
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organotrifluoroborates as attractive self-assembling systems: the case of bifunctional dipotassium phenylene-1,4-bis(trifluoroborate)†
Aurelia
Falcicchio‡
a,
Sten O.
Nilsson Lill‡
b,
Filippo M.
Perna
c,
Antonio
Salomone
c,
Donato I.
Coppi
c,
Corrado
Cuocci
a,
Dietmar
Stalke
d and
Vito
Capriati
*c
aIstituto di Cristallografia (IC-CNR), Via Amendola 122/o, I-70125 Bari, Italy
bPharmaceutical Innovation, Pharmaceutical Development, AstraZeneca, Pepparedsleden 1, SE-431 83, Mölndal, Sweden
cDipartimento di Farmacia-Scienze del Farmaco, Università di Bari “Aldo Moro”, Consorzio C.I.N.M.P.I.S., Via E. Orabona 4, I-70125, Bari, Italy. E-mail: vito.capriati@uniba.it
dInstitut für Anorganische Chemie, Universität Göttingen, Tammannstraße 4, D-37077, Göttingen, Germany
First published on 26th June 2015
Abstract
The first structure of an aromatic bis(trifluoroborate) dipotassium salt, elucidated by the combination of crystallography, DFT calculations, topological and non-covalent interaction analysis, discloses a 3D network undergoing spontaneous self-assembly thanks to the massive participation of weak intra- and intermolecular interactions for which fluorine atoms proved to play a leading role.
In recent years, growing attention has been paid to the chemistry of organoboron compounds as useful and valuable reagents for many transition-metal-catalysed1 and also “metal-free”2 C–C bond-forming reactions. Organotrifluoroborate salts, in particular, have been shown to be attractive and versatile boronic acid surrogates.3 This is either because of their exceptional stability toward oxygen and common reagents or because of their higher nucleophilicity and excellent functional group tolerance. Thus, it is no surprise that the recent burst in activity in their chemistry has seen them more and more as leading actors in a variety of chemical scenarios ranging from palladium-catalysed cross-coupling reactions3 and stereoselective rhodium-catalysed 1,2- and 1,4-addition reactions4 for the construction of complex organic frameworks.5
However, despite the exponential growth of papers dedicated to potassium organotrifluoroborates in the last 10 years, few investigators have, to date, concentrated their research on the structures of such salts.6 The few X-ray structure determinations reported so far for compounds containing a benzene ring carrying at least one –BF3K group7 reveal the singular and unique feature of each organotrifluoroborate salt in which large polarizable cations such as K+ (which are significantly better charge-delocalizing than Li+) permit extended crystal lattices to form because of their multihapto coordinations.8 Such molecular structures are often held together by weaker coordinations (e.g., C–H⋯π, K⋯π and π–π interactions) and, sometimes, also by long-range electrostatic forces. All these interactions contribute to define the final architecture of the packing.
This self-assembly aspect of organotrifluoroborates is surely amazing and worth investigating because the peculiar nature of each salt may promote self-organization and thus the obtainment of functional supramolecular architectures with potential interest in many fields (e.g., materials science, catalysis, sensing and separations) as has recently been ascertained in the case of boronic acids, which have been shown to undergo spontaneous supramolecular assembly either with themselves or with other N-donor compounds.9 Aromatic and heteroaromatic bis- and poly(trifluoroborate) compounds have also been successfully employed in the manufacture of novel conjugated polymers with excellent and interesting electron-transport properties.10 Thus, the design and synthesis of novel polyfunctional aromatic compounds carrying more than one trifluoroborate group onto the aromatic core is fascinating and challenging both from a crystallographic and a synthetic perspective in order to chemoselectively replace them in multistep organic synthesis and to obtain new functional structural motifs.
Recently, we have described the preparation of bifunctional dipotassium phenylene-1,4-bis(trifluoroborate) 2 from the corresponding commercially available bis(boronic acid) 1 (Scheme 1) and its successful employment in one-pot double Suzuki–Miyaura coupling reactions for the obtainment of conjugated tri(hetero)aryl derivatives.11
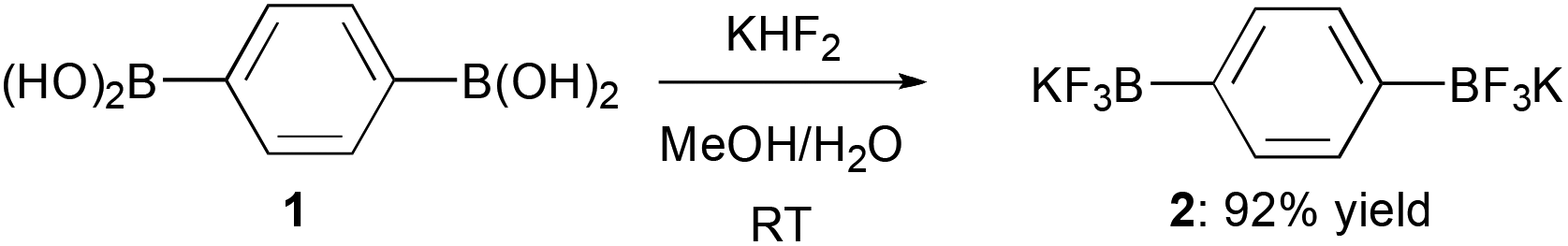 | ||
| Scheme 1 Synthesis of bis(trifluoroborate) 2 from bis(boronic acid) 1. | ||
In this paper, we present the first structural investigations on such a salt (2) which provide detailed insight on the presence of an impressive multitude of weak inter- and intramolecular interactions responsible for its self-assembly into a new 3D network.
Bis-trifluoroborate 2, directly obtained as a white free-flowing powder in 92% yield, has been crystallized at room temperature by slow evaporation from a solution of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetone/H2O (in ratio 2:1) and colourless needles (mp 342 °C) have been collected. Bis(trifluoroborate) 2, crystallizes in a tetragonal crystal system, in the space group I4l/acd with the cell parameters a = 17.597(9) Å and c = 40.203(9) Å. The asymmetric unit consists of 1.5 independent molecules of C6H4B2F6, 5 K atoms (one of them at the general position, and the rest at special positions with a site occupancy factor of 0.5) and half a molecule of H2O. A view of the refined crystal structure is shown in Fig. 1 where short K⋯F and K⋯O contacts are distinguishable. Of note, the H atom of the 0.5 H2O molecule points towards the benzene ring, the distance between the centroid of the aromatic ring and the H atom (labelled as H100) being 2.475 Å, thereby suggesting the presence of an aromatic OH⋯π interaction (vide infra).12
:1 acetone/H2O (in ratio 2:1) and colourless needles (mp 342 °C) have been collected. Bis(trifluoroborate) 2, crystallizes in a tetragonal crystal system, in the space group I4l/acd with the cell parameters a = 17.597(9) Å and c = 40.203(9) Å. The asymmetric unit consists of 1.5 independent molecules of C6H4B2F6, 5 K atoms (one of them at the general position, and the rest at special positions with a site occupancy factor of 0.5) and half a molecule of H2O. A view of the refined crystal structure is shown in Fig. 1 where short K⋯F and K⋯O contacts are distinguishable. Of note, the H atom of the 0.5 H2O molecule points towards the benzene ring, the distance between the centroid of the aromatic ring and the H atom (labelled as H100) being 2.475 Å, thereby suggesting the presence of an aromatic OH⋯π interaction (vide infra).12
 | ||
| Fig. 1 ORTEP-plot of the asymmetric unit of the title compound 2 with ellipsoids plotted at the 50% probability level. Short K⋯F, K⋯O contacts and OH⋯π interactions are drawn as dashed lines. Colour codes in the electronic version of the paper: carbon (blue), boron (light brown), fluorine (green), potassium (violet) and oxygen (red). Hydrogen atoms are also drawn as small spheres. | ||
The C–C bond lengths of the aromatic ring vary from 1.383(4) Å up to 1.397(4) Å and are thus consistent with π delocalization; the ring is, indeed, essentially planar. The B–F bond lengths range from 1.391(4) Å for B3–F9 to 1.447(3) Å for B1–F2 (Table S1, ESI†) and are in agreement with the values reported in the literature.7c,f All the three F atoms belonging to BF3 groups lie out of the plane of the aromatic ring.§ Analogous to what has been reported for other organotrifluoroborates,7f,10b,c the coordination geometry around all K+ cations is irregular and cannot be conveniently described by any polyhedron,13 K⋯F contacts ranging from 2.621(2) Å (K4⋯F8) to 3.382(3) Å (K3⋯F9). In particular, cations K1, K2 and K3 all have ten short contacts to F atoms (most of them less than 3.0 Å, see the ESI†), and two slightly longer contacts to B atoms (Table S1, ESI†). The environment of the K4 cation, which is depicted in Fig. 2, shows eight short contacts to F atoms in the range 2.621(2) Å–3.041(2) Å. In the case of a K5 cation, four interactions with F atoms less than 3.0 Å, one to oxygen 2.7247(17) Å and two distances to carbons C5 and C8 longer than 3.3 Å are present, instead.
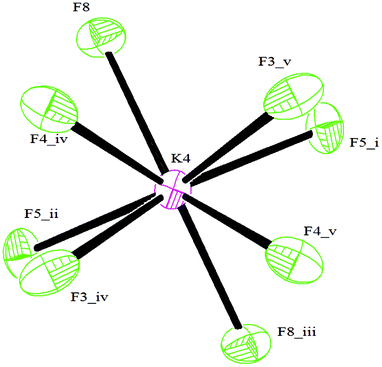 | ||
| Fig. 2 The environment of K4 cation. Symmetry codes: (i) −x + 1, −y + ½, z; (ii) −x + 1, −y, −z; (iii) x, y + ½, −z; (iv) −y + ¾, x − ¼, −z + ¼; (v) y + ¼, −x + ¾, −z + ¼. | ||
Fig. 3 depicts the packing of the title compound revealing the presence of K cation layers bridged by C6H4B2F6 and H2O molecules via K⋯F, K⋯O, OH⋯π and K⋯aryl interactions. Thus, despite its apparent structural and spectroscopic simplicity,¶ the overall 3D network of the salt 2 is made up of C6H4 layers coated with BF3 groups either side. The fluorinated walls leave enough space to coordinate the potassium cations of which one is slightly pulled towards the ring centre by the water molecule.
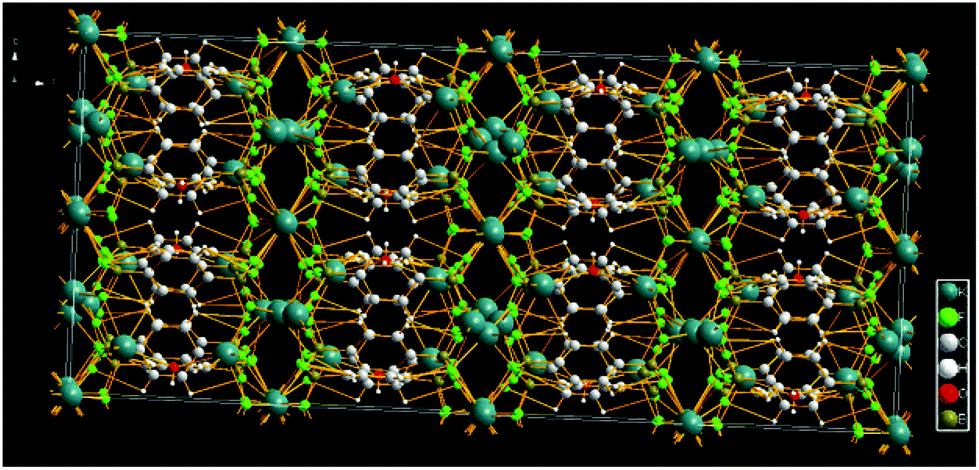 | ||
| Fig. 3 3D packing of the title compound 2. | ||
The study of non-covalent interactions, and in particular of the contribution of organic fluorine in the formation of different supramolecular motifs, is an expanding area of research amongst the scientific community and has recently gained much interest both in crystal engineering for the systematic design of advanced functional materials14,15 and in asymmetric synthesis.16 Thus, to gain further insights, the interactions within the crystal were investigated in detail by the use of an NCI (non-covalent interaction) analysis,17 which is an informative and illustrative tool to display hydrogen bonds and weak inter- and intramolecular interactions based on the calculated electron density. In addition, we have adopted Bader's atoms in molecules (AIM) concept for identifying a bond path and a bond critical point (BCP) from the electron density.18 In Fig. 4, the water OH⋯π interaction (vide supra) is clearly identified and visualized as an interaction surface. The hydrogen bond critical point is visualized as a light blue sphere and the hydrogen bond path is seen as a dotted line. The hydrogen bond strength was quantified to be ca. −8 kcal mol−1 by DFT calculations.19 It is also found that H2O interacts with the potassium cation K5, quantified to ca. −7 kcal mol−1. In addition, the two aromatic rings are stabilized by side-on dispersive CH⋯π interactions as visualized by the interaction surface and the two BCP spheres and bond paths. Interestingly, the fluorines in the BF3 group are observed to feature three distinctly different interactions (Fig. 4): F⋯F halogen bond interaction,15 CH⋯F interaction to an aromatic hydrogen, and in addition also to the potassium cation. A more detailed characterization of these interactions is found in Fig. S1 in the ESI.† The short distances between K⋯C were, by the BCP and bond path analysis, actually found to be due to agostic interactions20 between potassium and the aromatic CH hydrogens rather than due to the carbons. This conclusion was supported by an NBO-analysis, which showed electron delocalisation from the two aromatic C–H bonds to the potassium cations (for further details, see Fig. S2, ESI†). Thus, by the use of the NCI and BCP analysis we have been able to demonstrate the presence of K⋯F, K⋯O, K⋯HC, OH⋯π, CH⋯π, F⋯F and CH⋯F inter- and intramolecular interactions. It is thus manifested that the fluorines in the trifluoroborate group are of great importance for stabilization and formation of the crystal structure of compound 2.
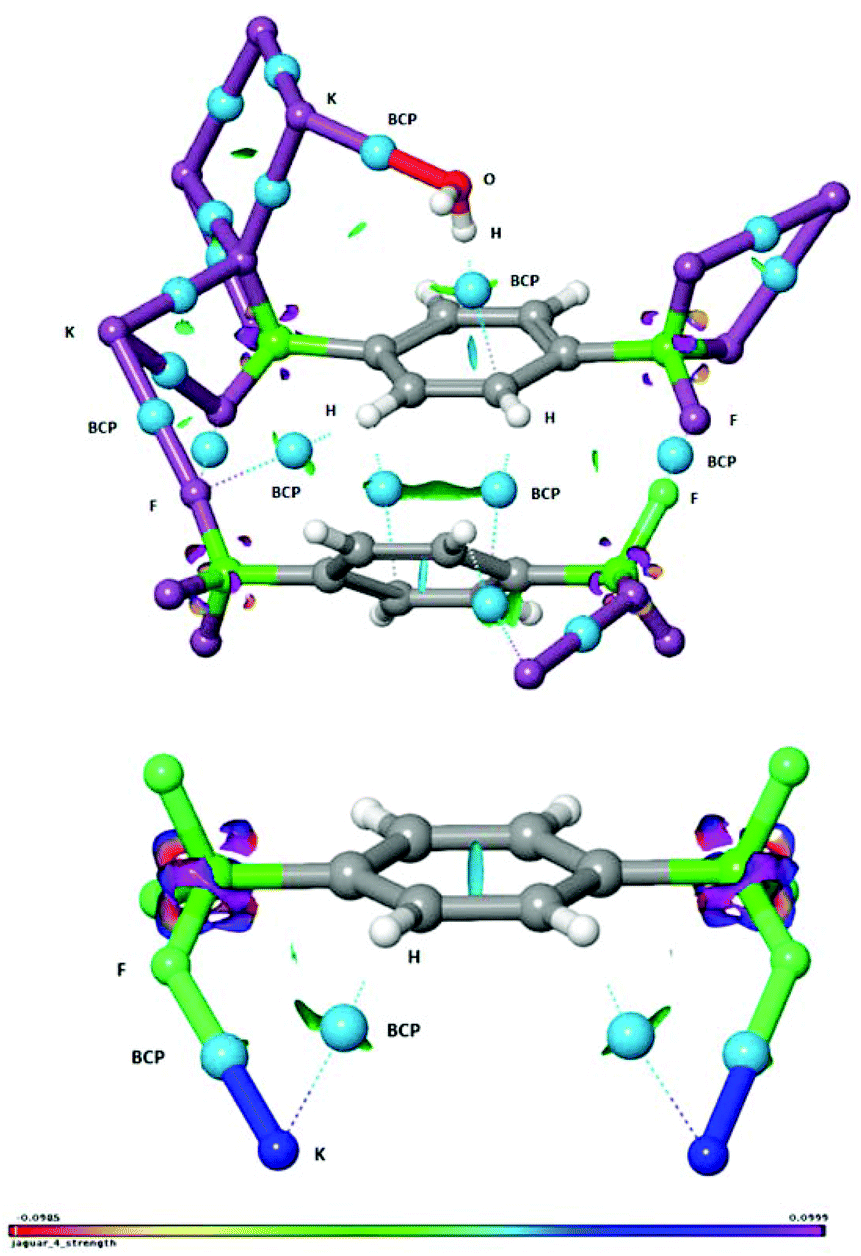 | ||
| Fig. 4 Non-covalent interaction (NCI) analysis of extracts of the crystal structure of compound 2. BCP = bond critical point marked with light-blue spheres and bond paths with dotted lines. | ||
Conclusions
In summary, single-crystal X-ray diffraction analysis of a structure containing a benzene ring with two –BF3 groups reveals an unprecedented 3D supramolecular network assembled by K⋯F, K⋯O, OH⋯π and K⋯aryl interactions. A large contribution to the stability of the packing, however, is offered by additional non-conventional weak inter- and intramolecular interactions (e.g., K⋯HC, CH⋯π, F⋯F and CH⋯F), as unveiled by the BCP and bond path analysis. It is worth mentioning that the participation of organic fluorine in the formation of intermolecular interactions has always been a matter of concern.15Acknowledgements
This work was financially supported by the University of Bari, the Interuniversities Consortium C.I.N.M.P.I.S., and by the German–Italian bilateral program Vigoni for the year 2011 (Code: E65E06000080001) funded by DAAD and the Italian Ateneo Italo-Tedesco. D. St. thanks the Danish National Research Foundation (DNRF93) funded Center for Materials Crystallography (CMC) for partially supporting this research project. The authors are also indebted to Mr Giuseppe Chita for collecting data and to Dr Cosimo Cardellicchio for stimulating discussion.Notes and references
- D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, ed. D. G. Hall, Wiley-VCH, Weinheim, 2005 Search PubMed.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494–499 CrossRef CAS PubMed.
- For leading reviews, see: (a) G. A. Molander and R. Figueroa, Aldrichimica Acta, 2005, 38, 49–56 CAS; (b) G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275–286 CrossRef CAS PubMed; (c) H. A. Stefani, R. Cella and A. S. Vieira, Tetrahedron, 2007, 63, 3263–3658 CrossRef PubMed; (d) S. Darses and J.-P. Genet, Chem. Rev., 2008, 108, 288–325 CrossRef CAS PubMed; (e) G. A. Molander and B. Canturk, Angew. Chem., Int. Ed., 2009, 48, 9240–9261 CrossRef CAS PubMed.
- (a) For a survey, see ref. 3c;; (b) A. Ros and V. K. Aggarwal, Angew. Chem., Int. Ed., 2009, 48, 6289–6292 CrossRef CAS PubMed.
- (a) G. A. Molander and F. Dehmel, J. Am. Chem. Soc., 2004, 126, 10313–10318 CrossRef CAS PubMed; (b) O. Skaff, K. A. Jolliffe and C. A. Hutton, J. Org. Chem., 2005, 70, 7353–7363 CrossRef CAS PubMed; (c) M. Lautens, S. G. Ouellet and S. Raeppel, Angew. Chem., Int. Ed., 2000, 39, 4079–4082 CrossRef CAS.
- In the case of compounds potentially being able to exist as internal salts, NMR spectroscopic data should always be treated with caution: the structure of aminomethyltrifluoroborates, for example, has recently been reinvestigated by Molander: J. Raushel, D. L. Sandrock, K. V. Josyula, D. Pakyz and G. A. Molander, J. Org. Chem., 2011, 76, 2762–2769 CrossRef CAS PubMed.
- (a) T. V. Ashworth, M. J. Nolte, R. H. Reimann and E. Singleton, J. Chem. Soc., Chem. Commun., 1977, 937–939 RSC; (b) P. A. Chase, L. D. Henderson, W. E. Piers, M. Parvez, W. Clegg and M. R. J. Elsegood, Organometallics, 2006, 25, 349–357 CrossRef CAS; (c) G. Conole, A. Clough and A. Whiting, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1056–1059 CrossRef; (d) T. W. Hudnall, J. F. Bondi and F. M. Gabbaï, Main Group Chem., 2006, 5, 319–327 CrossRef CAS PubMed; (e) L. F. Groux, T. Weiss, D. N. Reddy, P. A. Chase, W. E. Piers, T. Ziegler, M. Parvez and J. Benet-Buchholz, J. Am. Chem. Soc., 2005, 127, 1854–1869 CrossRef CAS PubMed; (f) D. Franz, M. Wagner, H.-W. Lerner and M. Bolte, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2010, 66, m152–m156 CAS.
- (a) J. D. Smith, Adv. Organomet. Chem., 1999, 43, 267–348 CrossRef; (b) H. Plenio, ChemBioChem, 2004, 5, 650–655 CrossRef CAS PubMed.
- For a recent feature article, see: R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1124–1150 RSC.
- (a) A. M. Richter, M. Herm and J. Schönewerk, DE102008052314A1, 2010 Search PubMed; (b) J.-K. Lee, M. C. Gwinner, R. Berger, C. Newby, R. Zentel, R. H. Friend, H. Sirringhaus and C. K. Ober, J. Am. Chem. Soc., 2011, 133, 9949–9951 CrossRef CAS PubMed; (c) J.-K. Lee, M. C. Gwinner, R. H. Friend, H. Sirringhaus and C. K. Ober, Polym. Prepr., 2011, 52, 978–979 CAS.
- A. Salomone, M. Petrera, D. I. Coppi, F. M. Perna, S. Florio and V. Capriati, Synlett, 2011, 1761–1765 CAS.
- For a comparison of aromatic NH⋯π, OH⋯π, and CH⋯π interactions, see: (a) J. Emsley, Chem. Soc. Rev., 1980, 9, 91–124 RSC; (b) G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer-Verlag, Berlin, 1991 Search PubMed; (c) G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed; (d) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS; (e) N. Mohan, K. P. Vijayalakshmi, N. Koga and C. H. Suresh, J. Comput. Chem., 2010, 31, 2874–2882 CAS; (f) E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CAS; (g) J. Overgaard and B. B. Iversen, Struct. Bonding, 2012, 146, 53–74 CrossRef CAS.
- Multihapto bonding is known to prevail with larger and less charge-localizing cations like K+; see: D. Hoffmann, W. Bauer, P. von R. Schleyer, U. Pieper and D. Stalke, Organometallics, 1993, 12, 1193–1200 CrossRef CAS.
- For leading reviews on organic fluorine compounds, see: (a) K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC; (b) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- D. Chopra and T. N. Row Guru, CrystEngComm, 2011, 13, 2175–2186 RSC.
- (a) C. Cardellicchio, M. A. M. Capozzi, A. Alvarez-Larena, J. F. Piniella and F. Capitelli, CrystEngComm, 2012, 14, 3972–3981 RSC; (b) M. A. M. Capozzi, F. Capitelli and C. Cardellicchio, Cryst. Growth Des., 2014, 14, 5442–5451 CrossRef CAS.
- (a) E. R. Johnson, S. Keinan, M.-P. Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed; (b) For previous examples visualizing non-covalent interactions, see: A. Salomone, F. M. Perna, A. Falcicchio, S. O. Nilsson Lill, A. Moliterni, R. Michel, S. Florio, D. Stalke and V. Capriati, Chem. Sci., 2014, 5, 528–538 RSC.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- Quantified by DFT calculations employing M06/LACVP+(d,p) as implemented in Jaguar 8.4, Schrödinger Inc. Further details are found in the ESI.†.
- For recent papers on this topic, see: (a) F. Feil and S. Harder, Organometallics, 2000, 19, 5010–5015 CrossRef CAS; (b) W. Scherer, V. Herz, A. Brück, C. Hauf, F. Reiner, S. Altmannshofer, D. Leusser and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 2845–2849 CrossRef CAS PubMed; (c) W. Scherer, A. C. Dunbar, J. E. Barquera-Lozada, D. Schmitz, G. Eickerling, D. Kratzert, D. Stalke, A. Lanza, P. Macchi, N. P. M. Casati, J. Ebad-Allah and C. Kuntscher, Angew. Chem., Int. Ed., 2015, 54, 2505–2509 CrossRef CAS PubMed; (d) W. Scherer and G. S. McGrady, Angew. Chem., Int. Ed., 2004, 43, 1782–1806 CrossRef CAS PubMed; (e) M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Structural characterization and crystallographic data of compound 2. Computational details and structural analysis. CCDC 1402900. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02020d |
| ‡ A. F. and S. O. N. L. contributed equally to this work. |
| § Torsion angles C4–C10–B1–F2 = −88.59°, C4–C10–B1–F5 = 153.67°, C4–C10–B1–F7 = 30.19°, C1–C2–B2–F1 = 86.21°, C1–C2–B2–F8 = −156.72°, C1–C2–B2–F6 = −33.34°, C8–C3–B3–F9 = −89.42°, C8–C3–B3–F4 = 150.34°, and C8–C3–B3–F3 = 32.36°. |
| ¶ The solution structure of 2 exhibits a single set of signals: 1H-NMR (400 MHz, DMSO-d6): δ = 7.04 (s, 4H); 13C-NMR (150 MHz, DMSO-d6): δ = 145.4 (br s); 19F-NMR (376 MHz, DMSO-d6): δ = −137.5 (br s); 11B-NMR (192 MHz, DMSO-d6): δ = 4.38 (br s). |
| This journal is © The Royal Society of Chemistry 2015 |