal products at 425 °C, but the main product observed is phthalic acid rather than terephthalic acid.109 The mix of products in the reaction system eventually disproportionates and rearranges to form the products, benzene, and the thermodynamic product 1,4-terephthalate.110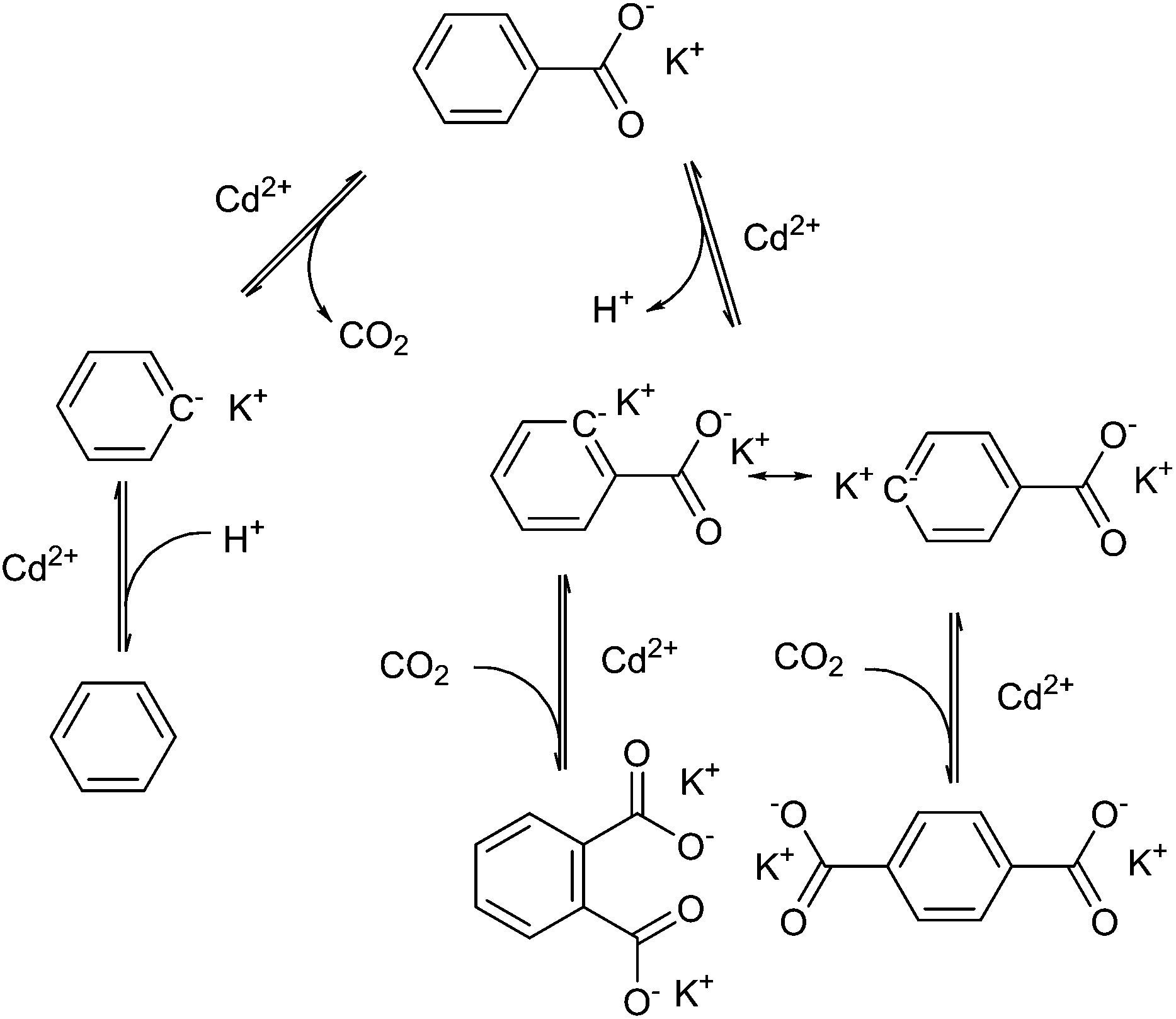 | ||
| Fig. 7 Radical rearrangement of Henkel reaction products. | ||
It is worth mentioning here that terephthalic acid can also be derived from other biomass sources such as via the oxidation of biomass-derived xylene.111 Xylene can also be formed by dimerization of isobutene, produced by glucose fermentation to isobutene112 or dehydrated bio-isobutano.113 Other methods include the oxidation of para-cymene,114 and the Diels–Alder reaction of muconic acid and ethylene followed by aromatisation.115 These are attractive processes for the production of ‘drop-in’ compounds from bio-based origins, however they are beyond the scope of this review.
Analogous to the Henkel route to terephthalic acid from benzoic acid, this reaction can be applied to furoic acid, derivable from furfural to form 2,5-furandicarboxylic acid (FDCA).116,117 Pan et al. showed an 86% selectivity for FDCA when this reaction was performed at 250 °C under 38 bar CO2 for 3 hours with zinc chloride as a catalyst, see Fig. 8.118 FDCA, when used in place of terephthalic acid to produce poly(ethylene furandicarboxylic acid) (PEF) can have similar physical properties to PET.119 FDCA can also be produced by the oxidation of 5-hydroxymethylfurfural, shown in section 2.1 to be able to be obtained from glucose by dehydration. This oxidation is reported to occur by the use of stoichiometric potassium permanganate,120 but recently has been found to occur readily with the use of gold nanoparticles on cerium oxide supports, giving as much as 99 mol% selectivity at 130 °C in water with air as an oxidant.121 One benefit of the Henkel reaction is that it produces furan as a side product, which itself can be hydrogenated to tetrahydrofuran or 1.4-butanediol. This was proposed by Pan et al. as a carbon-efficient means of producing polymers from furfural, using 1,4-butanediol and FDCA as precursors to produce poly(butyl furandicarboxylic acid), thus utilising 100% of the input carbon.118 The highest selectivity disproportionation was performed in a sealed reactor at 250 °C, using 0.1 g zinc chloride as a catalyst per gram of substrate, and in supercritical carbon dioxide. They report no activity when other metals such as cadmium, copper or nickel, were used, although a small amount of conversion was observed for lanthanum. They also show that chloride ions are a required counter ion, observing no activity when zinc oxide or zinc acetate were used instead.
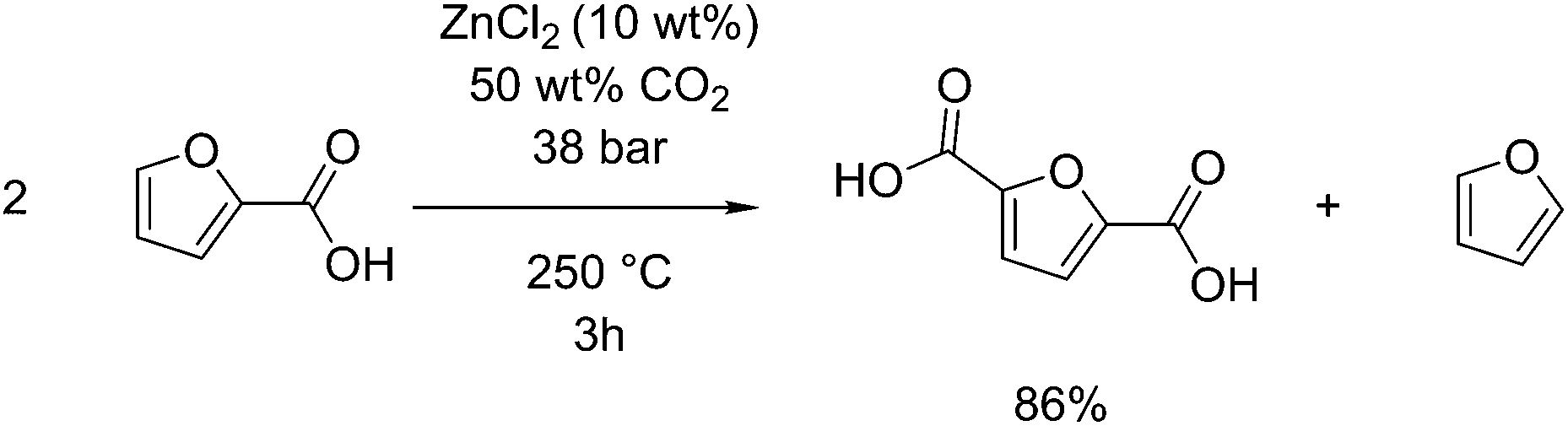 | ||
| Fig. 8 Henkel reaction performed on furoic acid. | ||
2.4. Kolbe reaction
The Kolbe reaction is a means of coupling two carbon chains together with subsequent oxidative decarboxylation. This electrochemical reaction is interesting from a technological point of view as the oxidation is carried out at the electrode, requiring no auxiliary compounds and thus, in theory, improving atom efficiency. This property has highlighted electro-synthetic reactions as particularly applicable to the aspects of Green Chemistry122 and several applications of this reaction can be seen in Fig. 9. However, the technical challenges of electro-synthetic reactions, especially the separation of the products from the solution and the necessary separation of anodic reactions from cathodic reactions, are significant.122 It was originally discovered as an electro-oxidation reaction,123 occurring on the anodic side of a reaction vessel, where oxidations take place. Mechanistically, this reaction is disagreed upon in its basic elements.124–126 The reaction however is generally agreed to proceed via the formation of a carboxyl radical and subsequent decarboxylation into an alkyl radical. Sufficiently stable radicals at this step can go on to form n-1 length alkanes, alkenes or alcohols, but with the correct substrate a radical coupling reaction can occur, creating 2n–2 length carbon backbones. This reaction can be directed by the use of pressure,127 in the case of pentanoic acid, 1.1 MPa was sufficient to boost selectivity for n-octane from 42% to 63% (scheme ii and iii, Fig. 9). As this reaction was originally observed for the formation of ethane from acetic acid, applications of this reaction are centred around the decarboxylative coupling of fatty acids, upgrading them to longer chains.128,129 Recent work has been shown to be able to use this technique for cross coupling nitrile terminated fragments in the production of adiponitrile, derived from glutamic acid.130 They show a yield of 78 mol%, indicating the selectivity of this reaction to carboxylate components. Adiponitrile has a global scale of around 1.5 million tonnes, and is selectively hydrogenated producing high-purity 1,6 hexanediamine for the production of nylon-6,6 (scheme i), Fig. 9).131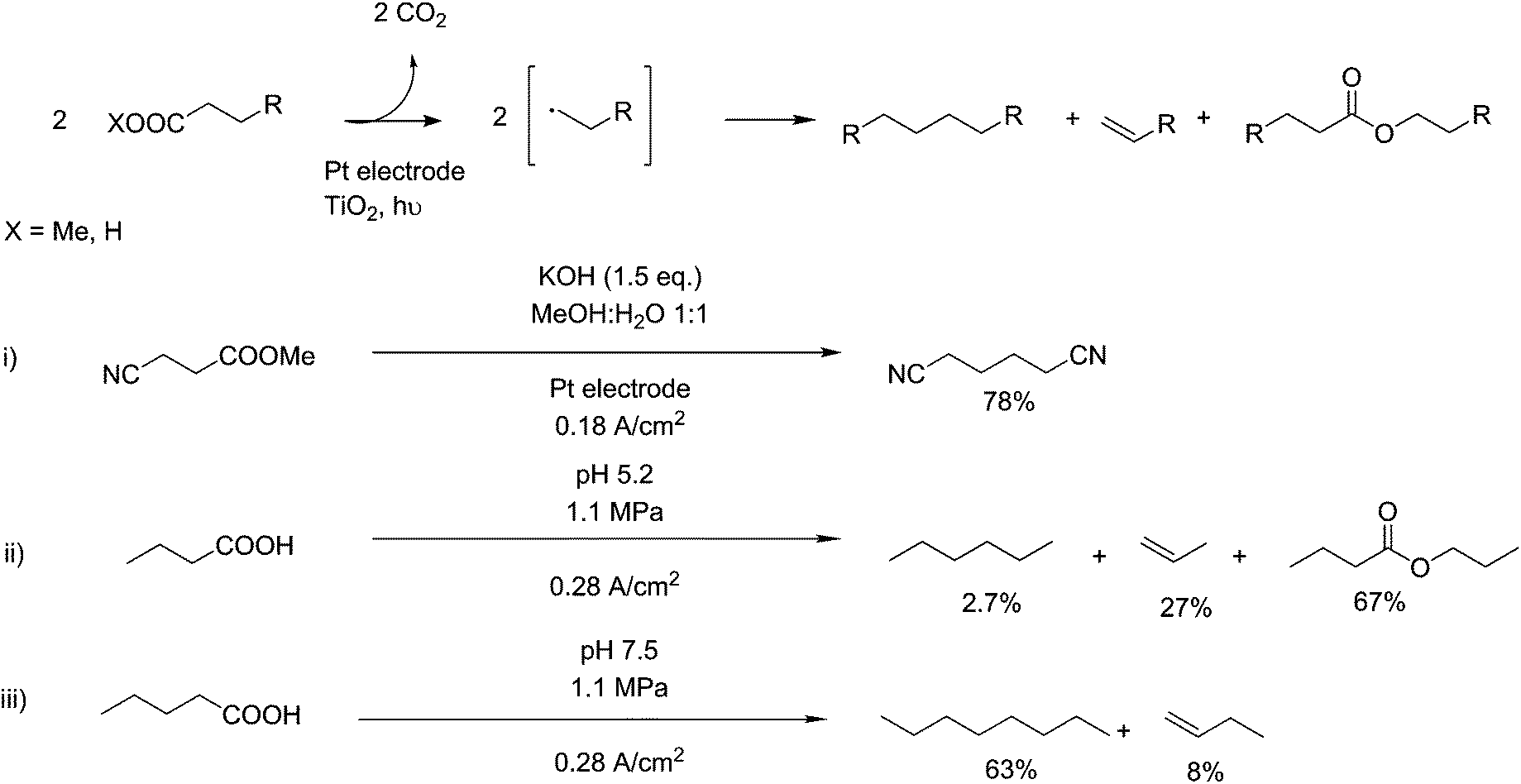 | ||
| Fig. 9 Kolbe reactions of different substrates. | ||
2.5. Bio- and organocatalysis
There are a number of potential sources of proteins and amino acids132,133 including feathers134 and vinasse (by products of sugar cane production)135 that can be used to produce a variety of interesting chemicals via deoxygenation. Enzymes are an attractive catalyst choice as they are capable of high selectivity for substrates in low concentrations in an aqueous environment. One of the main challenges in the use of amino acids is the ability to obtain pure amino acids for a synthetic route. However, as enzymes are substrate specific, this allows for potentially selective transformation in mixed streams, and potential separation as a result of the changed charge characteristics, for example by decarboxylation.136An alternative strategy in obtaining amino acids may be obtained using microorganisms. For example by the production of the storage protein, cyanophycin (multiarginyl-poly[L-aspartic acid]) produced by cyanobacteria.137 It has been shown that this well-defined molecule can be produced from complex amino acid containing aqueous mixtures.138 It is accumulated in the cell bodies and can be filtered out of the broth, allowing for simpler production of a concentrated stream of just two amino acids. When hydrolysed, only aspartic acid and arginine are produced.139 Arginine can be used to produce 1,4-diaminobutane via hydrolysis to ornithine and subsequent decarboxylation.46 1,4-Diaminobutane is a monomer for the production the polymer nylon-4,6 (Stanyl®) produced, by DSM. Aspartic acid can be alpha-decarboxylated to form beta-alanine by the use of aspartate alpha-decarboxylase, an intermediate that could be used for the synthesis of acrylamide.45 Other examples include the production of gamma-amino butyric acid from glutamic acid by enzymatic decarboxylation,44 which can be used for the production of for example N-methylpyrrolidone140 or N-vinyl pyrrolidone. Phenylalanine can be deaminated by the use of phenylalanine ammonia lyase (PAL) to obtain cinnamic acid that is subsequently converted by deoxygenation to styrene by either direct decarboxylation72 or to styrene and acrylic acid by ethenolysis.141 Other simple amines can be produced by the decarboxylation of amino acids, such as ethanolamine from serine, or ethylamine from alanine.74
Amino acids also be oxidatively decarboxylated to form nitriles, using NaOCl and NaBr to form 3-cyanopropanoic acid, and subsequently succinonitrile, from glutamic acid.142,143 Nitriles such as succinonitrile are used mostly for their ability to be selectively hydrogenated to diamines for the production of nylons. 3-Cyanopropanoic acid can be readily converted into acrylonitrile, a monomeric starting compound for the production of acrylonitrile-butadiene-styrene (ABS) and nitrile butadiene rubber (NBR). Acrylonitrile can also dimerised to from adiponitrile,144 or hydrated to form acrylamide.145 As such, the market scale for acrylonitrile reaches around 6 million tonnes.143 The reaction of methylglutamate to 3-cyanopropanoic methyl ester requires 3 equivalents of NaOCl and catalytic amounts of NaBr at 4 °C to give 70% isolated yield, and proceeds via the production of “Br+” equivalences, the activity of these is sufficient to decarboxylate glutamic acid. This reaction is not within the bounds of Green Chemistry as large amounts of waste salt are produced and the requirement for cooling makes for an expensive process,146 however this reaction remains interesting as enzymatic routes using vanadium bromoperoxidases and chloroperoxidases have been shown to oxidatively decarboxylate valine,147 and appear to produce the same “Br+” activity using hydrogen peroxide. Recently, vanadium chloroperoxidase has been used to perform the oxidative decarboxylation of glutamic acid using hydrogen peroxide as an oxidant and a catalytic amount of bromide salts, with a near 100% selectivity for 3-cyanopropanoic acid (Fig. 10).148
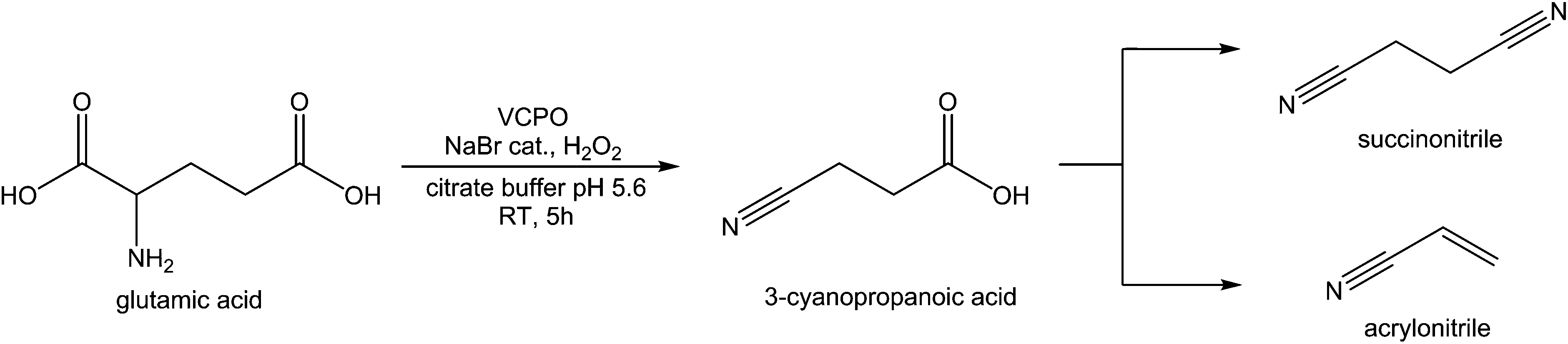 | ||
| Fig. 10 Production of succinonitrile from glutamic acid. | ||
Amino acids are not the only substrate that can be enzymatically deoxygenated, as the use of P450-based enzymes such as OleTJE have resulted in the enzymatic decarbonylation of stearic acid.149 They report a selectivity for 1-tridecene of >90% from myristic acid and hydrogen peroxide at 28 °C for 2 hours, with much of the byproduct being the beta-hydroxy alkane.
The decarboxylation of amino acids can be performed using non-metal based organocatalysts in an effective route to the formation of amines. The first paper on this topic shows amino acid transamination by the Strecker degradation,150 where alloxan is used as an electron and ammonia acceptor, turning into the purple murexide. Strong electrophiles such as ninhydrin151 also promotes decarboxylation, but are so active that amine hydrolysis is not possible and instead aldehydes are produced. Pyridoxal is a cofactor in many decarboxylase enzymes, but can also be used as a decarboxylation catalyst capable of converting about 50 mol% of phenylglycine in 30 minutes at 100 °C in water. It has the issue that it produces less than 50% yield of amines, preferring to transaminate amino acids to aldehydes.152 Both the activity and selectivity can be enhanced greatly by the use of an enzyme mimic to prevent amino acid hydrolysis.153 This can be as much as 16 times faster for the decarboxylation of phenylalanine in water. The same group goes on to show that they can perform selective transaminative decarboxylation using the same catalyst.154 Other catalysts that promote the decarboxylation of amino acids include benzaldehyde,155 or aromatic ketones, such as acetophenone and its derivatives,156 but significant quantities of Schiff bases are reported, sufficient for the authors to require boiling concentrated hydrochloric acid to hydrolyse to produce as much as 90% phenethylamine from phenylalanine, when o-methoxyacetophenone was used at equimolar concentrations, as shown in Table 4. A good selectivity was achieved for many of the amino acids used, however the selectivity for lysine conversion is reduced, possibly due to the second amine group on the molecule.
| Amino acid | Hydrolysis methodb | Yield amine product (mol%) | Yield transamination product (mol%) |
|---|---|---|---|
| a Reaction conditions: use of o-methoxyacetophenone (100 mol%), 100–240 °C. b A = 3 M NaOH, 100 °C, 3 h, B = conc. HCl, 100 °C, 3 h. c 200 mol% ketone used. | |||
| Phenylglycine | A | 80 | <1 |
| Phenylalanine | A | 90 | <1 |
| Tyrosine | B | 80 | 0 |
| Histidinec | B | 61–81 | 0 |
| Tryptophan | A | 50 | 0 |
| Leucine | A | 90 | 0 |
| Lysine | B | 25 | 0 |
3. Heterogeneous catalysis
3.1. Titanium dioxide photodecarboxylation
Titanium dioxide (TiO2) surfaces are known for their photoactive behaviour, and have been studied extensively for their ability to degrade organic compound, however it is a difficult process to make selective. Control of this process can be found by affecting the crystal structure i.e. by selecting between anatase, brookite and rutile phases. Further, adjusting the particle size, concentration of surface defects and applied gas pressure affects the photoactivity of titanium dioxide particles.157 anatase/rutile-TiO2 is active as a decarboxylation agent for levulinic acid, primarily producing methyl ethyl ketone before further degradation towards CO2 at higher concentrations (Fig. 11).158 The conversion of this feedstock is poor, but the selectivity for the desired product reaches 95%. It even shows a good activity of photodegradation of fatty acids such as valeric acid, resulting in a rapid (0.71 mmoles h−1) degradation to isobutane,159 implying it could be active on longer fatty acids also. Malic acid, a byproduct formed by the fermentation of sugars into lactic acid,160 can be photodegraded by TiO2 coated fibers, and produces malonic acid and other small intermediates such as pyruvic acid, fumaric acid and maleic acid, via a photo-Kolbe oxidation mechanism.161 Alternatively, TiO2 can be used to photo-deformylate tryptophan into kynurenine under black light with up to 90 mol% selectivity.162 Kynurenine can be used to produce alanine and, by further decarboxylation over zeolite catalysts, bio-based aniline.163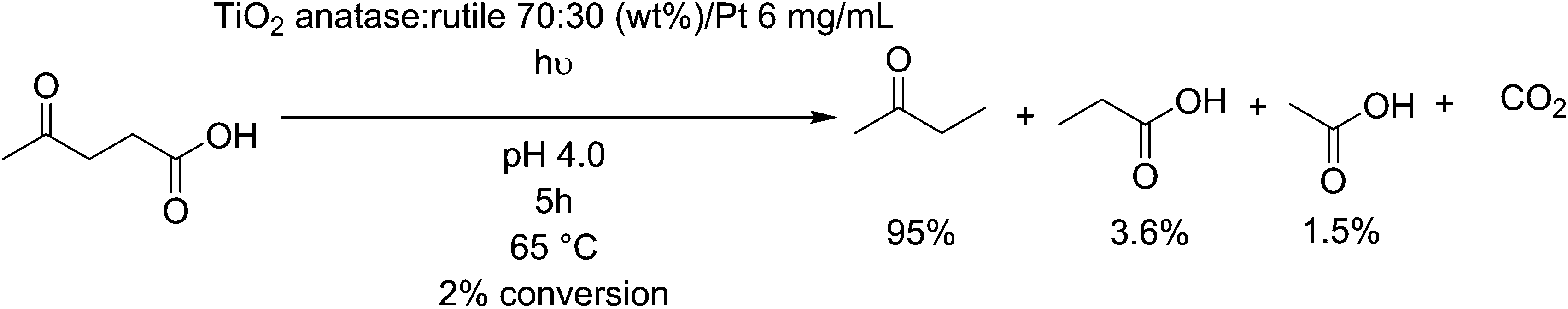 | ||
| Fig. 11 Photocatalytic degradation of levulinic acid. | ||
3.2. Platinum/palladium heterogeneous decarboxylation
In similar fashion to the use of palladium and rhodium164 in a homogeneous phase, significant work has been performed in using heterogeneous palladium165 or platinum on a solid support to decarboxylate fatty acids166 into fuel-compatible alkanes. Three competing reactions are at play on these metal surfaces: decarboxylation, decarbonylation, and ketone formation where two alkyl chains cross-couple (Table 5).167 The specificity of product formation appears to be controlled first by choice of metal: platinum produces more alkenes, palladium produces more alkanes.168 The use of the correct support is important: γ-alumina has been observed to convert canola oil into 77% light gases at 500 °C,169 but selectively produce stearone from stearic acid at 300 °C (Table 5, entries 1 and 2).170 Palladium or platinum on mesoporous carbon seems to show a much greater selectivity: palladium on carbon shows 92% selectivity for undecane formation from lauric acid at 270 °C (Table 5, entry 3),171 and platinum on carbon reaches 95% selectivity for decarboxylation of linoleic acid at 330 °C (Table 5, entry 4).172 This has been independently investigated by Ford et al., showing that palladium on carbon gives the greatest (96%) selectivity for decarboxylation, and palladium on silica (at 8.4% dispersion) gave the lowest (2.8%).173 Other supports are also successful: decarboxylative coupling can be used to form 5-nonanone from pentanoic acid (derived from levulinic acid via gamma-valerolactone) by the use of a palladium on niobium oxide catalyst (Table 5, entry 5).174 The addition of hydrogen gas into this reaction allows for a competing deoxygenation reaction to occur. In a similar mechanism, palladium on carbon gave 93% conversion of stearic acid at 300 °C,175 but the addition of hydrogen under argon continues to allow decarboxylation despite conditions for deoxygenation.176,177 In fact, more hydrogen appears to slow the reaction as carbon monoxide adsorbs onto palladium surfaces, selectively blocking them178 and promoting decarbonylation instead.179 palladium on carbon catalysts can also decarbonylate furfural,180 with a tendency to produce difuran and other polyfuranic species.| Entry | Fatty acid R = | Catalyst | Time (h) | Temp (°C) | Solvent | Yield alkane (mol%) | Yield 1- alkene (mol%) | Yield cross-coupled ketone (mol%) |
|---|---|---|---|---|---|---|---|---|
| a Gamma-valerolactone. b WHSV/h−1. c Also gave 6 mol% pentanoic acid. | ||||||||
| 1 | C15H31 | Pd/γ-Al2O3 | 24 | 250 | Dodecane | 100 | 0 | 0 |
| 2 | C15H31 | γ-Al2O3 | 24 | 250 | Dodecane | 0 | 0 | 100 |
| 3 | C9H19 | Pd/C | 0.5 | 270 | Dodecane | 92 | <1 | — |
| 4 | C15H31 | Pt/C | 2.5 | 330 | H2O | 95 | — | — |
| 5 | GVLa | Pd/Nb2O5 | 0.1b | 350 | H2O | — | — | 57c |
3.3. Hydrotreating catalysts in decarboxylation and decarbonylation
Hydrodeoxygenation (HDO) is a competing reaction type used on large scales where oxygen is removed from a molecule via the addition of hydrogen, yielding water and an alkane. Typically, these catalysts are unselective for which heteroatom they remove from a molecule, simultaneously performing hydrodenitrogenation and hydrodesulphurisation in crude oil feeds. Catalysts for this reaction are often molybdenum-based, such as Mo2C or NiMo alloys on alumina supports, but also Pd and Pt as described earlier.48 The main active sites of these catalysts are reported to be on the MoS2 crystal edges, formed after sulphidation of the catalyst.181 As these are particularly active on aryl compounds, the main biomass use for these catalysts is the conversion of lignin-derived phenols.182 Up to 35% of natural wood is lignin,183 and the separation of the lignocellulosic components has a strong history in the pulp and paper industries. There has been several alternative techniques investigated for the isolation of lignin.184,185 Depolymerisation of lignin would lead to a source of (oxygenated) aromatic ring structures. While some effort has been made, there are significant issues surrounding the isolation and depolymerisation of this feedstock.186–188 The desired products of benzene, toluene and xylenes (BTX) have a market scale of between 250 and 300 million tons.189 Lignin deoxygenation proceeds faster for more oxygenated molecules such as syringol rather than phenol, and is remarked to be rather unselective, producing a chain of intermediates before finally producing benzene and toluene, however cleavage of aryl groups was not observed.190 HDO catalysts suffer significantly from deactivation which is correlated to the acidity of the feedstock. Lignin derivatives are commonly phenol-based, which have a high pH, and can coke easily on catalysts.191HDO is also used for the deoxygenation of fatty acids, where both decarboxylation and hydrodeoxygenation reactions are taking place on the same catalyst. The two reactions however occur at different rates depending on the conditions. For example, considering the HDO of waste cooking oil rich in C18 fatty acids: after 40 hours at 350 °C, a CoMo/Al2O3 catalyst produced alkanes of which 20% were C17 decarboxylation product, but under the same conditions a NiW/Al2O3 catalyst produced up to 50% C17 alkanes. Because chain length shortening only occurs by decarboxylation or decarbonylation, this implies that the NiW/Al2O3 catalyst promotes these reactions better.192 Sulphided tungsten derivatives can also be effective HDO catalysts. The proportion of hydrogenation and decarboxylation can be tuned based on whether tungsten oxide (38% conversion, 91% decarboxylation) or tungsten carbide (81% conversion, 15% decarboxylation) are used as a catalyst. Both of these are supported on carbon nanofibres, and the conclusions are based on results from the HDO of stearic acid at 350 °C for 5 hours with 50 bar H2, 12.5 wt% catalyst, in dodecane.193 Jatropha is a hardy plant that grows in poor soil conditions, and produces an oil rich in C18 and C16 triglycerides.194 Hydrodeoxygenation of this oil over NiMo/SiO2 using 800 mL H2 at 4 MPa per mL solution and 250 °C for 10 h produced 82.9 wt% C11–20 alkanes and 4.3 wt% C1–4 alkanes, effectively hydrolysing and reducing the glycerol in one step (Fig. 12). Some decarboxylation and decarbonylation reactions were observed, accounting for the presence of C17/C15 molecules in the product distribution. Further investigation into the selectivity for hydrodeoxygenation versus hydrodecarboxylation has been reported, investigating the use of CoMo catalysts on several grades of Al2O3 support, using triglyceride-rich rapeseed oil as a substrate. Decarboxylation and decarbonylation are also avoided at higher temperatures, producing a 16:1 ratio of C18:C17 alkanes for CoMo on an organised mesoporous alumina support at 280 °C, compared to a 12:1 ratio at 250 °C, implying a negative relationship between decarboxylation and reaction temperature. Pressure plays a significant role, increasing the reaction pressure from 0.7 to 7 MPa changes the resulting alkanes from a 3:1 ratio of C18 to C17 to 16:1 at 280 °C.195,196 Conversely, when sunflower oil was used on a CoMo/Al2O3 catalyst with 40 bar H2,197 a reaction temperature of 380 °C produces 11.7% C17 alkanes and 70.3% C18 alkanes. At 300 °C the same conditions produced 2% C17 alkanes and 27% C18 alkanes, displaying a positive relationship between temperature and rate of decarboxylation.
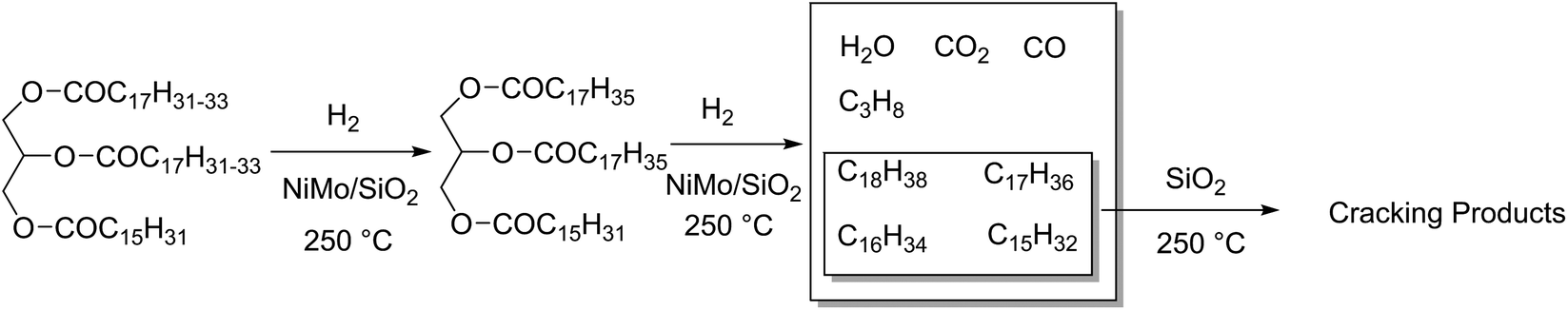 | ||
| Fig. 12 NiMo catalysed hydrotreatment of Jatropha oil to C18–15 alkanes and subsequent cracking products. | ||
4. Thermal decarboxylation
4.1. High temperature water treatment
Citric acid is produced fermentatively by Aspergillus niger at high productivities, over 70% theoretical yield,198 and has a market in the nutraceutical field of about 0.4 million tons per year.199,200 When citric acid is exposed to high temperature water (HTW) close to supercritical conditions (320 °C and 34.5 MPa), it is converted into 6% methacrylic acid and 35% itaconic acid, with a wide spectrum of intermediates such as citraconic acid, mesaconic acid and acetic acid, as shown in Fig. 13.201 When pure itaconic acid was subjected to the same reactor at 360 °C with 20 mol% NaOH, it produces 71% methacrylic acid, with the main byproducts being acetic acid, crotonic acid and hydroxyisobutyric acid. More recently it has been shown that methacrylic acid can be obtained from itaconic acid with yields up to 58% using 1 equiv. of NaOH at temperatures between 250 and 350 °C, 200 bar pressure, and a residence time of 520 seconds in continuous reactors.202 Itaconic acid is a novel molecule produced from biomass that has uses as a comonomer in plastic production,203 and as a surfactant, and is able to be transformed into molecules such as 3-methyl tetrahydrofuran, 3-methyl N-methylpyrrolidone, and 3-methylpyrrolidine. It has a current market scale of around 150000 tons per year globally,204 growing fast from 15000 tons in 2001. It is primarily produced fermentatively by fungi, however there are recent efforts in producing itaconic acid in other species such as plants.205 Methacrylic acid is a monomer produced at a scale of around 2.2 million tons per year for the manufacture of poly(methyl methacrylate), a polymer used for its high transparency.206
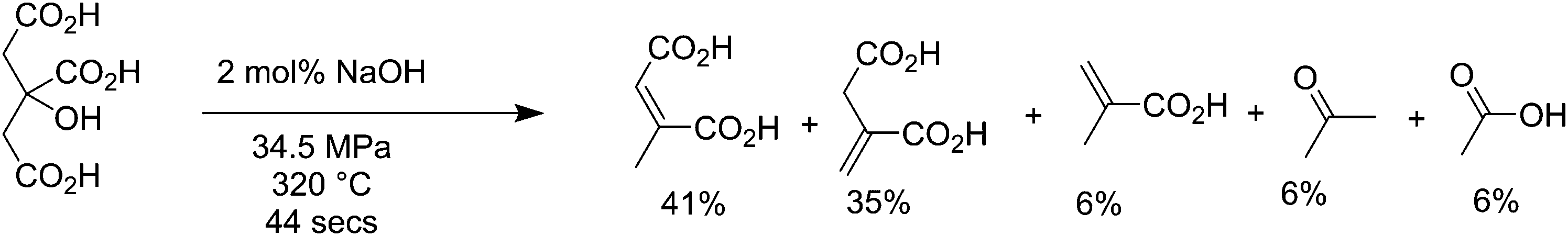 | ||
| Fig. 13 High temperature water decarboxylation of citric acid. | ||
HTW conditions also allow for controlled oxidation conditions with the addition of hydrogen peroxide, which can selectively remove carbon-rich molecules from a waste stream, without producing the oxides of sulphur and nitrogen that are associated with pyrolysis. This has been used on vanillic acid as a model compound for lignin to some success, showing catechol, cresol and phenol produced.207 Fatty acids have also been tested, and at 400 °C a wide range of diacids and ketones were formed.208 Glucose, when treated to similar conditions at 374 °C for 120 min with 4.5 mol% hydrogen peroxide in 100 mL gave a wide range of organic products (from propanoic acid to benzaldehyde), with 42% CO2 being produced at the same time.209
Amino acids and proteins have also been studied under HTW conditions as a means of reclaiming material from protein-rich side streams. An example is the reclamation of seafood waste, where HTW was used to hydrolyse the protein down, but only around 7% selectivity was observed at 250 °C.210 Investigation into this showed that significant non-selective decarboxylation and deamination reactions were occurring in the reaction conditions. A myriad of different products were formed, but deamination of aspartic acid was noted to be a predominant mechanism, forming fumaric acid, and serine underwent a retro-aldol condensation to form glycine.211 In the case of dipeptides, a significant amount of cyclisation also was found to occur.212 Phenylalanine is able to react with 1 eq. 2,4-decadienal at 180 °C in water at 9.8 atm for 60 min with a sodium phosphate buffer at pH 6, being converted to styrene by as much as 6 mol%.213 At 280 °C and 6.4 MPa in HTW for 60 minutes, phenylalanine conversion is at 97 mol%, producing 68 mol% phenethylamine and 4 mol% styrene, however 19 mol% of the mass is not accounted for.214 This process is shown diagrammatically in Fig. 14.
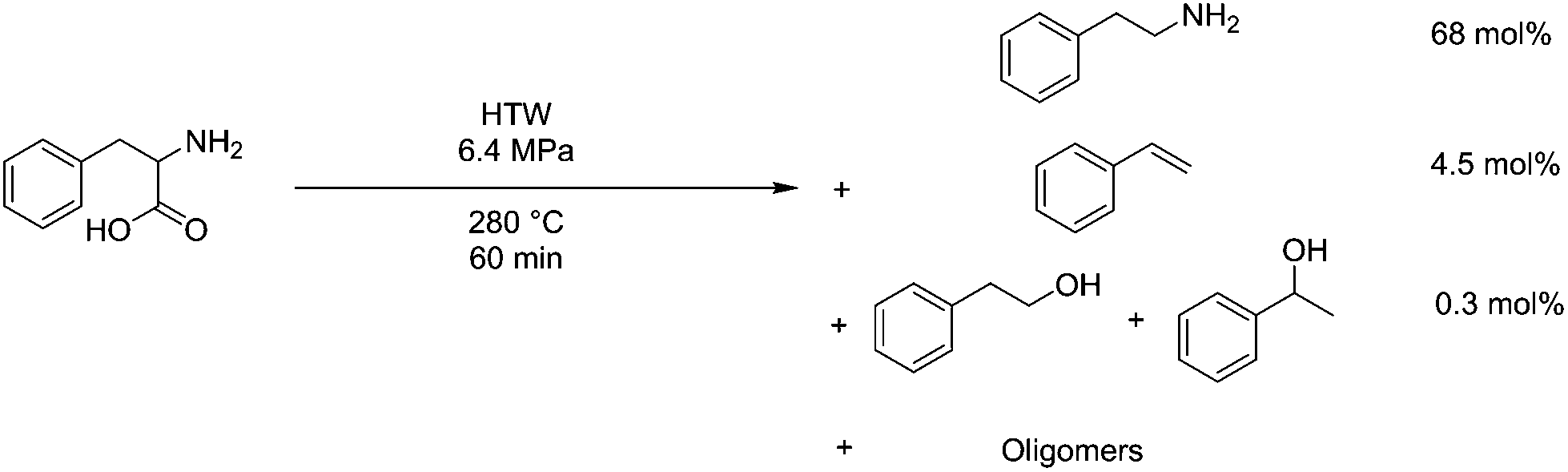 | ||
| Fig. 14 Degradation of phenylalanine in HTW conditions. | ||
4.2. Pyrolysis
By applying heat to biomass in an anaerobic environment, a significant amount of oxygen can be removed from the material, and liquefaction is achieved leading to what is called bio-oil.215 The liquefaction of soybean straw and vegetable oil has been attempted at 400 °C, where a significant amount of decarboxylation (88 wt%) takes place.216 In the gas phase, 3 wt% short alkanes (3 carbons or less) were formed, and at much as 50% yield of alkanes was shown, but only in the case where pure sunflower oil was used was no char formed, otherwise between 20–30 wt%. A wide range of compounds were formed in addition to char.The production of bio-oil from the high temperature liquefaction of lignocellulosic biomass is mainly performed over zeolite catalysts at a temperature of 600 °C. For example, a pine wood and methanol mix can be converted to 14 mol% aromatic molecules and 9.4 mol% olefins, with 26 mol% coke formation and 32 mol% carbon monoxide.217 Bio-oil is not a good replacement for petroleum, as it has a very high oxygen content (up to 60 wt%), is acidic, and often has a high water content.218 Pyrolysis has been used to stabilise a water soluble bio-oil by deoxygenation, but up to 32 mol% of char can be formed. A combined pyrolysis and hydrogenation technique using 4.8 g H2/100 g carbon at 52 bar over ruthenium on carbon at 125 °C succeeded in reducing char to 17.4 mol%, with a shift towards aromatic molecule production.
Instead of attempting to eliminate the production of char, it can be promoted by the use of hydrothermal carbonisation. Yields of 66% char from 72 hours under pressure at 200 °C have been reported for wood,219 and a marked reduction in O/C ratio from 0.58 to 0.17 has been achieved, alongside a reduction in H/C ratio, indicating aromatisation. Unfortunately, due to the wet nature of biomass and the requirement for water in this reaction, extracted organic compounds are always present and thus carbonisation does not proceed to completion.
5. Summary and outlook
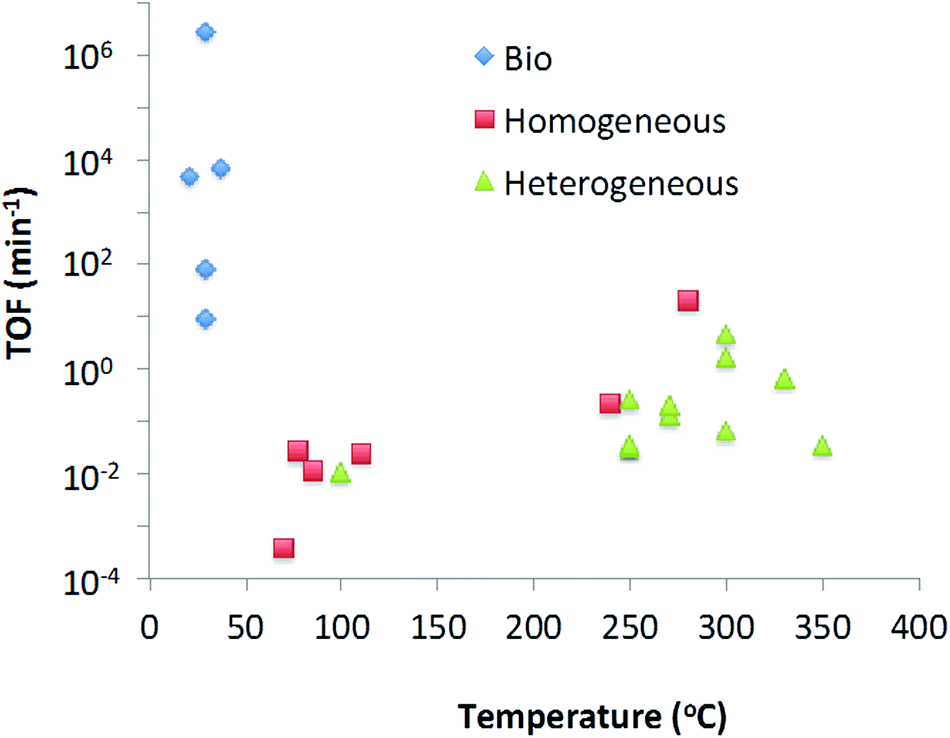
In this review we have looked at various methods of catalytic deoxygenation, using multiple Biobased substrates. In order to be able to draw conclusions on the suitability of certain catalysts with substrates for deoxygenation (in the form of decarboxylation and decarbonylation) a uniform entity should be defined. Due to the variability of the reaction conditions this is not trivial. For efficient industrial application the volumetric productivity would be a useful comparison. However volumetric productivity is linked to the maximum density of active sites per unit volume and the maximum turnover frequency of each active site. On laboratory scale, the maximum site density per unit volume is of less significance therefore making comparison of published data unrealistic. Instead, the turnover frequency (TOF) (moles substrate converted per mole catalyst per minute) has been selected as a suitable indication as to reactor productivity independent of type (plug flow, batch, etc.).From Table 6, it can be seen that the many reactions can be broken down to their fundamental reaction characteristics such as reaction rate, temperature and concentration based on literature data. The rates given have been calculated at a point where the reaction is close to linearity as possible. From this data we can see one very clear result, that all heterogeneous catalysts are tested at 250 °C and above, all homogeneous catalysts are between 280 and 70 °C, and all biocatalysts are 20–50 °C.
| Entry | Reactor design | Catalyst type | Catalyst | Substrate | Reaction temp (°C) | Reaction time (min) | Reaction pressure (atm) | Reaction solvent | Yield of desired product (mol%) | Rate (g min−1) | Active sites (mol) | Turnover frequency (mol conv./min/mol sites) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1147 | Batch | Bio | Bromoperoxidase and NaBr | Valine | 21 | 90 | 1 | Water | 0.9 | 0.30 × 10−4 | ||
| 245 | Batch | Bio | Aspartate Decarboxylase | Aspartic acid | 30 | 300 | 1 | Water | 0.0002 × 10−4 | 2 × 10−9 | 8.8 | |
| 344 | CSTR | Bio | Glutamic acid Decarboxylase | Glutamic acid | 30 | 3000 | 1 | — | 1 | 5 | 4 × 10−4 | 7.9 × 101 |
| 4148 | Batch | Bio | Vanadium Chloroperoxidase and NaBr | Glutamic acid and H2O2 | 21 | 240 | 1 | Water | 1 | 0.006 × 10−4 | 9 × 10−12 | 4.62 × 103 |
| 5136 | CSTR | Bio | Lysine Decarboxylase | Lysine | 37 | 1440 | 1 | Buffer | 1 | 0.8 × 10−4 | 2.5 × 10−8 | 6.5 × 103 |
| 646 | Batch | Bio | Ornithine Decarboxylase | Ornithine HCL | 30 | 8 | 1 | Buffer | 1 | 0.006 × 10−4 | 2 × 10−10 | 2.7 × 106 |
| 773 | Batch | Homo | Silver(I) picolinate | 2-(2-Phenylcyclopropyl)glycine | 70 | 60 | Water | 0.34 | 50 × 10−4 | 800 × 10−4 | 3.54 × 10−4 | |
| 858 | Batch | Homo | Silver picolinate | Pivalic acid | 85 | 720 | 1 | Acetonitrile–water | 0.7 | 22 × 10−4 | 20 × 10−4 | 1.1 × 10−2 |
| 992 | Batch | Homo | PdCl2 | Phenylbutyric acid | 110 | 960 | 1 | NMP | 0.7 | 1 × 10−4 | 0.3 × 10−4 | 2.4 × 10−2 |
| 1064 | Batch | Homo | Silver nitrate and copper nitrate | Palmitic acid | 78 | 20 | 1 | Water–acetonitrile 5:9 |
0.5 | 500 × 10−4 | 78 × 10−4 | 2.5 × 10−2 |
| 1196 | Batch | Homo | FeCl2 | Stearic anhydride | 240 | 180 | 20 | — | 0.74 | 23 × 10−4 | 0.2 × 10−4 | 2.1 × 10−1 |
| 12164 | Batch | Homo | Rhodium triphosphine | Stearic acid | 280 | 60 | 1 | — | 0.9 | 4260 × 10−4 | 0.77 × 10−4 | 1.9 × 101 |
| 1383 | Batch | Hetero | Silver nitrate | Levulinic acid | 100 | 30 | 1 | 100 ml buffer | 0.3 | 116 × 10−4 | 100 × 10−4 | 1 × 10−2 |
| 14165 | Batch | Hetero | Pd on non-oxidised CNF | Stearic acid | 250 | 20 | 5.92 | Dodecane | 0.05 | 25 × 10−4 | 2.8 × 10−4 | 3.2 × 10−2 |
| 15190 | Batch | Hetero | WO3 | Methyl stearate | 350 | 300 | 49.3 | Dodecane | 0.20 | 14 × 10−4 | 1 × 10−4 | 3.4 × 10−2 |
| 16167 | Batch | Hetero | Pd/Al2O3 | Stearic acid | 250 | 1440 | 6.91 | — | 0.1 | 6 × 10−4 | 0.6 × 10−4 | 3.4 × 10−2 |
| 17118 | Batch | Hetero | ZnCl2 | Furoic acid | 250 | 180 | 42.9 | CO2 (1 g) | 0.53 | 58 × 10−4 | 15 × 10−4 | 3.5 × 10−2 |
| 18170 | Batch | Hetero | Pd/C | Stearic acid | 300 | 240 | 2 | Dodecane | 0.96 | 64 × 10−4 | 3 × 10−4 | 6.7 × 10−2 |
| 19193 | PFR | Hetero | Ni on Al2O3 | Rapeseed oil | 270 | 120 | 34.5 | — | 0.78 | 2586 × 10−4 | 75 × 10−4 | 1.2 × 10−1 |
| 20168 | PFR | Hetero | Pd/C | Lauric acid | 270 | 75 | 9.86 | Mesitylene | 0.26 | 303 × 10−4 | 7.5 × 10−4 | 2.0 × 10−1 |
| 21165 | Batch | Hetero | Pd on oxidised CNF | Stearic acid | 250 | 20 | 5.92 | Dodecane | 0.4 | 200 × 10−4 | 2.8 × 10−4 | 2.5 × 10−1 |
| 22166 | Batch | Hetero | Pt/Al2O3 | Octanoic acid | 330 | 60 | 1 | Tetradecane | 0.89 | 685 × 10−4 | 7.5 × 10−4 | 6.3 × 10−1 |
| 23173 | PFR | Hetero | Pd/C | Stearic acid | 300 | 330 | — | Dodecane | 0.11 | 158 × 10−4 | 0.3 × 10−4 | 1.6 |
| 24165 | Batch | Hetero | Pd/C | Stearic acid | 300 | 360 | 5.92 | Dodecane | 0.95 | 119 × 10−4 | 12 × 10−4 | 4.6 |
| 25190 | Batch | Hetero | W2C | Methyl stearate | 350 | 300 | 49.3 | Dodecane | 0.93 | 62 × 10−4 | ||
| 26169 | Batch | Hetero | Pt/C | Stearic acid | 330 | 150 | — | Water | 0.85 | 3 × 10−4 | ||
| 2787 | Batch | Hetero | Palladium | Furfural | 200 | 1920 | 1 | Dibutyl phthalate 400 ml | 1 | 302 × 10−4 | ||
| 28121 | Batch | Hetero | Au–CeO2 | HMF | 65 | 480 | 9.89 | Water (20 ml) | 1 | 8 × 10−4 | ||
| 29158 | PFR | Hetero | TiO2 and 1% Pt | Levulinic acid | 65 | 300 | 1 | 15 ml water | 0.66 | 77 × 10−4 | ||
| 30174 | PFR? | Hetero | 1% Pt/Ce2O5 | Gamma valerolactone | 350 | 600 | 34.5 | Water | 0.46 | 269 × 10−4 |
In order to calculate the TOF, it is required to calculate the amount of active sites in contact with the substrate. For homogeneous and biocatalysts the concentration of catalyst in solution is used for this value. For heterogeneous catalysts, the active surface area of the catalyst surface area has been calculated from the BET data (for pure oxidic material) or particle size information (for metal). This method was not applied to mixed metal catalysts such as CoMo and NiMo HDO catalysts, due to the uncertainty in active site location – whether only the particle adjuncts are active for decarboxylation, or the Ni particles dramatically changes the calculated number of sites and thus the TOF. Finally, with the TOF data compiled, it is possible to show a trend of TOF with temperature in Chart 1.
 | ||
| Chart 1 TOF as a function of temperature for different classes of catalyst. | ||
From Table 6 and Chart 1 it can be concluded that, the substrate does not seem to have a significant effect on the TOF, e.g. the use of silver peroxydisulphate reaction on lauric acid and pivalic acid does not significantly affect the resultant TOF. As well as this it can be seen that there is a strong correlation between increasing specific activity (TOF) and increasing temperature for metal based catalysts either homogeneous or heterogeneous thus one is not more active than the other at the same temperature. Given that heterogeneous catalysts are more robust and easier to re-use, it could be considered that, at an industrial scale, heterogeneous catalysts are the most suitable choice for application.
However this trend is not followed by biocatalysts. Biocatalysts have significantly higher TOFs even at low temperatures. However the low volumetric productivities of biocatalytic processes lead to limitations. Reactor design will play an important role in overcoming this. One could consider increasing concentrations of substrate and enzyme. However in the case of exothermic reactions care needs to be taken as biocatalysts can often not be used at elevated temperatures without significantly affecting their stability. Biocatalysts are generally not as stable as traditional metal-based catalysts and often deactivate over time so developments to improve enzyme stability will be required. Most frequently they require an aqueous environment, which might be prohibitive for some desired reactions, although this has been shown to not always be true, and examples exist to ionic liquids,220 supercritical fluids221 and organic solvents222 have been reported.
In conclusion, in the deoxygenation by decarboxylation and decarbonylation of biomass-derived molecules the primary means of controlling the activity of a catalyst is by altering the reaction temperature. Depending on the reaction, substrate catalyst type and cost, this tool allows choices to be made for developing the process of choice. For biocatalysts this does not apply and although TOFs are high at low temperatures, improvements in enzyme stability and improvements in volumetric productivity should be made.
Acknowledgements
We are grateful to Catchbio for financial support.References
- EC, Europe 2020 Initiative, 2011; Available from: http://ec.europa.eu/europe2020/europe-2020-in-a-nutshell/targets/index_en.htm.
- A. Steinfeld and R. Palumbo, Solar Thermochemical Process Technology, in Encyclopedia of Physical Science and Technology, 2001, pp. 237–256 Search PubMed.
- J. W. Lund, D. H. Freeston and T. L. Boyd, Geothermics, 2011, 40, 159–180 CrossRef PubMed.
- SETIS, Hydropower Generation, 2011 [cited 2013 17-01-2013]; Available from: http://setis.ec.europa.eu/newsroom-items-folder/hydropower-generation.
- H. von Blottnitz and M. A. Curran, J. Cleaner Prod., 2007, 15, 607–619 CrossRef PubMed.
- NREL, What is a Biorefinery? 2009 [cited 2013 22/04/2013]; Available from: http://www.nrel.gov/biomass/biorefinery.html.
- A. Bioenergy, Abengoa Bioenergy Netherlands, 2011 [cited 2013 23/05/2013]; Available from: http://www.abengoabioenergy.com/web/en/acerca_de/oficinas_e_instalaciones/bioetanol/europa/netherland/.
- P. N. R. Vennestrøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef PubMed.
- Amyris, Amyris - Fuels, 2013 [cited 2013 20-07-2013]; Available from: http://www.amyris.com/Products/173/Fuels.
- R. K. Sukumaran, R. R. Singhania, G. M. Mathew and A. Pandey, Renewable Energy, 2009, 34, 421–424 CrossRef CAS PubMed.
- S. N. Naik, V. V. Goud, P. K. Rout and A. K. Dalai, Renewable Sustainable Energy Rev., 2010, 14, 578–597 CrossRef CAS PubMed.
- N. D. Hinman, D. J. Schell and C. J. Riley, Appl. Biochem. Biotechnol., 1992, 34, 639–649 CrossRef.
- M. O. S. Dias, T. L. Junqueira and M. P. Otávio Cavalett, Bioresour. Technol., 2012, 103, 152–161 CrossRef CAS PubMed.
- M. Ljunggren, O. Wallberg and G. Zacchi, Bioresour. Technol., 2011, 102, 9524–9531 CrossRef CAS PubMed.
- M. Stöcker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef PubMed.
- R. E. H. Sims, M. Warren, J. N. Saddler and M. Taylor, Bioresour. Technol., 2010, 101, 1570–1580 CrossRef CAS PubMed.
- Y. Nagasawa, Pure Appl. Chem., 1990, 62, 1441–1444 CrossRef.
- S. Takamatsu and T. Tosa, Bioprocess Technol., 1993, 16, 25 CAS.
- R. Gupta, Q. Beg and P. Lorenz, Appl. Microbiol. Biotechnol., 2002, 59, 15–32 CrossRef CAS PubMed.
- I. B.-B. Romdhane, A. Fendri, Y. Gargouri, A. Gargouri and H. Belghith, Biochem. Eng. J., 2010, 53, 112–120 CrossRef PubMed.
- S. Shanavas, G. Padmaja, S. N. Moorthy, M. S. Sajeev and J. T. Sheriff, Biomass Bioenergy, 201, 35, 901–909 CrossRef PubMed.
- J. Tao and J.-H. Xu, Curr. Opin. Chem. Biol., 2009, 13, 43–50 CrossRef CAS PubMed.
- C. A. Busacca, D. R. Fandrick, J. J. Song and C. H. Senanayake, Adv. Synth. Catal., 2011, 353, 1825–1864 CrossRef CAS.
- P. M. Könst, Ph.D. Thesis, 2011, Wageningen UR.
- M. C. R. Franssen, P. Steunenberg, E. L. Scott, H. Zuilhof and J. P. M. Sanders, Chem. Soc. Rev., 2013, 42, 6491–6533 RSC.
- D. J. M. de Vlieger, D. B. Thakur, L. Lefferts and K. Seshan, ChemCatChem, 2012, 4, 2068–2074 CrossRef CAS.
- A. Bridgwater, Fuel, 1995, 74, 631–653 CrossRef CAS.
- X. Jiang, L. Zhang, S. Wybornov, T. Staedler, D. Hein, F. Wiedenmann, W. Krumm, V. Rudnev and I. Lukiyanchuk, ACS Appl. Mater. Interfaces, 2012, 4, 4062–4066 CAS.
- A. R. de la Osa, A. De Lucas, A. Romero, J. L. Valverde and P. Sánchez, Catal. Today, 2011, 176, 298–302 CrossRef CAS PubMed.
- A. D. Klerk, Energy Environ. Sci., 2011, 4, 1177–1205 Search PubMed.
- H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef CAS PubMed.
- D. Knežević, W. van Swaaij and S. Kersten, Ind. Eng. Chem. Res., 2010, 49, 104–112 CrossRef.
- R. J. A. Gosselink, W. Teunissen, J. E. G. van Dam, E. de Jong, G. Gellerstedt, E. L. Scott and J. P. M. Sanders, Bioresour. Technol., 2012, 106, 173–177 CrossRef CAS PubMed.
- R. Sablong, R. Duchateau, C. E. Koning, G. de Wit, D. van Es, R. Koelewijn and J. van Haveren, Biomacromolecules, 2008, 9, 3090–3097 CrossRef CAS PubMed.
- M. Rose and R. Palkovits, ChemSusChem, 2012, 5, 167–176 CrossRef CAS PubMed.
- G. Liang, C. Wu, L. He, J. Ming, H. Cheng, L. Zhuo and F. Zhao, Green Chem., 2011, 13, 839–842 RSC.
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505–9514 CrossRef CAS.
- FDCA-based polyamides 2013 [cited 2013 20-07-2013]; Available from: http://avantium.com/yxy/products-applications/fdca/FDCA-based-polyamides.html.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- D. Y. C. Leung, X. Wu and M. K. H. Leung, Appl. Energy, 2010, 87, 1083–1095 CrossRef CAS PubMed.
- B. M. Bell, J. R. Briggs, R. M. Campbell, S. M. Chambers, P. D. Gaarenstroom, J. G. Hippler, B. D. Hook, K. Kearns, J. M. Kenney, W. J. Kruper, D. J. Schreck, C. N. Theriault and C. P. Wolfe, Clean: Soil, Air, Water, 2008, 36, 657–661 CrossRef CAS.
- Epicerol: A breakthrough in epichlorohydrin production 2013 [cited 2013 20-07-2013]; Available from: http://www.solvaychemicals.com/EN/Sustainability/Issues_Challenges/EPICEROL.aspx.
- B. K. Bower, US Pat, 5714552A, 1994 Search PubMed.
- T. M. Lammens, D. De Biase, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Green Chem., 2009, 11, 1562–1567 RSC.
- P. M. Könst, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Green Chem., 2009, 11, 1646–1652 RSC.
- P. M. Könst, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Green Chem., 2011, 13, 1167–1174 RSC.
- A. J. J. Straathof, Chem. Rev., 2014, 114, 1871–1908 CrossRef CAS PubMed.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- M. K. Lam, K. T. Lee and A. R. Mohamed, Biotechnol. Adv., 2010, 28, 500–518 CrossRef CAS PubMed.
- A. Behr, A. Westfechtel and J. Pérez Gomes, Chem. Eng. Technol., 2008, 31, 700–714 CrossRef CAS.
- P. T. J. Scheepers and R. P. Bos, Int. Arch. Occup. Environ. Health, 1992, 64, 149–161 CrossRef CAS.
- EIA, Monthly Biodiesel Production Report, 2013 [22-04-2013]; Available from: http://www.eia.gov/biofuels/biodiesel/production/.
- E. B. T. Platform, Fatty Acid Methyl Esters (FAME) Fact Sheet, 2013; Available from: http://www.biofuelstp.eu/factsheets/fame-fact-sheet.pdf.
- A. Srivastava and R. Prasad, Renewable Sustainable Energy Rev., 2000, 4, 111–133 CrossRef CAS.
- J. K. Kochi, R. A. Sheldon and S. S. Lande, Tetrahedron, 1969, 25, 1197–1207 CrossRef CAS.
- J. M. Anderson and J. K. Kochi, J. Am. Chem. Soc., 1970, 92, 1651–1659 CrossRef CAS.
- W. E. Fristad, M. A. Fry and J. A. Klang, J. Org. Chem., 1983, 48, 3575–3577 CrossRef CAS.
- J. M. Anderson and J. K. Kochi, J. Org. Chem., 1970, 35, 986–989 CrossRef CAS.
- D. A. House, Chem. Rev., 1962, 62, 185–203 CrossRef CAS.
- E. Mentasti, C. Baiocchi and J. S. Coe, Coord. Chem. Rev., 1984, 54, 131–157 CrossRef CAS.
- A. Kumar and P. Neta, J. Phys. Chem., 1979, 83, 3091–2095 CrossRef CAS.
- C. Walling and D. M. Camaioni, J. Org. Chem., 1978, 43, 3266–3271 CrossRef CAS.
- J. K. Kochi, J. A. Bemis and C. L. Jenkins, J. Am. Chem. Soc., 1968, 90, 4616–4625 CrossRef CAS.
- F. van der Klis, M. H. van den Hoorn, R. Blaauw, J. van Haveren and D. S. van Es, Eur. J. Lipid Sci. Technol., 2011, 113, 562–571 CrossRef CAS.
- R. W. Gosselink, S. A. W. Hollak, S.-W. Chang, J. van Haveren, K. P. de Jong, J. H. Bitter and D. S. van Es, ChemSusChem, 2013, 6, 1576–1594 CrossRef CAS PubMed.
- D. S. Seigler, Shikimic Acid Pathway, in Plant Secondary Metabolism, Springer, 1999, pp. 94–105 Search PubMed.
- I. Pastorova, C. G. de Koster and J. J. Boon, Phytochem. Anal., 1997, 8, 63–73 CrossRef CAS.
- P. E. Shaw, J. Agric. Food Chem., 1979, 27, 246–257 CrossRef CAS.
- N. A. Burges, H. M. Hurst and B. Walkden, Geochim. Cosmochim. Acta, 1964, 28, 1547–1554 CrossRef CAS.
- L. J. Gooßen, C. Linder, N. Rodríguez, P. P. Lange and A. Fromm, Chem. Commun., 2009, 7173–7175 RSC.
- L. J. Gooßen, C. Linder, N. Rodríguez, P. P. Lange and A. Fromm, ChemCatChem, 2010, 2, 430–442 CrossRef.
- US Pat 4,262,157, 1981 Search PubMed.
- Y. Zelechonok and R. B. Silverman, J. Org. Chem., 1992, 57, 5787–5790 CrossRef CAS.
- E. Scott, F. Peter and J. Sanders, Appl. Microbiol. Biotechnol., 2007, 75, 751–762 CrossRef CAS PubMed.
- J. L. O'Donoghue, S. R. Haworth, R. D. Curren, P. E. Kirby, T. Lawlor, E. J. Moran, R. D. Phillips, D. L. Putnam, A. M. Rogers-Back, R. S. Slesinski and A. Thilagar, Mater. Res., 1988, 206, 149–161 Search PubMed.
- D. W. Rackemann and W. O. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198–214 CrossRef CAS.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- F. M. A. Geilen, T. vom Stein, B. Engendahl, S. Winterle, M. A. Liauw, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2011, 50, 6831–6834 CrossRef CAS PubMed.
- V. E. Tarabanko, M. Y. Chernyak, S. V. Aralova and B. N. Kuznetsov, React. Kinet. Catal. Lett., 2002, 75, 117–126 CrossRef CAS.
- T. Werpy, G. Petersen, A. Aden, J. Bozell, J. Holladay, W. A. Manheim, D. Eliot, L. Lasure and S. Jones, Top Value Added Chemicals from Biomass Volume I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas, 2004, PNNL, NREL, EERE, http://oai.dtic.mil/oai/oai?verb=getRecord&metadataPrefix=html&identifier=ADA436528.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilskid and J. L. Jarnefelde, Resour., Conserv. Recycl, 2000, 28, 227–239 CrossRef.
- J.-P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS PubMed.
- Y. Gong and L. Lin, Molecules, 2011, 16, 2714–2725 CrossRef CAS PubMed.
- G. J. Sunley and D. J. Watson, Catal. Today, 2000, 58, 293–307 CrossRef CAS.
- J. H. Jones, Platinum Met. Rev., 2000, 44, 94–105 CAS.
- M. Beller, B. Cornils, C. D. Frohning and C. W. Kohlpaintner, J. Mol. Catal. A: Chem., 1995, 104, 17–85 CrossRef CAS.
- H. B. Copelin and D. I. Garnett, US Patent, 3,007,941, 1961 Search PubMed.
- D. T. Win, Aust. J. Technol., 2005, 8, 185–190 Search PubMed.
- N. D. Hinman, J. D. Wrigth, W. Hoagland and C. E. Wyman, Appl. Biochem. Biotechnol., 1989, 20–21, 391–401 CrossRef.
- B. P. Lavarack, G. J. Griffin and D. Rodman, Bioenergy, 2002, 23, 367–380 CAS.
- D. van Eylen, F. van Dongen, M. Kabel and J. de Bont, Bioresour. Technol., 2011, 102, 5995–6004 CrossRef CAS PubMed.
- L. J. Gooßen and N. Rodriguez, Chem. Commun., 2004, 724–725 RSC.
- L. J. Gooßen and J. Paetzold, Angew. Chem., Int. Ed., 2004, 43, 1095–1098 CrossRef PubMed.
- L. J. Gooßen, K. Gooßen, N. Rodríguez, M. Blanchot, C. Linder and B. Zimmermann, Pure Appl. Chem., 2008, 80, 1725–1733 CrossRef.
- J. Le Nôtre, E. L. Scott, M. C. R. Franssen and J. P. M. Sanders, Tetrahedron Lett., 2010, 51, 3712–3715 CrossRef PubMed.
- S. Maetani, T. Fukuyama, N. Suzuki, D. Ishihara and I. Ryu, Chem. Commun., 2012, 48, 2552–2554 RSC.
- M. O. Miranda, A. Pietrangelo, M. A. Hillmyer and W. B. Tolman, Green Chem., 2012, 14, 490–494 RSC.
- S. Maetani, T. Fukuyama, N. Suzuki, D. Ishihara and I. Ryu, Organometallics, 2011, 30, 1389–1394 CrossRef CAS.
- W. I. Dzik, P. P. Lange and L. J. Gooßen, Chem. Sci., 2012, 3, 2671–2678 RSC.
- L. J. Gooßen, G. Deng and L. M. Levy, Science, 2006, 313, 662–664 CrossRef PubMed.
- A. G. Myers, D. Tanaka and M. R. Mannion, J. Am. Chem. Soc., 2002, 124, 11250–11251 CrossRef CAS PubMed.
- Z. Fu, et al. , Org. Lett., 2010, 12, 4992–4995 CrossRef CAS PubMed.
- L. Goossens and G.-J. Deng, WO Pat, 2006136135, 2006 Search PubMed.
- L. J. Gooßen, N. Rodríguez, B. Melzer, C. Linder, G. Deng and L. M. Levy, J. Am. Chem. Soc., 2007, 129, 824–4833 CrossRef PubMed.
- L. J. Gooßen, N. Rodríguez and P. P. Lange, Chem. – Eur. J., 2009, 15, 9336–9349 CrossRef PubMed.
- C. Wang, I. Piel and F. Glorius, J. Am. Chem. Soc., 2009, 131, 4194–4195 CrossRef CAS PubMed.
- B. Raecke and B. Blaser, US Pat, 2, 863, 913, 1953 Search PubMed.
- Terephthalic Acid (TPA): 2010 World Market Outlook and Forecast Been Recently Released by MarketPublishers.com, 2010 [cited 2013; Available from: http://www.thefreelibrary.com/Terephthalic%20Acid%20(TPA):%202010%20World%20Market%20Outlook%20and%20Forecast%20Been…a0221893067.
- R. Dabestani, P. F. Britt and A. C. Buchanan, Energy Fuels, 2005, 19, 365–373 CrossRef CAS.
- E. McNelis, J. Org. Chem., 1965, 30, 1209–1213 CrossRef CAS.
- I. Ryoji, Y. Shinobu and O. Hirohito, US Pat, 5847256, 1998 Search PubMed.
- P. Marlier, WO Pat, 2010001078, 2010 Search PubMed.
- M. W. Peters, J. D. Taylor, M. Jenni, L. E. Manzer and D. E. Henton, US Pat, 20110087000, 2011 Search PubMed.
- M. Colonna, C. Berti, M. Fiorini, E. Binassi, M. Mazzacurati, M. Vannini and S. Karanam, Green Chem., 2011, 13, 2543–2548 RSC.
- R. R. Schmidt, Acc. Chem. Res., 1986, 19, 250–259 CrossRef CAS.
- D. S. van Es, J. Renewable Mater., 2012, 1, 61–72 CrossRef.
- J. van Haveren, S. Thiyagarajan and A. T. Morita, WO Pat, 2013096998, 2013 Search PubMed.
- T. Pan, J. Deng, Q. Xu, Y. Zuo, Q. X. Guo and Y. Fu, ChemSusChem, 2013, 6, 47–50 CrossRef CAS PubMed.
- A. Gandini, A. J. D. Silvestre, C. P. Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., Part A: Polym. Chem., 2008, 47, 295–298 CrossRef.
- T. Miura, H. Kakinuma, T. Kawano and H. Matsuhisa, US Pat, 7411078, 2008 Search PubMed.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- H. Kolbe, Justus Liebigs Ann. Chem., 1848, 64, 339–341 CrossRef.
- K. Sasaki, K. Uneyama and S. Nagaura, Electrochim. Acta, 1966, 11, 891–894 CrossRef CAS.
- L. Eberson, Electrochim. Acta, 1967, 12, 1473–1478 CrossRef CAS.
- A. K. Vijh and B. E. Conway, Chem. Rev., 1967, 67, 623–664 CrossRef CAS.
- J. E. Sanderson, P. F. Levy, L. K. Cheng and G. W. Barnard, J. Electrochem. Soc., 1983, 130, 1844–1848 CrossRef CAS PubMed.
- D. Bradin, WO Pat, 2007095215, 2007 Search PubMed.
- D. Bradin and G. L. Grune, WO Pat, 2008123925 A2 20081016, 2008 Search PubMed.
- J.-J. Dai, Y.-B. Huang, C. Fang, Q.-X. Guo and Y. Fu, ChemSusChem, 2012, 5, 617–620 CrossRef CAS PubMed.
- C. De Bellefon and P. Fouilloux, Catal. Rev., 1994, 36, 459–506 CAS.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- T. M. Lammens, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Biomass Bioenergy, 2012, 44, 168–181 CrossRef CAS PubMed.
- A. Grazziotin, F. A. Pimentel, E. V. de Jong and A. Brandelli, Anim. Feed Sci. Technol., 2006, 126, 135–144 CrossRef CAS PubMed.
- E. Cibis, A. Ryznar-Luty, M. Krzywonos, K. Lutosławski and T. Miśkiewicz, J. Environ. Manage., 2011, 92, 1733–1739 CrossRef CAS PubMed.
- Y. Teng, E. L. Scott, A. N. T. van Zeeland and J. P. M. Sanders, Green Chem., 2011, 13, 624–630 RSC.
- H. Mooibroek, N. Oosterhuis, M. Giuseppin, M. Toonen, H. Franssen, E. L. Scott, J. P. M. Sanders and A. Steinbüchel, Appl. Microbiol. Biotechnol., 2007, 77, 257–267 CrossRef CAS PubMed.
- Y. Elbahloul, J. P. M. Sanders, E. L. Scott, H. Mooibroek, M. Obst and A. Steinbüchel, WO Pat, 200693411, 2006 Search PubMed.
- P. M. Könst, E. L. Scott, M. C. R. Franssen and J. P. M. Sanders, J. Biobased Mater. Bioenergy, 2011, 5, 102–108 CrossRef PubMed.
- T. M. Lammens, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Green Chem., 2010, 12, 1430–1436 RSC.
- J. Spekreijse, J. Le Nôtre, J. van Haveren, E. L. Scott and J. P. M. Sanders, Green Chem., 2012, 14, 2747–2751 RSC.
- T. M. Lammens, J. Le Nôtre, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, ChemSusChem, 2011, 4, 785–791 CrossRef CAS PubMed.
- J. Le Nôtre, E. L. Scott, M. C. R. Franssen and J. P. M. Sanders, Green Chem., 2011, 13, 807–809 RSC.
- K. Kashiwagi, R. Sugise, T. Shimakawa, T. Matuura, M. Shirai, F. Kakiuchi and S. Murai, Organometallics, 1997, 16, 2233–2235 CrossRef CAS.
- L. J. Mersinger, E. C. Hann, F. B. Cooling, J. E. Gavagan, A. Ben-Bassat, S. Wu, K. L. Petrillo, M. S. Payne and R. DiCosimo, Adv. Synth. Catal., 2005, 347, 1125–1131 CrossRef CAS.
- T. M. Lammens, S. Gangarapu, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, Biofuels, Bioprod. Biorefin., 2012, 6, 177–187 CrossRef CAS.
- M. Nieder and L. Hager, Arch. Biochem. Biophys., 1985, 240, 121–127 CrossRef CAS.
- A. But, J. Le Nôtre, E. L. Scott, R. Wever and J. P. M. Sanders, ChemSusChem, 2012, 5, 1199–1202 CrossRef CAS PubMed.
- Y. Liu, C. Wang, J. Yan, W. Zhang, W. Guan, X. Lu and S. Li, Biotechnol. Biofuels, 2014, 7, 28 CrossRef PubMed.
- A. Strecker, Justus Liebigs Ann. Chem., 1862, 123, 363–365 CrossRef.
- A. Schönberg, R. Moubasher and A. Mostafa, J. Chem. Soc., 1948, 176–182 RSC.
- G. D. Kalyankar and E. E. Snell, Biochemistry, 1962, 1, 594–600 CrossRef CAS.
- L. Liu and R. Breslow, Bioorg. Med. Chem., 2004, 12, 3277–3287 CrossRef CAS PubMed.
- L. Liu, W. Zhou, J. Chruma and R. Breslow, J. Am. Chem. Soc., 2004, 126, 8136–8137 CrossRef CAS PubMed.
- K. Dose, Nature, 1957, 179, 734–735 CrossRef CAS.
- A. F. Al-Sayyab and A. Lawson, J. Chem. Soc. C, 1968, 406–410 RSC.
- O. Carp, C. L. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32, 33–177 CrossRef CAS PubMed.
- H. L. Chum, M. Ratcliff, F. L. Posey, J. A. Turner and A. J. Nozik, J. Phys. Chem., 1983, 87, 3089–3093 CrossRef CAS.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 5985–5992 CrossRef CAS.
- Z. Y. Zhang, B. Jin and J. M. Kelly, World J. Microbiol. Biotechnol., 2007, 23, 229–236 CrossRef CAS.
- A. Danion, J. Disdier, C. Guillard and N. Jaffrezic-Renault, J. Photochem. Photobiol., A, 2007, 190, 135–140 CrossRef CAS PubMed.
- M. S. Hamdy, E. L. Scott, R. H. Carr and J. P. M. Sanders, Catal. Lett., 2012, 142, 338–344 CrossRef CAS.
- Y. Takemura, A. Nakamura, H. Taguchi and K. Ouchi, Ind. Eng. Chem. Prod. Res. Dev., 1985, 24, 213–215 CrossRef CAS.
- T. A. Foglia and P. A. Barr, J. Am. Oil Chem. Soc., 1976, 53, 737–741 CrossRef CAS.
- R. W. Gosselink, W. Xia, M. Muhler, K. P. de Jong and J. H. Bitter, ACS Catal., 2013, 3(10), 2397–2402 CrossRef CAS.
- P. T. Do, M. Chaippero, L. L. Lobban and D. E. Resasco, Catal. Lett., 2009, 130, 9–18 CrossRef CAS.
- M. Snåre, I. Kubickova, P. Mäki-Arvela, K. Eränen, J. Wärnå and D. Yu. Murzin, Chem. Eng. J., 2007, 134, 29–34 CrossRef PubMed.
- M. Snåre and D. Yu. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708–5715 CrossRef.
- R. O. Idem, S. P. Katikaneni and N. N. Bakhshi, Fuel Process. Technol., 1997, 51, 101–125 CrossRef CAS.
- S. A. W. Hollak, J. H. Bitter, J. van Haveren, D. S. van Es and K. P. de Jong, R. Soc. Chem. Adv., 2012, 2, 9387–9391 CAS.
- P. Mäki-Arvela, M. Snåre, K. Eränen, J. Myllyoja and D. Y. Murzin, Fuel, 2008, 87, 3543–3549 CrossRef PubMed.
- J. Fu, X. Lu and P. E. Savage, ChemSusChem, 2011, 4, 481–486 CrossRef CAS PubMed.
- J. Ford, J. Immer and H. H. Lamb, Top. Catal., 2012, 55, 175–184 CrossRef CAS.
- J. C. Serrano-Ruiz, D. Wang and J. A. Dumesic, Green Chem., 2010, 12, 574–577 RSC.
- I. Simakova, O. Simakova, P. Mäki-Arvela and D. Yu. Murzin, Catal. Today, 2010, 150, 28–31 CrossRef CAS PubMed.
- I. Simakova, B. Rozmyslowicz, O. Simakova, P. Mäki-Arvela, A. Simakoz and D. Yu. Murzin, Top. Catal., 2011, 54, 460–466 CrossRef CAS.
- J. G. Immer, M. J. Kelly and H. H. Lamb, Appl. Catal., A, 2010, 375, 134–139 CrossRef CAS PubMed.
- A. T. Madsen, E. H. Ahmed, C. H. Christensen, R. Fehrmann and A. Riisager, Fuel, 2011, 90, 3433–3438 CrossRef CAS PubMed.
- J. G. Immer and H. H. Lamb, Energy Fuels, 2010, 24, 5291–5299 CrossRef CAS.
- K. J. Jung, A. Gaset and J. Molinier, Biomass, 1988, 16, 63–76 CrossRef CAS.
- M. Badawi, J. F. Paul, S. Cristol, E. Payen, Y. Romero, F. Richard, S. Brunet, D. Lambert, X. Portier, A. Popov, E. Kondratieva, J. M. Goupil, J. El Fallah, J. P. Gilson, L. Mariey, A. Travert and F. Maugé, J. Catal., 2011, 282, 155–164 CrossRef CAS PubMed.
- G. de la Puente, A. Gil, J. J. Pis and P. Grange, Langmuir, 1999, 15, 5800–5806 CrossRef CAS.
- R. J. A. Gosselink, Lignin as a renewable aromatic resource for the chemical industry, Thesis, 2011, Wageningen UR Search PubMed.
- R. J. A. Gosselink, A. Abacherli, H. Semke, R. Malherbe, P. Kauper, A. Nadif and J. E. G. van Dam, Ind. Crops Prod., 2004, 19, 271–281 CrossRef CAS PubMed.
- L. Szabó, A. Soria Ramirez, J. Forsstrom, J. Keranen and E. Hytonen, Environ. Sci. Policy, 2009, 12, 257–269 CrossRef PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS PubMed.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- P. de Wild, R. Van der Laan, A. Kloekhorst and E. Heeres, Environ. Prog. Sustainable Energy, 2009, 28, 461–469 CrossRef CAS.
- J. van Haveren, E. L. Scott and J. P. M. Sanders, Biofuels, Bioprod. Biorefin., 2008, 2, 41–57 CrossRef CAS.
- A. L. Jongerius, R. Jastrzebski, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2012, 285, 315–323 CrossRef CAS PubMed.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS PubMed.
- M. Toba, Y. Abe, H. Kuramochi, M. Osako, T. Mochizuki and Y. Yoshimura, Catal. Today, 2011, 164, 533–537 CrossRef CAS PubMed.
- R. W. Gosselink, D. R. Stellwagen and J. H. Bitter, Angew. Chem., Int. Ed., 2013, 52, 5089–5092 CrossRef CAS PubMed.
- Y. Liu, R. Sotelo-Boyás, K. Murata, T. Minowa and K. Sakanishi, Chem. Lett., 2009, 38, 552–553 CrossRef CAS.
- D. Kubička, P. Šimáček and N. Žilková, Top. Catal., 2009, 52, 161–168 CrossRef.
- D. Kubička and L. Kaluža, Appl. Catal., A, 2010, 372, 199–208 CrossRef PubMed.
- M. Krár, S. Kovács, D. Kalló and J. Hancsók, Bioresour. Technol., 2010, 101, 9287–9293 CrossRef PubMed.
- M. Papagianni, Biotechnol. Adv., 2007, 25, 244–263 CrossRef CAS PubMed.
- H. S. Grewal and K. L. Kalra, Biotechnol. Adv., 1995, 13, 209–234 CrossRef CAS.
- C. R. Soccol, L. P. S. Van den berghe, C. Rodrigues and A. Pandey, Food Technol. Biotechnol., 2006, 44, 141 CAS.
- M. Carlsson, L. C. Kam, M. J. Antal, N. Bian, R. J. Cunningham and M. Jones, Ind. Eng. Chem. Res., 1994, 33, 1989–1996 CrossRef CAS.
- D. W. Johnson, G. R. Eastman, M. Poliakoff and T. A. Huddle, EU Pat, 20110791623, 2011 Search PubMed.
- T. Willke and K. D. Vorlop, Appl. Microbiol. Biotechnol., 2001, 56, 289–295 CrossRef CAS.
- S. Ma, X. Liu, Y. Jiang, Z. Tang, C. Zhang and J. Zhu, Green Chem., 2013, 15, 245–254 RSC.
- A. J. Koops, H. L. de Graaf, I. van der Meer and W. A. M. van der Berg, WO Pat, 2009102205, 2009 Search PubMed.
- K. Zhang, A. P. Woodruff, M. Xiong, J. Zhou and Y. K. Dhande, ChemSusChem, 2011, 4, 1068–1070 CrossRef CAS PubMed.
- G. González, J. Salvadó and D. Montané, J. Supercrit. Fluids, 2004, 31, 57–66 CrossRef PubMed.
- F. Jin, J. Cao, H. Enomoto and T. Moriya, J. Supercrit. Fluids, 2006, 39, 80–88 CrossRef CAS PubMed.
- P. T. Williams and J. Onwudili, Ind. Eng. Chem. Res., 2005, 44, 8739–8749 CrossRef CAS.
- A. T. Quitain, N. Sato, H. Daimon and K. Fujie, Ind. Eng. Chem. Res., 2001, 40, 5885–5888 CrossRef CAS.
- N. Sato, A. T. Quitain, K. Kang, H. Daimon and K. Fujie, Ind. Eng. Chem. Res., 2004, 43, 3217–3222 CrossRef CAS.
- M. Faisal, N. Sato, A. T. Quitain, H. Daimon and K. Fujie, Eng. Chem. Res., 2005, 44, 5472–5477 CrossRef CAS.
- F. J. Hidalgo and R. Zamora, J. Agric. Food Chem., 2007, 55, 4902–2906 CrossRef CAS PubMed.
- S. Changi, M. Zhu and P. E. Savage, ChemSusChem, 2012, 5, 1743–1757 CAS.
- S. Lestari, P. Mäki-Arvela, J. Beltramini, G. Q. Max Lu and D. Y. Murzin, ChemSusChem, 2009, 2, 1109–1119 CrossRef CAS PubMed.
- Y. Chen, C. Wang, W. Lu and Z. Yang, Bioresour. Technol., 2010, 101, 4600–4607 CrossRef CAS PubMed.
- H. Zhang, T. R. Carlson, R. Xiao and G. W. Huber, Green Chem., 2012, 14, 98–110 RSC.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- A. Funke and F. Ziegler, Biofuels, Bioprod. Biorefin., 2010, 4, 160–177 CrossRef CAS.
- U. Kragl, M. Eckstein and N. Kaftzik, Curr. Opin. Biotechnol., 2002, 13, 565–571 CrossRef CAS.
- T. W. Randolph, H. W. Blanch, J. M. Prausnitz and C. R. Wilke, Biotechnol. Lett., 1985, 7, 325–328 CrossRef CAS.
- A. M. Klibanov, Trends Biochem. Sci., 1989, 14, 141–144 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |