
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical analysis of multicellular tumour spheroids
L. E.
Jamieson
a,
D. J.
Harrison
b and
C. J.
Campbell
*a
aEaStCHEM, School of Chemistry, University of Edinburgh, Edinburgh, EH9 3JJ, UK. E-mail: colin.campbell@ed.ac.uk
bMedical and Biological Sciences Building, University of St Andrews, North Haugh, St Andrews, Fife KY16 9TF, UK
First published on 23rd April 2015
Abstract
Conventional two dimensional (2D) monolayer cell culture has been considered the ‘gold standard’ technique for in vitro cellular experiments. However, the need for a model that better mimics the three dimensional (3D) architecture of tissue in vivo has led to the development of Multicellular Tumour Spheroids (MTS) as a 3D tissue culture model. To some extent MTS mimic the environment of in vivo tumours where, for example, oxygen and nutrient gradients develop, protein expression changes and cells form a spherical structure with regions of proliferation, senescence and necrosis. This review focuses on the development of techniques for chemical analysis of MTS as a tool for understanding in vivo tumours and a platform for more effective drug and therapy discovery. While traditional monolayer techniques can be translated to 3D models, these often fail to provide the desired spatial resolution and z-penetration for live cell imaging. More recently developed techniques for overcoming these problems will be discussed with particular reference to advances in instrument technology for achieving the increased spatial resolution and imaging depth required.
 L. E. Jamieson | Lauren Jamieson graduated with a BSc (Hons) in Chemistry with Medicinal Chemistry in 2012 from the University of St Andrews. She subsequently started her PhD at the University of Edinburgh under the supervision of Dr Colin Campbell and Prof. David Harrison. Her research focuses on using SERS nanosensors to measure redox potential and pH in multicellular tumour spheroids (MTS). |
 D. J. Harrison | David Harrison is a clinician with an interest in cellular determinants of response to injury and quantitative systems biology application in pathology. He is Professor of Pathology in University of St Andrews, with an honorary professorship in University of Edinburgh, and he is Director of Laboratory Medicine in Edinburgh overseeing clinical provision in four hospitals. He chairs the steering group of the Framework 7 concerted action CASyM, drawing up the roadmap for implementation of systems medicine in Europe. |
 C. J. Campbell | Colin Campbell is Director of the EPSRC and MRC CDT in Optical Medical Imaging and a Senior Lecturer in the School of Chemistry at the University of Edinburgh. He works exclusively at the interface with biology and medicine. His interest is primarily in understanding the role of redox processes in determining phenotype and his research primarily focuses on developing new biophysical techniques to measure cellular redox states and developing new approaches to analyse, visualize and interpret redox biology in cells and tissues. |
Introduction
Cell culture is used to study all aspects of cellular characteristics and function and for in vitro testing of drug candidates and therapies. Conventionally cells are cultured on a flat surface in a petri dish or flask where their primary physical contacts are with the substrate on which they are growing and the culture medium surrounding them, with very little cell–cell interaction.1 This suffers from the major drawback that the cells are not in a realistic physiological environment. In vivo, cells are in a 3D environment with the major physical contact being cell–cell interactions with surrounding cells, and cell–matrix interactions with the extracellular matrix (ECM). This 3D environment introduces complexity that is not observed in traditional monolayer cell culture. As a result, drugs and disease therapies that prove effective in the monolayer cell culture models often fail to carry this efficacy forward into in vivo trials.2 While tissue explants grown in culture provide a step between monolayer cell culture and the in vivo environment, access to such samples is limited. There is therefore a need for an in vitro model of tumour biology that mimics better the 3D environment that exists in vivo without the added ethics, health, safety, cost and availability limitations encountered when using animal models or explanted tissue.Multicellular tumour spheroids (MTS) have become increasingly used for the study of cell function and testing of drugs as they satisfy the requirement for a more complex and physiologically relevant cell culture model. Attempts have been made to incorporate cells of different types, for example by mixing cancer cell lines with non-malignant fibroblast or other stromal cells.2,3 In particular MTS are used as an ex vivo model of cancer. Importantly, the blood supply to many cancers in vivo is poor due to rapidly proliferating cells producing cellular masses where the centre regions may be far from the well organised vasculature of the body. Cancer cells are capable of signalling for the formation of new blood vessels via proteins such as vascular endothelial growth factor (VEGF),4 however the resulting vasculature is disorganised and leaky so does not effectively deliver oxygen and nutrients to the tumour. As a result, concentration gradients develop in factors including oxygen, nutrients and pH (Fig. 1).5 The concentration gradients developed in vivo are mimicked by MTS where a radial structure develops with a core that is frequently necrotic, surrounding quiescent cells and an outer proliferating layer (Fig. 1).6
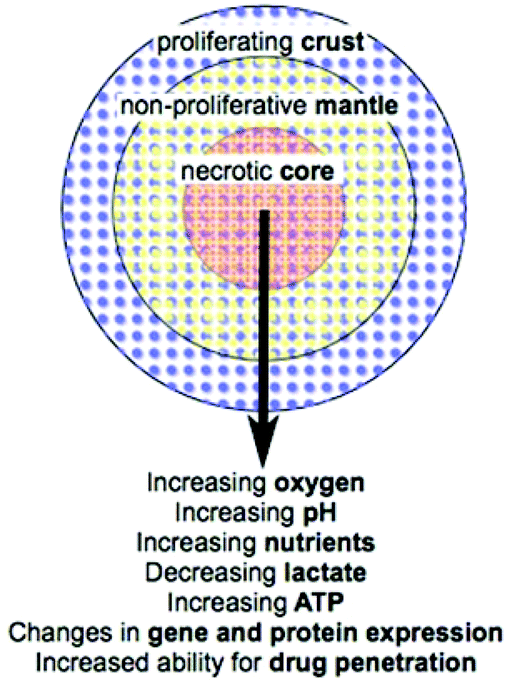 | ||
| Fig. 1 MTS organisation and radial changes that are established or predicted (adapted from Lin R. et al.7). | ||
The microenvironments4 that develop in cancer give rise to local heterogeneity in aspects including gene expression and regulation; cell differentiation, proliferation, viability and death; drug metabolism; and response to stimuli (Fig. 1).8 This combination of factors leads to increased complexity when considering drug and therapy treatments in vivo compared to simple monolayer cell culture assays. For example, increased resistance to cytotoxic drugs and ionising radiation in vivo may be a direct consequence of the microenvironment that develops.5,9 This makes the use of MTS in drug and therapy development increasingly important as they mimic better the in vivo environment in comparison to traditional monolayer culture models. This review brings together the current techniques and enabling technologies for chemical analysis of various characteristics of MTS. It summarises the latest technologies being used to improve analysis of MTS as an important model of tumour biology highlighting where there is room for improvement in chemical, spatial and temporal resolution.
Methods for growing MTS
A variety of techniques have been developed for the formation of MTS. The basic requirement for MTS formation is that adhesions between cells is stronger than that between cells and the substrate they are grown on.7 The first examples of multicellular spheroid growth were performed by Holtfreter10 who was studying gastrulation, and Moscona and Moscona11 who demonstrated the aggregation of isolated chick chondrogenic and myogenic cells.5 These initial studies formed non-tumour multicellular spheroids with the advantage that they better mimic the in vivo 3D environment. While this review focuses on MTS as an in vitro cancer tumour model, techniques discussed are equally applicable to non-tumour spheroids and organoids.Depending on the culture technique and cell type employed, spheroids are formed in varying quantities, over various time periods and with varying degrees of homogeneity and the particular technique used is likely to be guided by the particular application being employed. For example, using spinner flasks allows large quantities of MTS to be formed but these often lack homogeneity while using the hanging drop technique allows much more homogenous and uniform MTS formation but is difficult to perform on a large scale.1,7
Fig. 2 illustrates some of the techniques employed for MTS formation.
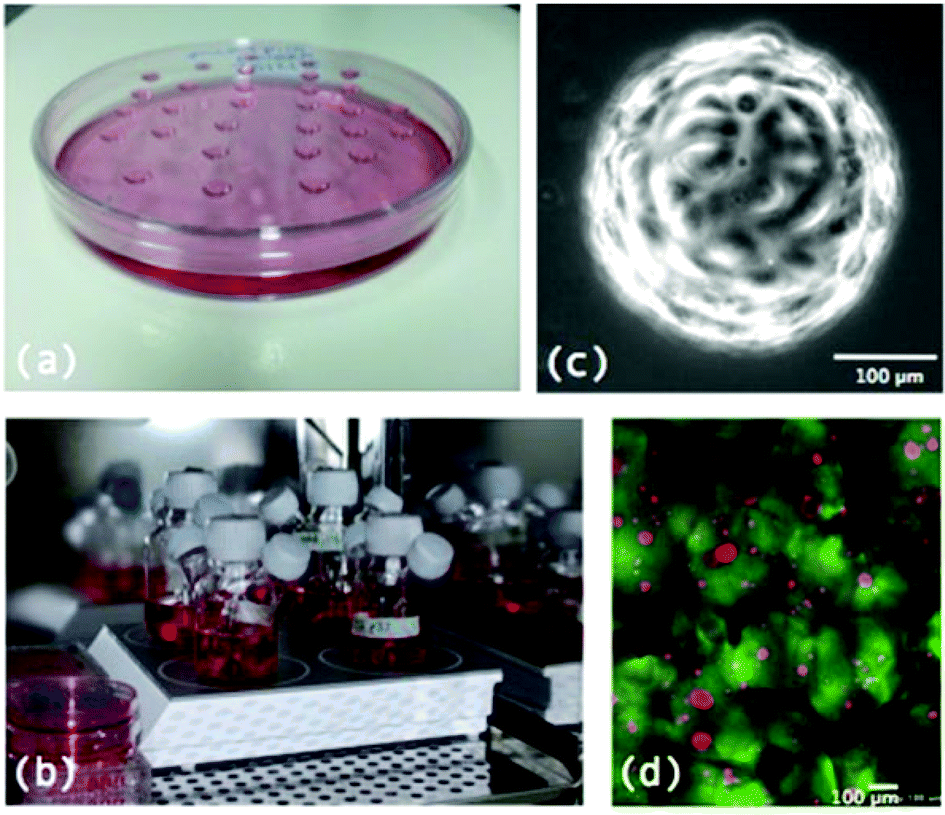 | ||
| Fig. 2 Examples of methods of MTS formation: (a) MTS being grown using the basic hanging drop technique, (b) MTS growing in spinner flasks, (c) MTS being grown on microcarrier beads and (d) cells (red) growing in a scaffold of alginate gel (green), an example of the start of MTS growth in a scaffold of NovaMatrix-3D™. | ||
Spontaneous aggregation
The simplest method for the formation of MTS is spontaneous aggregation, where the cells spontaneously cluster to form cell aggregates that grow in 3D. Technically these are cell aggregates rather than MTS and only a few cell lines grow in this way – MDA-MB-435 human breast cancer cells being one example.1 Due to these limitations, a variety of other more complex methods have been developed.Hanging drop
In its simplest form, the hanging drop methods involves growing MTS from drops of cell suspension in culture media suspended from the lid of a petri dish (Fig. 2a). Cells aggregate in the drops which remain in place by surface tension and then grow into MTS.7 More recent technologies have developed multiwell plates for the growth of MTS using the hanging drop method including the Perfecta3D™ Hanging Drop Plates12 where the new design allows for easier treatment, media exchange and handling of the MTS. Growing MTS using this method allows for the formation of MTS of uniform size although it is difficult to culture large numbers of MTS in this way.13Gyratory methods and spinner flasks
A popular method for formation of large numbers of MTS is the spinner flask method (Fig. 2b). This involves putting a suspension of cells in media into a flask which is constantly kept stirring. Due to the constant motion, cells are not given the chance to adhere to surfaces in the flask and therefore aggregate together and grow to form MTS. Similarly gyratory methods involve placing the cell suspension in flasks that are put into incubators where they are rotated by gyratory motions. Both of these techniques allow for larger quantities of MTS to be formed however the drawback is that the MTS formed are heterogeneous in size and shape and the constant rotary motion could mechanically alter the MTS.1,7Non adhesive plates/liquid overlay
Another popular method for MTS formation involves growing cells in plates with substrates that limit cell adhesion. This relies on the cell–cell adhesion being stronger than the cell–substrate adhesion, encouraging cells to aggregate and form MTS rather than adhere to the substrate in the plate. Using substrates such as agarose,14 polyethylene glycol (PEG) and polydimethylsiloxane (PDMS) to coat the bottom of plates allows for this type of MTS formation.15Microcarrier beads
Microcarrier beads are small 100–400 μM diameter1 solid beads on which cells are grown in 3D (Fig. 2c). They can be produced from various materials including dextran and cellulose. Microcarrier beads are available at a low cost commercially and tend to allow spheroid formation from primary cells and transformed cell lines as well as cell types which are normally difficult to grow such as endothelial cells. As the beads form the central regions of these cultures there is less likelihood that a necrotic core will develop and therefore these models are more representative of healthy cells growing in vivo as opposed to cancer tumour cell lines.16Scaffolds
Scaffolds are extracellular matrix (ECM) mimics that can either be synthetic e.g. polyglycolic acid or naturally derived e.g. type I collagen and Matrigel® (Fig. 2d). They are highly porous 3D matrices which allow cells to attach, proliferate and differentiate and guide cells to produce 3D MTS structures.2 3D Biotek have developed culture kits based on the biodegradable scaffold poly(γ-caprolactone) (PCL).17Microwells and microfluidics
A relatively recent advancement in the growth of uniform MTS on a large scale is the use of microwells and microfluidics, enabled by the development of microprinting techniques including photolithography and soft lithography.7 Arrays of wells are printed into materials such as polyethylene glycol (PEG), polydimethylsiloxane (PDMS) and agarose which are nonadherent to cells.18 Thousands of wells can be printed in a single substrate with a defined diameter. When a cell suspension is added to such a printed substrate in a plate, cells settle into the wells such that each well contains an equal number of cells that then form MTS homogenous in size and shape. Size and geometry of MTS formed can be controlled by well shape and size. Recently many systems have been developed on microfluidic chips patterned with microwells and microchannels such that MTS can be formed in arrays and, for example, drugs can be delivered to these systems.2,15Chemical analysis of MTS – understanding tumour physiology
A variety of techniques have been developed to investigate the physiology of MTS and their changes in response to drugs and therapy. These techniques include advances in instrument technology to allow imaging in real time, in 3D; a variety of techniques to allow observation of variation in oxygen, hypoxia, pH and nutrients; and methods to measure protein expression as well as general growth characteristics and morphology.Advances in instrument technology to analyse MTS
In the simplest form standard histological methods are used to visualise MTS but this involves fixing and sectioning the samples, which introduces artefacts and does not allow the observation of dynamic processes. Conventional histological dyes also have low spatial resolution.8 New instrumentation has allowed MTS to be analysed in situ.Confocal microscopy can be used to view MTS with z resolution but penetration depths are limited and may not allow full z penetration through MTS. Efforts have been made to improve resolution and penetration depth of confocal microscopy to achieve the best images including the use of borosilicate glass bottoms on dishes as opposed to polystyrene, a water immersion microscope objective instead of a dry objective and increased laser power/light output at deeper image depths.19 While better resolution and increased z penetration was observed, the z penetration was still limited to 350 μM with this technique meaning it was unable to visualise the entire depth of a MTS.
Two and multi photon microscopy have been used for imaging 3D samples as they can give a two to three fold increase in penetration depth in comparison to one photon confocal microscopy.20 Samples are probed with laser pulses of longer wavelengths but high intensities to stimulate multi photon excitation.21 High intensities are required to increase the chance of non-linear multi photon excitations and the laser is generally operated by using long wavelength femto-second pulses of high intensity to decrease phototoxicity and photobleaching.22 Increased z penetration and contrast are achieved and only the point of interest is subjected to illumination so photobleaching and phototoxicity are minimised in the surrounding sample.20–23
Recent advances in microscopy to view MTS involve the use of optical coherence tomography (OCT) (Fig. 3b), optical projection tomography (OPT) (Fig. 3c) and light sheet microscopy (LSM) (Fig. 3d) to map MTS. In OCT back scattered or back reflected light is detected to provide cross sectional images through a 3D sample.24 In OPT the sample is rotated and straight line projections are taken through it to generate 3D images.25,26 The advantage of OCT and OPT are that much larger sample volumes can be imaged but resolution is much lower than that which can be obtained from sectioned samples viewed through standard high resolution microscopes or by using confocal microscopy.27
 | ||
| Fig. 3 (a) MTS imaged using a light microscope; (b) a cross section through the centre of an MTS using OCT; (c) an OPT image of an MTS derived from a primary cancer specimen where blue core = implanted MTS, green = outgrowth into culture in collagen and red = positive immunoreactivity for Her2; and (d) LSM image of a MTS.8 | ||
A more recent and very useful microscopy advancement for the analysis of MTS is LSM or single plane illumination microscopy (SPIM). This involves using a light sheet, a plane of light, to illuminate a sample as opposed to a single point. As well as providing an image of a whole specimen slice at once and therefore reducing acquisition time, this technique reduces the light exposure of areas of the sample and therefore reduces photobleaching and phototoxicity.8 It is an ideal technique for analysis of MTS as it allows whole sections through MTS to be observed at once and z stacks to be built into a 3D image with better z resolution than confocal microscopy (Fig. 3d). A disadvantage of this technique is the general need to embed samples in a transparent gel material, usually agarose or low melting point agarose. Embedding in such materials can impact the growth, placing mechanical restraints on specimens and making growth time course experiments inaccurate. Recently, Desmaison, A. et al.28 developed a sample holder allowing MTS to be mounted in hydrogel and a time course experiment of MTS growth to be performed.
Further advances have recently been made to LSM including the use of self-reconstructing Bessel beams and two-photon fluorescence excitation which saw an increase of nearly two fold in axial resolution and 5–10 fold increase in contrast in comparison to linear Bessel beam excitation and 3–5 fold increase in z penetration depth in comparison to Gaussian beam linear excitation.29,30 Very recently LSM has been performed using an Airy beam to provide further advances in particular allowing a larger field of view with high contrast and high resolution.31
It is worth mentioning some additional microscopy techniques that can reveal morphological characteristics of MTS compared to monolayer culture. Transmission electron microscopy (TEM) has been used to visualise cell–cell32 and cell–ECM33 interactions in MTS while scanning electron microscopy (SEM) reveals high magnification and high resolution information about the surface characteristics of MTS including visualisation of cell–cell interactions and the morphology of cells.34–36 Both techniques require cells to be fixed and processed and SEM only reveals surface topology, with samples often coated in gold removing a lot of surface detail. Helium ion microscopy (HIM) removes the need for coating and provides increased resolution compared to SEM.37Fig. 4 compares morphology of monolayer cultured cells to MTS using HIM. Table 1 summarises the microscopy techniques used when analysing MTS along with their advantages and limitations.
 | ||
| Fig. 4 comparison of the morphology of cells growing in (a) monolayer culture to (b) cells growing as MTS and (c) a magnified area of cells growing as MTS. | ||
| Microscopy technique | Advantages | Limitations |
|---|---|---|
| Standard | Simple | No z depth |
| Primarily fixed and sectioned samples | ||
| Confocal | z Depth possible | Limited to a maximum of 350 μM z depth |
| Two and multi-photon | Increased z depth and contrast | z Depth still limited |
| Photobleaching and phototoxicity to surrounding sample minimised | ||
| OCT | Can image whole MTS | Resolution is poorer |
| OPT | Can image whole MTS | Resolution is poorer |
| LSM | Can image whole MTS | Samples typically embedded in a transparent gel which can impact growth |
| Acquisition time reduced | ||
| Photobleaching and phototoxicity reduced | ||
| TEM | High resolution to visualise cell–cell and cell–ECM interactions | Limited field of view |
| Involves fixing and processing samples | ||
| SEM | High resolution and high magnification of surface characteristics | Involves fixing samples |
| Only reveals surface information | ||
| Samples often gold coated which removes surface detail | ||
| HIM | Increased resolution compared to SEM for revealing surface characteristics | Involves fixing samples |
| No requirement for coating | Only reveals surface information |
Measuring oxygen concentration and hypoxia
A significant characteristic of MTS in comparison to monolayer culture is variation in oxygen concentration mimicking in vivo tumours. As tumours and MTS grow the central regions become increasingly depleted in oxygen (and hypoxic) and eventually cells may die creating a necrotic core.38 Hypoxia in tumours is incredibly significant as it can induce pro-survival signalling, leading to metabolic changes and resistance to therapy.4,5,39,40It is desirable to chemically analyse variations in oxygen concentration and hypoxia through MTS. Oxygen concentration and hypoxia are highly interlinked but not identical characteristics. Oxygen concentration is singularly a measurement of the level of oxygen present while hypoxia is a more complex characteristic which can be defined in several ways including nitroreductase activity,41 HIF activation42 and expression of HIF associated genes43 to name a few. This distinction is important since a hypoxic phenotype (HIF stabilisation, glycolytic metabolism) is a characteristic of many cancers irrespective of oxygen concentration (Warburg Effect).44 When considering chemical analysis of MTS some techniques have been developed specifically to measure hypoxia as a function of, for example, nitroreductase activity, while other techniques directly measure oxygen concentration.
One of the first methods developed for measuring hypoxia uses small molecules such as 2-nitroimidazole that are metabolised under hypoxic conditions and subsequently detected.45 An example is the commercially available Hypoxyprobe-1™ (PimonidazoleHCl)46, where the metabolised product in hypoxic regions is detected by an antibody and subsequently visualized by either a fluorescent tag47 or colorimetric detection (Fig. 5). This technique is limited to distinguishing cells below and above a hypoxic limit without giving truly quantitative information. Autoradiography was used in initial detection methods for these probes and extension to magnetic resonance spectroscopy (MRS) and positron emission tomography (PET) allowed non-invasive detection of hypoxia in living systems.48–54 Hypoxyprobe-1™ has been used to visualise distributions of hypoxia in MTS, giving good spatial resolution, but its use is limited to fixed sections.
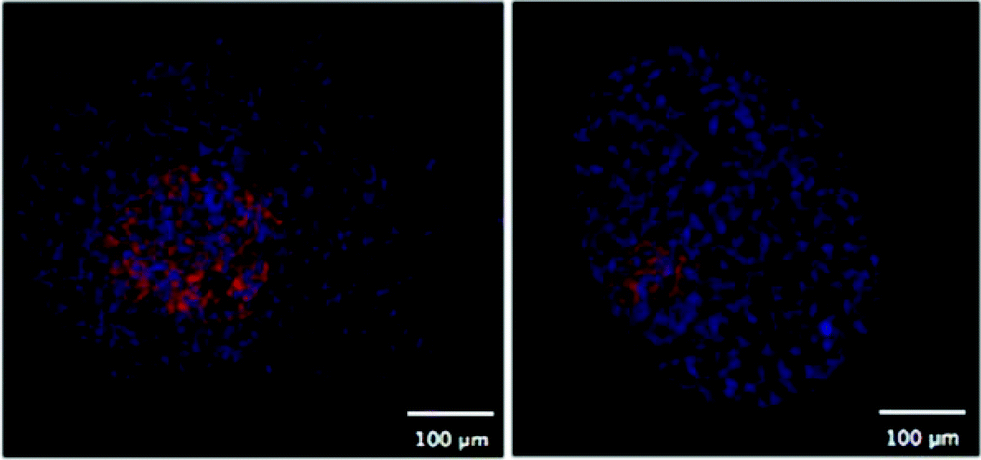 | ||
| Fig. 5 Two fluorescent images of MTS stained with Hypoxyprobe-1™ (pink) where cell nuclei are stained blue. | ||
Direct measurement of oxygen concentration can be achieved using oxygen microelectrodes. These have been miniaturised to give increased spatial resolution of oxygen measurements both in vivo and ex vivo. The main disadvantage is that this is an invasive technique with the risk that the oxygen electrodes perturb their immediate environment and thus distort values obtained.41,55
Recent advances have been made to develop increasingly efficient O2 sensing probes such as new cell-penetrating phosphorescent probes based on conjugates of Pt(II)-tetrakis(pentafluorophenyl)porphine (PtPFPP) dyes.56 The Pt probe was conjugated with glucose which minimised aggregation and self-quenching for measurements in aqueous media and was used to perform high resolution phosphorescence lifetime based O2 imaging (PLIM) in PC12 MTS. Nanoprobes have also been used in vivo to measure reactive oxygen species (ROS) concentrations and hypoxia and these techniques could potentially be applied to MTS models and, with the use of more advanced instrument technologies, allow live 3D imaging through the MTS.57
pH and nutrient concentration
Lactate accumulation is common in areas of low oxygen due to increased glycolysis, utilisation of the pentose phosphate pathway and decreased oxidative phosphorylation.58 Therefore decreasing oxygen concentration and hypoxia tends to correlate with an increase in lactate accumulation and decreasing pH. Gradients in nutrient concentration including glucose and ATP that are intimately linked to changes in metabolic pathways can also correlate with pH and oxygen gradients.Methods for measuring pH and nutrient concentrations in MTS are limited and primarily based on monolayer techniques. Significantly, studies of pH in tumour cells have revealed that intracellular and extracellular pH differ significantly, with intracellular pH becoming neutral to alkaline and extracellular pH acidic.5,59 This intra- and extracellular trend is attributed to ion pumps exporting protons from inside to outside cells which can become activated by certain growth factors which promote tumour angiogenesis, often associated with requirement for increased blood supply in areas of lower oxygen. This gradient of lower extracellular to intracellular pH is the opposite of that observed under normal tissue conditions. Detection of differences between intra- and extracellular environment requires very high resolution techniques which are currently limited.
Most methods to detect pH in culture employ fluorescence and can also be used in MTS, for example the intracellular monitoring dye BCECF.60 One of the commonly used techniques to monitor nutrient and pH variations in MTS is bioluminescence imaging. Walenta, S. et al.61 used the technique of bioluminescence of cryosectioned non-proliferating Rat 1 aggregates and MR1 MTS to determine variations in ATP, glucose and lactate. While bioluminescence imaging reveals detailed information about nutrient gradients in MTS with high spatial resolution, the resolution is not sufficient to distinguish intra- and extra-cellular compartments.
The more recent use of nanoprobes for pH imaging in vivo also has potential in live MTS. This technique is based on pH sensitive fluorescent probes conjugated to nanoprobes that can also be antibody-targeted to specific cells. Live cell imaging is possible but there is the drawback of limited range of pH detection.57 Surface enhanced Raman scattering (SERS) has been used to measure intracellular pH using gold nanoshells functionalised with pH active molecules in monolayer culture.62–66 This technique could be extended to MTS and has the potential for multiplexing to allow numerous characteristics, such as pH and redox, to be measured simultaneously.67
1H magnetic resonance chemical shift imaging of MTS incubated with imidazole was used to provide information on pH variations in MTS with 32 × 32 μm resolution. While this resolution is not enough to distinguish intra- and extracellular pH variations it revealed a 0.6 pH unit difference between central and peripheral regions of the MTS.68
Spectroscopic imaging techniques have been used to map changes in chemical, elemental and nutrient concentrations in MTS. DLD-1 MTS have been analysed by scanning transmission ion microscopy (STIM), proton-induced X-ray emission (PIXE) mapping, scanning X-ray fluorescence microscopy (SXFM) and Fourier transform infrared (FT-IR). STIM revealed useful information regarding cell density where lower density was observed in central regions relating to the necrotic core. PIXE revealed elemental maps giving information of element density of P, S, Cl, K and Fe. Cu and Zn distribution were mapped using SXFM while FT-IR revealed detailed information regarding the distribution of different biomolecules such as proteins, lipids and DNA as well as lactate.69
Proteins
Primarily as a result of the gradients of, for example, oxygen that develop in MTS, protein expression is often altered. Standard techniques for measuring protein expression such as Western Blotting can be difficult to perform with MTS samples due to the requirement for large numbers of cells, although this problem has been addressed to some extent by techniques such as in-cell westerns.2 To overcome this problem multiple MTS for the same conditions can be used. An example study using western blotting shows that the multidrug resistance (MDR1) gene is hypoxia responsive and regulated by Hypoxia Inducible Factor-1 (HIF-1).70 Both immunohistochemistry and immunofluorescence can be used to detect and quantify protein expression using specific antibodies. For example, proteins involved in cell–cell and cell–ECM interactions and components of ECM such as cadherins, integrins and collagens respectively can be immunochemically stained. It is also possible to perform quantitative immunofluorescence, for example using Automated Quantitative Analysis (AQUA), which allows quantitation of a target signal e.g. Her2 in Fig. 6 in cells stained for cytokeratin (tumour cells).71 While spatial information can be acquired using these techniques there is the need to fix samples. A similar technique that is also often used and based on fluorescence is flow cytometry of trypsinised samples. While this is simple and can provide quantitative data, it removes the majority of spatial information.72,73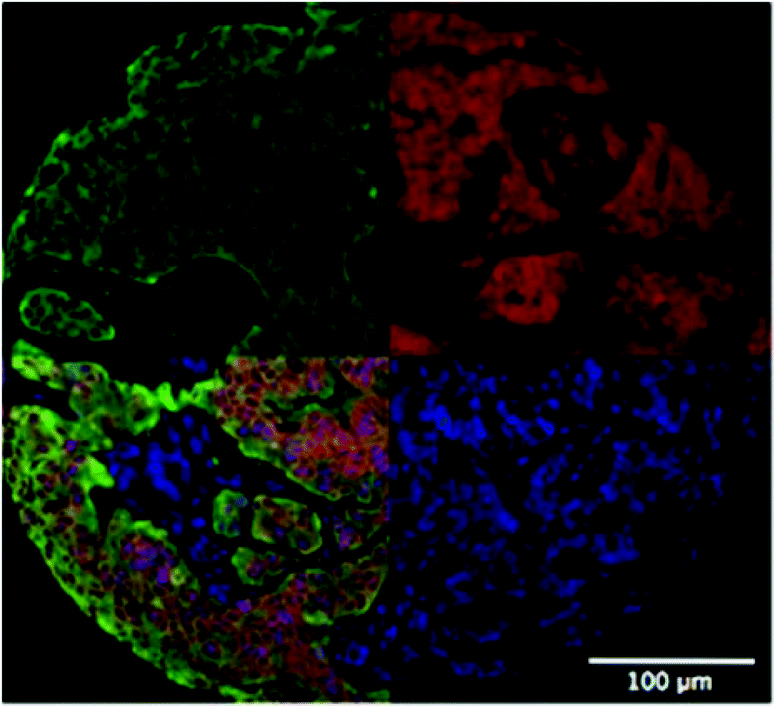 | ||
| Fig. 6 Quantitative immunofluorescence using AQUA to quantify target signal (Her2, red, top right) in cells marked for cytokeratin (i.e. tumour cells, green, top left) in MTS. Nuclei are stained with DAPI (blue, bottom right). All fluorescent signals are combined in the bottom left image. | ||
Bioluminescence has also been used to detect protein expression, protein–protein interactions and enzyme activity by employing different versions of the luciferase bioluminescence assay.74,75 This is a promising technique as it has the potential to provide spatial information by live imaging of intact MTS. In comparison to fluorescence, bioluminescence has the advantage of a lower background and therefore better sensitivity, a larger range over which quantitative signal can be obtained and less perturbation by coloured compounds.2
Nanoprobes can be used for the detection of particular enzymes to analyse their expression. For example, a nanoprobe was developed to detect the activity of caspase-3, an enzyme involved in apoptosis, where nanoparticles were conjugated with a caspase-3 substrate and scattering colour varied as individual nanoparticles were released by the enzyme acting to digest the substrate.57 This technique was used in vivo but has the potential for spatial analysis of differences in enzyme expression in MTS.
A novel demonstration of spatially resolved protein expression information was performed by McMahon,76 where MTS layers were progressively trypsinised to isolate the outer proliferating layer, middle peri-necrotic layer and necrotic core. Mass spectrometry using an iTRAQ approach with MALDI-TOF-TOF and ESI-Q-TOF was used to analyse the protein content of each layer and compare and contrast. This new approach was also complemented with traditional techniques including immunohistological staining using GLUT-1 and CAIX antibodies. While this technique does not provide highly resolved spatial information it is the first technique to demonstrate spatial changes in protein expression through MTS.
Another recent and promising technique based on mass spectrometry that provides spatial information on proteins in MTS is mass spectrometry imaging (MSI). Matrix-assisted laser desorption/ionization mass spectrometry (MALDI) has been used to analyse frozen or fixed tissue sections for spatial imaging of protein and peptides as well as other species such as lipids and small molecules.77 Typically frozen tissue sections are analysed,78 however, this technique has also been performed on formalin-fixed, paraffin-embedded tissue sections.79 To limit analyte spreading during preparation, matrix-free MSI has also been developed.80 In addition to tissue sections MALDI imaging has been performed on sectioned MTS embedded in collagen to give information on protein variations through the structure. Proteins were identified using MALDI-TOF-TOF and nanoflow-LC-MS/MS of lysates from MTS and cytochrome C and histone H were both detected as two of the species mapped. This technique is useful as it provides spatially resolved information without the need for specific stains such as those used in immunohistochemistry and immunofluorescence. However it still requires samples to be fixed or frozen and sectioned and therefore live samples cannot be analysed.
Another emerging and promising technique for chemical analysis of MTS is Raman imaging. This technique is a label free and non-invasive technique for probing differences in chemical composition. Raman imaging probes the vibrational modes of molecules giving information on the chemical composition of the area being probed in a spatially resolved manner. This technique has been predominantly investigated in monolayer culture but recently it has been shown to be useful for the analysis of 3D MTS culture also.81 While still in its initial stages of research, this technique is very promising for live and label free chemical imaging of MTS.
The techniques of MSI and confocal Raman imaging (CRI) have been combined to provide enhanced chemical information, combining information on integrity of cells and protein levels using CRI and small molecule location using MSI.82 In this example, MTS were fixed, processed and sectioned for analysis. While CRI has potential for live cell imaging of whole MTS, MSI is only possible on sectioned samples.
Localisation of and cellular response to drugs
The development of the MTS model and techniques to chemically analyse and characterise MTS has provided a more realistic ex vivo model of cancer. In addition to increasing insight into physiological characteristics of MTS, this provides a platform for more effective testing of drug candidates and therapies.83 Screening of drug candidates on MTS gives more reliable results saving time and money in later stages of drug trials. Recent developments in high throughput screening have allowed MTS to be used in place of monolayer culture, allowing drug candidates to be better selected for efficacy.13,34,84The interesting technique of biodynamic imaging has been used to analyse cellular response to drugs toxic to mitochondria and B-Raf inhibitors.85 This is an optical technique which monitors dynamic changes in cells over time and how these changes vary when treated with a particular drug. Biodynamic imaging detects dynamic light scattering using low-coherence digital holography to elucidate information on intracellular motions. Such motions include dynamic processes in cells including organelle movement and cytoskeletal changes that are often affected by drug treatment. The effect of drugs affecting the mitochondria and Raf kinase inhibitors on two colon cancer cell lines was investigated using this biodynamic imaging technique.
Nanomedicine is a rapidly advancing area of cancer research. It is apparent that when developing promising nanoparticle based techniques for cancer therapy, imaging technologies capable of successful visualising the 3D MTS structure are essential. As already discussed the advances in instrument technology, primarily with OCT, OPT and LSM, allows successful 3D images of MTS to be formed. Fluorescent LSM would be particularly useful for monitoring nanoparticle uptake if nanoparticles could be fluorescently labelled. Studies have investigated the use of techniques such as fluorescence lifetime imaging microscopy (FLIM) to examine the distribution of doxorubicin (DOX) functionalised iron oxide nanoparticles where native DOX released from the nanoparticles had a different fluorescence lifetime to DOX conjugated to the nanoparticles allowing distinction between released and conjugated DOX to be investigated.86 In another study 3D multiphoton fluorescence microscopy was successfully employed to investigate the uptake of quantum dots and DOX encapsulated in micelles.36
MALDI imaging has also been used to monitor drug uptake in MTS. Both the drug irinotecan and its metabolites were imaged by MALDI in HCT 116 colon cancer MTS at various time points to investigate uptake and metabolism of the drug.87 Raman imaging also has potential for use in drug uptake studies in MTS.81
Conclusions
MTS are now established as a valuable in vitro tool for investigation of tumour physiology and response to drugs and therapy. There is a need for these models due to the limitations of monolayer culture in displaying the complex characteristics found in vivo as a result of their 2D rather than 3D nature. This includes characteristics such as lack of cell–cell and cell–ECM interactions and failure to develop the gradients found in tumours where central regions become deprived of oxygen and nutrients – a very important characteristic as limited penetration to these areas by drugs and oxygen leads to resistance to many therapies. However, the increasing use of the more physiologically relevant MTS model has led to the requirement of more sophisticated functional imaging, in real time and in three dimensions, and chemical analysis techniques to characterise the physiological environment of these MTS and monitor responses to drugs and therapy, particularly for use in high throughput screening methods.The fundamental consideration when chemically analysing MTS is choice of a suitable technique and instrumentation to obtain the desired spatial, chemical and 3D resolution. Conventional light and confocal fluorescence microscopy techniques can be used to successfully image whole MTS with limited z depth, whilst immunohistochemistry and immunofluorescence can be used to image processed and sectioned samples. Improvements to confocal microscopy along with the use of two- and multiphoton techniques allows increasing z penetration depth. The techniques of OCT, OPT and particularly LSM have allowed whole MTS samples to be viewed and advanced our understanding of MTS.
There is still huge potential for development in analysis of MTS as many of the techniques currently used are based on analysing lysed MTS which removes spatial information or fixing and processing MTS which can introduce artefacts and does not allow for real time imaging. Variants of traditional techniques including flow cytometry, immunohistochemistry, immunofluorescence, bioluminescence imaging and western blotting are currently the main techniques employed for monitoring various chemical characteristics of MTS. There is constant development of new techniques to allow real time analysis of live MTS and perhaps the most promising areas for development is in the use of nanoprobes and Raman imaging. The combination of new probes, new imaging techniques and improved culture techniques will help drive MTS use into the mainstream of drug discovery. This review has highlighted these latest areas pushing the limits of chemical analysis of MTS and enabling technologies for imaging results. Table 2 summarises the techniques discussed for chemical analysis of MTS.
| Enabling technology | Use | Limitations |
|---|---|---|
| Hypoxia probes e.g. Hypoxyprobe™ | Hypoxia detection | Sample has to be fixed and sectioned |
| Only detects above/below a certain hypoxic threshold | ||
| Oxygen microelectrodes | Measure oxygen concentration | Invasive so can perturb environment |
| Cell penetrating phosphorescent probes | O2 sensing | Delivery of probes to cells |
| Nanoprobes | ROS detection pH detection Enzyme activity | Delivery of nanoprobes to cells |
| Fluorescent dyes | pH detection | Limited pH detection range |
| Bioluminescence imaging | pH and nutrient detection | Intra- and extra-cellular compartments cannot be distinguished |
| Protein expression protein–protein interactions | ||
| Enzyme activity | ||
| 1H magnetic resonance chemical shift imaging | pH detection | Intra- and extra-cellular compartments cannot be distinguished |
| Spectroscopic imaging | Cell density (STIM) | Samples have to be frozen and sectioned |
| Element density (PIXE and SXFM) | ||
| Protein, lipid, DNA and lactate distribution (FT-IR) | ||
| Western blotting | Protein expression | Spatial information lost |
| Immunohistochemistry and immunofluorescence | Protein expression | Samples have to be fixed and processed |
| Flow cytometry | Protein expression | Spatial information limited |
| Mass spectrometry | Protein expression | Spatial resolution possible but limited |
| Mass spectrometry imaging | Protein expression | Samples have to be frozen and sectioned |
| Lipid and small molecule expression | ||
| Drug uptake and metabolism | ||
| Raman imaging | Chemical composition Molecule distribution | Promising but limited research so far |
| SERS | pH and redox potential measurement | Delivery of nanoprobes to cells |
| Biodynamic imaging | Monitoring dynamic changes in response to drug | Monitors motions only |
| FLIM | Drug release | Promising but limited use so far |
Acknowledgements
The authors gratefully acknowledge Peter Mullen for providing the image in Fig. 2b and Fig. 5; Dr A Leeper for use of the image in Fig. 3c; Dr Pierre Bagnaninchi for acquisition of the OCT image in Fig. 3b; Dr S Langdon for use of the image in Fig. 2d; and Prof. Ernst Stelzer for permission to use the image I Fig. 3d. This research received support from the QNano Project http://www.qnano-ri.eu which is financed by the European Community Research Infrastructures under the FP7 Capacities Programme (grant no. INFRA-2010-262163), and its partner Trinity College Dublin.Notes and references
- H. Page, P. Flood and E. G. Reynaud, Cell Tissue Res., 2013, 352, 123–131 CrossRef PubMed.
- G. Mehta, A. Y. Hsiao, M. Ingram, G. D. Luker and S. Takayama, J. Controlled Release, 2012, 164, 192–204 CrossRef CAS PubMed.
- L. A. Kunz-Schughart, P. Heyder, J. Schroeder and R. Knuechel, Exp. Cell Res., 2001, 266, 74–86 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- P. Vaupel, Semin. Radiat. Oncol., 2004, 14, 198–206 CrossRef PubMed.
- A. S. Mikhail, S. Eetezadi, S. N. Ekdawi, J. Stewart and C. Allen, Int. J. Pharm., 2014, 464, 168–177 CrossRef CAS PubMed.
- R.-Z. Lin and H.-Y. Chang, Biotechnol. J., 2008, 3, 1172–1184 CrossRef CAS PubMed.
- F. Pampaloni, N. Ansari and E. H. K. Stelzer, Cell Tissue Res., 2013, 352, 161–177 CrossRef PubMed.
- H.-G. Kang, J. M. Jenabi, J. Zhang, N. Keshelava, H. Shimada, W. A. May, T. Ng, C. P. Reynolds, T. J. Triche and P. H. B. Sorensen, Cancer Res., 2007, 67, 3094–3105 CrossRef CAS PubMed.
- J. Holtfreter, J. Exp. Zool., 1944, 95, 171–212 CrossRef.
- A. Moscona and H. Moscona, J. Anat., 1952, 86, 287–301 CAS.
- Using Perfecta3D™ Hanging Drop Plates to Assess Chemosensitivity (White Paper), 3D Biomatrix™, https://3dbiomatrix.com/wp-content/uploads/downloads/2011/11/3D-Biomatrix-White-Paper-Hanging-Drop-Plate.pdf (accessed March 2015).
- Y.-C. Tung, A. Y. Hsiao, S. G. Allen, Y. Torisawa, M. Ho and S. Takayama, Analyst, 2011, 136, 473–478 RSC.
- G. Hamilton, Cancer Lett., 1998, 131, 29–34 CrossRef CAS PubMed.
- F. Hirschhaeuser, H. Menne, C. Dittfeld, J. West, W. Mueller-Klieser and L. A. Kunz-Schughart, J. Biotechnol., 2010, 148, 3–15 CrossRef CAS PubMed.
- A. L. van Wezel, Nature, 1967, 216, 64–65 CrossRef CAS PubMed.
- J. Comley, Drug Discovery World, 2010,(summer), 25–41 Search PubMed.
- H. Hardelauf, J.-P. Frimat, J. D. Stewart, W. Schormann, Y.-Y. Chiang, P. Lampen, J. Franzke, J. G. Hengstler, C. Cadenas, L. A. Kunz-Schughart and J. West, Lab Chip, 2011, 11, 419–428 RSC.
- L. le Roux, D. Schellingerhout, A. Volgin, D. Maxwell, K. Ishihara and J. Gelovani, Microsc. Microanal., 2008, 14, 734–735 CrossRef.
- R. M. Williams, W. R. Zipfel and W. W. Webb, Curr. Opin. Chem. Biol., 2001, 5, 603–608 CrossRef CAS PubMed.
- W. R. Zipfel, R. M. Williams and W. W. Webb, Nat. Biotechnol., 2003, 21, 1369–1377 CrossRef CAS PubMed.
- V. E. Centonze and J. G. White, Biophys. J., 1998, 75, 2015–2024 CrossRef CAS PubMed.
- K. König, A. Uchugonova and E. Gorjup, Microsc. Res. Tech., 2011, 74, 9–17 CrossRef PubMed.
- J. G. Fujimoto, C. Pitris, S. A. Boppart and M. E. Brezinski, Neoplasia, 2000, 2, 9–25 CrossRef CAS PubMed.
- J. Sharpe, Annu. Rev. Biomed. Eng., 2004, 6, 209–228 CrossRef CAS PubMed.
- J. Sharpe, U. Ahlgren, P. Perry, B. Hill, A. Ross, J. Hecksher-Sørensen, R. Baldock and D. Davidson, Science, 2002, 296, 541–545 CrossRef CAS PubMed.
- M. Sharma, Y. Verma, K. D. Rao, R. Nair and P. K. Gupta, Biotechnol. Lett., 2007, 29, 273–278 CrossRef CAS PubMed.
- A. Desmaison, C. Lorenzo, J. Rouquette, B. Ducommun and V. Lobjois, J. Microsc., 2013, 251, 128–132 CrossRef CAS PubMed.
- F. O. Fahrbach, V. Gurchenkov, K. Alessandri, P. Nassoy and A. Rohrbach, Opt. Express, 2013, 21, 13824–13839 CrossRef CAS PubMed.
- O. E. Olarte, J. Andilla, R. Jorand, B. Ducommun, C. Lorenzo and P. Loza-Alvarez, http://www.focusonmicroscopy.org/2013/PDF/392_Olarte.pdf (accessed March 2015).
- T. Vettenburg, H. I. C. Dalgarno, J. Nylk, C. Coll-Lladó, D. E. K. Ferrier, T. Čižmár, F. J. Gunn-Moore and K. Dholakia, Nat. Methods, 2014, 11, 541–544 CrossRef CAS PubMed.
- R. M. Sutherland, J. A. McCredie and W. R. Inch, J. Natl. Cancer Inst., 1971, 46, 113–120 CAS.
- T. Nederman, B. Norling, B. Glimelius, J. Carlsson and U. Brunk, Cancer Res., 1984, 44, 3090–3097 CAS.
- W. Y. Ho, S. K. Yeap, C. L. Ho, R. A. Rahim and N. B. Alitheen, PLoS One, 2012, 7, e44640 CAS.
- M. T. Santini, G. Rainaldi and P. L. Indovina, Crit. Rev. Oncol. Hematol., 2000, 36, 75–87 CrossRef CAS PubMed.
- H. Ma, Q. Jiang, S. Han, Y. Wu, J. C. Tomshine, D. Wang, Y. Gan, G. Zou and X.-J. Liang, Mol. Imaging, 2012, 11, 487–498 CAS.
- G. Hlawacek, V. Veligura, R. van Gastel and B. Poelsema, J. Vac. Sci. Technol., B, 2014, 32, 020801 Search PubMed.
- T. Anada, J. Fukuda, Y. Sai and O. Suzuki, Biomaterials, 2012, 33, 8430–8441 CrossRef CAS PubMed.
- A. Hunter, A. Hendrikse, M. Renan and R. Abratt, Apoptosis, 2006, 11, 1727–1735 CrossRef PubMed.
- H. Harada, J. Radiat. Res., 2011, 52, 545–556 CrossRef CAS PubMed.
- K. A. Krohn, J. M. Link and R. P. Mason, J. Nucl. Med., 2008, 49, 129S–148S CrossRef CAS PubMed.
- M. W. Dewhirst, Y. Cao and B. Moeller, Nat. Rev. Cancer, 2008, 8, 425–437 CrossRef CAS PubMed.
- G. L. Semenza, Nat. Rev. Cancer, 2003, 3, 721–732 CrossRef CAS PubMed.
- W. H. Koppenol, P. L. Bounds and C. V. Dang, Nat. Rev. Cancer, 2011, 11, 325–337 CrossRef CAS PubMed.
- A. J. Varghese, S. Gulyas and J. K. Mohindra, Cancer Res., 1976, 36, 3761–3765 CAS.
- HypoxyprobeTM-1 Plus Kit for the Detection of Tissue Hypoxia, Chemicon International, http://webcache.googleusercontent.com/search?q=cache:8bxU7yy_OUoJ:www.ibrarian.net/navon/paper/HypoxyprobeTM_1_Plus_Kit_for_the_Detection_of_Tis.pdf%3Fpaperid%3D4633961+&cd=5&hl=en&ct=clnk&gl=uk (accessed March 2015).
- J. A. Raleigh, G. G. Miller, A. J. Franko, C. J. Koch, A. F. Fuciarelli and D. A. Kelly, Br. J. Cancer, 1987, 56, 395–400 CrossRef CAS PubMed.
- J. D. Chapman, A. J. Franko and J. Sharplin, Br. J. Cancer, 1981, 43, 546–550 CrossRef CAS PubMed.
- J. D. Chapman, K. Baer and J. Lee, Cancer Res., 1983, 43, 1523–1528 CAS.
- A. J. Franko and C. J. Koch, Int. J. Radiat. Oncol. Biol. Phys., 1984, 10, 1333–1336 CrossRef CAS.
- A. J. Varghese and G. F. Whitmore, Int. J. Radiat. Oncol. Biol. Phys., 1984, 10, 1341–1345 CrossRef CAS.
- J. A. Raleigh, A. J. Franko, C. J. Koch and J. L. Born, Br. J. Cancer, 1985, 51, 229–235 CrossRef CAS PubMed.
- R. C. Urtasun, C. J. Koch, A. J. Franko, J. A. Raleigh and J. D. Chapman, Br. J. Cancer, 1986, 54, 453–457 CrossRef CAS PubMed.
- J. A. Raleigh, D. P. Calkins-Adams, L. H. Rinker, C. A. Ballenger, M. C. Weissler, W. C. Fowler, D. B. Novotny and M. A. Varia, Cancer Res., 1998, 58, 3765–3768 CAS.
- B. Bourrat-Floeck, K. Groebe and W. Mueller-Klieser, Int. J. Cancer, 1991, 47, 792–799 CrossRef CAS.
- R. I. Dmitriev, A. V. Kondrashina, K. Koren, I. Klimant, A. V. Zhdanov, J. M. P. Pakan, K. W. McDermott and D. B. Papkovsky, Biomater. Sci., 2014, 2, 853–866 RSC.
- H. Koo, M. S. Huh, J. H. Ryu, D.-E. Lee, I.-C. Sun, K. Choi, K. Kim and I. C. Kwon, Nano Today, 2011, 6, 204–220 CrossRef CAS.
- C. Riganti, E. Gazzano, M. Polimeni, E. Aldieri and D. Ghigo, Free Radicals Biol. Med., 2012, 53, 421–436 CrossRef CAS PubMed.
- M. K. Danquah, X. A. Zhang and R. I. Mahato, Adv. Drug Delivery Rev., 2011, 63, 623–639 CrossRef PubMed.
- D. Rotin, D. Steele-Norwood, S. Grinstein and I. Tannock, Cancer Res., 1989, 49, 205–211 CAS.
- S. Walenta, J. Doetsch, W. Mueller-Klieser and L. A. Kunz-Schughart, J. Histochem. Cytochem., 2000, 48, 509–522 CrossRef CAS PubMed.
- A. Jaworska, L. E. Jamieson, K. Malek, C. J. Campbell, J. Choo, S. Chlopicki and M. Baranska, Analyst, 2015, 140, 2321 RSC.
- S. W. Bishnoi, C. J. Rozell, C. S. Levin, M. K. Gheith, B. R. Johnson, D. H. Johnson and N. J. Halas, Nano Lett., 2006, 6, 1687–1692 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, Nano Lett., 2007, 7, 2819–2823 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, J. Phys. Chem. C, 2010, 114, 7421–7426 CAS.
- M. A. Ochsenkuehn, P. R. T. Jess, H. Stoquert, K. Dholakia and C. J. Campbell, ACS Nano, 2009, 3, 3613–3621 CrossRef PubMed.
- L. E. Jamieson, A. Jaworska, J. Jiang, M. Baranska, D. J. Harrison and C. J. Campbell, Analyst, 2015, 140, 2330 RSC.
- J. Alvarez-Pérez, P. Ballesteros and S. Cerdán, MAGMA, 2005, 18, 293–301 CrossRef PubMed.
- J. Z. Zhang, N. S. Bryce, R. Siegele, E. A. Carter, D. Paterson, M. D. de Jonge, D. L. Howard, C. G. Ryan and T. W. Hambley, Integr. Biol., 2012, 4, 1072–1080 RSC.
- K. M. Comerford, T. J. Wallace, J. Karhausen, N. A. Louis, M. C. Montalto and S. P. Colgan, Cancer Res., 2002, 62, 3387–3394 CAS.
- R. L. Camp, G. G. Chung and D. L. Rimm, Nat. Med., 2002, 8, 1323–1328 CrossRef CAS PubMed.
- N.-C. Cheng, S. Wang and T.-H. Young, Biomaterials, 2012, 33, 1748–1758 CrossRef CAS PubMed.
- J. M. Lee, P. Mhawech-Fauceglia, N. Lee, L. C. Parsanian, Y. G. Lin, S. A. Gayther and K. Lawrenson, Lab. Invest., 2013, 93, 528–542 CrossRef PubMed.
- P. M. Tiwari, K. Vig, V. A. Dennis and S. R. Singh, Nanomaterials, 2011, 1, 31–63 CrossRef CAS.
- E. Salomonnson, A. C. Stacer, A. Ehrlich, K. E. Luker and G. D. Luker, PLoS One, 2013, 8, e51500 CAS.
- K. Mcmahon, PhD Thesis, University of Bradford, 2011 Search PubMed.
- R. J. A. Goodwin, S. R. Pennington and A. R. Pitt, Proteomics, 2008, 8, 3785–3800 CrossRef CAS PubMed.
- M. Stoeckli, P. Chaurand, D. E. Hallahan and R. M. Caprioli, Nat. Med., 2001, 7, 493–496 CrossRef CAS PubMed.
- M. C. Djidja, E. Claude, M. F. Snel, P. Scriven, S. Francese, V. Carolan and M. R. Clench, J. Proteome Res., 2009, 8, 4876–4884 CrossRef CAS PubMed.
- R. J. A. Goodwin, A. R. Pitt, D. Harrison, S. K. Weidt, P. R. R. Langridge-Smith, M. P. Barrett and C. L. MacKay, Rapid Commun. Mass Spectrom., 2011, 25, 969–972 CrossRef CAS PubMed.
- E. Zuser, PhD Thesis, Northeastern University, 2013 Search PubMed.
- D. R. Ahlf, R. N. Masyuko, A. B. Hummon and P. W. Bohn, Analyst, 2014, 139, 4578–4585 RSC.
- W. Fayad, L. Rickardson, C. Haglund, M. H. Olofsson, P. D'Arcy, R. Larsson, S. Linder and M. Fryknäs, Chem. Biol. Drug Des., 2011, 78, 547–557 CAS.
- B. G. Reid, T. Jerjian, P. Patel, Q. Zhou, B. H. Yoo, P. Kabos, C. A. Sartorius and D. V. LaBarbera, Curr. Chem. Genomics Transl. Med., 2014, 8, 27–35 CrossRef PubMed.
- R. An, D. Merrill, L. Avramova, J. Sturgis, M. Tsiper, J. P. Robinson, J. Turek and D. D. Nolte, J. Biomol. Screening, 2014, 19, 526–537 CrossRef CAS PubMed.
- J. S. Basuki, H. T. T. Duong, A. Macmillan, R. B. Erlich, L. Esser, M. C. Akerfeldt, R. M. Whan, M. Kavallaris, C. Boyer and T. P. Davis, ACS Nano, 2013, 7, 10175–10189 CrossRef CAS PubMed.
- X. Liu, E. M. Weaver and A. B. Hummon, Anal. Chem., 2013, 85, 6295–6302 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |