Solvent-free synthesis of a microporous metal–organic framework†
Anne
Pichon
,
Ana
Lazuen-Garay
and
Stuart L.
James
*
Centre for the Theory and Application of Catalysis, School of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Road, Belfast, UK BT9 5AG. Fax: +44 (0) 28 90972117; Tel: +44 (0)28 90975419
First published on 6th February 2006
Abstract
We describe the first solvent-free mechanochemical synthesis of a microporous metal–organic framework [Cu(INA)2] (INA = isonicotinic acid); the product has robust 3-dimensional connectivity and is obtained quantitatively by grinding together copper acetate and isonicotinic acid for 10 minutes without any applied heating—the high efficiency of the synthesis suggests that mechanochemical synthesis should be further investigated as a convenient method for the preparation of microporous metal–organic frameworks (MOFs).
Solvent-free synthesis1 of coordination polymers or metal–organic frameworks (MOFs)2 is of interest for several reasons. For example, it could give insight into the roles of solvent molecules in templating microporous structures, give access to green large scale production processes and even provide more convenient lab-scale preparative methods. There have been several reports recently on the use of mechanochemical methods (grinding) to produce discrete coordination complexes,3 and three reports of the synthesis of 1-dimensional coordination polymers.4–6 However, the coordination polymers obtained previously are not porous, and it has not previously been demonstrated, to our knowledge, that grinding can produce the higher-dimensional connectivity which is generally required to support permanent open porosity. Porous MOFs are of great interest for their sorption properties and potentially for applications.2 Solvent-free synthesis of porous MOFs would therefore be a significant advance. We describe here our observations of the quantitative and rapid synthesis of a microporous framework which is crystalline and has 3-dimensional connectivity, in particular [Cu(INA)2] (INA = isonicotinate). The reaction occurs within minutes of grinding together copper acetate and isonicotinic acid, and the material can then simply be heated to remove the water and acetic acid byproducts to give the empty, crystalline, porous framework in quantitative yield.
A ball mill was used to grind together copper acetate monohydrate, Cu(O2CCH3)2·H2O, and isonicotinic acid, NC5H4-4-CO2H (INAH).‡ Typical conditions involved a 20 ml steel vessel containing a steel ball bearing and ca. 0.5 g of reactants, at an oscillation rate of 25 Hz maintained for 10 minutes. The formation of a reaction product, material 1, was indicated by a change in colour from green to blue and the characteristic odour of acetic acid, released as a by-product (see Fig. 1 for a reaction scheme). X-Ray powder diffraction showed the product to be highly crystalline, and also revealed that the reaction was quantitative, since no starting materials were detected, as shown in Fig. 2, top.
![Solventless reaction between Cu(OAc)2·H2O and isonicotinic acid (INAH) to give the 3-dimensional framework [Cu(INA)2]. The initial form of the product (material 1) still contains some water and acetic acid by-products. These can be driven off by heating to give material 2, which is the empty porous framework.](/image/article/2006/CE/b513750k/b513750k-f1.gif) | ||
| Fig. 1 Solventless reaction between Cu(OAc)2·H2O and isonicotinic acid (INAH) to give the 3-dimensional framework [Cu(INA)2]. The initial form of the product (material 1) still contains some water and acetic acid by-products. These can be driven off by heating to give material 2, which is the empty porous framework. | ||
![Top: comparison of XRPD patterns of starting materials Cu(OAc)2·H2O, isonicotinic acid, and the product material 1. Middle: comparison of XRPD pattern of material 1 and the pattern for [Cu(INA)2]·2H2O calculated from its single-crystal diffraction data (CCD code BAHGUN). Bottom: comparison of XRPD patterns of material 2 and the calculated pattern for [Cu(INA)2]
(CCD code UFUMUD).](/image/article/2006/CE/b513750k/b513750k-f2.gif) | ||
| Fig. 2 Top: comparison of XRPD patterns of starting materials Cu(OAc)2·H2O, isonicotinic acid, and the product material 1. Middle: comparison of XRPD pattern of material 1 and the pattern for [Cu(INA)2]·2H2O calculated from its single-crystal diffraction data (CCD code BAHGUN). Bottom: comparison of XRPD patterns of material 2 and the calculated pattern for [Cu(INA)2] (CCD code UFUMUD). | ||
The X-ray diffraction pattern of material 1 was similar, but not identical, to that calculated for a previously characterised metal–organic framework dihydrate, in particular [Cu(INA)2]·2H2O7a,b (INA = isonicotinate) (Fig. 2, middle). We obtained the single crystal diffraction data for [Cu(INA)2]·2H2O from the Cambridge Crystallographic Database (CCD). The CCD letter code for this structure is BAHGUN. Its XRPD pattern was simulated from the single crystal data using Mercury 1.3 software. The BAHGUN single crystal struture was previously reported by Liu et al.7a They synthesised crystals of this dihydrated compound in a solvothermal reaction between 4-cyanopyridine, NC5H4-4-CN, and copper chloride, in a mixture of water and ethanol solvents, by heating to 150 °C under autogenous pressure for 48 hours. The structure they reported consists of isonicotinate ligands which link together square-pyramidal Cu(II) centres to form a continuous three-dimensional network. The structure also exhibits channels which contain the water molecules (see Fig. 3, top). It is closely related to iron,8 cobalt9 and manganese10 [M(INA)2] structures, but based on five-coordinate rather than octahedral metal centres. The similarity between the XRPD pattern of material 1, and that simulated for [Cu(INA)2]·2H2O (BAHGUN) indicates that they have similar structures. However, there are some differences between the two patterns, for example between 2-θ values of 17 and 26° (see Fig. 2, middle, for a comparison of the two XRPD patterns). These differences could arise from the inclusion of acetic acid in the channels 1, which is a by-product of the solvent-free reaction, as opposed to the water present in BAHGUN, or conceivably conformational differences between the frameworks due to their different methods of preparation.
![X-Ray crystal structures of [Cu(INA)2]·2H2O (CCD code BAHGUN)7a and desolvated [Cu(INA)2]
(CCD code UFUMUD).7b](/image/article/2006/CE/b513750k/b513750k-f3.gif) | ||
| Fig. 3 X-Ray crystal structures of [Cu(INA)2]·2H2O (CCD code BAHGUN)7a and desolvated [Cu(INA)2] (CCD code UFUMUD).7b | ||
Other groups have also reported on the [Cu(INA)2] framework structure in various solvated forms. The group of Lu and Babb have reported the synthesis of single crystals of the [Cu(INA)2]·solvent, where solvent = 2H2O, MeOH, EtOH, and n-PrOH.7b Further, they reported that the dihydrate could be dehydrated by heating to 200 °C for three hours, to leave the empty porous host [Cu(INA)2]. They also described the single crystal structure of this dehydrated product (CCD letter code UFUMUD). This dehydrated product has identical framework connectivity to the starting dihydrate, but has no water molecules in the channels. Therefore, the Cu(INA)2 framework structure has been shown to be sufficiently robust that it can support open cavities. Also, they showed that the desolvated material exhibited selective sorption behaviour with regard to organic guests. The ethanol solvate [Cu(INA)2]·EtOH has also been reported by the group of Lin et al.7c It was obtained from isonicotinic acid and copper nitrate by heating for five days in a mixture of ethanol, acetonitrile and water.
It should be noted that during our solvent-free synthesis of material 1, no evidence of a liquid, melt phase was observed even if the grinding was stopped at various intervals to inspect the contents of the reaction vessel. In addition, grinding Cu(OAc)2·H2O and INAH by hand produced an indentical colour change as did use of the ball mill, indicating the formation of material 1, but once again no liquid phase was observed. These observations make an interesting comparison with certain reactions between organic compounds for which liquid phases have been clearly observed.1d
Strikingly, we also found that in the solvent-free synthesis, grinding was actually only required to initiate the reaction, and it was not necessary to continue grinding to drive it to completion. In particular, if grinding was applied for only one minute, the reaction still proceeded in quantitative yield to give 1 although the reaction was slower to go to completion, requiring 6 hours overall. The progress of this reaction, as monitored by XRPD is illustrated in Fig. 4. It seems that the essential role of the grinding is to finely divide and intimately mix the two reactants, and that once this has happened the reaction can proceed unaided, although it is accelerated by continued grinding.
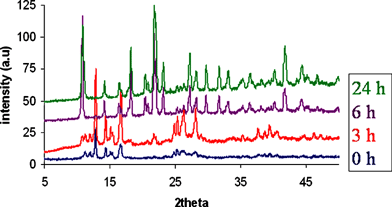 | ||
| Fig. 4 XRPD patterns showing progress of the solvent-free reaction with standing time, after initiating the reaction by grinding for 1 minute. | ||
Also interesting is that different microscopic morphologies are exhibited by samples prepared by grinding for 1 minute, compared to those prepared by grinding for longer times, such as 5 minutes. In particular, the SEM images shown in Fig. 5, which were taken after two such samples had been left to stand for one week, show that the 1-minute sample consists of crystals which are larger and have more clearly defined faces and edges than those of the 5-minute sample. Clearly, these different morphologies suggest that grinding time during the reaction may be important in determining the bulk properties of MOFs prepared by the solvent-free method. The XRPD patterns of a samples prepared by 1 minute of grinding also showed more intense peaks at higher 2-θ values (see ESI†).
 | ||
| Fig. 5 SEM images of samples prepared by grinding for 1 minute or for 5 minutes. The images were obtained after a standing time of one week. | ||
We next investigated whether samples of material 1 could be converted to the empty structure Cu(INA)2, the single crystal structure of which was reported previously by Lu et al.,7b as described above. We observed that, after removal from the reaction vessel, microcrystalline samples of material 1, which had been prepared mechanochemically by 10 minutes grinding, lost between 15–20% by weight on standing in air over several days, due to partial or complete loss of acetic acid and water (as supported by the reduction of the O–H stretching band in their IR spectra). After six days, such a sample was analysed by thermogravimetric analysis to determine if any water or acetic acid remained. This showed a further 5% weight loss between 25 °C and 150 °C. This weight change is most likely due to loss of remaining acetic acid, or one equivalent of water (loss of water from [Cu(INA)2]·H2O corresponds to 4.2% weight loss).
We fully desolvated a bulk sample of material 1 (which had been prepared by 10 minutes grinding) by heating it to 200 °C for 3 hours. This indeed gave a second crystalline material, material 2, as indicated by a change in its XRPD pattern. The XRPD pattern of material 2 can be compared with that simulated from the single crystal data for the empty framework host [Cu(INA)2] (CCD letter code UFUMUD) as reported by Lu and Babb.7b The comparison, shown in Fig. 2 (bottom) reveals a very close match, confirming that the crystal structures are essentially identical. For reference, the single crystal structure determined by Lu and Babb for the empty structure (UFUMUD) is shown in Fig. 3 (bottom).
We have also begun to investigate whether other MOFs can also be prepared under similar solvent-free conditions. Preliminary results show that it is indeed possible to prepare a second microporous MOF, in particular Cu3(BTC)2 (BTC = 1,3,5-benzenetricarboxylate),11 by 10 minutes grinding under the same conditions as used for material 1. Cu3(BTC)2 has larger pores than Cu(INA)2 and is of interest for gas separation applications.11b We will report in more detail on this and other solventless MOF syntheses in due course.
In summary, grinding together metal salts and bridging organic ligands is demonstrated here as a convenient and effective preparative method for a robust, 3-dimensional microporous metal–organic framework. The method is quick and gives a quantitative yield, without the need for solvents or external heating. Clearly, it can present higher efficiency in terms of materials, energy and time compared to solvothermal methods.
Acknowledgements
We are grateful to Dr Sam Motherwell of the Cambridge Crystallographic Data Centre for valuable input, and to the EPSRC (grant no. GR/T23145) for financial support.Notes and references
- (a) G. Kaupp, CrystEngComm, 2003, 5, 117 RSC; (b) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS; (c) G. W. V. Cave, C. L. Raston and J. L. Scott, Chem. Commun., 2001, 2159 RSC; (d) G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701 CrossRef CAS.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC; C. Janiak, Dalton Trans., 2003, 2781 RSC; S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS; M. J. Rosseinsky, Microporous Mesoporous Mater., 2004, 73, 15 CrossRef CAS; J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3 CrossRef CAS; B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS; B. Moulton and M. J. Zaworotko, Curr. Opin. Solid State Mater. Sci., 2002, 6, 117 CrossRef CAS; M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS.
- V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, Chem. Commun., 2002, 1606 RSC; A. Orita, L. Jiang, T. Nakano, N. Ma and J. Otera, Chem. Commun., 2002, 1362 RSC; M. Tsuchimoto, G. Hoshina, N. Yoshioka, H. Inoue, K. Nakajima, M. Kamishima, M. Kojima and S. Ohba, J. Solid State Chem., 2000, 153, 9 CrossRef CAS; P. J. Nichols, C. L. Raston and J. W. Steed, Chem. Commun., 2001, 1062 RSC.
- W. J. Belcher, C. A. Longstaff, M. R. Neckening and J. W. Steed, Chem. Commun., 2002, 1602 RSC.
- D. Braga, S. L. Giaffreda, F. Grepioni and M. Polito, CrystEngComm, 2004, 6, 458 Search PubMed.
- D. Braga, M. Curzi, F. Grepioni and M. Polito, Chem. Commun., 2005, 2915 RSC.
- (a) Y.-H. Liu, Y.-L. Lu, H.-L. Tsai, J.-C. Wang and K. L. Lu, J. Solid State Chem., 2001, 158, 315 CrossRef CAS; (b) J. Y. Lu and A. M. Babb, Chem. Commun., 2002, 1340 RSC; (c) M. E. Chapman, P. Ayyappan, B. M. Foxman, G. T. Lee and W. Lin, Cryst. Crowth Des., 2001, 1, 159 Search PubMed.
- R.-G. Xiong, S. R. Wilson and W. B. Lin, J. Chem. Soc., Dalton Trans., 1998, 4089 RSC.
- Q. Wei, M. Nieuwenhuyzen, F. Meunier, C. Hardacre and S. L. James, Dalton Trans., 2004, 1807 RSC.
- Q. Wei, M. Nieuwenhuyzen and S. L. James, Microporous Mesoporous Mater., 2004, 73, 97 CrossRef CAS.
- (a) S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS; (b) Q. M. Wang, D. M. Shen, M. Bulow, M. L. Lau, S. G. Deng, F. R. Fitch, N. O. Lemcoff and J. Semanscin, Microporous Mesoporous Mater., 2002, 55, 217 CrossRef CAS; (c) A. Vishnyakov, P. I. Ravikovitch, A. V. Neimakr, M. Bulow and Q. M. Wang, Nano Lett., 2003, 3, 713 CrossRef CAS; (d) K. A. I. Skoulidas, J. Am. Chem. Soc., 2004, 126, 1356 CrossRef; (e) K. Schlichte, T. Kratzke and S. Kaskel, Micoporous Mesoporous Mater., 2004, 73, 81 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Comparison of XRPD patterns from samples of material 1 prepared by 1 and 10 minutes grinding. See DOI: 10.1039/b513750k |
| ‡ Reagents were purchased from Aldrich and used as supplied. Synthesis of 1: A 20 cm3 steel vessel was charged with Cu(OAc)2·H2O (0.203 g, 1.0 mmol), isonicotinic acid (0.252 g, 2.0 mmol) and a steel ball bearing, and shaken with a Retsch MM200 mixer mill for 10 minutes at 25 Hz. Analysis calculated for Cu(INA)2·½H2O∶ C, 45.51; H, 2.84; N, 8.85; Cu, 20.07. Found for 2 : C, 45.45; H, 3.07; N, 8.64; Cu, 20.34%. The partial hydration of material 2 may have occurred on standing in ambient conditions prior to analysis. |
| This journal is © The Royal Society of Chemistry 2006 |