 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The chemistry of cationic polyphosphorus cages – syntheses, structure and reactivity
Michael H.
Holthausen
a and
Jan J.
Weigand
*b
aDepartment of Chemistry, University of Toronto, Toronto, Canada. E-mail: m.holthausen@utoronto.ca
bFachrichtung Chemie und Lebensmittelchemie, TU Dresden, Professur für Koordinationschemie, Dresden, Germany. E-mail: jan.weigand@tu-dresden.de; Tel: +49 (351) 468-42800
First published on 17th April 2014
Abstract
The aim of this review is to provide a comprehensive view of the chemistry of cationic polyphosphorus cages. The synthetic protocols established for their preparation, which are all based on the functionalization of P4, and their intriguing follow-up chemistry are highlighted. In addition, this review intends to foster the interest of the inorganic, organic, catalytic and material oriented chemical communities in the versatile field of polyphosphorus cage compounds. In the long term, this is envisioned to contribute to the development of new synthetic procedures for the functionalization of P4 and its transformation into (organo-)phosphorus compounds and materials of added value.
 Michael H. Holthausen | Michael H. Holthausen received his diploma degree from the WWU Münster at the end of 2009 and his Dr rer nat. in March 2013. As a PhD student in the Weigand group he was supported by a scholarship of the Fond der Chemischen Industrie. He studied the reactivity of cationic polyphosphorus cages and developed protocols for their preparation based on the activation of elemental white phosphorus by phosphenium ions. After a brief postdoctoral stay at the TU Dresden he recently joined the research group of Prof. Douglas Stephan at the University of Toronto in Canada and obtained a Feodor Lynen Scholarship for postdoctoral researchers. |
 Jan J. Weigand | Jan J. Weigand obtained his diploma in chemistry in 2002 and his Dr rer nat. in 2005 from the LMU in Munich (Germany). He was awarded in 2005 the Bavarian culture prize and obtained a Lynen Scholarship from the AvH foundation for postdoctoral research at Dalhousie University in Halifax (Canada). He returned to Germany with a Lynen Return Fellowship and started his habilitation at the WWU Münster at the end of 2007 under the supervision of Prof. Hahn. He was awarded shortly after the Liebig scholarship of the FCI which allowed him in 2008 to start his independent career. In April 2010 he became a fellow of the Emmy Noether research program awarded by the DFG and obtained recently the Wöhler research award for young scientists. In 2012, he obtained an ERC starting grant from the European Council. Since 2013 he has been Professor at the TU University Dresden. |
1. Introduction
Discovering novel pathways for the activation and transformation of white phosphorus (P4) is important for the ongoing search for new, systematic entries to polyphosphorus and organo-phosphorus compounds. Especially in the realm of polyphosphorus cations methods for the preparation of species featuring a high P to substituent ratio are rare. In contrast, a systematic access to highly substituted cations RnPm (n > m) is achieved with synthetic protocols mainly based on the utilization of neutral catena or cyclic polyphosphanes RnPm.1 Protocols for phosphorus-rich cations RnPm (n < m) often involve P1-precursors and are based on the reduction of either P–Cl2 or P–H bonds.3 Multiple P–P bonds are formed in these reactions giving access to elaborate P–P bonded frameworks. However, in most cases the reaction outcome is unpredictable which hampers the targeted preparation of polyphosphorus cations. Thus, a synthetic approach that takes advantage of the tetrahedral P4 framework should allow for a targeted and systematic assembly of phosphorus-rich cations RnPm (n < m). Additionally, the application of P4 in such conversions is of high interest, since it constitutes an important raw material in industrial chemistry and is produced on a megaton-scale nowadays.4 The desire to develop synthetic protocols for the more sustainable production of P-containing bulk chemicals has sparked significant academic and industrial research efforts within the last decades. Progress in the areas of transition metal5 and main group6 mediated P4 activation has been reviewed several times. However, no account was given so far on the importance of P4 as a starting material for the preparation of polyphosphorus cations.The aim of this review is to provide a comprehensive view of the chemistry of cationic polyphosphorus cages.
The synthetic protocols established for their preparation, which are all based on the functionalization of P4, and their intriguing follow-up chemistry are highlighted. In addition, this review intends to foster the interest of the inorganic, organic, catalytic and material oriented chemical communities in the versatile field of polyphosphorus cage compounds. In the long term, this is envisioned to contribute to the development of new synthetic procedures for the functionalization of P4 and its transformation into (organo-)phosphorus compounds and materials of added value.
In the following, black dots denote P atoms in order to provide easily comprehensible drawings of complex polyphosphorus frameworks for the reader. These frameworks may give rise to complicated, sometimes higher order, spin systems in their 31P NMR spectra. Their designation is derived by assigning letters in alphabetical order starting with the resonance at the highest field. The spin systems were considered to be higher order and consecutive letters are assigned if Δδ(PiPii)/nJ(PiPii) < 10. For Δδ(PiPii)/nJ(PiPii) > 10, the spin system is considered to be pseudo first order and the assigned letters are separated. However, if a group of similar compounds is discussed, only one spin system is mentioned for the sake of clarity. All cationic polyphosphorus cages presented here are obtained by functionalization of P4. Mostly, phosphenium ions or cationic phosphorus species which formally serve as a phosphenium ion source are used for this functionalization. It is of high importance for the reader to be aware of the general reactivity pattern of P4 and the general characteristics of phosphenium ions. Thus, a brief insight into both fields is given in the first two sections.
2. P4 activation pathways
In order to gain an in depth understanding of the reactions of P4 and main group element compounds, it is crucial to understand the properties of the P4 tetrahedron. The bonding in P4 is almost “cluster-like”, strongly delocalized and mostly effected through 3p atomic orbitals. Interestingly, P4 shows spherical aromaticity and is virtually unstrained despite acute bond angles of 60°.7 Generalized reactions of P4 with nucleophiles (Nu−), electrophiles (El+) and ambiphiles (Ab) are shown in Fig. 1. Radical reactions involving P4 are excluded. A nucleophile (Nu−) interacts with the LUMO of P4 (−1.8 eV),7 which leads to the rupture of a P–P bond giving butterfly-type bicyclo[1.1.0]tetraphosphane A (Fig. 1I). The reactions of P4 with nucleophiles were intensely investigated using an array of organo-alkali and organo-alkali earth reagents.6 However, in many cases the formation of a derivative of A only constitutes the first step of a reaction sequence which ultimately leads to the degradation of P4 to P1-compounds.6 Only a few reactions involving a selective cleavage of only a single bond in the P4 tetrahedron are reported.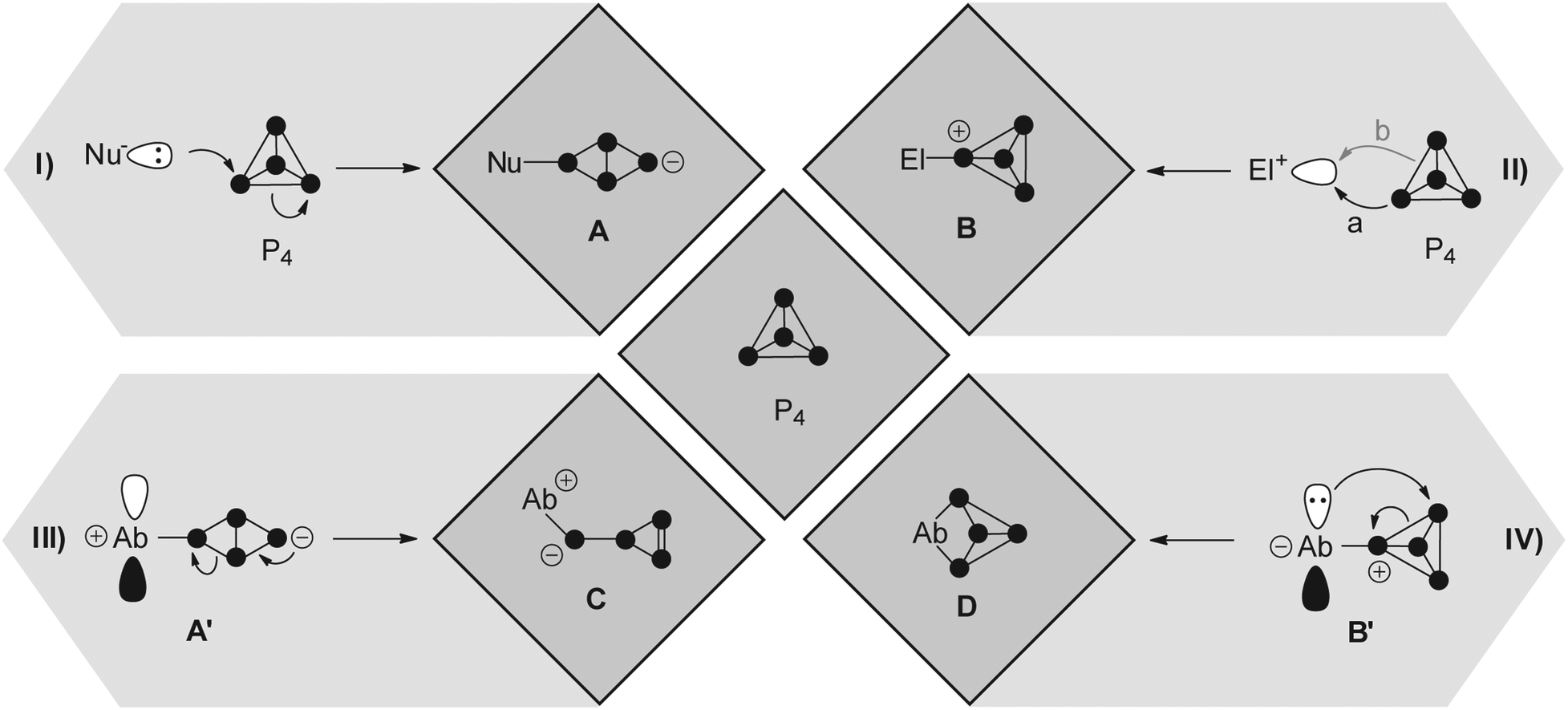 | ||
| Fig. 1 Generalized reactions of P4 with nucleophiles (I, Nu−), electrophiles (II, El+), predominantly nucleophilic ambiphiles (III, Ab), and predominantly electrophilic ambiphiles (IV, Ab); A–D illustrate structural motifs obtained after reaction with the aforementioned species. | ||
One is the reaction of Mes*Li (Mes* = 2,4,6-tri-tert-butylphenyl) with one equivalent of P4 yielding a tetraphosphanide intermediate of type A. Subsequent reaction with Mes*Br yields the butterfly-type species 1 (Fig. 2).8 Further degradation of 1 is prevented by the sterically demanding Mes*-groups. Nucleophiles based on silicon, main group 5 or main group 6 elements were also employed.6 An electrophile may attack at a non-bonding orbital of lone pair character (HOMO − 6, −7.5 eV)7 which results in the formation of compounds of type B (Fig. 1II a). Alternatively, an electrophile may attack at a bonding orbital at one of the edges of the tetrahedron (HOMO, −6.7 eV; Fig. 1II b). However, this mode of attack is commonly less productive for main group element centered electrophiles and is not depicted. In total, only very few reactions with electrophiles were reported due to the low nucleophilicity of P4.9 One example constitutes the reaction of P4 with two equivalents of the sterically encumbered Lewis acid Ga(t-Bu)3. This yields compound 2; however, mechanistic details regarding its formation were not reported (Fig. 2).10
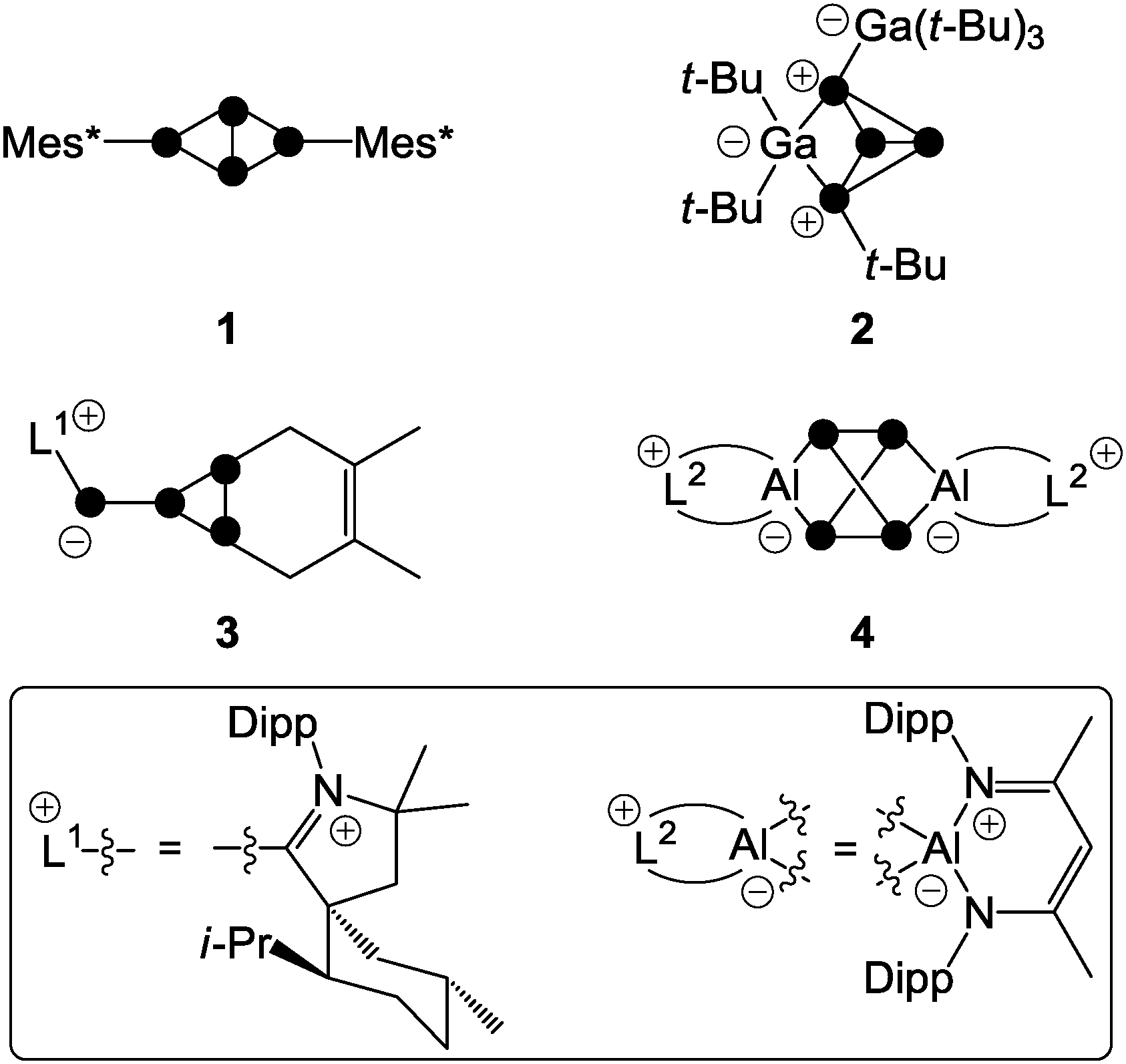 | ||
| Fig. 2 Examples of polyphosphorus compounds obtained by the functionalization of P4 by nucleophiles (1), electrophiles (2), predominantly nucleophilic ambiphiles (3), and predominantly electrophilic ambiphiles (4). | ||
The utilization of ambiphilic main group element compounds (Ab) for the activation of P4 represents a rather new synthetic approach. Reactions of P4 with ambiphiles can be divided into two categories assuming an asynchronous process with two consecutive steps. The first category comprises reactions of P4 with predominantly nucleophilic ambiphiles. Similar to the reactions of P4 and nucleophiles, intermediate A′ is obtained in the first step of the reaction. Subsequently, A′ rearranges to cyclo-triphosphirene derivative C (Fig. 1III). The rearrangement is attributed to the propensity of Ab to accept electron density from the adjacent P atom which formally leads to the formation of an Ab–P double bond. Carbenes are ambiphiles with a predominantly nucleophilic character.11 Two types of carbenes, i.e. N-heterocyclic carbenes (NHC) and cyclic or acyclic alkyl amino carbenes (cAAC or aAAC), were investigated in reactions with P4 by the research group of Bertrand.12 The formation of an intermediate of type A′ was confirmed by DFT calculations12 and of type C by trapping experiments with 2,3-dimethylbutadiene yielding [2+4] cyclo-addition product 3 (Fig. 2, e.g. L1 = cAAC).
The second category comprises reactions of P4 with predominantly electrophilic ambiphiles. By analogy with the reactions involving electrophiles, the first step of the reaction is an electrophilic attack of Ab yielding an intermediate B′ (Fig. 1). Subsequently, B′ rearranges to a bicyclo[1.1.0]tetraphosphane D featuring a bridging Ab moiety (IV). This reaction sequence equals the formal insertion of the ambiphile in one of the P–P bonds of the P4 tetrahedron. P4 functionalization involving a predominantly electrophilic ambiphile is an experimentally more widespread approach. Monovalent group 13 element compounds with the oxidation state +I are a class of substances that are widely used in such transformations.13 The first type of such a structural motif was achieved by Roesky and coworkers by reacting P4 with two equivalents of Al(I) compound AlL2 (L2 = CH{(CMe)(2,6-i-Pr2C6H3N)}2).13 The formal insertion of AlL2 into one P–P bond of P4 is assumed to give an intermediate of type B′ in the first step. However, the insertion of a second equivalent of AlL2 into the opposing P–P bond of the P4 tetrahedron occurs rapidly yielding the two-fold insertion product 4 (Fig. 2). In addition, P4 activation by predominantly electrophilic silylenes,14 disilenes,15 phosphasilenes,16 and a bis(stannylene)17 was reported. Reactions of P4 with phosphenium cations (R2P+) are also classified as P4 functionalization with predominantly electrophilic ambiphiles. They will be thoroughly discussed within this review from an experimental as well as a mechanistic point of view.
3. Syntheses and characteristics of phosphenium ions
The term phosphenium ion describes a cation featuring a di-coordinated, positively polarized P atom.18 Phosphenium ions reveal a lone pair of electrons and a formally vacant p-type orbital, and thus, they constitute carbene analogues.11 The stability of phosphenium ions strongly depends on their substituents. While aryl- or alkylphosphenium ions R2P+ (7+, Fig. 3) are strongly electrophilic and generally elusive, a large series of phosphenium ions bearing amino-substituents (R2N)2P+ (R = alkyl, aryl) are known.18 Three methods for their preparation are mainly reported throughout the literature. Halide abstraction from the corresponding halo-phosphane precursor is the most commonly used synthetic protocol.18 Further methods constitute the protolysis of P–N single bonds by Brønsted acids and the coordination of strong Lewis acids to P–N double bonds.18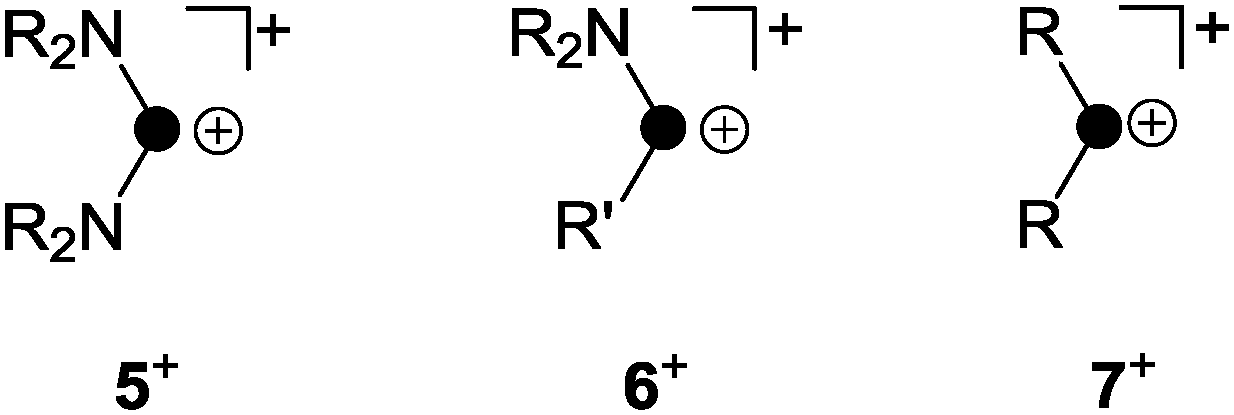 | ||
| Fig. 3 Distinct types of phosphenium ions featuring two (5+) or one (6+) stabilizing amino-substituents and elusive, non-stabilized phosphenium ion 7+ (R, R′ = alkyl, aryl). | ||
The increased stability of phosphenium ions (R2N)2P+ (5+, Fig. 3) stems from a lowered electrophilicity due to donation of π-electron density from the lone pair of electrons at the nitrogen atoms to the vacant p-type orbital at the P atom.19 Phosphenium ions of type 6+ featuring one amino-substituent are borderline cases between both of the aforementioned types and are only scarcely investigated. Only a few fully characterized derivatives are reported to date bearing either (pseudo-) halogens20 or sterically demanding aryl-moieties21 as the second substituent R′ on phosphorus (Fig. 3).
A phosphenium ion bearing only alkyl- or aryl-substituents has not been isolated to date.18 The reaction of phosphanes bearing organo- and chloro-groups RnPCl(3−n) (n = 1, 2) and a halide abstracting agent (e.g. Me3SiOTf, GaCl3, or AlCl3) in the appropriate stoichiometry usually results in the formation of phosphanylphosphonium ions.1 This is best exemplified by the reaction of Ph2PCl and GaCl3 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry which yields 8[GaCl4] (Scheme 1).22
:1 stoichiometry which yields 8[GaCl4] (Scheme 1).22
 | ||
| Scheme 1 Synthesis of 8[GaCl4] by the reaction of Ph2PCl and GaCl3 in 2:1 stoichiometry and possible reaction sequences giving 8[GaCl4] either via free Ph2P+-phosphenium ion (red) or zwitterion 9 (blue). | ||
Two mechanisms for the formation of 8+ are conceivable. Firstly, Ph2PCl reacts with GaCl3 as a halide abstracting agent giving a transient Ph2P+-phosphenium ion. This reacts with the second equivalent of Ph2PCl yielding 8+. The second and in the author's opinion more likely mechanism proceeds via the zwitterionic intermediate 9 which features a Ph2PCl molecule donating electron density from its lone pair of electrons to the lobes of the antibonding σ*(P–Cl) orbital of a second molecule of Ph2PCl. Subsequently, chloride abstraction by GaCl3 yields 8+ without an intermediary formation of a free Ph2P+-phosphenium ion. The phosphoniumyl-moiety in 8+ is easily substituted when 8+ is reacted with phosphanes of higher basicity than the leaving group.1 This is illustrated by the reaction of 8+ with Ph3P yielding 10+ and Ph2PCl (Scheme 2, left).23 Other Lewis bases are also suitable as nucleophiles. This is illustrated by the reaction of 8+ with 1,3-di-iso-propyl-4,5-dimethylimidazol-2-ylidene (L3) which yields the imidazoliumyl-substituted phosphane 11+.23
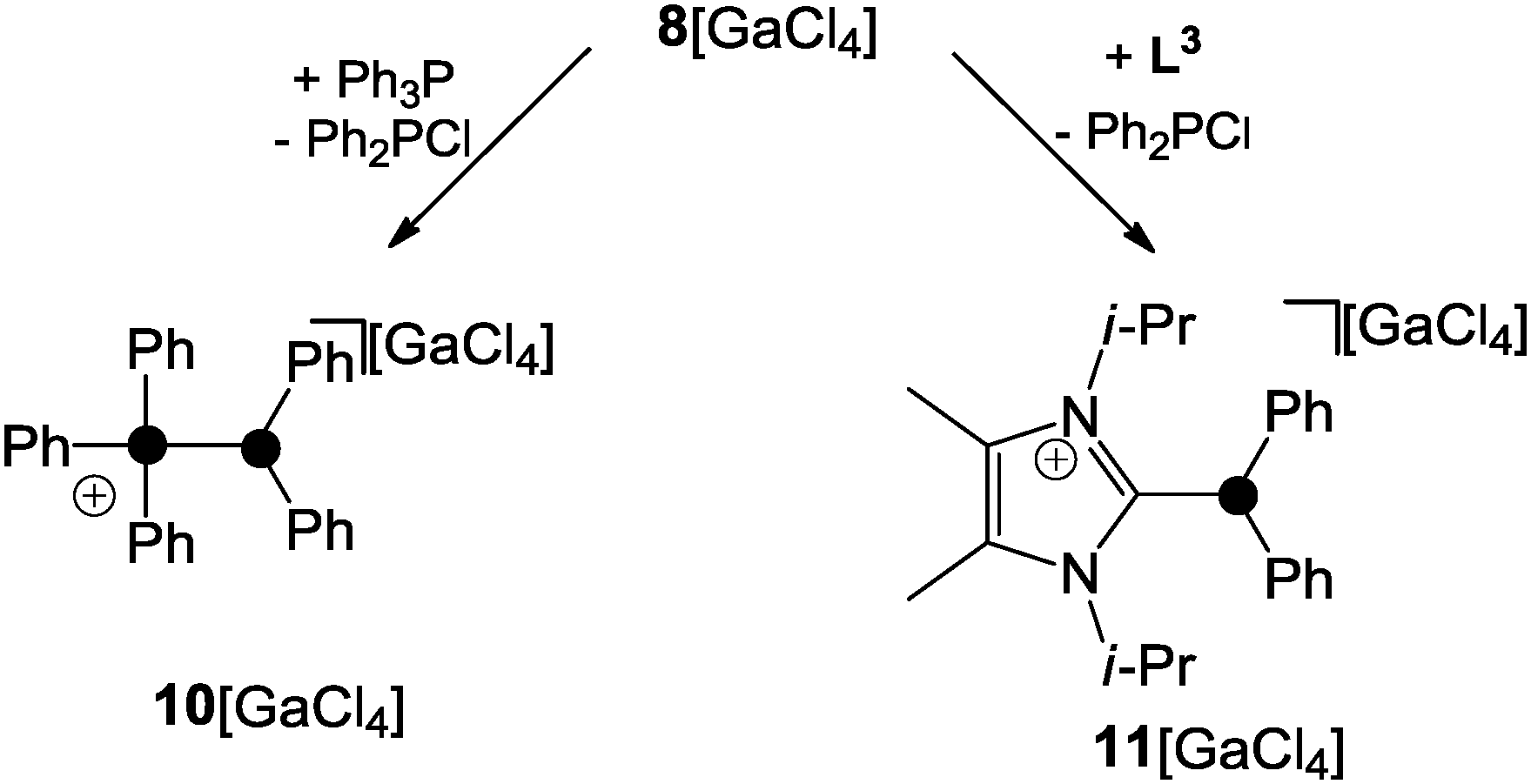 | ||
| Scheme 2 Substitution of the phosphoniumyl-moiety of 8+ by Lewis-bases. | ||
Detailed investigations of mixtures of phosphanyl-phosphonium ion 12+ and Ph3P revealed second-order kinetics for the exchange process of Ph3P consistent with a SN2-type pathway (Scheme 3).24
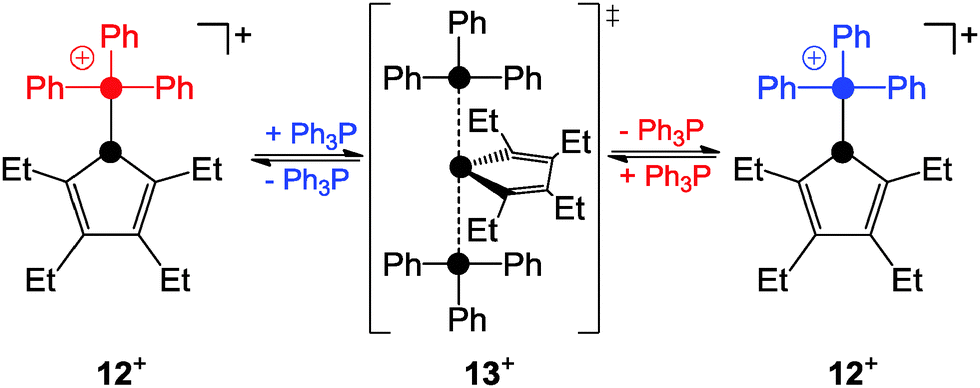 | ||
| Scheme 3 SN2-type substitution of the phosphoniumyl-moiety in 12+. | ||
This was further supported by quantum chemical calculations which suggested the phosphoranide-type transition state 13+ for the substitution process.24 In contrast, the phosphanyl-phosphonium ion 14+, which is formed via the reaction of phosphenium ion 15+ and PMe3, was reported to favour a dissociative SN1-type reaction pathway in substitution reactions (Scheme 4).25
 | ||
| Scheme 4 SN1-type dissociation of phosphanylphosphonium ion 14+. | ||
For phosphanylphosphonium ions such as those described above the term “ligand stabilized phosphenium ions” is frequently used in the literature while the described substitution reactions are also called “ligand exchange” reactions.1 Independent of any such controversy, however, these distinct points of view are based on the labile P–P bond observed in phosphanylphosphonium ions. This allows for the transfer of R2P+-moieties (formally phosphenium ions) between distinct Lewis bases (e.g. phosphanes, carbenes or P4). Thus, for reasons of simplification, phosphanylphosphonium ions will be regarded as “sources of phosphenium ions”1 throughout this review.
Phosphanylphosphonium ions were frequently used as phosphenium ion sources. The reaction of a mixture of Me2PCl and Me3SiOTf with diphosphane (Ph2P)2 gave diphosphanylphosphonium ion 16+ as a triflate salt (Fig. 4).26 Species 16+ is formally derived from the insertion of a Me2P+-phosphenium ion into the P–P bond of the diphosphane (Ph2P)2. Mixtures of Ph2PCl and Me3SiOTf with the cyclo-phosphanes (PhP)4 or (PhP)5 give in both cases the cyclo-tetraphosphanylphosphonium ion 17+.
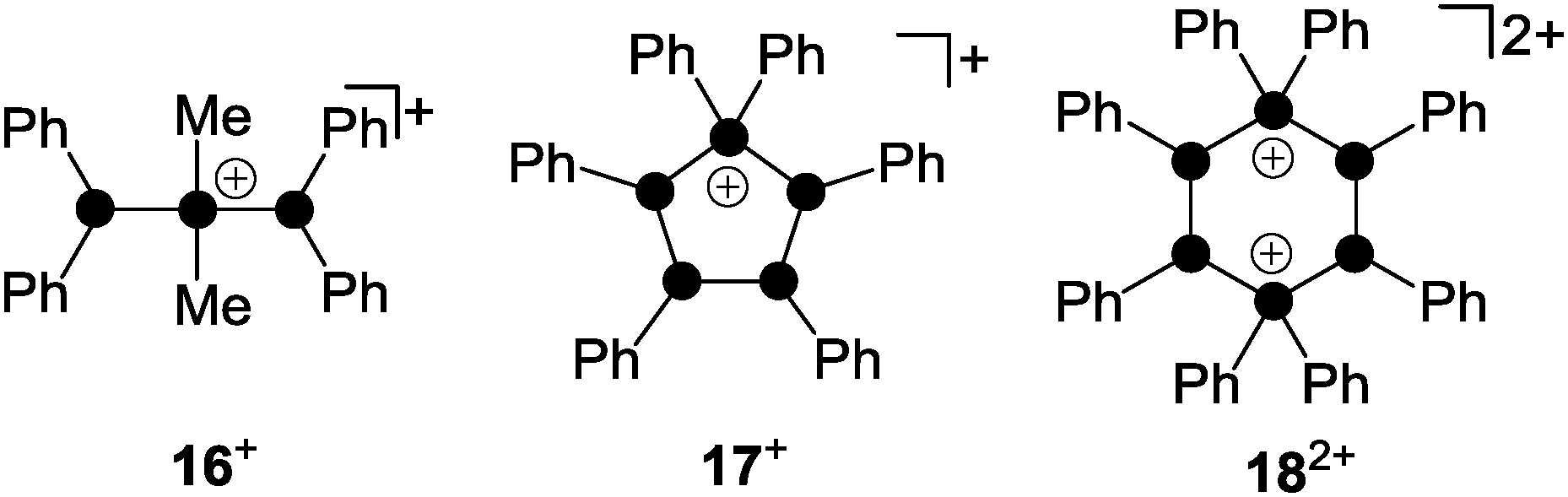 | ||
| Fig. 4 Polyphosphorus cations 16+, 17+ and 182+ obtained via the formal insertion of R2P+-phosphenium ions (R = Me, Ph) into the P–P bond of (Ph2P)2, (PhP)4 and (PhP)5. | ||
A ring expansion is observed in the reaction with (PhP)4 whereas a 5-membered ring is retained in the reaction involving (PhP)5via an unknown redistribution process.26 Both reactions proceed via the formal transfer of a Ph2P+-phosphenium ion from the intermediary formed phosphanylphosphonium ion 8+. In both cases 17+ is exclusively formed which demonstrates the thermodynamic preference of the five-membered ring over the six-membered alternative. The highly reactive, cyclic six-membered dication 182+ is only obtained by employing a melt approach.27 Solvent-free mixtures of Ph2PCl and GaCl3 provide room temperature molten media. These melts represent a powerful source of phosphenium ions Ph2P+.28
4. Cationic homoleptic polyphosphorus cages
For decades the investigation of homoleptic polyphosphorus cations was limited to mass spectroscopy29 and quantum chemical calculation30 in the gas phase. Homoleptic Pn+ cations are paramagnetic if the number of P atoms n is even. In the case of an odd number of P atoms the respective cation is diamagnetic. In general, the paramagnetic series of polyphosphorus cations is less stable. In the odd-membered series, the smaller Pn+ cations 19+ (n = 5) and 20+ (n = 7) may be described as electron-deficient Wade clusters whereas larger Pn+-cages (n ≥ 9) feature electron-precise Zintl-type structures. According to Wade's rules, a square pyramidal structure is anticipated for cation 19+ (Fig. 5, nido-cluster). Such a structure was confirmed as the most stable isomer by means of quantum chemical calculations.30a The structural motif of the second most stable isomer 19′+ (34.7 kcal mol−1 higher in energy) does not follow Wade's rules and shows a di-coordinated P atom. The most stable isomer of P7+-cage 20+ is a tricapped trigonal prism that is missing two of the capping vertices (arachno-cluster). A second isomer, which is only slightly higher in energy (20′+, 2.0 kcal mol−1), shows the P5-cage motif of 19′+ and a three-membered P ring which are both fused by a bridging phosphonium moiety. The P9+-cage 21+, which is composed of two P4-moieties fused by a phosphonium moiety, is one of the most stable homoleptic polyphosphorus cations according to quantum chemical calculations (Fig. 5).30b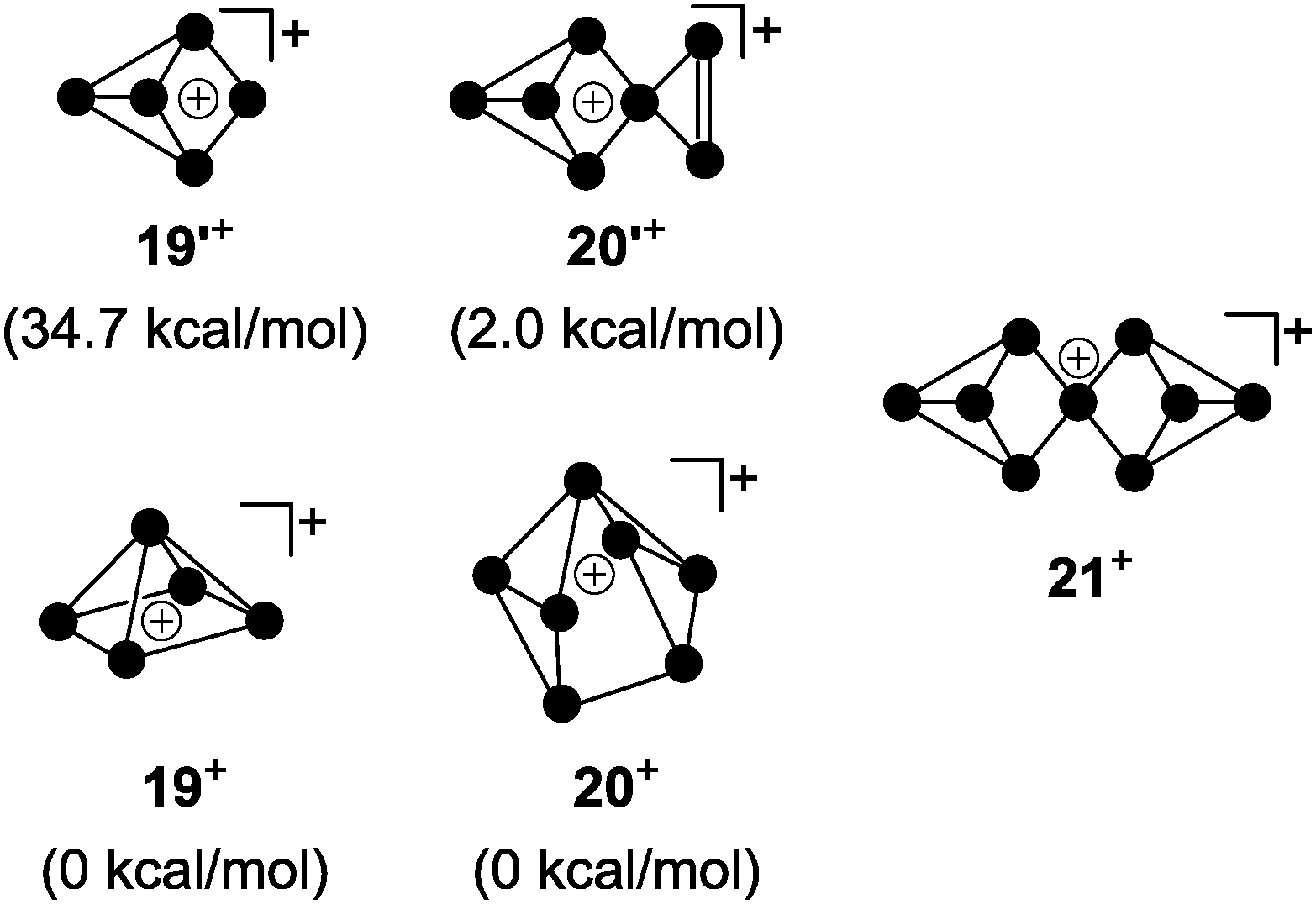 | ||
| Fig. 5 Anticipated structures of homoleptic, diamagnetic polyphosphorus cations 19+, 20+ and 21+. | ||
Krossing and co-workers were the first to report evidence for the existence of homoleptic polyphosphorus cations in the condensed phase.31 The attempted oxidation of P4 with I2 or Br2 in the presence of Ag(CH2Cl2)[A] (A = Al(OC(CF3)3)4) was suggested to proceed via the intermediary formation of P5+-cage cation 19+ (Scheme 5).32 However, cation 19+ is highly reactive and reacts with the solvent to give phosphonium ion 22+ as one of the main products. Cation 22+ forms via elimination of P4 and two-fold insertion into C–Cl bonds of CDCl3 molecules which was used as solvent. In the case of I2 as oxidant, P4 reacts partially to give PI3 which was suggested to react with intermediate 19+ to give P4 and the bis(phosphanyl)-substituted phosphonium ion 23+. Experimental evidence confirming the presence of 19+ in the reaction mixtures was not obtained; however, the suggested reaction pathways are in accordance with quantum chemical calculations.32 The nitrosonium salt [NO][A] (A = Al(OC(CF3)3)4) was also investigated as a possible one electron oxidant. However, the reaction of P4 with [NO][A] yields P4NO+-cage compound 24[A] via insertion of the nitrosonium cation into a P–P bond (Scheme 6).33
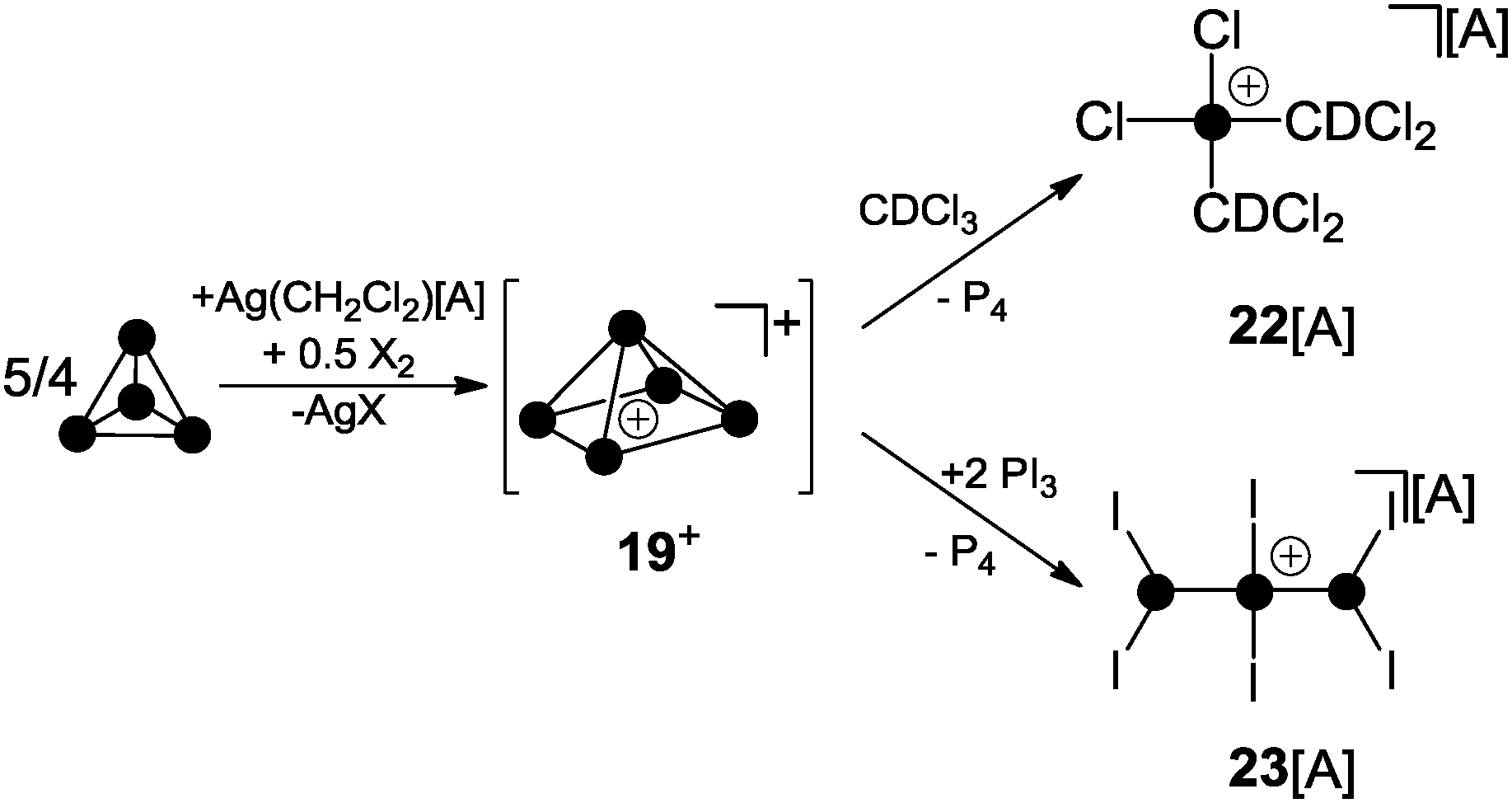 | ||
| Scheme 5 Oxidation of P4 with I2 or Br2 in the presence of Ag(CH2Cl2)[A] via P5+-cage intermediate 19+; A = Al(OC(CF3)3)4. | ||
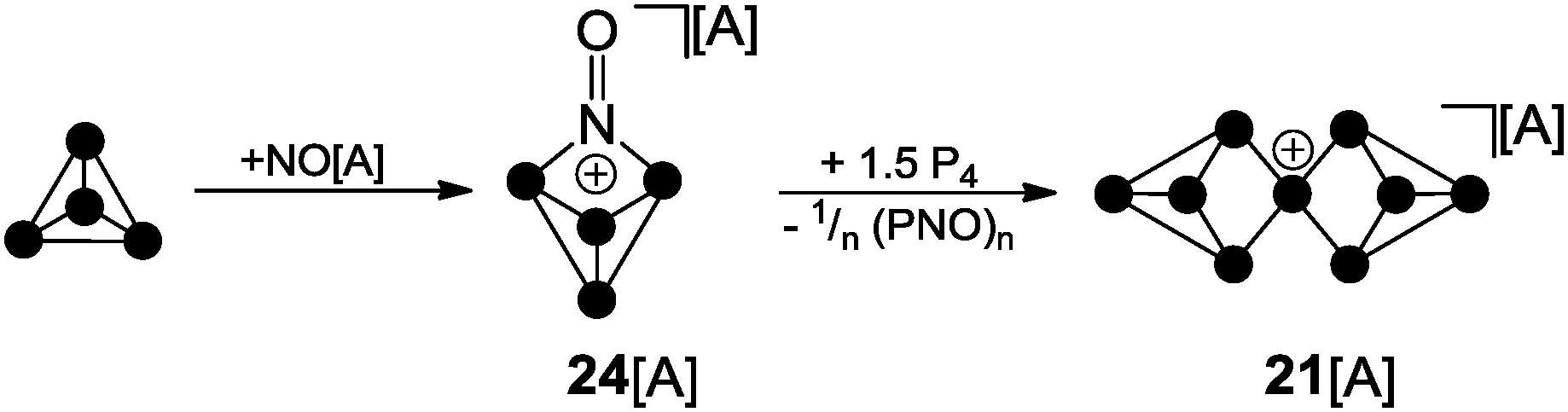 | ||
| Scheme 6 Reaction of P4 and NO[A] yielding P4NO+-cage 24[A] and subsequent reaction with P4 yielding P9+-cage compound 21[A]; A = Al(OC(CF3)3)4. | ||
Although X-ray structure determination of compound 24[A] was not successful, the molecular structure is confirmed by spectroscopic data and computational investigations. The theoretical investigations suggested a two-step mechanism indicating the HOMO of P4 and a π*-type LUMO at NO+ as the interacting frontier orbitals (II b, Fig. 1).33 Similar results were obtained utilizing the carborate salt NO[HCB11Cl11].34 The reaction of P4NO+-cage compound 24[A] with additional 1.5 equivalents of P4 was reported to yield P9+-cage compound 21[A] which is the first isolated salt of a homoleptic phosphorus cation (Scheme 6).35
The reaction proceeds very likely via extrusion of 1/n (PNO)n and intermediary formation of a P3+-species. The P9+ cation 21+ is obtained upon reaction of the latter with 1.5 equivalents of P4via an unknown reaction mechanism. The 31P NMR spectrum of cation 21+ shows a characteristic A2A′2BC2C′2 spin system which confirms the D2d symmetric Zintl-type structure. Despite the electron precise Lewis formula of eight neutral, three-coordinated and one cationic, four-coordinated P atom the charge is almost evenly distributed over all nine atoms according to quantum chemical calculations.35
5. Cationic polyphosphorus cages featuring halogen-substituents
The oxidation of Ag(I) complex 25[A] (A = Al(OC(CF3)3)4) featuring two intact P4 ligands with elemental iodine at low temperatures gives rise to interesting binary PI cations. The P5I2+-cage 26a+ was observed in the reaction mixture at −78 °C together with PI3 and P4 (Scheme 7).36 However, on raising the temperature above −40 °C, decomposition of 26a+ was observed, leading to the formation of P3I6+ (23+) and unidentified by-products. A proposed reaction mechanism indicates the partial oxidation of the P4 ligands in 25+ by I2 to give PI3.36 The latter reacts with Ag[A] (A = Al(OC(CF3)3)4) via halide abstraction to give AgI and formally the phosphenium ion PI2+. This highly reactive, predominantly electrophilic ambiphile reacts with white phosphorus via insertion in one of the P–P bonds of the P4 tetrahedron yielding the P5I2+-cage 26a+. Likewise, according to the observations described in Section 3, a mechanism involving the formation of phosphanylphosphonium ion P2I5+ can also be considered. Here, P2I5+ is assumed to transfer a PI2+ phosphenium ion to P4 and, thus, serves as a phosphenium ion source. Upon warming the reaction mixture, the excess of PI3 reacts with P4 to yield diphosphane P2I4 in a conproportionation reaction. The diphosphane reacts with 26a+via transfer of the phosphenium ion PI2+. This gives P4 and the P3I6+ cation 23+ which is formed upon insertion of the PI2+ ion into the P–P bond of P2I4. On the basis of these observations, a synthetic protocol for the targeted preparation of P5X2+-cages was developed (Scheme 7).36,37 Thus, white phosphorus reacts with PX3 (X = I, Br) in the presence of Ag(CH2Cl2)[A] as a halide abstracting agent and salts of cage cations 26a+ and 26b+ can be isolated in good yield. However, utilizing PCl3, the formation of the respective cation 26c+ was observed only in trace amounts since it readily decomposes in the reaction mixture.38 The molecular structure of 26b+ is shown in Scheme 7. The structural motif of the P5-core of the P5X2+-cage was unprecedented and was not previously observed as part of the many known polyphosphides and organo-polyphosphanes.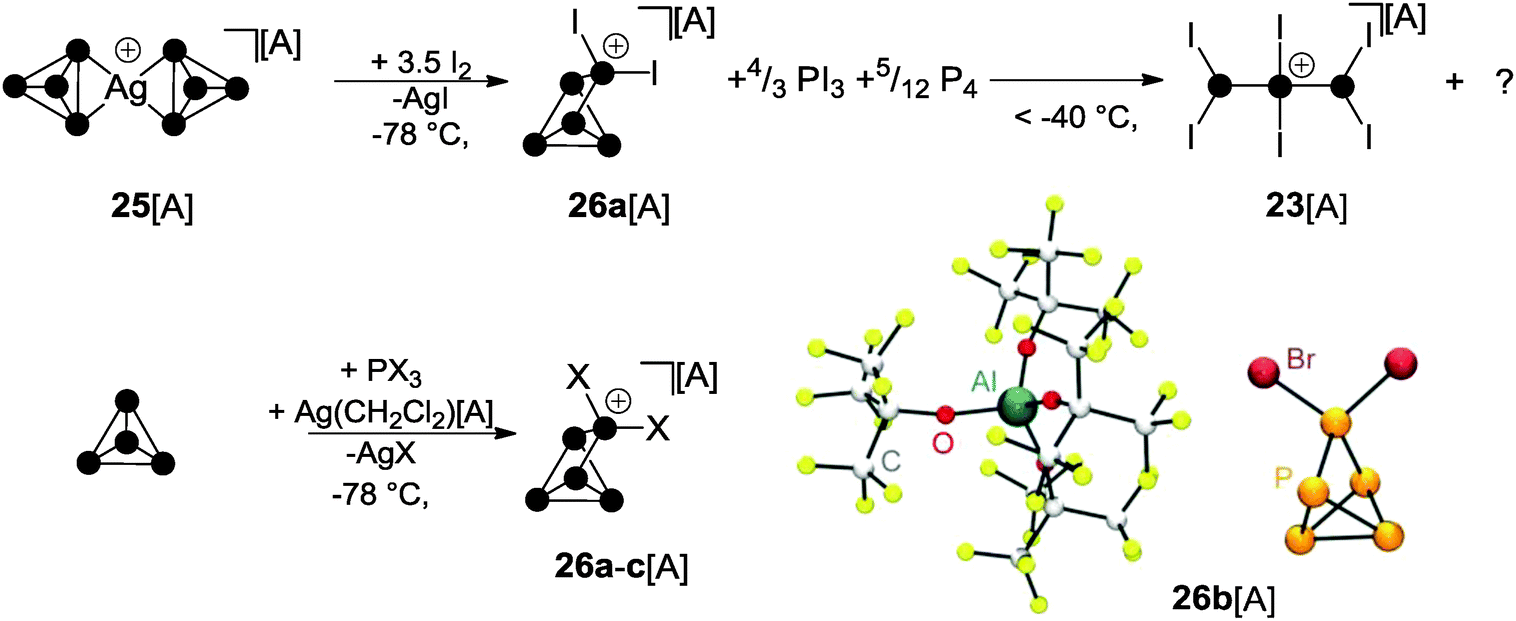 | ||
| Scheme 7 Oxidation of Ag(I) complex 25[A] with I2 at low temperatures giving intermediary P5I2+-cage 26a+ (top), targeted syntheses of P5X2+-cages 26a–c+ (X = I, Br, Cl) by the reaction of P4, PX3 and Ag(CH2Cl2)[A] and molecular structure of 26b[A] (bottom); A = Al(OC(CF3)3)4. | ||
6. Cationic polyphosphorus cages featuring alkyl- and aryl-groups
A versatile approach to cationic polyphosphorus cages featuring alkyl- and aryl-groups represents the utilization of dichlorophosphanes RPCl2 (R = alkyl, aryl) instead of PX3 (X = I, Br, Cl).39 Mixtures of dichlorophosphanes RPCl2 and a strong Lewis acid (GaCl3, AlCl3) as a halide abstracting reagent can be utilized as the source for the phosphenium ion RPCl+. In the presence of P4, insertion into one of the P–P bonds takes place, giving access to a series of RP5Cl+-cages featuring distinct substituents R.39 Mixtures of dichlorophosphanes and AlCl3 were previously utilized for the in situ formation of phosphenium ion salts [RPCl][AlCl4] and subsequent syntheses of various phosphorus heterocycles.40 However, neither free phosphenium ions nor respective phosphenium ion sources could be verified. In some cases, the formation of Lewis acid–base complexes of the type mRPCl2·nAlCl3 (n = 1, 2; m = 1, 2) was suggested.41 Detailed investigations of mixtures of mono- and dichlorophosphanes in the presence of Lewis acids revealed the formation of chlorophosphanylchlorophosphonium ions of type 27+ (Fig. 6).42 In most cases, characteristic 1J(PP) coupling constants were observed by 31P NMR spectroscopy at ambient temperature. | ||
| Fig. 6 Phosphanylphosphonium ion derivatives 27+–29+ and Lewis acid–base adduct 30 (classical) and 30′ (non-classical); R = alkyl, aryl. | ||
However, the spectra of mixtures of dichlorophosphanes and Lewis acids in CH2Cl2 were less informative and showed in most cases only broad resonances.42 A systematic study based on Raman and 31P NMR spectroscopy of mixtures of RPCl2 and GaCl3 in fluorobenzene applying varying stoichiometries gave important insight into these reactions.39 Depending on the ratio of the reactants and the substituent R in RPCl2, mixtures of the structurally distinct species 28+, 29+ and 30 were formed (Fig. 6).
The classical Lewis acid–base adducts of type 30 are only formed in reaction mixtures involving dichlorophosphanes RPCl2 featuring alkyl-substituents R. The formation of non-classical adducts of type 30′ is not observed and is unlikely according to quantum chemical calculations.39 This is further supported by the isolation and structural characterization of 30a (R = t-Bu), which was proven to form a classical Lewis acid–base adduct. An increasing amount of phosphanylphosphonium ions of type 28+ is formed with decreasing steric demand of the substituent R (t-Bu > Cy > i-Pr). The formation of cations of type 29+ is observed when the basicity and the steric requirements of the dichlorophosphanes are further reduced (R = Et, Me, Ph). Such cations are the result of adduct formation between GaCl3 and the phosphane moiety of phosphanylphosphonium ions of type 28+. Most mixtures show dynamic exchange indicating a possible interconversion of species 28+, 29+ and 30.39 The exchange rates of these processes strongly depends on the concentration of GaCl3. In the reaction mixtures equilibrium dissociation of the GaCl4− anion to free GaCl3 and Cl− occurs. The dynamic exchange is linked to these chloride anions which nucleophilically attack phosphanylphosphonium species yielding the phosphane starting materials in a back reaction. By using an excess of GaCl3 the GaCl4− forms higher gallates (Ga2Cl7− or Ga3Cl10−) and the concentration of free chloride anions is reduced.43
Quantum chemical calculations were carried out to determine which of the observed species serves as the phosphenium ion source in a reaction with P4. According to these results,44 the formation of RP5Cl+-cages via a free phosphenium ion RPCl+ can be excluded. Attempts to calculate a feasible reaction mechanism from adducts 30 or 30′ as sources of phosphenium ions were not successful. Thus, the reaction of P4 with methyl-substituted phosphanylphosphonium derivative 28e+ was investigated (Fig. 7). A single step insertion of the phosphenium moiety into a P–P bond of the P4 tetrahedron is viable and the calculated energy profile of the reaction path is denoted in black. In addition, a two-step reaction pathway is feasible as well (energy profile is shown in red).
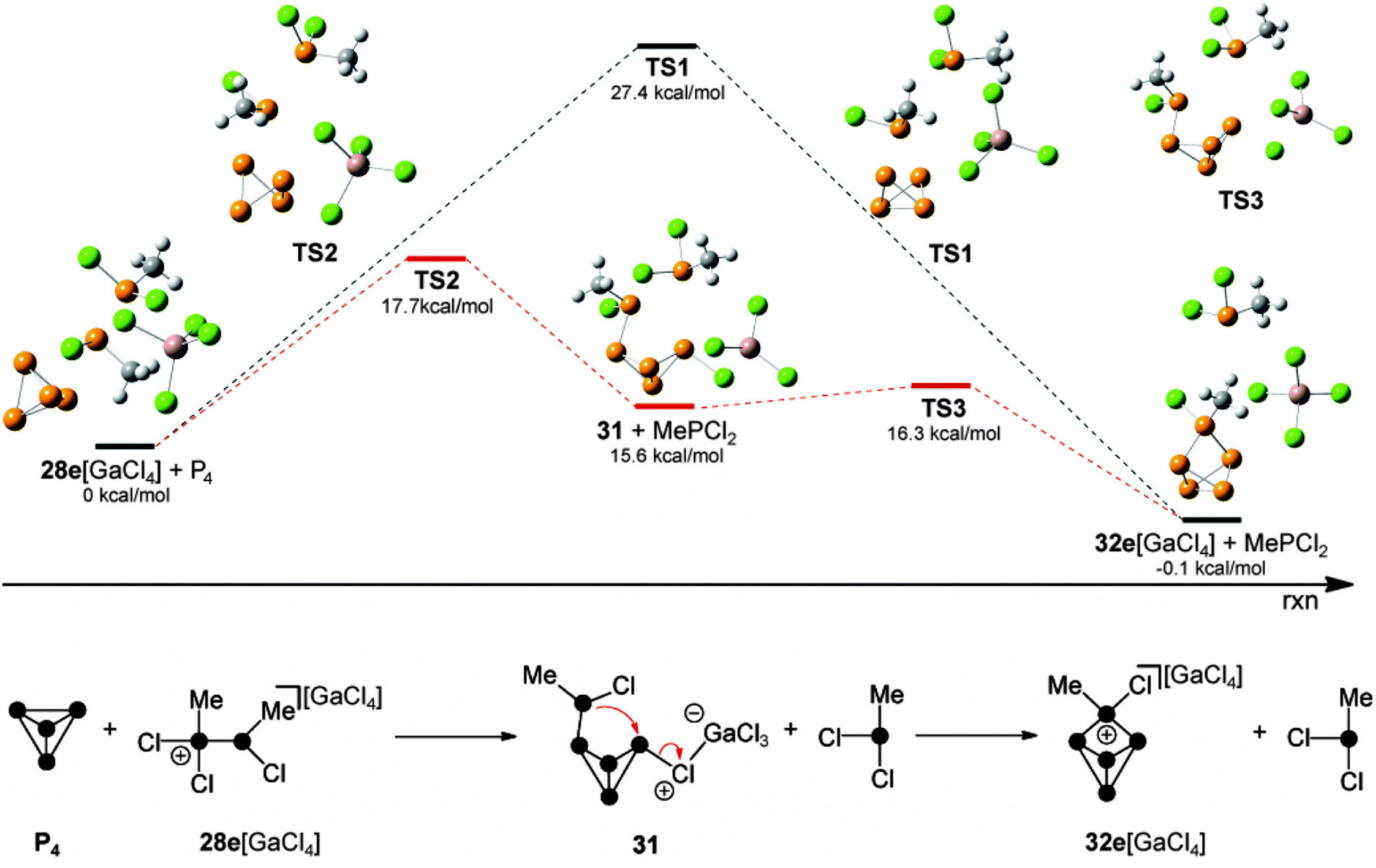 | ||
| Fig. 7 Calculated reaction pathways for the reaction of 28e[GaCl4] and P4; calculated differences of the enthalpies at 298.15 K (ΔH298) are given for the optimized structures of MP2/6-31G(d) and the optimized structures of 28e[GaCl4] + P4 were defined as 0 kcal mol−1. | ||
The two step reaction pathway proceeds via butterfly-type compound 31 as an intermediate (bottom, Fig. 7). The single step transfer of the phosphenium moiety in 28e[GaCl4] and insertion thereof into a P–P bond of P4 shows an energy barrier of 27.4 kcal mol−1 (TS1) and is energetically viable. In the light of recent mechanistic studies on the reaction of isoelectronic silylenes with P4,7a this is best understood as a combined electrophilic and nucleophilic attack of the phosphenium moiety. On the one hand the P–P bond of P4 (HOMO) nucleophilically attacks the p-type orbital of the phosphenium moiety. On the other hand the lone pair of electrons of the phosphenium moiety donates electron density to the LUMO of the P4 tetrahedron which corresponds to p-orbitals situated perpendicular to the P4 lone pairs.7 It was found that a lower barrier reaction pathway is possible if 28e+ does not act as a nucleophile. Instead, a chloro-substituent of the GaCl4− anion nucleophilically attacks the P4 tetrahedron along with the electrophilic attack of the phosphenium moiety of 28e+ on P4. This leads to the slightly endothermic formation of the intermediate 31 (15.6 kcal mol−1) via transition state TS2 (17.7 kcal mol−1). Compound 31 reveals a butterfly-type structure featuring a chloro-substituent in an exo-position and a phosphanyl-substituent in an endo-position. Finally, 31 reacts via TS3 (16.3 kcal mol−1), which shows only a very low energy barrier. This step proceeds via the intramolecular nucleophilic attack of the phosphanyl-substituent on the chloro-substituted P atom. This eliminates the GaCl4− anion and leads to the formation of the P5+-cage cation 32e+.
Despite their different compositions 1:1 mixtures of RPCl2 and a Lewis acid ECl3 (E = Al, Ga) in fluorobenzene are potent sources of reactive phosphenium ion RPCl+ equivalents, which insert formally into P–P bonds of P4.39 Dissolution of P4 in these mixtures yielded white to yellowish precipitates of the corresponding RP5Cl+-cage salts for a large range of different alkyl- and aryl-substituents R (32a–h[GaCl4], Scheme 8).
 | ||
| Scheme 8 Preparation of compounds 32a–h[ECl4] from P4, RPCl2 and ECl3 (E = Ga, Al; R = alkyl, aryl) in fluorobenzene. | ||
All compounds are obtained in almost quantitative yield and high purity. In contrast to the halogen-substituted species 26a–c[A], they are stable in the solid state or when dissolved in non-coordinating solvents at ambient temperature.36,37 The cations 32a–h+ show characteristic 31P NMR spectra. Iterative line shape analysis of the observed spin systems gave chemical shifts and coupling constants in accordance with CS symmetric RP5Cl+-cages with four chemically non-equivalent phosphorus nuclei. All cages possess a mirror plane which includes the tetra-coordinated P atom and both P atoms opposing the former. Due to the reduced symmetry compared to the C2V-symmetric P5X2+-cages 26a–c+ an ABM2X spin system is observed for 32a–d+ and an ABMX2 spin system for 32e–h+. Due to the similar geometry of the P5+-core in all cations, the respective 1J(PP) and 2J(PP) coupling constants deviate only marginally. However, the chemical shifts are strongly dependent on the substituent R attached to the RP5Cl+-cage (Fig. 8). The PA and PB atoms exhibit characteristic low field resonances at approximately −275 ppm. The assignment of the A and B part to the respective P nuclei is based on the observed coupling pattern. First, the non-symmetrically substituted P5+-cage is divided by a plane spanned by the tetra-coordinated and both adjacent P atoms into a HCl- and HR-hemisphere (Fig. 9).
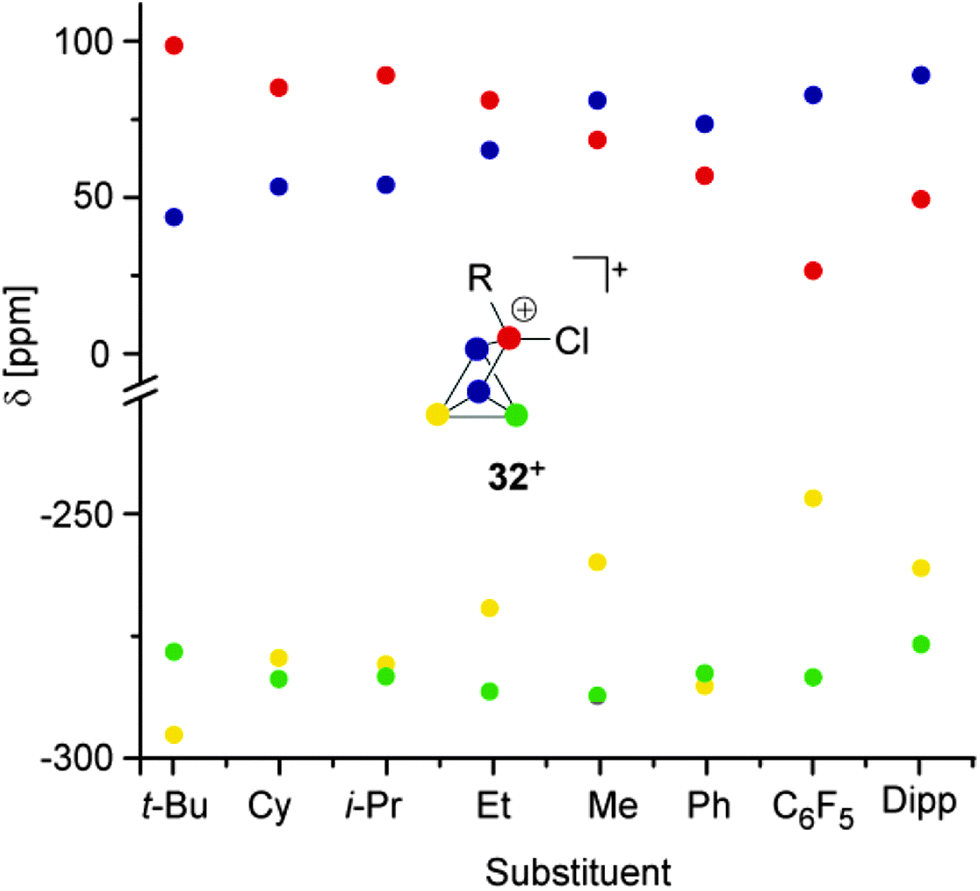 | ||
| Fig. 8 Plot of the 31P NMR chemical shifts of RP5Cl+-cages 32a-h+versus their alkyl- or aryl-substituent. | ||
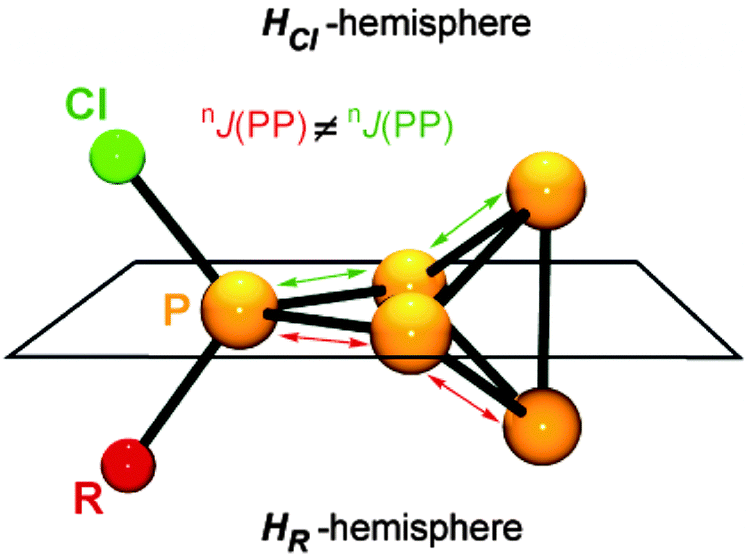 | ||
| Fig. 9 Definition of the HCl- and HR-hemisphere in cations 32a–h+. The tetra-coordinated and the adjacent P atoms span a plane. The tri-coordinated P atom above the plane is within the HCl-hemisphere, and the P atom below the plane is in the HR-hemisphere. | ||
The HCl-hemisphere contains the chloro-substituent and the HR-hemisphere the alkyl- or aryl-substituent. Within the series of cations 32a–h+ the P atom located in the HCl-hemisphere shows values of 1J(PP) and 2J(PP) coupling constants which are reminiscent of those of P5X2+-cages 26a–c+.36,37 Accordingly, the P atom located in the HR-hemisphere reveals one- and two-bond P–P coupling constants similar to the values observed for the respective R2P5+-cages. In addition, the former group of P atoms experiences the spatial proximity of the chloro-group, and, therefore, shows similar chemical shifts (marked in green, Fig. 8). In contrast, the P atoms in the HR-hemisphere show resonances in a much larger chemical shift range. This is attributed to the distinct electronic parameters of the substituents. They affect the chemical shifts of the P atoms most likely through “cross-ring through space” interactions of the lone pairs on P atoms and the respective group R.45 For RP5Cl+-cages featuring alkyl-substituents R (32a–e+) the resonances of the P atoms adjacent to the phosphonium moiety (marked in blue, Fig. 8) are shifted stepwise to lower field with a decreasing positive inductive effect of the substituent (from 44 ppm (32a+) to 81 ppm (32e+)). This is in agreement with the increased shielding of a P atom caused by additional alkyl-moieties in the γ-position relative to the P nuclei. This trend was previously termed γ-effect.46 In contrast, the chemical shifts of tetra-coordinated P atoms (marked in red, Fig. 8) exhibit an almost inverse trend (from 99 ppm (32a+) to 69 ppm (32e+)). This high-field shift correlates with an increasing number of hydrogen atoms at the α-carbon atom of the substituent. This constitutes a characteristic feature of phosphonium moieties and was termed a-effect.47 Overall, these influences are reflected in a change of the spin system between 32e–h+ featuring aryl- and methyl-substituents (ABMX2 spin system) and those bearing alkyl-substituents 32a–d+ (ABM2X spin system).
Employing dichlorophosphanes R2NPCl2 (R = Cy, i-Pr) in combination with GaCl3 in reactions with P4 gave distinct results. In mixtures of R2NPCl2 (R = Cy, i-Pr) and GaCl3 the corresponding phosphenium ions 33a,b+ are the only observable species.20a Indicative of their formation is a resonance in the 31P NMR which is shifted to remarkable low field.18 It is highly influenced by the nature of the respective anion (compare 33a[GaCl4]: δ = 310 ppm, 33a[Ga2Cl7]: δ = 350 ppm). The GaCl4− salt of 33a+ can be isolated and constitutes a rare example of a structurally characterized mono-amino substituted phosphenium ion (Scheme 9). Upon reacting phosphenium ions 33a,b+ with P4 insertion into a P–P bond is observed giving the CS-symmetric RP5Cl+-cage cations 34a,b+. However, these cages are in equilibrium with the respective free phosphenium ions and P4 which hampers the isolation of pure compounds 34a,b[GaCl4]. The observation of an equilibrium can be attributed to the relative stability of free 33a,b+. A similar reversibility of the phosphenium ion insertion was observed in the case of RP5Cl+ compounds. The addition of coordinating solvents like acetonitrile to solutions of 32[ECl4] (E = Ga, Al) decomposes the respective metallate anion via chloride liberation. Nucleophilic attack of free chloride anions on 32+ yields mainly the starting materials P4 and RPCl2 (R = alkyl, aryl) in a back reaction.
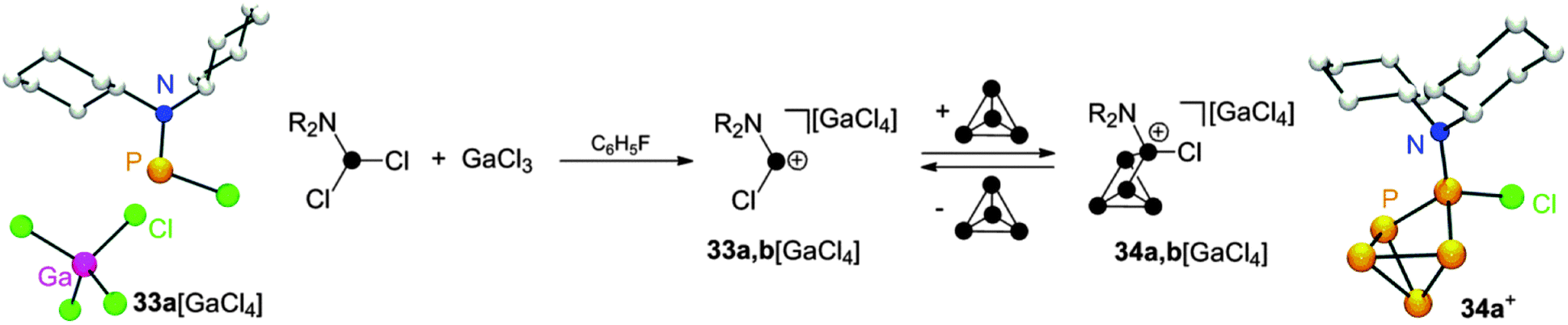 | ||
| Scheme 9 Reaction of R2NPCl2 (R = Cy, i-Pr) with GaCl3 and P4 and equilibrium of 34a,b+ with 33a,b+ and P4 (middle) and molecular structure of 33a[GaCl4] (left) and 34a+ (right). | ||
It is interesting to note that a reaction between the two-fold amino-substituted phosphenium ion [(i-Pr2N)2P]+ (35+) and P4 was not observed.20a This is attributed to a significantly lowered electrophilicity of 35+ compared to 33a,b+.19 Also, diamino-phosphenium ions of type 35+ reveal frontier orbitals comparable to those of allyl-anions48 with the HOMO mainly located at the N atoms, and, thus, are not ambiphilic at the P moiety.
R2P5[GaCl4] cage compounds 36[GaCl4] featuring two alkyl- or aryl-substituents R are obtained in high yield via the reaction of chlorophosphanes R2PCl, GaCl3 and P4 (Scheme 10).49
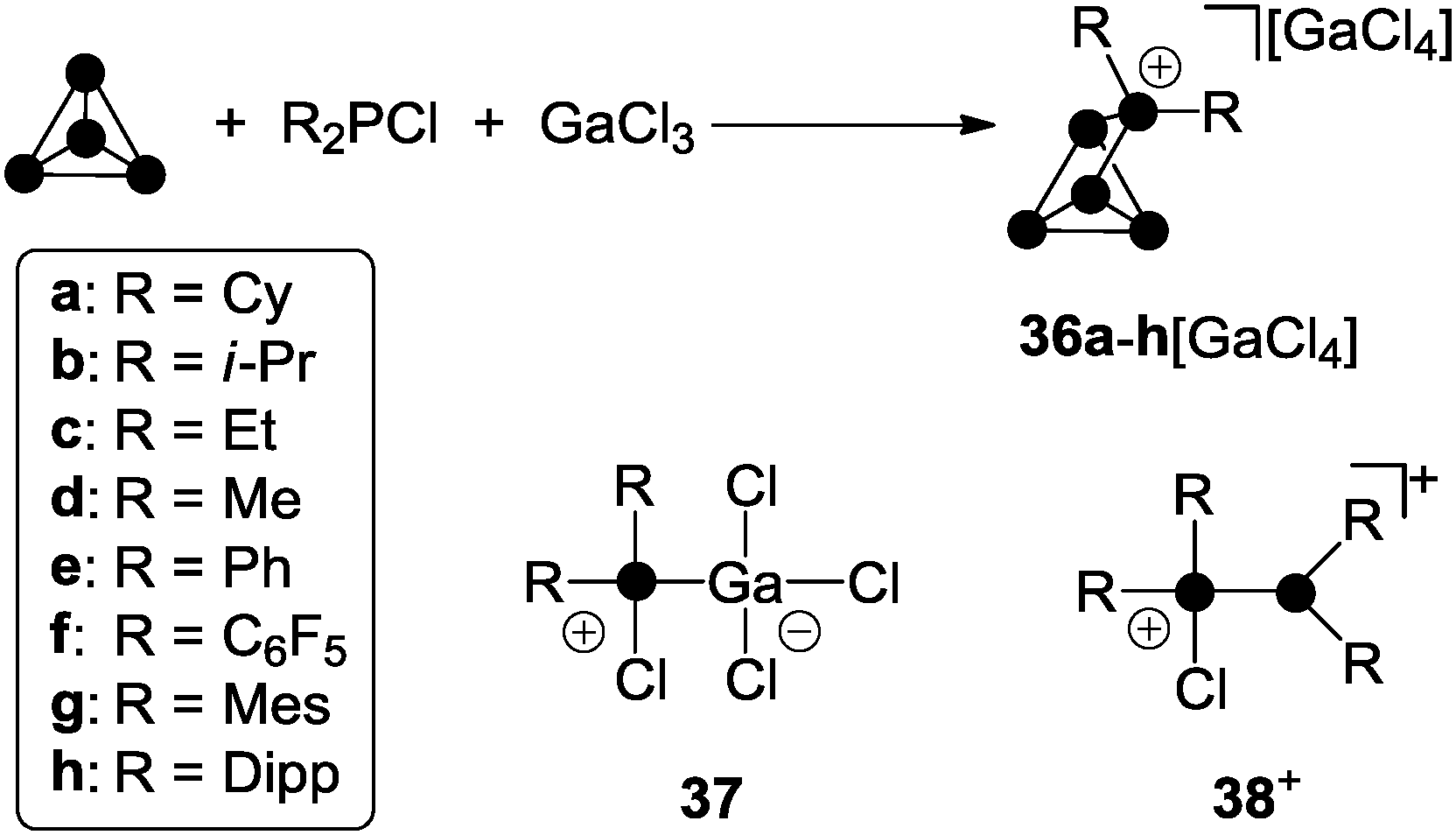 | ||
| Scheme 10 Preparation of compounds 36a–h[GaCl4] from P4, R2PCl and GaCl3 in fluorobenzene (R = aryl) or according to a solvent free approach (R = alkyl) and species 37 and 38+ commonly observed in mixtures of R2PCl and GaCl3. | ||
The Lewis acid–base adduct 37 and phosphanylphosphonium ion 38+ are commonly formed in mixtures of chlorophosphanes and GaCl3 in various stoichiometries.22b Both convert into each other via equilibria involving free R2PCl and GaCl3.49,22b Cations of type 38+ serve as phosphenium ion sources in the presence of P4 allowing for the formation of R2P5+-cage cations 36+. Most likely, this proceeds in analogy to quantum chemical calculations on the mechanism of the formation of MeP5Cl+ cage 32e+.39 In contrast to dichlorophosphanes, however, the reaction conditions for the formation of R2P5+-cages 36a–h+ depend strongly on the substituent R. In the case of chlorophosphanes featuring aryl substituents R, the reactions proceeds smoothly at ambient temperature in fluorobenzene solution. A significant decrease in reaction time is observed with increasing steric bulk of the substituents (Dipp > Mes > C6F5 > Ph). For the preparation of R2P5+-cages 36a–d+ featuring alkyl-substituents R, solvent-free conditions are necessary. Mixtures of R2PCl (R = Cy, i-Pr, Et, Me) and GaCl3 in a 1:1 stoichiometry form melts at ambient temperature.28 Upon addition of P4 to these melts, the formation of the corresponding cage compounds 36a–d[GaCl4] is observed. With increasing steric demand of the substituents R (Cy > i-Pr > Et > Me) extended reaction times and higher temperatures (100 °C to 150 °C) are required. The different reactivity of alkyl- and aryl-substituted phosphanes in the synthesis of R2P5-cage compounds of type 36[GaCl4] can be rationalized in terms of the different Lewis acidities of the corresponding phosphenium ions. The Lewis acidity is reflected e.g. by their distinct fluoride ion affinities (e.g. Me2P+: FIA = 959 kJ mol−1, Ph2P+: FIA = 838 kJ mol−1).19,50 This necessitates a more Lewis acidic environment for the transfer of a phosphenium ion featuring alkyl-groups, which is realized in a solvent free medium.
The molecular structures of all compounds of the series 36a–h[GaCl4] were determined by single crystal X-ray structure determination. This allowed for an in-depth evaluation of the influence of substituents of distinct steric demand on the structural parameters of the P5-cage in the solid state (Fig. 10).
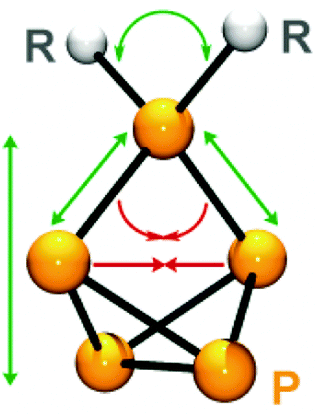 | ||
| Fig. 10 Influence of the steric demand of substituents R on the structural parameters of the P5+-cage core in cations of type 36+; upon increasing the steric demand of R green arrows indicate increasing angles and distances and red arrows indicate declining angles and distances. | ||
The phosphonium P atoms of cations of type 36+ show a distorted tetrahedral environment. If the steric demand of the substituent R is increased a stepwise increase in the corresponding C–P–C angle is observed from the sterically very bulky substituted Dipp2P5+ (36h+) to the methyl-substituted derivative 36e+. This is accompanied by a decreasing P–P–P angle at the phosphonium moiety and stepwise increase in P–P bond lengths involving the phosphonium P atom.
As a consequence, the tetraphosphabicyclo[1.1.0]butane moieties display a more pronounced folding (distance between both P atoms adjacent to the phosphonium P atom decreases) and the P5+-cages are stretched (distance between the bridgehead P–P bond and the phosphonium P atom increases).
The 31P NMR spectra of cage cations 36+ show A2M2X or A2MX2 spin systems in accordance with their C2V symmetry and are comparable to those observed for the P5X2+ cages 26a–c+ (Fig. 11). The observation of two different spin systems for R2P5+-cages of type 36+ may be explained in terms of different steric and electronic influences of the alkyl- or aryl-substituent R. In the series of alkyl-substituted R2P5+-cages (36a+ to 36d+) the resonances of the phosphonium P atoms are shifted to higher field and the resonances of the adjacent P atoms are shifted to lower field. This can be explained in terms of a combination of a-effect and γ-effect (vide infra).46,47 The resonances of the tetra-coordinated P atoms in aryl-substituted cations 36e–h+ are shifted to higher field compared to those of the corresponding P atoms in cages 36a–d+. This is due to a positive mesomeric effect, namely the donation of π-electron density from the aryl substituents to the lobes of the anti-bonding σ*(P–P) orbitals at the phosphonium moiety.47a Some main group centered, predominantly electrophilic ambiphiles react with P4via multiple insertions into P–P bonds of the P4 tetrahedron. This is exemplified by SiP4-cage compound 40, which is obtained by the reaction of P4 with zwitterionic silylene 39. This compound reacts with a second equivalent of 39 to give the Si2P4-cage compound 41 (Scheme 11).14 The second insertion takes place at a P–P bond opposing the initially inserted main group element. The related product 4 was obtained by the reaction of P4 with a low valent Al(I) species (Fig. 2).13
 | ||
| Fig. 11 Plot of the 31P NMR chemical shifts of R2P5+-cages 36a–h+versus their alkyl- or aryl-substituent. | ||
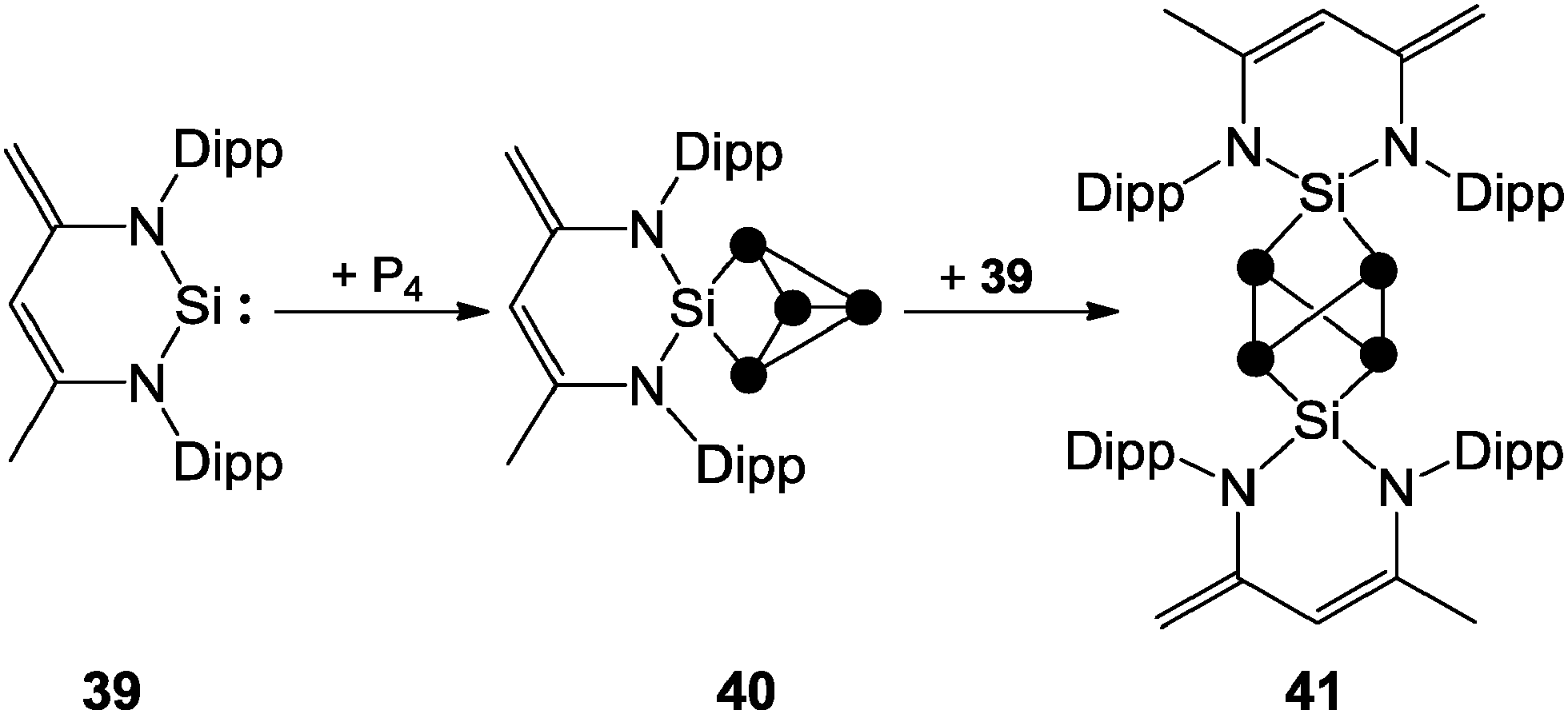 | ||
| Scheme 11 Stepwise insertion of zwitterionic silylene 39 into P–P bonds of P4 yielding SiP4-cage compound 40 and Si2P4-cage compound 41. | ||
Distinct results were obtained in the investigation of multiple insertions of phosphenium ions into P–P bonds of P4. In this context, solvent-free mixtures of P4, Ph2PCl and GaCl3 in various stoichiometries and at different temperatures were investigated. A 1:1:1 mixture yields quantitatively the Ph2P5+-cage compound 36f[GaCl4] after 45 min at 60 °C (Scheme 12).51
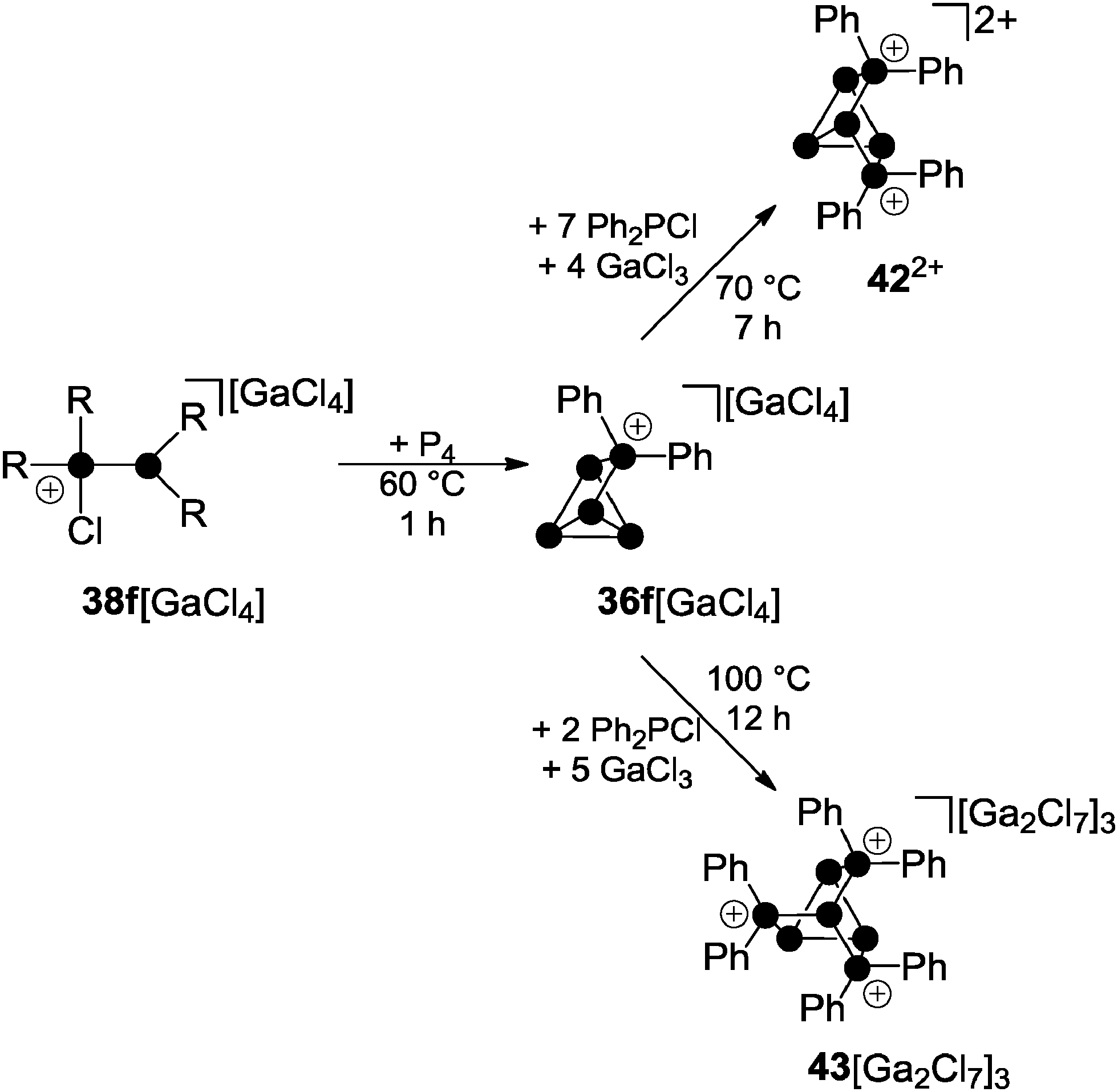 | ||
| Scheme 12 Stepwise insertion of Ph2P+-phosphenium ions into P–P bonds of P4 yielding Ph4P62+-cage cation 422+ and Ph3P73+-cage compound 43[Ga2Cl7]3. | ||
The Ph4P62+-cage cation 422+ is observed in a mixture of 1:8:5 stoichiometry (P4:Ph2PCl:GaCl3) as the main product after a reaction time of seven hours at 70 °C. The 31P NMR spectrum of 422+ shows a characteristic ABMM′XX′ spin system which is in accordance with the insertion of a second Ph2P+-phosphenium ion into a P–P bond adjacent to the phosphonium moiety in 36f+. Two second-order resonances corresponding to an AA′XX′X′′X′′′ spin system are expected for the isomer of 422+ formed via formal insertion into two opposing P–P bonds of P4. Such a species is not formed in the melt reaction. The formation of dication 422+ can only be observed if the ratio of Ph2PCl and GaCl3 is higher than 0.5. In these mixtures, the dominant gallium species is GaCl4−; hence, the melt can be considered as basic medium. In a more Lewis acidic melt, composed of P4, Ph2PCl and GaCl3 in a 1:3:6 stoichiometry, the tricationic Ph6P73+-cage 433+ is formed exclusively. Large single crystals of 433+ as a heptachlorodigallate salt are formed in the respective melt after 12 h at 100 °C. Cation 433+ features a nortricyclane-type (tricyclo[2.2.1.02.6]heptane) framework. It is composed of a basal ring of three-coordinated P atoms, three tetra-coordinated P atoms at the bridging positions and a three-coordinated P atom at the apex of the cage. This skeleton is reminiscent of the trianionic phosphide P73−,52 several polyphosphanes R3P753 and many polyphosphorus-chalcogenides like e.g. P4S3.54 The 31P NMR spectrum of 433+ shows an AA′A′′BXX′X′′ spin system resulting from the C3 symmetry of the cage. A 2J or 3J P–P bond coupling to the apex of the cage is not observed which might be a result of the adjacent phosphonium P atoms. This leads to a first-order quartet resonance for the apical P atom. The highly electrophilic cation 433+ is stable only in the presence of excess GaCl3. This prevents the detrimental presence of chloride anions which decompose 433+ by nucleophilic attack and subsequent degradation via422+ to 36f+. This illustrates that the consecutive insertion of up to three Ph2P+-moieties into P–P bonds of P4 is directed by the Lewis acidity of the reaction mixture.
7. Cationic polyphosphorus cages featuring four-membered heterocycles
Cyclic diaminohalophosphanes are important precursors for the preparation of cyclic phosphenium ions via halide abstraction.55 Within this class of compounds, phosphazanes, like the diphosphadiazane 44, are of particular interest (Scheme 13). These compounds feature two chloro-substituted P moieties and, thus, offer a versatile reactivity.56 The diphosphadiazenium ion 45+ is generated from 44 upon chloride abstraction with GaCl3. Solutions of 45+ are characterized by a bright red colour and the 31P NMR spectrum shows a broad resonance at characteristic low field (δ = 242.3 ppm) indicating the formation of a di-coordinated P moiety. Subsequent addition of P4 to this solution leads to discolouration and quantitative formation of the P5+-cage compound 46[GaCl4].57 The molecular structure of cation 46+ shows a planar four-membered (NP)2 ring and an almost orthogonal oriented P–Cl bond (Scheme 13). This arrangement is also reflected by the A2MVXZ spin system observed in the 31P NMR spectrum of CS-symmetric cation 46+. Interestingly, the P5+-cage does not couple with the chloro-substituted P atom resulting in the observation of a singlet resonance for the latter. This P–Cl functionality was used for the in situ generation of a phosphenium ion upon addition of three equivalents of GaCl3 to the reaction mixture. The resulting dicationic intermediate was not detected. However, upon addition of P4, the formation of the corresponding insertion product 472+ is observed. The 31P NMR spectrum of 472+ shows an A2MX2 spin system which is consistent with two C2V-symmtric P5+-cages bridged by two imido-groups. The dication can be isolated as heptachlorodigallate salt 47[Ga2Cl7]2 and the molecular structure of the N2P10-cage was confirmed by single crystal structure determination (Scheme 13). This illustrates that the stepwise insertion of the disguised bifunctional Lewis acid [DippNP]22+ into P–P bonds of two P4 tetrahedra can be mediated by the Lewis acidity of the reaction mixture. The utilization of an excess of GaCl3 allows for the preparation of the more electrophilic, higher charged species 472+, similar to the reaction sequence yielding 433+ (Scheme 12).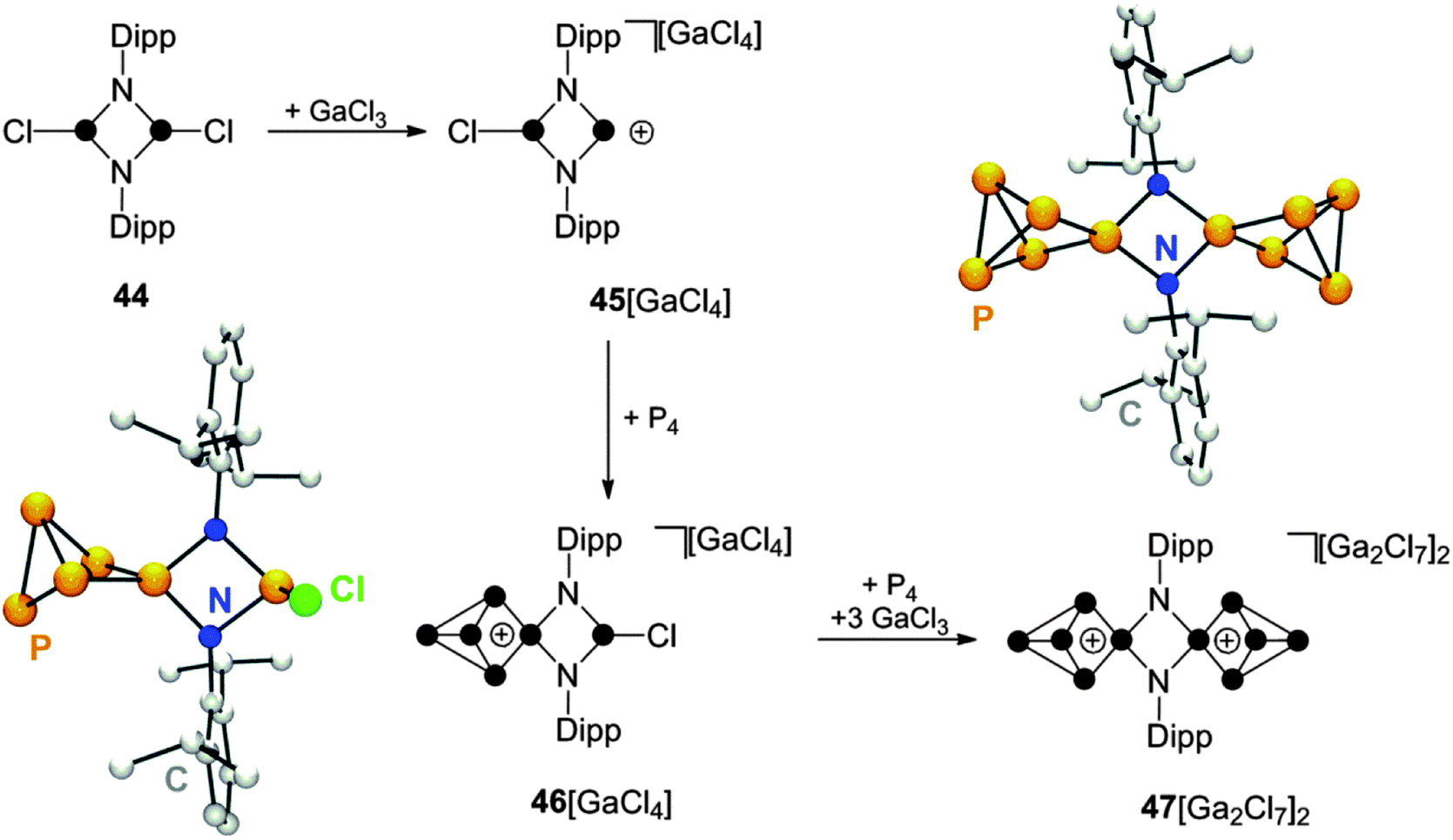 | ||
| Scheme 13 Stepwise synthesis of N2P10-cage compound 47[Ga2Cl7]2via insertion of phosphenium ions generated in situ by the reaction of diphosphadiazane 44 with GaCl3. | ||
It is interesting to note that related NHC analogues, five-membered 1,3,2-diazaphospholenium ions, do not react with P4 under various reaction conditions58 similar to acyclic, diamino-phosphenium ion (i-Pr2N)2P+ (vide infra). This indicates that the strained four-membered ring geometry present in diphosphadiazenium ions is crucial for its reactivity towards P4.
Other cyclic, four-membered phosphorus containing heterocycles can be employed in reactions with P4 as well.59 The cyclic chlorophosphane 48, featuring a SiCl2-backbone,60 reacts with GaCl3 to give the corresponding Lewis acid–base adduct 49 (Scheme 14). The formation of related phosphenium ion 50+ is observed only upon addition of a second equivalent of GaCl3. This can be explained by the suppression of detrimental concentrations of nucleophilic chloride anions through the formation of Ga2Cl7−.
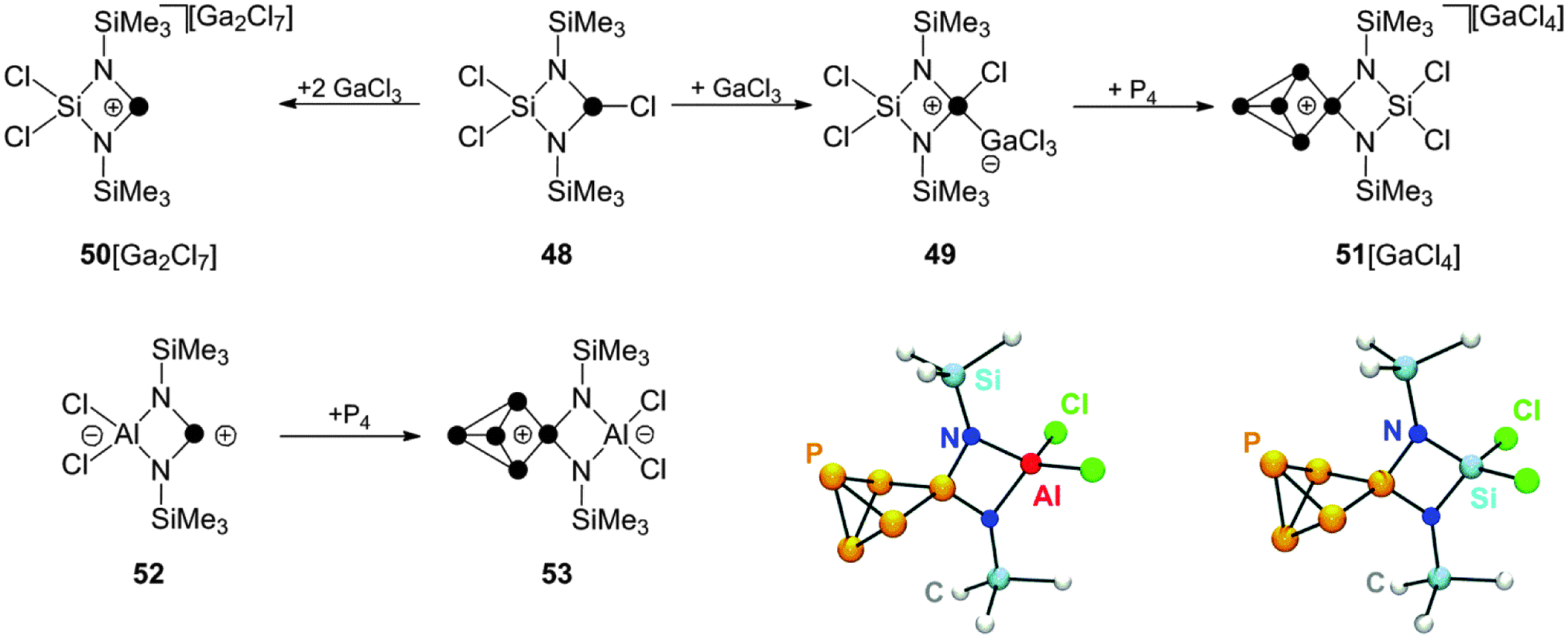 | ||
| Scheme 14 Preparation of P5+-cage cation 51+ from P4, GaCl3 and chlorophosphane 48 (top) and preparation of zwitterionic P5-cage compound 53 from P4 and zwitterionic phosphenium ion 52. | ||
Cation 50+ is not stable in solution and decomposes via Lewis acid mediated Me3SiCl elimination. However, the insertion reaction with P4 requires only the use of one equivalent of GaCl3. In 1:1:1 mixtures of 48, GaCl3 and P4 the corresponding P5+-cage compound 51[GaCl4] is formed slowly within four days presumably due to the presence of small amounts of 50+ formed from 49 in a series of equilibrium reactions.59 The related zwitterionic phosphenium ion 52 features a formally anionic AlCl2-backbone.60 It reacts with P4 in toluene giving the formally neutral P5-cage compound 53. A conversion of only 30% to the respective product is observed in the reaction mixture, presumably due to the low electrophilicity of 52. However, the developed synthetic protocol includes removal of unreacted starting materials 52 and P4 by sublimation which can be used in additional synthetic cycles increasing the overall isolated yield.
8. Cationic polyphosphorus-chalcogen cages
A multitude of phosphorus-chalcogenides have been characterized to date and many of their structural motifs are displayed even in undergraduate textbooks.61 However, until recently, only very few examples of polyphosphorus-chalcogen cations were known which was due to the lack of established synthetic routes for their preparation. To the best of our knowledge only three distinct protocols have been reported so far. The first is based on the reaction of P4S3 with in situ generated phosphenium ion PI2+ (Scheme 15).44b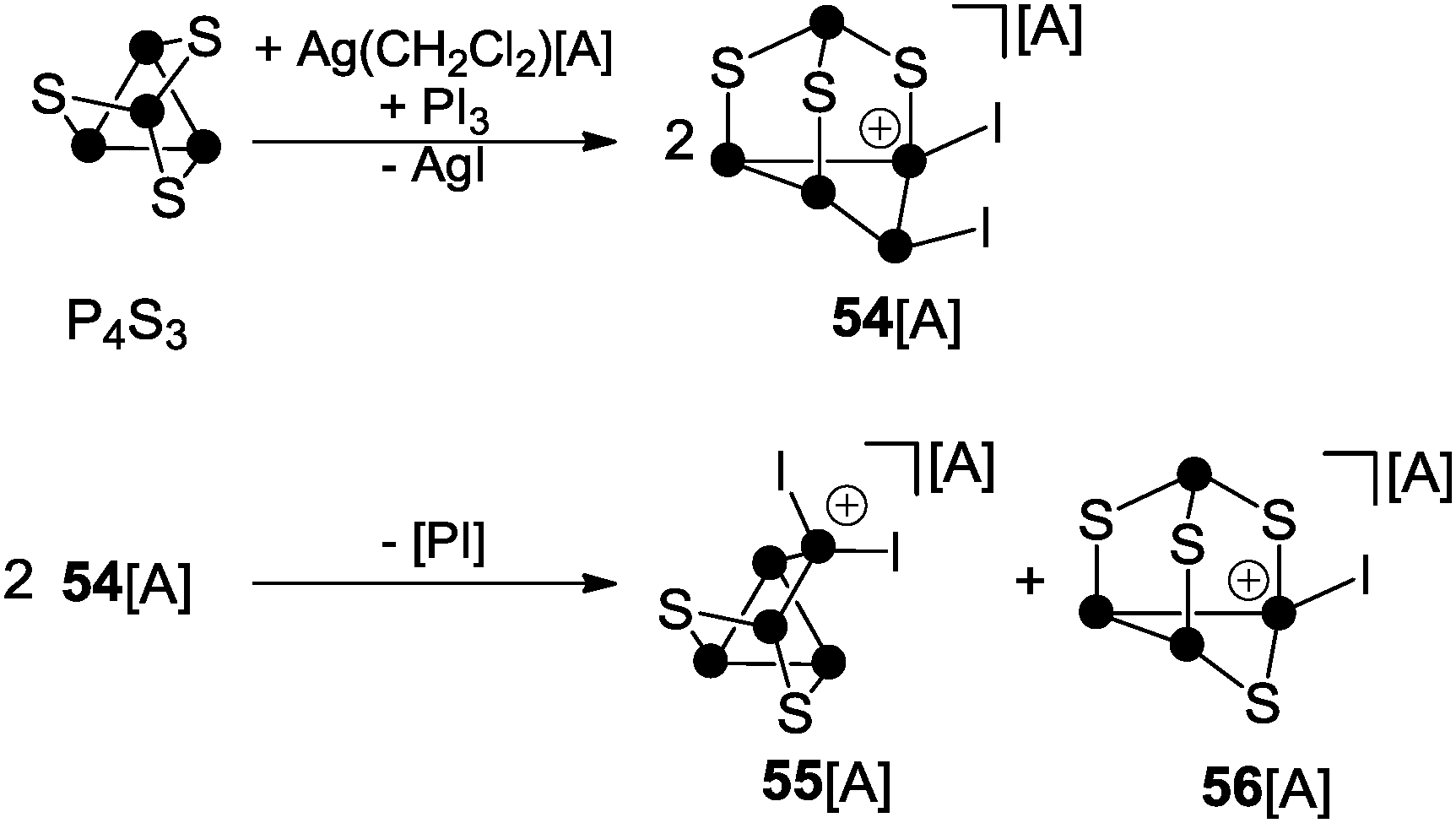 | ||
| Scheme 15 Reaction of P4S3 with in situ generated phosphenium ion PI2+; A = Al(OC(CF3)3)4. | ||
The phosphenium ion formally inserts into a P–P bond of the basal P3-ring accompanied by migration of one of the iodo-substituents to an adjacent P atom giving cation 54+. However, 54+ is not stable and subsequently disproportionates via an unknown reaction pathway to form 55+ and 56+. This process involves the extrusion of a very reactive iodo-phosphinidene [PI] and redistribution of the sulfur atoms.
The second protocol is based on halide abstraction from α-P4S3I2 with Ag(CH2Cl2)[Al(OC(CF3)3)4] and yields the spiro-cyclic cage cation 58+ (Scheme 16).62 The initial step involves the formation of 57+via iodide abstraction from α-P4S3I2. Cation 57+ subsequently reacts with a second equivalent of α-P4S3I2 and this in association with the formal extrusion of phosphinidene [PI] gives rise to spiro-cyclic cage 58+. However, detailed information on the mechanism of the formation of 58+ was not gained. The structural motif of this cation is unprecedented and contains the first tetra-coordinated P atom exclusively bonded to P and S atoms.62
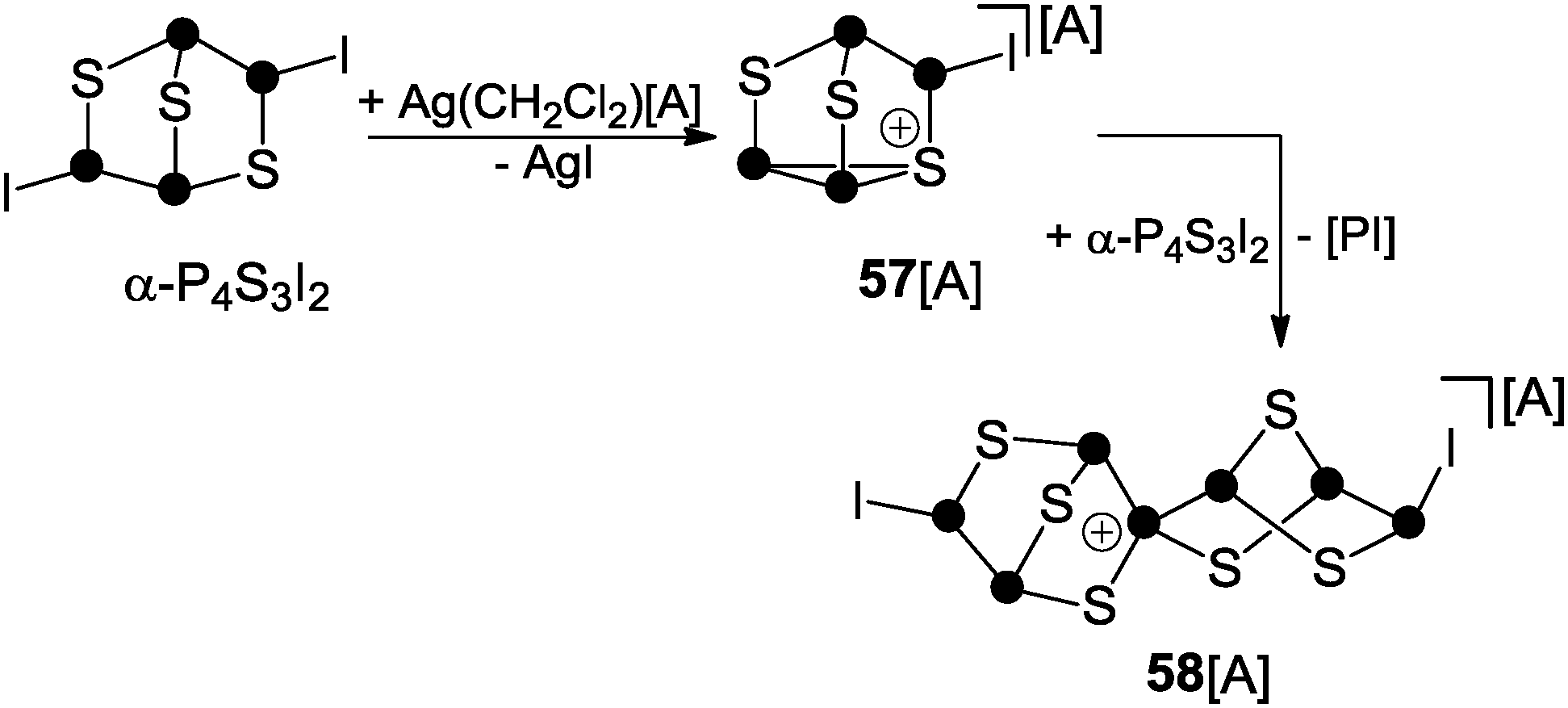 | ||
| Scheme 16 Reaction of α-P4S3I2 with Ag(CH2Cl2)[A], A = Al(OC(CF3)3)4. | ||
Recently, a third approach garnered interest which is based on using cationic polyphosphorus cages as starting materials for the preparation of cationic polyphosphorus-chalcogen cages. They constitute potentially versatile reagents due to the multitude of distinctly substituted derivatives which are all conveniently obtained in one step procedures from white phosphorus.49 Chalcogenation reactions of R2P5+-cage compounds 36a[GaCl4] and 36f[GaCl4] with elemental grey selenium yield the corresponding polyphosphorus-selenium cages 59a[GaCl4] and 59f[GaCl4] (Scheme 17). Both are obtained at elevated temperatures (110−150 °C) following a solvent-free protocol. In some cases, the addition of one equivalent of GaCl3 is beneficial since it lowers the melting point of the respective melt. Both cations are formed upon insertion of two selenium atoms into two P–P bonds adjacent to the phosphonium moieties in 36a,f+.
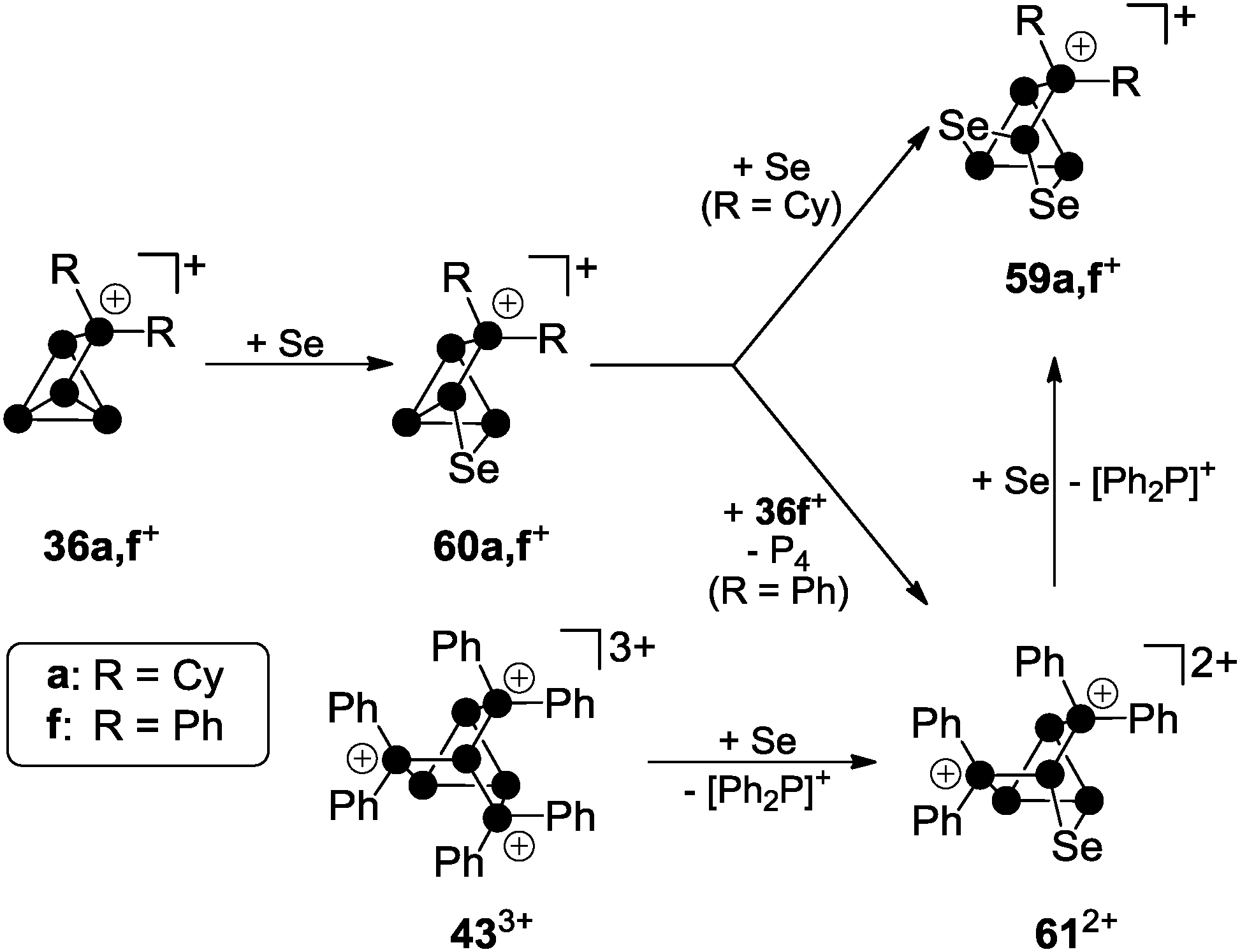 | ||
| Scheme 17 Stepwise insertion of selenium atoms into P–P bonds of 36a,f+ and stepwise substitution of [Ph2P]+-moieties in 433+ by selenium atoms giving the nortricyclane-type polyphosphorus-chalcogen cage cations 59a,f+ and 612+. | ||
Their structural motif resembles that of nortricyclane, with a basal P3-ring, the tetra-coordinated P atom and the selenium atoms occupying the bridging positions, and one P atom defining the apex of the cage. This class of compounds feature interesting 31P and 77Se NMR characteristics. Cages 59a,f+ reveal an AM2OX spin system for the CS-symmetric isotopomer without a 77Se nucleus. These resonances are superimposed by the C1-symmetric isotopomer featuring one 77Se atom in one of the bridging positions. This isotopomer gives rise to an AMNOXZ spin system which is highly influenced by higher order effects. However, in the case of 59a+, the spin systems of both isotopomers were successfully simulated allowing for the exact determination of chemical shifts and coupling constants. A series of experiments employing varying temperatures, reaction times and stoichiometries gave meaningful insights into the mechanism of the chalcogenation. These experiments indicate that the insertion of Se atoms into P–P bonds of 36a,f+ proceeds in a stepwise manner via the intermediates 60a,f+. In the case of alkyl-substituted cage 36a+ the insertion of a second equivalent grey selenium is fast, yielding the respective product 59a+ quantitatively. If the aryl-substituted starting material 36f+ is employed, the intermediate formation of dication 612+ is observed. This species forms via the transfer of a [Ph2P]+ moiety from a second equivalent of 36f+ to the reactive intermediate 60f+. Due to the higher stability of the corresponding phosphenium ion Ph2P+,19,50 this transfer is faster than the insertion of the second selenium atom. Subsequently, one of the [Ph2P]+-moieties of 612+ is substituted by a selenium atom giving rise to 59f+. The formally liberated Ph2P+-phosphenium ion is not stable and reacts with a GaCl4− anion to give the Lewis acid–base adduct 37 (Ph2PCl–GaCl3). This is in accordance with the observation of only 50% conversion and the quantitative formation of P4 and 37 or the respective oxidation product Ph2P(Se)Cl–GaCl3 in the case of reactions involving 36f+ as a starting material. The targeted preparation of 612+ as GaCl4− salt is achieved by utilizing a 2:1 stoichiometry of 36f+ and grey selenium. Another synthetic approach for the preparation of 612+ is the targeted substitution of one [Ph2P]+-moiety in the tricationic cage 433+. This is achieved by reacting 433+ with grey selenium under solvent-free conditions (Scheme 17).49 Dication 612+ was comprehensively characterized by X-ray crystallography (Fig. 12) as well as 31P and 77Se NMR spectroscopy. The 31P NMR spectrum reveals a characteristic AA′MOXX′-spin system for the isotopomer without a 77Se nucleus which is superimposed by the respective AA′MOXX′Z-spin system of the 77Se containing species.
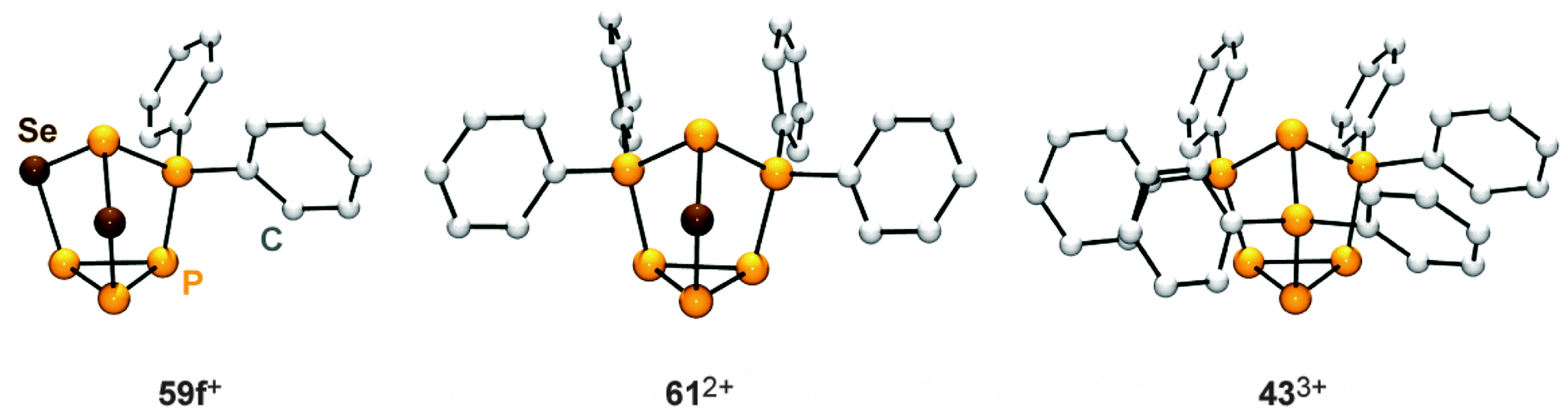 | ||
| Fig. 12 Nortricyclane type molecular structures of the related, polyphosphorus cations 59f+, 612+ and 433+. | ||
A similar reactivity was observed for reactions of the P5+-cage 36a+ or the P73+-cage 433+ with elemental α-S8.49 The polyphosphorus cation 433+ and cationic polyphosphorus-chalcogen cages 612+ and 59f+ are formally derived from the stepwise isolobal exchange of [Se] atoms by [Ph2P]+ units in the bridging positions of the nortricyclane-type structure of P4Se3. This allows for an in-depth study of the 31P NMR characteristics of the whole series of compounds and a correlation with the observed structural features in the solid state. Fig. 13 shows the dependence of the chemical shifts of 433+, 612+ and 59f+, the related sulfur-containing cages 62a+ and 632+, and P4Ch3 (Ch = Se, S)63 on the number of chalcogen atoms in the corresponding molecules. The stepwise exchange of tetra-coordinated P atoms in 433+ by Se or S atoms is accompanied by a high-field shift of the resonances of the P atoms of the basal three-membered ring. The chemical shifts of basal P atoms in nortricyclane-type cages are influenced by the exocyclic angles of the P3-ring.63 The observed high-field shift correlates well with decreasing exocyclic angles observed in the solid state structures of the respective compounds. The resonances of apical P atoms exhibit the widest range of chemical shifts and reveal a stepwise down-field shift upon the substitution of tetra-coordinated P atoms by chalcogen atoms. This is consistent with different electronegativities of directly bonded phosphorus or chalcogen atoms. Moreover, apical P atoms show a high dependency of their chemical shift on elongation or compression of the nortricyclane framework.64 Elongation is accompanied by a decrease in the P–P–P angles involving the apical P atom. This increases the s-orbital contribution to the lone pair of electrons and leads to an upfield shift of the corresponding resonance in the 31P NMR spectrum.65 On this basis, the observed downfield shift indicates a stepwise elongation of the cages which is observed in the respective molecular structures in the solid state.
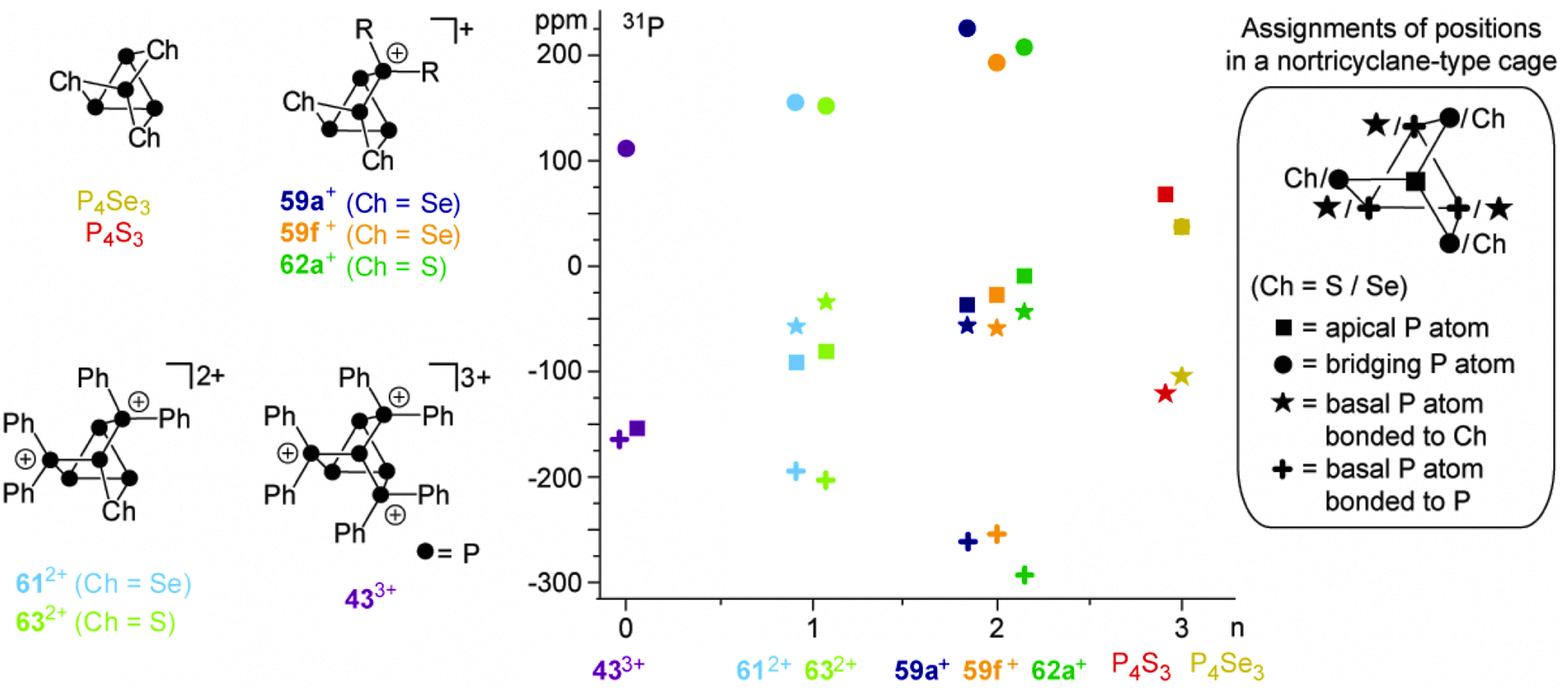 | ||
| Fig. 13 Family of cationic polyphosphorus-chalcogen cages formally derived from stepwise isolobal exchange of [Ch] by [R2P]+ units in P4Ch3 (Ch = Se, S, left), their 31P NMR shifts versus the number of chalcogen atoms (n, middle) and the assignment of P or Ch atoms to the positions of a nortricyclane-type cage (right). | ||
9. Nucleophilic fragmentation of cationic polyphosphorus cages
The activation of white phosphorus with carbenes, which belong to the class of predominantly nucleophilic ambiphiles, displays one of the most diverse fields of P4 chemistry.6 The cyclo-triphosphirene derivative C constitutes a key intermediate in all transformations, independent of the characteristic of the respective carbene employed (Fig. 1). However, intermediate C is elusive and distinct reaction pathways occur depending on the electronic and steric features of carbene L (Scheme 18). Bertrand and co-workers reacted P4 with carbenes L1 and L3 in a 1:2 stoichiometry and obtained E/Z isomers 64a,bvia an intermediate of type C.12 Bicyclic species 65 is the result of a cyclo-addition reaction involving the phosphorus double bond of an intermediate of type C and the alkyl amino carbene L4.66 Compound 66 results from a ring-opening reaction of an intermediate C with two equivalents of L5.66 This reaction is accompanied by the formation of 67 as a side product. This P2-species is formed by the formal [2+2] fragmentation of P4 by carbene L5. A [3+1]-fragmentation of the P4 tetrahedron was achieved using the sterically less demanding carbene L6 in a reaction with P4 in a 3:1 stoichiometry.66 The P1-fragment was identified as 68+ and isolated as chloride salt. The presence of chloride anions is explained by the decomposition of CHCl3 solvent molecules.
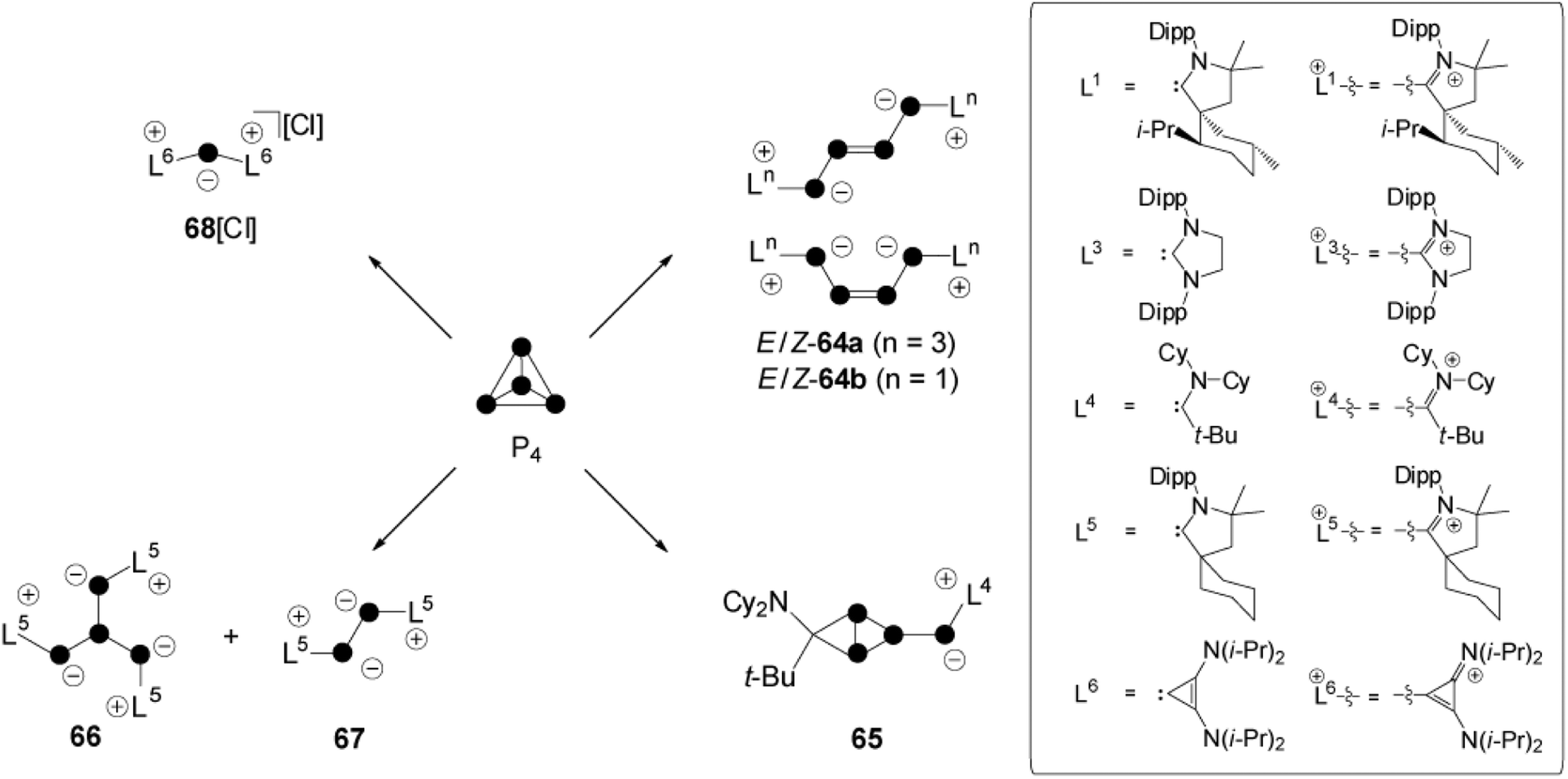 | ||
| Scheme 18 Carbene-induced transformation and fragmentation reactions of P4. | ||
A compound of unknown constitution featuring a P3 moiety was indicated in the 31P NMR spectrum of the respective reaction mixture.66
A combination of phosphenium ion and carbene mediated P4 activation constitutes a novel, potentially versatile approach for the preparation of cationic polyphosphorus cages. This strategy allows for the preparation of polyphosphorus cations featuring imidazoliumyl-substituents. These substituents are valuable for two purposes. First, they serve well for the stabilization of cations by delocalization of the positive charge.70 Second, they stabilize low-coordinated P moieties by reducing the nucleophilicity of directly bonded P atoms.71 The reaction of P5+-cage compound 32h[GaCl4] with carbene L7 in a 1:1 stoichiometry yields the bicyclo[1.1.0]tetraphosphane 69[GaCl4] (Scheme 19).67 The bicyclic framework is substituted with an imidazoliumyl-group in an exo-position and a phosphanyl-group in an endo-position. This is reminiscent of the intermediate 31 observed in the formation of RP5Cl+-cages. Cation 69+ features an ACEMX spin system indicating a non-symmetrical molecular structure due to hindered rotation around the P–P bond involving the Dipp-substituted P atom. The endo,exo-substitution of 69+ causes a short intermolecular distance between the Dipp- and the imidazoliumyl-substituted P atoms in the solid state (see molecular structure in Scheme 19). This spatial proximity is also indicated in solution by an extraordinarily large 3J(PP) coupling constant of 244.6 Hz in the 31P NMR spectrum.
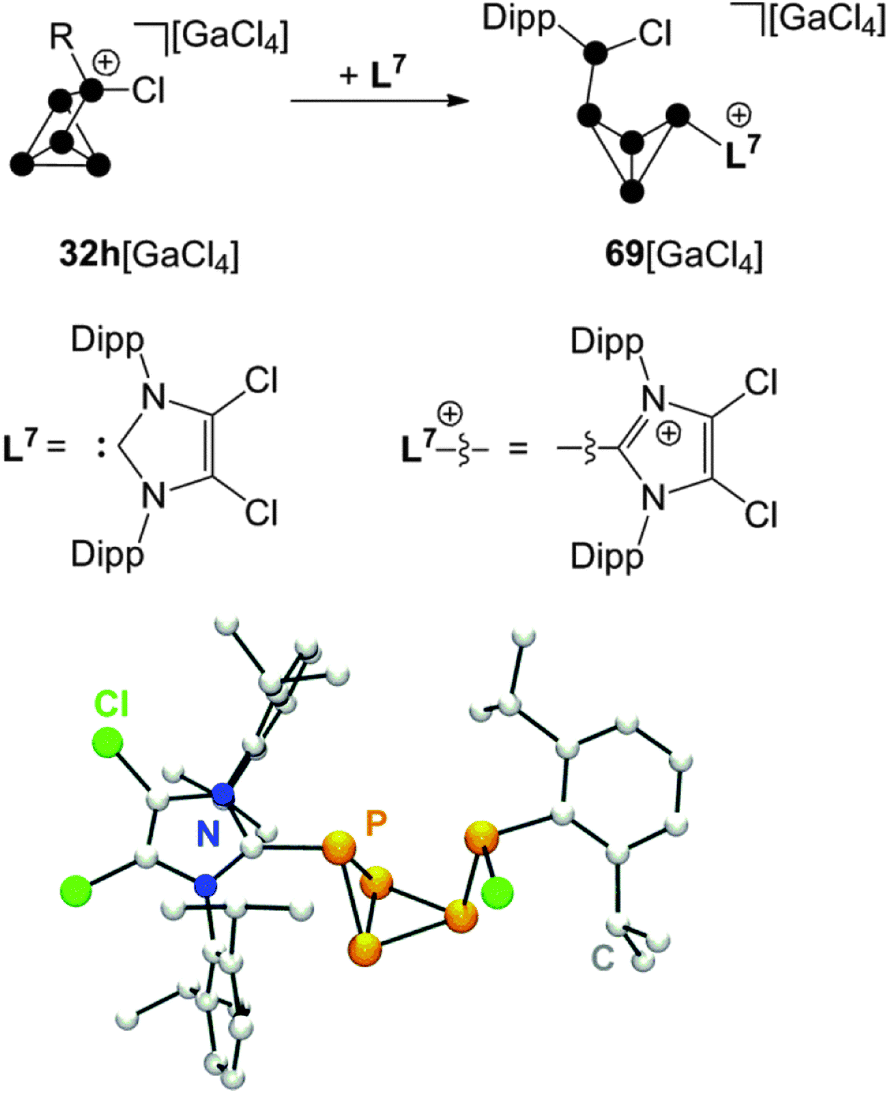 | ||
| Scheme 19 Reaction of 32h[GaCl4] with carbene L7 in a 1:1 stoichiometry (top) and molecular structure of cation 69+ (bottom). | ||
The reaction of 32h[GaCl4] with carbene L7 in a 1:3 stoichiometry proceeds via a quantitative [3+2]-fragmentation of the P5+-cage (Scheme 20).67
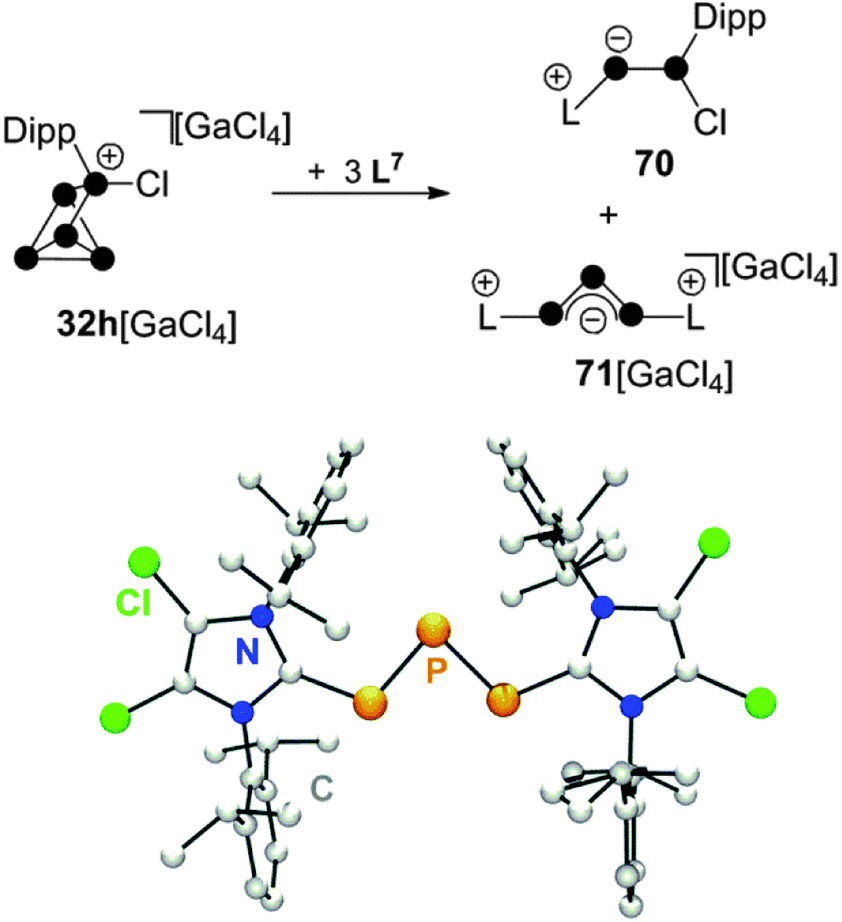 | ||
| Scheme 20 Reaction of 32h[GaCl4] with carbene L7 in a 1:3 stoichiometry (top) and molecular structure of cation 71+ (bottom). | ||
The P2 fragment was identified as the neutral P2 species 70 featuring an inversely-polarized68 phosphaalkene moiety. The di-coordinated P atom bears a phosphanyl-substituent which originates from the tetra-coordinated P atom of starting material 32h+. The P3 fragment was identified as GaCl4− salt of cation 71+ which features a chain of three di-coordinated P atoms terminated by two imidazoliumyl-substituents. This compound is characterized by a deep green colour that results from n → π* and π → π* transitions similar to those observed in diphosphenes.69 Quantum chemical calculations elucidated the bonding in 71+.67 The frontier orbital arrangement of the cation is closely related to the classical π-system of the C3-allyl anion. Thus, 71+ features a local triphosphaallylanion moiety substituted with imidazoliumyl-groups. The mechanism of the [3+2] fragmentation is explained by the reaction sequence in Scheme 21 on the basis of experimental evidence and quantum chemical calculations.67 The reaction of 32h+ with the first equivalent of L7 yields the experimentally verified species 69+. The nucleophilic attack of L7 occurs at a P atom adjacent to the phosphonium moiety in 32h+ and initiates a P–P bond cleavage. This reaction step is the reverse of the last step in the formation of RP5Cl+-cages (Fig. 7) and is in accordance with the observed reversibility of phosphenium ion insertion into P–P bonds of P4 (vide infra). The nucleophilic attack of a second carbene L7 occurs at the endo-substituted P atom of 69+ and initiates a P–P bond cleavage in the respective P3-ring.
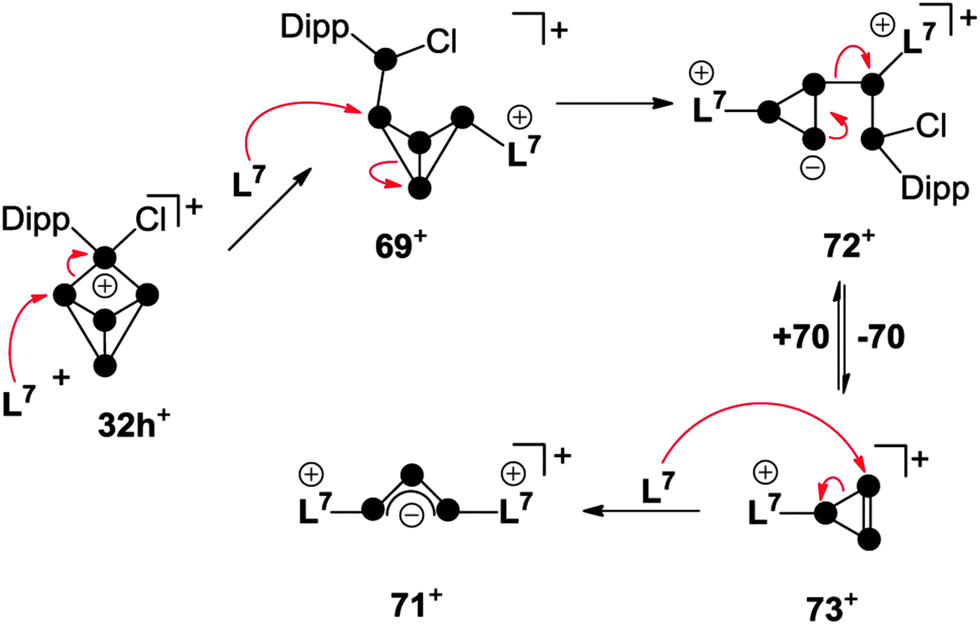 | ||
| Scheme 21 Reaction sequence for the carbene-induced [3+2]-fragmentation of P5+-cage 32h+. | ||
This yields intermediate 72+ according to quantum chemical calculations.67 Subsequently, this intermediate intramolecularly eliminates the P2 fragment 70. This yields the elusive triphosphirene derivative 73+ which is related to the key intermediate C (Fig. 1) of carbene-induced P4 activation.12,66 The nucleophilic attack of a third carbene L7 on the PP double bond of 73+ initiates a ring-opening and yields the second fragment 71+. The ease of fragmentation (high yields, multi-gram scale) together with the facile accessibility of cationic phosphorus cages from P4 and the multitude of carbenes available render this approach suitable for the preparation of a plethora of interesting polyphosphorus compounds.
Abbreviations
| aAAC | Acyclic alkyl amino carbene |
| Ab | Ambiphile |
| Ch | Chalcogen atom (Se or S) |
| El | Electrophile |
| Et | Ethyl |
| cAAC | Cyclic alkyl amino carbene |
| Cy | Cyclo-hexyl |
| Dipp | 2,6-Di-iso-propylphenyl |
| FIA | Fluoride ion affinity |
| HOMO | Highest occupied molecular orbital |
| i-Pr | Iso-propyl |
| LUMO | Lowest unoccupied molecular orbital |
| Me | Methyl |
| Mes* | 2,4,6-Tri-tert-butylphenyl |
| NHC | N-Heterocyclic carbene |
| Nu | Nucleophile |
| OTf | Triflate, trifluoromethylsulfonate |
| t-Bu | tert-Butyl |
Acknowledgements
This work was supported by the Fonds der Chemischen Industrie (FCI, scholarship for M.H.H.), the German Science Foundation (DFG, WE 4621/2-1), and the ERC (SynPhos 307616).References
- C. A. Dyker and N. Burford, Chem. – Asian. J., 2008, 3, 28 CrossRef CAS PubMed.
- M. Donath, E. Conrad, P. Jerabek, G. Frenking, R. Fröhlich, N. Burford and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 2964 CrossRef CAS PubMed.
- K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 7545 CrossRef CAS PubMed.
- D. E. Corbridge, C Phosphorus – an Outline of its Chemistry and Technology, Elsevier, Amsterdam, 5th edn, 1995 Search PubMed.
- (a) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164 CrossRef CAS PubMed; (b) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4235 CrossRef PubMed.
- (a) M. Scheer, G. Balazs and A. Seitz, Chem. Rev., 2010, 110, 4236 CrossRef CAS PubMed; (b) N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342 CrossRef CAS PubMed; (c) S. Khan, S. S. Sen and H. W. Roesky, Chem. Commun., 2012, 48, 2169 RSC.
- (a) W. W. Schoeller, Phys. Chem. Chem. Phys., 2009, 11, 5273 RSC; (b) W. W. Schoeller, V. Staemmler, P. Rademacher and E. Niecke, Inorg. Chem., 1986, 25, 4382 CrossRef CAS; (c) R. O. Jones and D. Hohl, J. Chem. Phys., 1990, 11, 6710 CrossRef PubMed; (d) V. G. Tsierelson, N. P. Tarasova, M. F. Bobrov and Y. V. Smetannikov, Heteroat. Chem., 2006, 17, 572 CrossRef; (e) C. R. C. R. Brundle, N. A. Kuebler, M. B. Robin and H. Basch, Inorg. Chem., 1972, 11, 20 CrossRef CAS; (f) S. S. Evans, P. J. Joachim, A. F. Orchard and D. W. Turner, Int. J. Mass Spectrom. Ion Phys., 1972, 9, 41 CrossRef CAS; (g) R. R. Hart, M. B. Robin and N. A. Kuebler, J. Chem. Phys., 1965, 42, 3631 CrossRef CAS PubMed; (h) M. Driess and H. Nöth, Molecular Clusters of the Main Group Elements, Wiley-VCH, Weinheim, 1st edn, 2004 Search PubMed; (i) I. Krossing, Homoatomic Cages and Clusters of the Heavier Group 15 Elements: Neutral Species and Cations, 2004, pp. 209–229 Search PubMed.
- (a) R. Riedel, H.-D. Hausen and E. Fluck, Angew. Chem., Int. Ed. Engl., 1985, 24, 1056 CrossRef; (b) E. Fluck, R. Riedel, H.-D. Hausen and G. Heckmann, Z. Anorg. Allg. Chem., 1987, 46, 7052 Search PubMed.
- E. Fluck, C. M. E. Pavlidou and R. Janoscheck, Phosphorus Sulfur Relat. Elem., 1979, 6, 469 CAS.
- M. B. Power and A. R. Barron, Angew. Chem., Int. Ed. Engl., 1991, 30, 1353 CrossRef.
- H. M. Tuononen, R. Roesler, J. L. Dutton and P. J. Ragogna, Inorg. Chem., 2007, 46, 10693 CrossRef CAS PubMed.
- (a) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052 CrossRef CAS PubMed; (b) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180 CrossRef CAS PubMed.
- (a) Y. Peng, H. Fan, H. Zhu, H. W. Roesky, J. Magull and C. E. Hughes, Angew. Chem., Int. Ed., 2004, 43, 3443 CrossRef CAS PubMed; (b) G. Prabusankar, A. Doddi, C. Gemel, M. Winter and R. A. Fischer, Inorg. Chem., 2010, 49, 7976 CrossRef CAS PubMed; (c) W. Uhl and M. Benter, Chem. Commun., 1999, 771 RSC; (d) C. Dohmeier, H. Schnöckel, C. Robl, U. Schneider and R. Ahlrichs, Angew. Chem., Int. Ed. Engl., 1994, 33, 199 CrossRef.
- (a) Y. Xiong, S. Yao, M. Brym and M. Driess, Angew. Chem., Int. Ed., 2007, 46, 4511 CrossRef CAS PubMed; (b) S. S. Sen, S. Khan, H. W. Roesky, D. Kratzert, K. Meindl, J. Henn, D. Stalke, J.-P. Demers and A. Lange, Angew. Chem., Int. Ed., 2011, 50, 2322 CrossRef CAS PubMed; (c) S. Khan, R. Michel, S. S. Sen, H.-W. Roesky and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 11786 CrossRef CAS PubMed.
- (a) M. Driess, A. D. Fanta, D. Powell and R. West, Angew. Chem., Int. Ed. Engl., 1989, 28, 1038 CrossRef; (b) A. D. Fanta, R. P. Tan, N. M. Comerlato, M. Driess, D. R. Powell and R. West, Inorg. Chim. Acta, 1992, 198, 733 CrossRef.
- M. Driess, Angew. Chem., Int. Ed. Engl., 1991, 30, 1022 CrossRef.
- S. Khan, R. Michel, J. M. Dieterich, R. A. Mata, H. W. Roesky, J.-P. Demers, A. Lange and D. Stalke, J. Am. Chem. Soc., 2011, 133, 17889 CrossRef CAS PubMed.
- (a) A. H. Cowley and R. A. Kemp, Chem. Rev., 1985, 85, 367 CrossRef CAS; (b) D. Gudat, Coord. Chem. Rev., 1997, 163, 71 CrossRef CAS.
- J. M. Slattery and S. Hussein, Dalton Trans., 2012, 41, 1808 RSC.
- (a) M. H. Holthausen and J. J. Weigand, Z. Anorg. Allg. Chem., 2012, 638, 1103 CrossRef CAS; (b) C. Hering, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2012, 51, 6241 CrossRef CAS PubMed; (c) C. Hering, A. Schulz and A. Villinger, Inorg. Chem., 2013, 52, 5214 CrossRef CAS PubMed.
- (a) R. W. Reed, Z. Xie and C. A. Reed, Organometallics, 1995, 14, 5002 CrossRef CAS; (b) A. Dumitrescu, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Eur. J. Inorg. Chem., 2002, 1953 CrossRef CAS.
- (a) F. S. Shagvaleev, T. V. Zykova, R. I. Tarasova, T. S. Sitdikova and V. V. Moskva, Zh. Obshch. Khim., 1990, 60, 1775 CAS; (b) N. Burford, T. S. Cameron, D. J. LeBlanc, P. Losier, S. Sereda and G. Wu, Organometallics, 1997, 16, 4712 CrossRef CAS.
- N. Burford, T. S. Cameron and P. J. Ragogna, J. Am. Chem. Soc., 2001, 123, 7947 CrossRef CAS.
- J. M. Slattery, C. Fish, M. Green, T. N. Hooper, J. C. Jeffery, R. J. Kilby, J. M. Lynam, J. E. McGrady, D. A. Pantazis, C. A. Russel and C. E. Williams, Chem. – Eur. J., 2007, 13, 6967 CrossRef CAS PubMed.
- M. B. Abrams, B. L. Scott and R. T. Baker, Organometallics, 2000, 19, 4944 CrossRef CAS.
- N. Burford, C. A. Dyker and A. Decken, Angew. Chem., Int. Ed., 2005, 44, 2364 CrossRef CAS PubMed.
- J. J. Weigand, N. Burford, M. D. Lumsden and A. Decken, Angew. Chem., Int. Ed., 2006, 45, 6733 CrossRef CAS PubMed.
- J. J. Weigand, N. Burford and A. Decken, Eur. J. Inorg. Chem., 2008, 4343 CrossRef CAS.
- T. P. Martin, Z. Phys. D Atom. Mol. Cl., 1986, 3, 211 CrossRef CAS.
- (a) M. D. Chen, R. B. Huang, L. S. Zheng, Q. E. Zhang and C. T. Au, Chem. Phys. Lett., 2000, 325, 22 CrossRef CAS; (b) T. Xue, J. Luo, S. Shen, F. Li and J. Zhao, Chem. Phys. Lett., 2010, 485, 26 CrossRef CAS PubMed.
- T. A. Engesser and I. Krossing, Coord. Chem. Rev., 2013, 257, 946 CrossRef CAS PubMed.
- I. Krossing, J. Chem. Soc., Dalton Trans., 2002, 500 RSC.
- T. Köchner, S. Riedel, A. J. Lehner, H. Scherer, I. Raabe, T. A. Engesser, F. W. Scholz, U. Gellrich, P. Eiden, R. A. Paz Schmidt, D. A. Plattner and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 8139 CrossRef PubMed.
- C. Bolli, T. Köchner and C. Knapp, Z. Anorg. Allg. Chem., 2012, 638, 559 CrossRef CAS.
- T. Köchner, T. A. Engesser, H. Scherer, A. D. Plattner, A. Steffani and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6529 CrossRef PubMed.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2001, 40, 4406 CrossRef CAS.
- (a) M. Gonsior, I. Krossing, L. Müller, I. Raabe, M. Jansen and L. van Wüllen, Chem. – Eur. J., 2002, 8, 4475 CrossRef CAS; (b) I. Krossing, J. Chem. Soc., Dalton Trans., 2002, 500 RSC.
- A. Bihlmeier, M. Gonsior, I. Raabe, N. Trapp and I. Krossing, Chem. – Eur. J., 2004, 10, 5041 CrossRef CAS PubMed.
- M. H. Holthausen, K.-O. Feldmann, S. Schulz, A. Hepp and J. J. Weigand, Inorg. Chem., 2012, 51, 3374 CrossRef CAS PubMed.
- (a) Y. Kashman, Y. Menachem and E. Benary, Tetrahedron, 1973, 29, 4279 CrossRef CAS; (b) P. Crews, J. Org. Chem., 1975, 40, 1170 CrossRef CAS; (c) Y. Kashman and A. Rudi, Tetrahedron Lett., 1976, 32, 2819 CrossRef; (d) A. Rudi and Y. Kashman, Tetrahedron Lett., 1978, 25, 2209 CrossRef; (e) A. H. Cowley, C. A. Stewart, B. R. Whittlesey and T. C. Wright, Tetrahedron Lett., 1984, 25, 815 CrossRef CAS.
- C. Symmes and L. D. Quin, J. Org. Chem., 1978, 43, 1250 CrossRef CAS.
- Y. Carpenter, N. Burford, M. D. Lumsden and R. McDonald, Inorg. Chem., 2011, 50, 3342 CrossRef CAS PubMed.
- S. Ulvenlund, A. Whaetley and L. A. Bengtsson, J. Chem. Soc., Dalton Trans., 1995, 245 RSC.
- (a) D. Gudat, Eur. J. Inorg. Chem., 1998, 1087 CrossRef CAS; (b) M. Gonsior, I. Krossing and E. Matern, Chem. – Eur. J., 2006, 12, 1703 CrossRef CAS PubMed.
- (a) M. Baudler, C. Adamek, S. Opiela, H. Budzikiewicz and D. Ouzounis, Angew. Chem., Int. Ed., 1988, 27, 1059 CrossRef; (b) P. Jutzi and U. Meyer, J. Organomet. Chem., 1987, 333, C18 CrossRef CAS.
- (a) R. K. Harris, E. M. Norval and M. Fild, J. Chem. Soc., Dalton Trans., 1979, 826 RSC; (b) J. J. Weigand, S. D. Riegel, N. Burford and A. Decken, J. Am. Chem. Soc., 2007, 129, 7969 CrossRef CAS PubMed.
- (a) S. O. Grim, W. McFarlane, E. F. Davidoff and T. J. Marks, J. Phys. Chem., 1966, 70, 581 CrossRef CAS; (b) S. O. Grim and W. McFarlane, Can. J. Chem., 1968, 46, 2071 CrossRef CAS.
- (a) G. Trinquler and M.-R. Marre, J. Phys. Chem., 1983, 87, 1903 CrossRef; (b) A. H. Cowley, M. C. Crushner, M. Lattman, M. L. McKee, J. S. Szobota and J. C. Wilburn, Pure Appl. Chem., 1980, 52, 789 CrossRef CAS; (c) W. W. Schoeller and U. Tubbesing, THEOCHEM, 1995, 343, 49 CrossRef CAS.
- M. H. Holthausen, A. Hepp and J. J. Weigand, Chem. – Eur. J., 2013, 19, 9895 CrossRef CAS PubMed.
- B. D. Ellis, P. J. Ragogna and C. L. B. McDonald, Inorg. Chem., 2004, 43, 7857 CrossRef CAS PubMed.
- J. J. Weigand, M. H. Holthausen and R. Fröhlich, Angew. Chem., Int. Ed., 2009, 48, 295 CrossRef CAS PubMed.
- (a) M. Baudler, W. Faber and J. Hahn, Z. Anorg. Allg. Chem., 1980, 469, 15 CrossRef CAS; (b) G. Fritz, H. Rothmann and E. Matern, Z. Anorg. Allg. Chem., 1992, 610, 33 CrossRef CAS; (c) I. Kovacs, G. Baum, G. Fritz, D. Fenske, N. Wiberg, H. Schuster and K. Karaghiosoff, Z. Anorg. Allg. Chem., 1993, 619, 453 CrossRef CAS; (d) S. Charles, J. C. Fettinger and B. W. Eichorn, J. Am. Chem. Soc., 1995, 117, 5303 CrossRef CAS.
- (a) G. Fritz and K. D. Hoppe, J. Organomet. Chem., 1983, 249, 63 CrossRef CAS; (b) V. A. Milyukov, A. V. Kataev, E. Hey-Hawkins and O. G. Sinyshin, Russ. Chem. Bull., 2007, 56, 298 CrossRef CAS PubMed; (c) M. Baudler and R. Riekenhof-Böhmer, Z. Naturforsch., B: J. Chem. Sci., 1985, 40, 1424 Search PubMed; (d) M. Baudler and T. Pontzen, Z. Naturforsch., B: J. Chem. Sci., 1983, 38, 955 Search PubMed; (e) G. Fritz and W. Hölderich, Naturwissenschaften, 1975, 62, 573 CrossRef CAS.
- Y. C. Leung, J. Waser, v. S. Houten, A. Vos, G. A. Wiegers and E. H. Wiebenga, Acta Crystallogr., 1957, 10, 574 CrossRef CAS.
- D. Gudat, Top. Heterocycl. Chem., 2010, 21, 63 CAS.
- (a) G. David, E. Niecke, M. Nieger, V. von der Gönna and W. W. Schoeller, Chem. Ber., 1993, 126, 1513 CrossRef CAS; (b) N. Burford, T. S. Cameron, K. D. Conroy, B. Ellis, M. Lumsden, C. L. B. Mcdonald, R. McDonald, A. D. Philips, P. J. Ragogna, R. W. Schurko, D. Walsh and R. E. Wasylishen, J. Am. Chem. Soc., 2002, 124, 14012 CrossRef CAS PubMed; (c) J. R. Davidson, J. J. Weigand, N. Burford, T. S. Cameron, A. Decken and U. Werner-Zwanziger, Chem. Commun., 2007, 4671 RSC; (d) D. Michalik, A. Schulz, A. Villinger and N. Wedling, Angew. Chem., Int. Ed., 2008, 47, 6465 CrossRef CAS PubMed.
- M. H. Holthausen and J. J. Weigand, J. Am. Chem. Soc., 2009, 131, 14210 CrossRef CAS PubMed.
- Unpublished results.
- M. H. Holthausen, C. Richter, A. Hepp and J. J. Weigand, Chem. Commun., 2010, 46, 6921 RSC.
- E. Niecke and R. Kröher, Angew. Chem., Int. Ed. Engl., 1976, 15, 692 CrossRef.
- A. F. Hollemann and E. Wiberg, Lehrbuch der Anorganischen Chemie, W. d. Gruyter, Berlin, New York, 102nd edn, 2007 Search PubMed.
- M. Gonsior, I. Krossing and E. Matern, Chem. – Eur. J., 2006, 12, 1986 CrossRef CAS PubMed.
- B. W. Tattershall, J. Chem. Soc., Dalton Trans., 1988, 2055 RSC.
- W. Hönle and H. G. v. Schnering, Z. Anorg. Allg. Chem., 1978, 440, 171 CrossRef.
- M. Baudler, Angew. Chem., Int. Ed. Engl., 1987, 26, 419 CrossRef.
- O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530 CrossRef CAS PubMed.
- M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078 CrossRef CAS PubMed.
- L. Weber, Eur. J. Inorg. Chem., 2000, 2425 CrossRef CAS ; a P3 chain similar to fragmentation product 71+ was reported shortly after this review was completed, see A. M. Tondreau, Z. Benkő, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545 RSC.
- L. Weber, Chem. Rev., 1992, 92, 1839 CrossRef CAS.
- (a) O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369 CrossRef CAS PubMed; (b) J. J. Weigand, K.-O. Feldmann and F. D. Henne, J. Am. Chem. Soc., 2010, 132, 16321 CrossRef CAS PubMed.
- (a) F. D. Henne, E.-M. Schnökelborg, K.-O. Feldmann, J. Grunenberg, R. Wolf and J. J. Weigand, Organometallics, 2013, 32, 6674 CrossRef CAS; (b) K. Schwedtmann, M. H. Holthausen, K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |