
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New methods for the synthesis of naphthyl amines; application to the synthesis of dihydrosanguinarine, sanguinarine, oxysanguinarine and (±)-maclekarpines B and C†
Matthew R.
Tatton
a,
Iain
Simpson
b and
Timothy J.
Donohoe
*a
aDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: timothy.donohoe@chem.ox.ac.uk
bAstraZeneca Pharmaceuticals, Mereside, Alderley Park, Macclesfield, Cheshire, SK10 4TG, UK
First published on 13th August 2014
Abstract
A new method for preparing naphthyl amines from 1,5 unsaturated dicarbonyl precursors is described; the utility of this new method was proven in the syntheses of several natural products, all containing the benzo[c]phenanthridine core and enabled by a radical promoted cyclisation of the naphthyl amine products formed in the key cyclisation.
We have recently reported the results of a programme of research aimed at expanding methods for the de novo construction of aromatic rings from linear precursors.1 When we consider the opportunities that exist for functionalisation of the non-aromatic intermediates produced en-route to an arene, and the potential for cross coupling and C–H activation on the aromatic products, then an attractive situation emerges.
One of our previous endeavours focussed on preparing benzenoid compounds from unsaturated 1,5 dicarbonyls A and amines by using enamine chemistry (Scheme 1, the geometry and position of the alkene between the carbonyls is unimportant).2 We consider that both Mannich and electrocyclic based mechanisms are feasible for this transformation. Recently, we wanted to examine if this method might be extended to allow the formation of naphthyl amines via reaction of a keto-aldehyde containing an aromatic spacer, see B. As an additional objective, we also wanted to explore the possibility of adding extra functionality onto the cyclisation product and forming another ring onto the new aromatic system. This tactic is expected to greatly increase the utility of the method in the area of natural products synthesis.
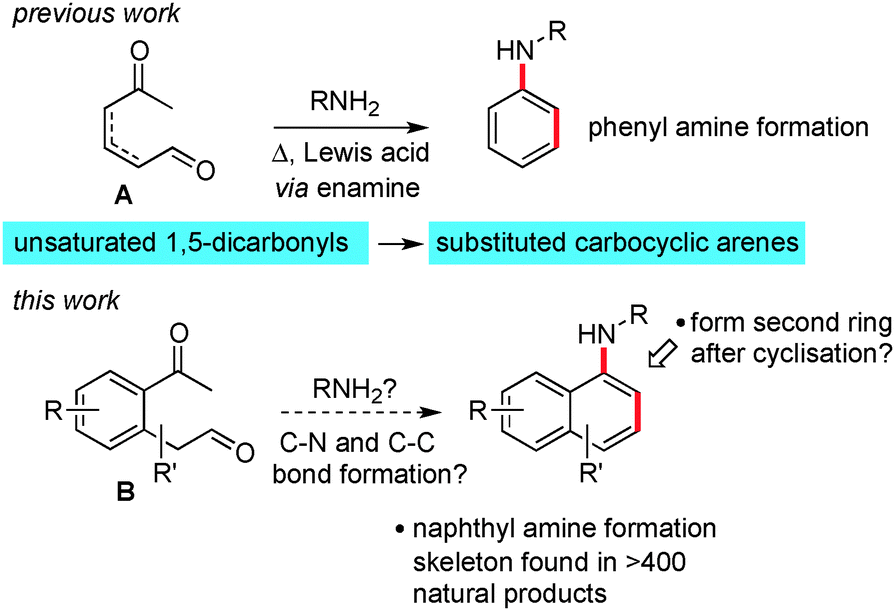 | ||
| Scheme 1 De novo route to substituted naphthyl amines. | ||
In the first instance, we used a combination of enolate arylbromide cross coupling reactions3 and alkene oxidative cleavage to prepare a set of three keto-aldehyde precursors (1–3) on which to test our hypotheses, Scheme 2. The cyclisation of these three substrates was examined under a series of acidic and Lewis acidic conditions, and we found that the most general set of conditions involved heating in toluene with acetic acid in the presence of 5 equivalents of a primary or secondary amine. Pleasingly, this sequence gave rise to the naphthyl amine core with three sets of substitution patterns in reasonable yields (see compounds 4–9). We also found that reaction with an amine in the presence of zinc chloride at 55 °C provided a milder alternative set of conditions (providing comparable yields for 8 (55%), 6 (68%) and 7 (58%)). At this point we choose not to speculate on the mechanism of this cyclisation, beyond our previously presented hypotheses for the synthesis of phenyl amine compounds and which involve pericyclic and/or Mannich type processes.2a
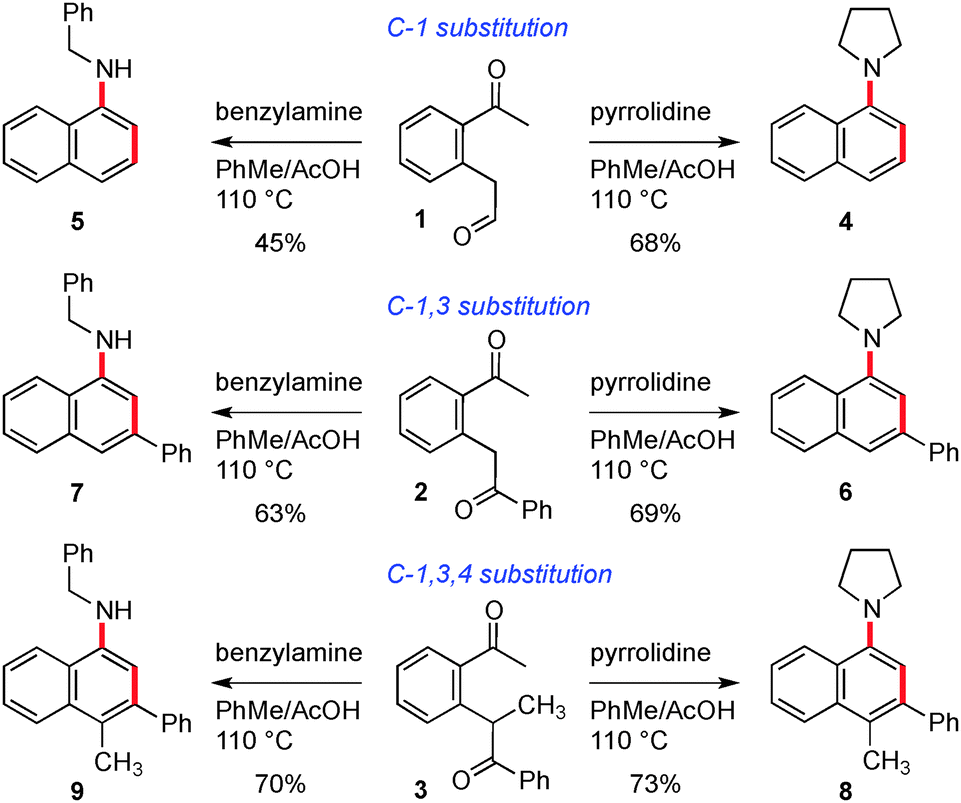 | ||
| Scheme 2 Amine promoted cyclisation of dicarbonyl compounds. | ||
Having proven the viability of the basic methodology and promoted the synthesis of substituted naphthyl amines under these new conditions, we wished to apply the chemistry to the synthesis of a set of natural products. As mentioned earlier, we also wanted to use this synthesis to explore the formation of new rings adjacent to the newly formed naphthyl amine. Therefore, a set of natural product targets based on the fused hexacyclic ring structure (benzo[c]phenanthridine) of dihydrosanguinarine4 was identified to provide an interesting and challenging test of the methodology.
It is appropriate to note here that other synthetic approaches to the benzo[c]phenanthridine alkaloids have been published. In early work, Bailey reported a successful synthesis of related alkaloids utilising a key Friedel–Crafts acylation.5 Ninomiya reported the use of an enamide photocyclisation in their alkaloid synthesis.6 Diels–Alder cycloaddition reactions have also been used effectively in both formal and completed syntheses of the benzo[c]phenanthridine alkaloids.7 Cushman reported a key condensation of a Schiff base with an anhydride to prepare chelidonine.8 Cheng and co-workers utilised their nickel catalysed cyclisation methodology in the synthesis of three benzo[c]phenanthridine alkaloids.9 Xu and co-workers completed their alkaloid synthesis using a key palladium-catalyzed ring-opening coupling reaction,10 while Hibino constructed an aza-triene moiety which underwent a microwave assisted 6π-electrocyclisation to give the core scaffold.11
Our synthesis of dihydrosanguinarine began from commercially available ketone 10, which was subjected to addition/elimination (11) and oxidative cleavage to provide the substituted keto-aldehyde precursor 12 for the key cyclisation, Scheme 3. Next, the requisite amine partner 14 was prepared in two steps from commercially available aldehyde 13, via a reductive amination sequence. For cyclisation, the application of a range of conditions was examined and we found that naphthyl amine 15 could be produced in 61% yield using 5 equivalents of amine 14 and zinc chloride as a promoter. Reduced yields of 15 (20%) were obtained with the more vigorous acetic acid conditions. We also decided to utilise a lower number of equivalents of valuable amine 14, and optimisation of a reaction using 1.5 equivalents and addition of the dicarbonyl via a syringe pump, was able to produce naphthyl amine 15 in 54% yield.
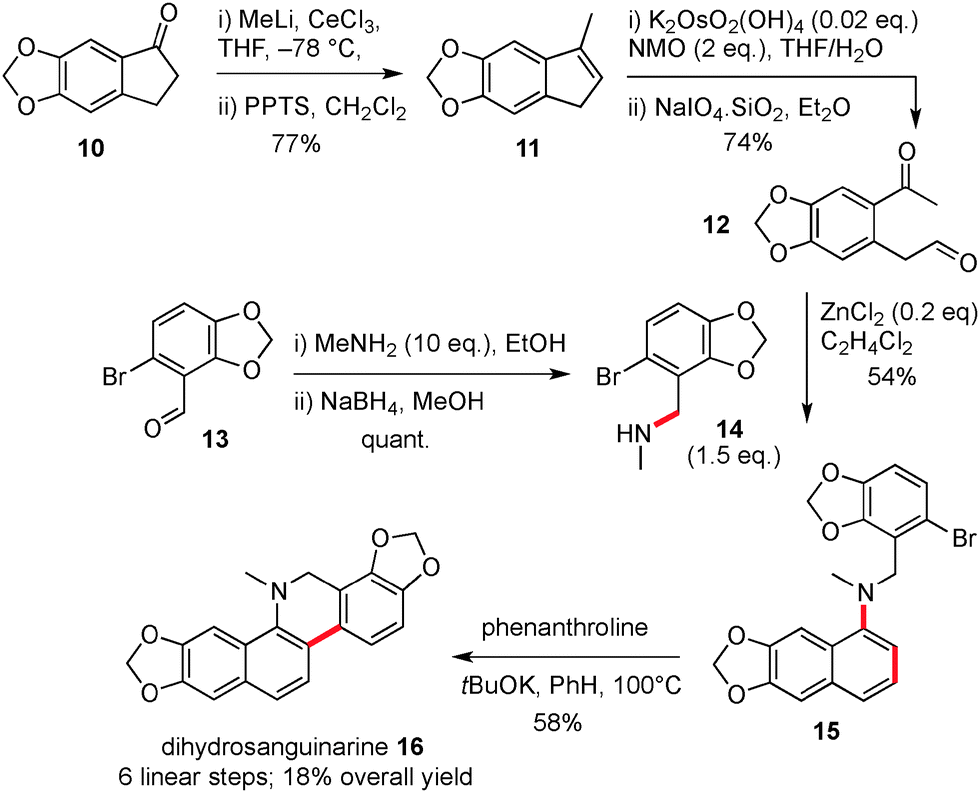 | ||
| Scheme 3 The synthesis of dihydrosanguinarine. | ||
The second key step in the sequence involved the formation of a six membered ring attached to the naphthyl amine moiety. This was accomplished by using base promoted cyclisation conditions as reported by Shi,12 and which furnished the natural product dihydrosanguinarine 16 in 58% yield by forming a new C–C bond. In our hands the use of C–H activation methods or tin hydride induced radical cyclisation were not effective for this cyclisation.13 To summarise the route so far, the synthesis of dihydrosanguinarine was competed in 6 linear steps and 18% overall yield.
Our next objective was the synthesis of the more complex targets maclekarpine B and C, which contain the dihydrosanguinarine core but with an exocyclic unsaturated butyrolactone ring attached α-to the nitrogen.14 We set about preparing a suitable amine precursor 20 to test in cyclisation reactions, Scheme 4.
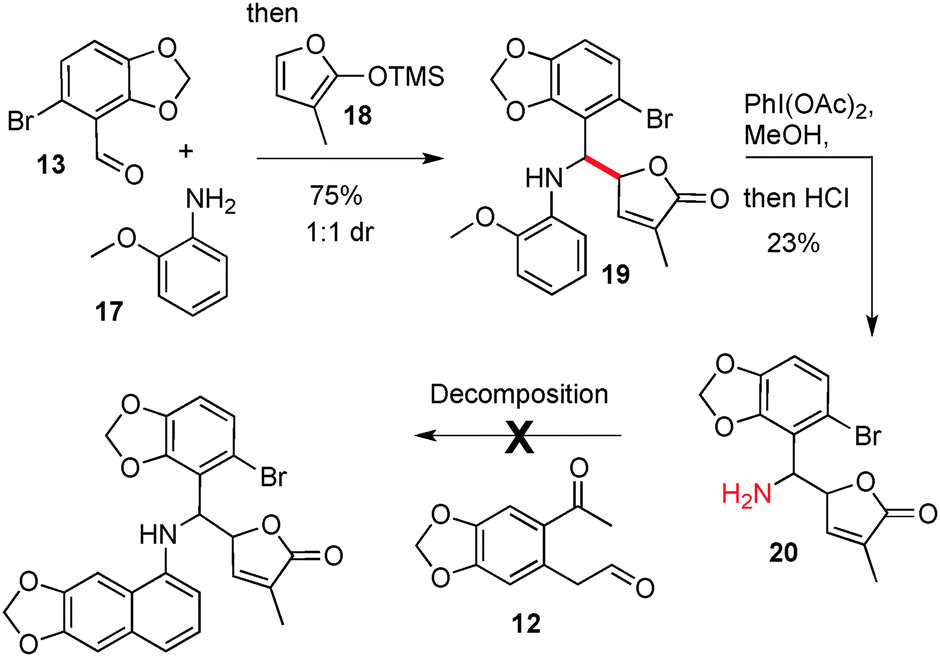 | ||
| Scheme 4 Attempted cyclisation of a complex amine towards a synthesis of maclekarpines B and C. | ||
This was accomplished by the addition of TMS–furan 18 into the imine formed by reaction of 13 and 17, followed by oxidative deprotection (20).15 The yields and diastereoselectivity (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) of 19 and 20 remained unoptimised as we quickly sought to test a significantly more bulky amine in the cyclisation regimen. Unfortunately, all cyclisation conditions that were examined between amine 20 and dicarbonyl 12 led to either a lack of reactivity or decomposition of the substrates; so it was concluded that this route to the maclekarpines was not a viable one.
:1 dr) of 19 and 20 remained unoptimised as we quickly sought to test a significantly more bulky amine in the cyclisation regimen. Unfortunately, all cyclisation conditions that were examined between amine 20 and dicarbonyl 12 led to either a lack of reactivity or decomposition of the substrates; so it was concluded that this route to the maclekarpines was not a viable one.
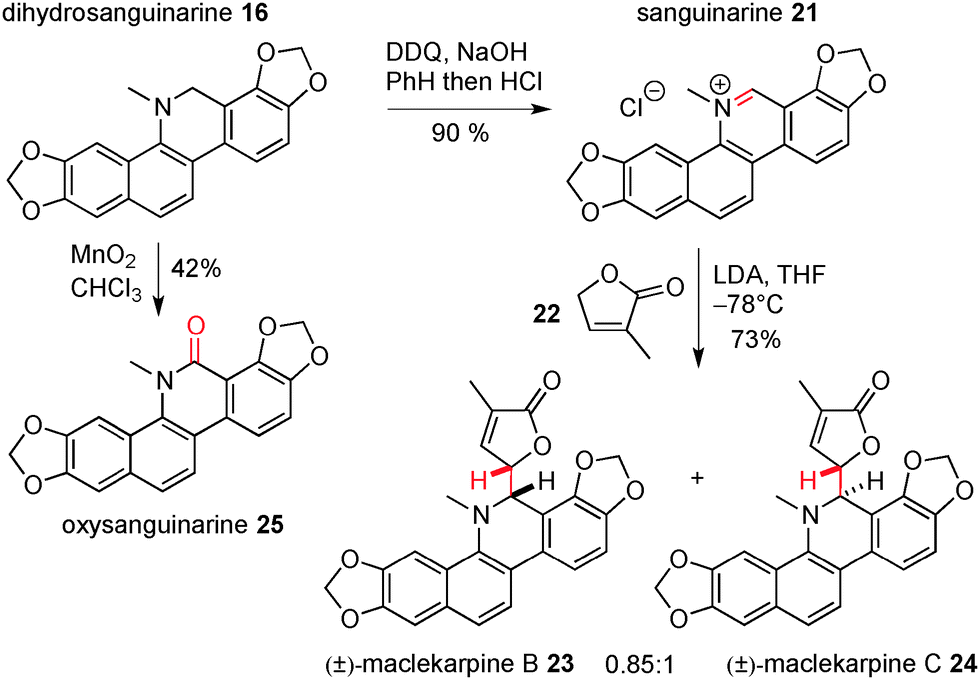
However, we were able to progress towards these targets by altering the sequence of events and installing the lactone in the final step, Scheme 5. Oxidation of 16 with DDQ gave another natural product, sanguinarine (21),16 in 90% yield; the pyridinium functionality in this molecule was then subject to regioselective nucleophilic attack by the extended enolate formed from reaction of lactone 22 with LDA. The addition products were formed as a 0.85:1 mixture of diastereoisomers in 73% yield (these were inseparable by silica gel chromatography). The NMR data for the two compounds matched exactly with that reported for the natural products maclekarpine B (23) and C (24), and this allowed us to confirm the first synthesis of these two targets, albeit as a mixture. Finally, and as part of our studies towards the oxidation of dihydrosanguinarine, we also found that it could be transformed into another natural product, namely oxysanguinarine (25), by reaction with manganese dioxide.17 The data for all natural products reported herein was a very good match with that in the literature.
 | ||
| Scheme 5 Completion of the syntheses of (±)-maclekarpines B and C, and of sanguinarine and oxysanguinarine. | ||
To conclude, we have developed a new piece of methodology that extends the scope of aromatic ring formation in a de novo fashion from acyclic precursors. The scope of the method to prepare naphthyl amines was explored and it was then tested in the successful and efficient syntheses of several natural products.
We thank the EPSRC and AstraZeneca for support.
Notes and references
- (a) T. J. Donohoe, J. F. Bower and L. K. M. Chan, Org. Biomol. Chem., 2012, 10, 1322 Search PubMed; (b) T. J. Donohoe, L. P. Fishlock and P. A. Procopiou, Chem. – Eur. J., 2008, 14, 5716 CrossRef CAS PubMed; (c) T. J. Donohoe, C. R. Jones and L. C. A. Barbosa, J. Am. Chem. Soc., 2011, 133, 16418 Search PubMed.
- (a) M. R. Tatton, I. Simpson and T. J. Donohoe, Org. Lett., 2014, 16, 1920 CrossRef CAS PubMed ; for related earlier work see: ; (b) I. W. Davies, J. Marcoux, J. D. O. Taylor and P. G. Dormer, Org. Lett., 2002, 4, 439 CrossRef CAS PubMed; (c) T. Y. Lam, Y. Wang and R. L. Danheiser, J. Org. Chem., 2013, 78, 9396 Search PubMed; (d) T. P. Willumstad, O. Haze, X. Y. Mak, T. Y. Lam, Y. Wang and R. L. Danheiser, J. Org. Chem., 2013, 78, 11450 CrossRef CAS PubMed; (e) X. Y. Mak, A. L. Crombie and R. L. Danheiser, J. Org. Chem., 2011, 76, 1852 CrossRef CAS PubMed; (f) M. Arambasic, J. F. Hooper and M. C. Willis, Org. Lett., 2013, 15, 5162 Search PubMed.
- For a review on enolate cross coupling: (a) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676 Search PubMed ; for related studies see: ; (b) T. J. Donohoe, B. S. Pilgrim, G. R. Jones and J. A. Bassuto, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11605 CrossRef CAS PubMed; (c) B. S. Pilgrim, A. E. Gatland, C. T. McTernan, P. A. Procopiou and T. J. Donohoe, Org. Lett., 2013, 15, 6190 CrossRef CAS PubMed.
- (a) R. D. Williams and B. E. Ellis, Phytochemistry, 1993, 32, 719 CrossRef CAS; (b) G. Zhang, G. Rucker, E. Breitmaier and R. Mayer, Phytochemistry, 1995, 40, 1813 Search PubMed.
- A. S. Bailey and C. R. Worthing, J. Chem. Soc., 1956, 4535 RSC.
- I. Ninomiya, T. Naito, H. Ishii, T. Ishida, M. Ueda and K. Harada, J. Chem. Soc., Perkin Trans. 1, 1975, 762 RSC.
- (a) D. Perez, E. Guitian and L. Castedo, J. Org. Chem., 1992, 57, 5911 Search PubMed; (b) R. D. Clark and Jahangir, J. Org. Chem., 1988, 53, 2378 CrossRef CAS; (c) T. N. Le, S. G. Gang and W. J. Cho, J. Org. Chem., 2004, 69, 2768 CrossRef CAS PubMed.
- M. Cushman, T. C. Choong, J. T. Valko and M. P. Koleck, J. Org. Chem., 1980, 45, 5067 CrossRef CAS.
- C. Cheng and R. P. Korivi, Chem. – Eur. J., 2010, 16, 282 CrossRef PubMed.
- P. Lv, K. Huang, L. Xie and X. Xu, Org. Biomol. Chem., 2011, 9, 3133 Search PubMed.
- Y. Ishihara, S. Azuma, T. Choshi, K. Kohno, K. Ono, H. Tsutsumi, T. Ishizu and S. Hibino, Tetrahedron, 2011, 67, 1320 CrossRef CAS PubMed.
- (a) C. Sin, Y. Gu, W. Huany and Z. Shi, Chem. Commun., 2011, 47, 9813 RSC; (b) S. De, S. Mishra, B. N. Kakde, D. Dey and A. Bisai, J. Org. Chem., 2013, 78, 7823 CrossRef CAS PubMed ; for a review on tBuO-mediated arylation see: ; (c) S. Yanagisawa and K. Itami, ChemCatChem, 2011, 3, 827 CrossRef CAS PubMed.
- (a) L. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581 Search PubMed; (b) T. Nakanishi, M. Suzuki, A. Mashiba, K. Ishikawa and T. Yokotsuka, J. Org. Chem., 1998, 63, 4235 CrossRef CAS.
- A. Deng and H. Qin, Phytochemistry, 2010, 71, 816 CrossRef CAS PubMed.
- G. Landelle, A. Claraz, S. Oudeyer and V. Levacher, Tetrahedron Lett., 2012, 53, 2414 Search PubMed.
- (a) M. Hanaoka, W. J. Wo, S. Yoshida, T. Fueki and C. Mukai, Chem. Pharm. Bull., 1990, 38, 3335 CrossRef CAS; (b) P. Lv, K. Huang, L. Xie and X. Xu, Org. Biomol. Chem., 2011, 9, 3133 RSC.
- R. SanMartin, E. M. Marigorta, I. Moreno and E. Dominguez, Heterocycles, 1997, 45, 757 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data for all novel compounds. See DOI: 10.1039/c4cc05209a |
| This journal is © The Royal Society of Chemistry 2014 |