
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoselective homologative preparation of trisubstituted alkenyl halides from carbonyls and carbenoids†
Margherita
Miele
 *a,
Davide
Castiglione
a,
Wolfgang
Holzer
b,
Laura
Castoldi
*c and
Vittorio
Pace
*ab
*a,
Davide
Castiglione
a,
Wolfgang
Holzer
b,
Laura
Castoldi
*c and
Vittorio
Pace
*ab
aUniversity of Turin – Department of Chemistry, Via Giuria 7, 10125, Turin, Italy. E-mail: margherita.miele@unito.it; vittorio.pace@univie.ac.at; vittorio.pace@unito.it
bDepartment of Pharmaceutical Sciences, Division of Pharmaceutical Chemistry, Josef-Holaubek-Platz 2, 1090, Vienna, Austria
cUniversity of Milan – Department of Pharmaceutical Sciences, General and Organic Chemisty Section “A. Marchesini” – Via Venezian 21, 20133 Milan, Italy. E-mail: laura.castoldi@unimi.it
First published on 6th December 2024
Abstract
The chemoselective synthesis of trisubstituted alkenyl halides (Cl, Br, F, I) starting from ketones and aldehydes and lithium halocarbenoids is reported. Upon forming the corresponding tetrahedral intermediate adduct, followed by the addition of thionyl chloride, a selective E2-type elimination is triggered, furnishing the targeted motifs. The transformation takes place under full chemocontrol: various sensitive functionalities (e.g. ester, nitrile, nitro, or halogen groups) can be placed on the starting materials, thus documenting a wide reaction scope, as well as the application of the technique to biologically active substances.
The interest in the alkenyl halide moiety encompasses distinct areas of organic synthesis; in fact, it represents a versatile motif well suited for undergoing further elaboration through conceptually different regimes.1 As a consequence of the unique electronic environment imparted by the halogen, the material can be advantageously employed in C–C or C–heteroatom bond formation (either via polar metalation or transition-metal-catalyzed sequences – Scheme 1).2 Moreover, it is expressed in some natural products (mainly in the form of vinyl-type chlorides) and can also be present in medicinally relevant structures or agrochemicals.3
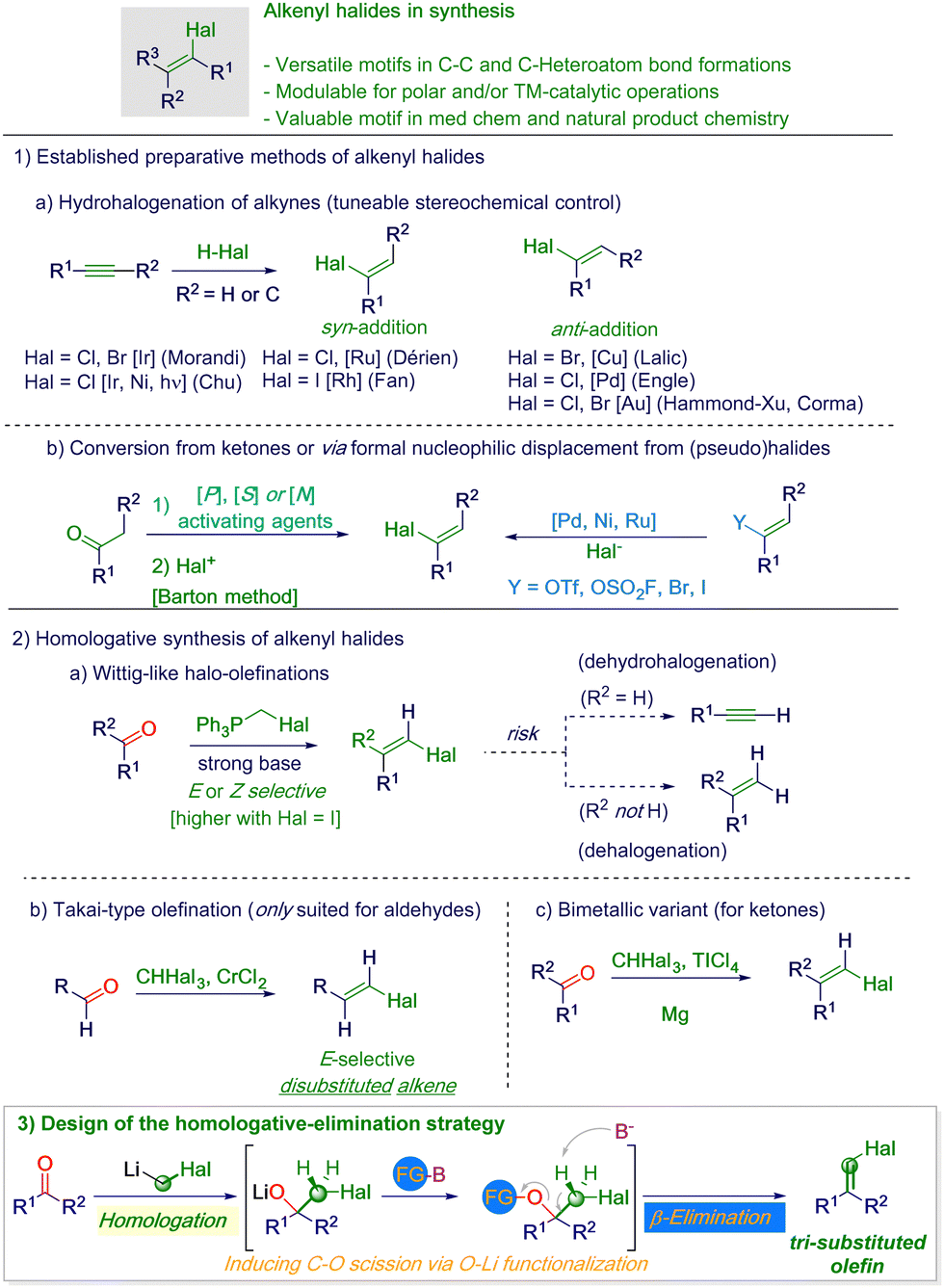 | ||
| Scheme 1 General context of the presented work. | ||
The desired substitution pattern–and consequently the stereochemistry–of the alkenyl cluster dictate the rationale for selecting the most suitable preparative route. Accordingly, the hydrohalogenation of alkynes offers a valuable strategy, which is also tuneable in both syn- and anti-Markovnikov-type additions when organic halides replace pure mineral acids (HX – Scheme 1, path 1a).4 Although various transition-metal-catalyzed approaches have been developed, procedures leading to alkenyl fragments featuring the four halogens have rarely been described.5 Nevertheless, previously considered elusive internal alkynes can nowadays be used as competent starting materials.5c,6 More traditional methods are based on the conversion of carbonyl groups into vinyl halides, well illustrated by the so-called Barton synthesis, which unfortunately shows high substrate sensitivity and may require the preparation of capricious intermediate species (e.g. hydrazones – Scheme 1, path 1b left).7 In this context, although Prati's modification addresses these difficulties, it still remains challenging for common materials such as benzophenones.8 Alternatively, vinylic-type nucleophilic substitutions conducted on functionalized alkenes in transition-metal-catalyzed reactions have been also reported (Scheme 1, path 1b right).9 From a specular perspective, the same carbonyl moiety constitutes a placeholder for a halo-alkene through a formal homologative event. Wittig-like methodologies with α-halosubstituted phosphorous ylides can furnish either E- or Z-olefins,10 but due to the highly basic conditions required, undesirable elimination to alkynes or reduction to methylene units could affect the chemoselectivity (Scheme 1, path 2a).11 In general, gaining remarkable control during these processes is somewhat counterbalanced by the restriction of the protocol to more electrophilic carbonyls (aldehydes). In this sense, the venerable Takai halo-olefination furnishes disubstituted alkenes starting from aldehydes and a haloform in the presence of problematic Cr(II) salts (Scheme 1, path 2b).12 However, engaging ketones proved to be more challenging and usually needed the employment of bimetallic species (e.g. Mg–TiCl4) to generate an adequately reactive nucleophile (Scheme 1, path 2c).13 Significantly, the inconvenient manipulability of fluoroform (bp −82 °C)14 precluded the adoption of these protocols for the preparation of alkenyl fluorides.
Cognizant of the excellent reactivity of lithiated halomethanes towards carbonyls (ketones and aldehydes) generating tetrahedral nucleophilic addition intermediates,15 we reasoned that the induction of a proper eliminative event would deliver the desired halo olefins (Scheme 1, path 3). To make the concept productive, it became critical to identify a suitable reactant (FG-B) that is able to assist the β-elimination directly on the tetrahedral intermediate during C–O bond scission. Herein, we report a robust and flexible protocol harnessed to a sequential homologation–elimination realized on a carbonyl precursor, furnishing alkenyl halides in high yield and stereo-preference for the more stable configurational isomer.
4-Bromoacetophenone (1) was selected as a suitable model compound susceptible to base-mediated enolate formation in the reaction with the basic carbenoid LiCH2Cl (Table 1). The constitutive presence of the exchangeable bromine atom would permit assessment of the chemocontrol over the transformation in the presence of the lithiated carbanion-type species. Based on our previous studies on homologative/deoxygenative sequences,16 1.4 equiv. of carbenoid–generated from 1.5 equiv. of ClCH2I and 1.4 equiv. of MeLi–LiBr at −78 °C in THF–we could verify the complete conversion (upon acidic quenching) of the ketone into chlorohydrine 1b (90% yield). With this confirmation in hand, we studied the subsequent E2-type elimination run on the lithiated chlorohydrin 1a triggered by an external agent. Thus, by slowly adding SOCl2 (1.5 equiv.) at −78 °C and stirring for 10 min followed by 30 min at ambient temperature, the desired E-chloroalkene (2) was obtained in 86% yield and E/Z ratio > 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, as deduced from NOESY experiments (entry 1). Some points merit mention: (a) Adding SOCl2 to the lithiated intermediate cooled at 0 °C had a detrimental effect, since the formation of impurities was observed in the 1H-NMR of the reaction crude and the E/Z ratio sensitively diminished (entry 2). (b) Continuing stirring at −78 °C did not induce any elimination and the corresponding chlorosulfite intermediates could be isolated (entry 3 - vide infra). (c) The addition of bases such as DIPEA or pyridine, although it enabled the formation of the alkene, was not comparable to the process carried out with SOCl2 alone, suggesting the chloride ion (released during the formation of the chlorosulfite ester) to be the active base for boosting elimination on intermediate 1c (entries 4 and 5). (d) Replacing SOCl2 with SOBr2 reduced the chemical yield (entry 6), as did the use of analogous (COCl)2 and phosphorous-based electrophiles (POCl3, POBr3, PCl3, entries 7–10). Plausibly, the release of gaseous SO2 at the end of the eliminative sequence accounts for the high efficiency of the transformation.
:1, as deduced from NOESY experiments (entry 1). Some points merit mention: (a) Adding SOCl2 to the lithiated intermediate cooled at 0 °C had a detrimental effect, since the formation of impurities was observed in the 1H-NMR of the reaction crude and the E/Z ratio sensitively diminished (entry 2). (b) Continuing stirring at −78 °C did not induce any elimination and the corresponding chlorosulfite intermediates could be isolated (entry 3 - vide infra). (c) The addition of bases such as DIPEA or pyridine, although it enabled the formation of the alkene, was not comparable to the process carried out with SOCl2 alone, suggesting the chloride ion (released during the formation of the chlorosulfite ester) to be the active base for boosting elimination on intermediate 1c (entries 4 and 5). (d) Replacing SOCl2 with SOBr2 reduced the chemical yield (entry 6), as did the use of analogous (COCl)2 and phosphorous-based electrophiles (POCl3, POBr3, PCl3, entries 7–10). Plausibly, the release of gaseous SO2 at the end of the eliminative sequence accounts for the high efficiency of the transformation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Eliminating agent (equiv.) | Temperature [°C] | Reaction timea (h) | Yield of 2b (%) | E/Z ratio of 2 |
| a Reaction time refers to the stirring of the mixture after removing the cooling bath. b Isolated yield. c See Scheme 3 for isolation of chlorosulfite intermediates. d DIPEA (1.3 equiv.) was added after concluding the addition of SOCl2. e Pyridine (1.3 equiv.) was added after concluding the addition of SOCl2. | |||||
| 1 | SOCl2 (1.5) | −78 to rt | 0.5 | 95 | >99:1 |
| 2 | SOCl2 (1.5) | 0 to rt | 0.5 | 67 | 93:7 |
| 3c | SOCl2 (1.5) | −78 | 2 | — | — |
| 4d | SOCl2 (1.5) | −78 to rt | 0.5 | 75 | 98:2 |
| 5e | SOCl2 (1.5) | −78 to rt | 0.5 | 58 | 98:2 |
| 6 | SOBr2 (1.5) | −78 to rt | 0.5 | 82 | >99:1 |
| 7 | (COCl)2 (1.5) | −78 to rt | 0.5 | 63 | 98:2 |
| 8 | POCl3 (1.5) | −78 to rt | 0.5 | 54 | 99:1 |
| 9 | POBr3 (1.5) | −78 to rt | 0.5 | 62 | 99:1 |
| 10 | PCl3 (1.5) | −78 to rt | 0.5 | 48 | >99:1 |

Having demonstrated that SOCl2 induces an E2-type elimination conducted on the nucleophilic addition intermediates (alkoxides)–without furnishing any (OH → Cl) substitution products17–we then studied the scope of the method (Scheme 2). Variously aryl-substituted acetophenones were all amenable substrates, giving the corresponding trisubstituted chloro-alkenes in very high chemical yield and E-selectivity. Not only could the whole series of halogens (2–5)–including the trifluoromethyl analogue (6)–be conveniently placed on the aromatic ring but substrates decorated with nitrile (7), ether (10) or ester (13) functionalities also worked equally well, furnishing in all cases halo-olefins in E/Z ratio > 99:1. The simple acetophenone (8) and the more sterically hindered o-methyl analogues (9) underwent the transformation with comparable efficiency. While using the (linear) propiophenone gave compound 11 in E/Z ratio > 99:1, switching to the highly sterically demanding t-butyl-phenyl ketone (12) reversed the ratio in favour of the Z-isomer. The employment of di(hetero)arylketones [benzophenones (14–17) the bis-thienyl-(18), the ferrocenyl-(19) and the bis-cyclohexyl (20) analogues] guaranteed access to (mainly) (Z)-halo-alkenes in comparable yields. Extending the protocol to bi- and tri-cyclic systems was possible, as observed in the cases of 1-indanone (21), 9H-fluoren-9-one (22), 9H-thioxanthen-9-one (23) and (the more elaborate) fluoro-substituted chromeno[2,3-c]pyrazol-4(1H)-one (24). Pure carbocyclic analogues, including octan-1-one (25) and the expanded 15-membered ketone (26) gave the trisubstituted alkenes in high yield (also when scaling up to 20 mmol), as well as, the sterically hindered adamant-1-one (27). The chemoselective methodology was not restricted to the use of LiCH2Cl as the C1-donor but was also applicable to the preparation of bromo-(28) iodo-(29) and fluoro-(30–31) olefins with LiCH2Br,18 LiCH2I19 and LiCH2F,20 respectively, showing remarkable synthetic versatility. Conjugated haloalkenes could be easily prepared in the cases of both cyclic (32) and acyclic (33) materials. It is interesting to highlight the success of the transformation conducted on the anthelmintic natural product Santonin (32) whose constitutive lactone moieties were inert under the reaction conditions. As a consequence of the higher thermodynamic stability of endocyclic double bonds (compared to exocyclic ones),21 vinyl–allyl halide isomerization could take place, yielding structures 34–37 during the usual work-up. Again, the application of the method to the narcotic drug codeinone (37) further illustrates the significance of the protocol for the elaboration of medicinally relevant substances. Finally, switching to aromatic aldehydes as electrophilic partners for carbenoids–coeteris paribus–provided a smooth route to β-halostyrenes (38–40), validating the protocol for obtaining (E)-haloalkenes.22
 | ||
| Scheme 2 Scope of the sequential carbonyl homologation/elimination en route to haloalkenes. | ||
As mentioned in the optimization study (Table 1), by keeping the temperature at −78 °C after the addition of SOCl2, it was possible to unambiguously demonstrate the genesis of chlorosulfite esters as the pertinent intermediates (Scheme 3). Nevertheless, subsequent treatment with a solution of LiCl in THF at room temperature yielded the corresponding halo-alkene 44, albeit with poorer stereocontrol (presumably due to conducting the reaction at high temperature).
 | ||
| Scheme 3 Trapping chlorosulfite esters for mechanistic analysis. | ||
In summary, we have reported an effective synthesis for trisubstituted halo-alkenes from ketones through a sequential homologation with lithium halocarbenoids (LiCH2Cl, LiCH2Br, LiCH2I and LiCH2F–generated from dihalomethanes and MeLi–LiBr under Barbier-type conditions at −78 °C), followed by an E2-type elimination triggered on the tetrahedral intermediate with thionyl chloride. The protocol exhibits remarkable chemocontrol, as indicated by reacting ketones presenting a wide range of chemical functionalities (e.g. halogen, azido, nitrile, ferrocenyl, ether, ester, lactone, conjugated olefin, or amine), which, in principle, may interfere with the carbenoids during the homologative event. Not only are simple alkl-aryl ketones amenable for the process, but (hetero)aryl-(hetero)aryl,(alkyl)–(alkyl) including macrocycles and a series of biologically active substrates featuring the ketone group could also be equally employed. The application of the method to aromatic aldehydes furnishes β-halostyrenes, whereas aliphatic analogues are currently under investigation and the results will be reported in due course.
We thank the University of Vienna, the University of Turin and All4Labels Group (Hamburg, Germany) for generous funding. The support from Project CH4.0 under MUR (Italian Ministry for the University) program “Dipartimenti di Eccellenza 2023–2027 (CUP: D13C22003520001) and PRIN 2022 (ref. 20228W9TBL) are gratefully acknowledged.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, S. Brase and M. Oestreich, Wiley-VCH, Weinheim, 2014 Search PubMed.
- (a) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003 CrossRef CAS PubMed; (b) P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302 CrossRef CAS PubMed; (c) Organometallics in Synthesis, ed. M. Schlosser, John Wiley and Sons, Hoboken, Third Manual, 2013 Search PubMed.
- G. W. Gribble, Acc. Chem. Res., 1998, 31, 141 CrossRef CAS.
- (a) P. Yu, A. Bismuto and B. Morandi, Angew. Chem., Int. Ed., 2020, 59, 2904 CrossRef CAS PubMed; (b) S. Liang, R. Ebule, G. B. Hammond and B. Xu, Org. Lett., 2017, 19, 4524 CrossRef CAS PubMed; (c) J. A. Mulder, K. C. M. Kurtz, R. P. Hsung, H. Coverdale, M. O. Frederick, L. Shen and C. A. Zificsak, Org. Lett., 2003, 5, 1547 CrossRef CAS PubMed; (d) D. Shen, D. Cao, R. Zhang, P. Bai and Z. Liu, Org. Biomol. Chem., 2024, 22, 4062 RSC; (e) D. Shen, D. Cao, P. Bai and Z. Liu, J. Org. Chem., 2024, 89, 11818 CrossRef CAS PubMed; (f) H. Asahara, Y. Mukaijo, K. Muragishi, K. Iwai, A. Ito and N. Nishiwaki, Eur. J. Org. Chem., 2021, 5747 CrossRef CAS; (g) W. Chen, J. C. L. Walker and M. Oestreich, J. Am. Chem. Soc., 2019, 141, 1135 CrossRef CAS PubMed; (h) D. A. Petrone, I. Franzoni, J. Ye, J. F. Rodríguez, A. I. Poblador-Bahamonde and M. Lautens, J. Am. Chem. Soc., 2017, 139, 3546 CrossRef CAS PubMed; (i) B. Wang, J.-l Jiang, H.-z Yu and Y. Fu, Organometallics, 2017, 36, 523 CrossRef CAS.
- (a) M. R. Uehling, R. P. Rucker and G. Lalic, J. Am. Chem. Soc., 2014, 136, 8799 CrossRef CAS PubMed; (b) R. Ebule, S. Liang, G. B. Hammond and B. Xu, ACS Catal., 2017, 7, 6798 CrossRef CAS PubMed; (c) J. Derosa, A. L. Cantu, M. N. Boulous, M. L. O’Duill, J. L. Turnbull, Z. Liu, D. M. De La Torre and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 5183 CrossRef CAS PubMed; (d) S. Dérien, H. Klein and C. Bruneau, Angew. Chem., Int. Ed., 2015, 54, 12112 CrossRef PubMed; (e) J. Chen, W.-T. Wei, Z. Li and Z. Lu, Chem. Soc. Rev., 2024, 53, 7566 RSC; (f) K. Iwai and N. Nishiwaki, Adv. Synth. Catal., 2024, 366, 1950 CrossRef CAS; (g) Y. Bai, Z. Lin, Z. Ye, D. Dong, J. Wang, L. Chen, F. Xie, Y. Li, P. H. Dixneuf and M. Zhang, Org. Lett., 2022, 24, 7988 CrossRef CAS PubMed; (h) J. Li, R. Devi Laishram, J. Chen, D. Xu, G. Shi, H. Lv, R. Fan and B. Fan, Asian J. Org. Chem., 2020, 9, 73 CrossRef CAS; (i) J. Oliver-Meseguer, A. Doménech-Carbó, M. Boronat, A. Leyva-Pérez and A. Corma, Angew. Chem., Int. Ed., 2017, 56, 6435 CrossRef CAS PubMed.
- L. Huo, X. Li, Y. Zhao, L. Li and L. Chu, J. Am. Chem. Soc., 2023, 145, 9876 CrossRef CAS PubMed.
- (a) D. H. R. Barton, R. E. O'Brien and S. Sternhell, J. Chem. Soc., 1962, 470 RSC; (b) M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 5436 CrossRef CAS PubMed; (c) D. P. Ojha and K. R. Prabhu, Org. Lett., 2015, 17, 18 CrossRef CAS PubMed; (d) M. A. Saputra, L. Ngo and R. Kartika, J. Org. Chem., 2015, 80, 8815 CrossRef CAS PubMed; (e) J. Kagan, S. K. Arora, M. Bryzgis, S. N. Dhawan, K. Reid, S. P. Singh and L. Tow, J. Org. Chem., 1983, 48, 703 CrossRef CAS; (f) W. Su and C. Jin, Org. Lett., 2007, 9, 993 CrossRef CAS PubMed; (g) K. Kamei, N. Maeda and T. Tatsuoka, Tetrahedron Lett., 2005, 46, 229 CrossRef CAS.
- A. Spaggiari, D. Vaccari, P. Davoli, G. Torre and F. Prati, J. Org. Chem., 2007, 72, 2216 CrossRef CAS PubMed.
- (a) J. L. Hofstra, K. E. Poremba, A. M. Shimozono and S. E. Reisman, Angew. Chem., Int. Ed., 2019, 58, 14901 CrossRef CAS PubMed; (b) A. Nitelet and G. Evano, Org. Lett., 2016, 18, 1904 CrossRef CAS PubMed; (c) X. Shen, A. M. Hyde and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14076 CrossRef CAS PubMed; (d) Y. Imazaki, E. Shirakawa, R. Ueno and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 14760 CrossRef CAS PubMed; (e) M.-Y. Chen, S. Charvet, P.-A. Payard, M.-E. L. Perrin and J. C. Vantourout, Angew. Chem., Int. Ed., 2024, 63, e202311165 CrossRef CAS PubMed.
- (a) X.-p Zhang and M. Schlosser, Tetrahedron Lett., 1993, 34, 1925 CrossRef CAS; (b) G. Stork and K. Zhao, Tetrahedron Lett., 1989, 30, 2173 CrossRef CAS; (c) M.-E. Lebrun, P. Le Marquand and C. Berthelette, J. Org. Chem., 2006, 71, 2009 CrossRef CAS PubMed.
- S. Miyano, Y. Izumi, K. Fujii, Y. Ohno and H. Hashimoto, Bull. Chem. Soc. Jpn., 2006, 52, 1197 CrossRef.
- (a) K. Takai, K. Nitta and K. Utimoto, J. Am. Chem. Soc., 1986, 108, 7408 CrossRef CAS; (b) K. Takai, T. Ichiguchi and S. Hikasa, Synlett, 1999, 1268 CrossRef CAS; (c) J. M. Concellón, P. L. Bernad and C. Méjica, Tetrahedron Lett., 2005, 46, 569 CrossRef.
- (a) C.-C. Tsai, C.-T. Chien, Y.-C. Chang, H.-C. Lin and T.-H. Yan, J. Org. Chem., 2005, 70, 5745 CrossRef CAS PubMed; (b) Y. R. Bhorge, S.-H. Chang, C.-T. Chang and T.-H. Yan, Tetrahedron, 2012, 68, 4846 CrossRef CAS; (c) T.-H. Yan and M.-Y. Chen, J. Chin. Chem. Soc., 2017, 64, 390 CrossRef CAS.
- S. Kyasa, Synlett, 2015, 1911 CAS.
- (a) V. Pace, L. Castoldi, E. Mazzeo, M. Rui, T. Langer and W. Holzer, Angew. Chem., Int. Ed., 2017, 56, 12677 CrossRef CAS PubMed; (b) V. Pace, L. Castoldi, A. D. Mamuye, T. Langer and W. Holzer, Adv. Synth. Catal., 2016, 358, 172 CrossRef CAS; (c) L. Castoldi, S. Monticelli, R. Senatore, L. Ielo and V. Pace, Chem. Commun., 2018, 54, 6692 RSC; (d) S. Monticelli, L. Castoldi, S. Touqeer, M. Miele, E. Urban and V. Pace, Chem. Heterocycl. Compd., 2018, 54, 389 CrossRef CAS; (e) L. Ielo, V. Pillari, M. Miele, D. Castiglione and V. Pace, Synlett, 2021, 551 CrossRef CAS; (f) L. Degennaro, F. Fanelli, A. Giovine and R. Luisi, Adv. Synth. Catal., 2015, 357, 21 CrossRef CAS.
- (a) M. Miele, A. Citarella, T. Langer, E. Urban, M. Zehl, W. Holzer, L. Ielo and V. Pace, Org. Lett., 2020, 22, 7629 CrossRef CAS PubMed; (b) L. Ielo, M. Miele, V. Pillari, R. Senatore, S. Mirabile, R. Gitto, W. Holzer, A. R. Alcántara and V. Pace, Org. Biomol. Chem., 2021, 19, 2038 RSC.
- G. A. Olah, H. Wu and O. Farooq, J. Org. Chem., 1989, 54, 1375 CrossRef.
- (a) L. Ielo, V. Pillari, M. Miele, W. Holzer and V. Pace, Adv. Synth. Catal., 2020, 362, 5444 CrossRef CAS; (b) M. Miele, L. Castoldi, X. Simeone, W. Holzer and V. Pace, Chem. Commun., 2022, 58, 5761 RSC.
- L. Ielo, L. Castoldi, S. Touqeer, J. Lombino, A. Roller, C. Prandi, W. Holzer and V. Pace, Angew. Chem., Int. Ed., 2020, 59, 20852 Search PubMed.
- (a) G. Parisi, M. Colella, S. Monticelli, G. Romanazzi, W. Holzer, T. Langer, L. Degennaro, V. Pace and R. Luisi, J. Am. Chem. Soc., 2017, 139, 13648 CrossRef CAS PubMed; (b) M. Colella, P. Musci, M. Andresini, M. Spennacchio, L. Degennaro and R. Luisi, Org. Biomol. Chem., 2022, 20, 4669 RSC; (c) M. Colella, A. Tota, Y. Takahashi, R. Higuma, S. Ishikawa, L. Degennaro, R. Luisi and A. Nagaki, Angew. Chem., Int. Ed., 2020, 59, 10924 CrossRef CAS PubMed; (d) M. Miele, R. D’Orsi, V. Sridharan, W. Holzer and V. Pace, Chem. Commun., 2019, 55, 12960 RSC.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley-VCH, Weinheim, 5th edn, 2001 Search PubMed.
- J. A. Bull, J. J. Mousseau and A. B. Charette, Org. Lett., 2008, 10, 5485 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05937a |
| This journal is © The Royal Society of Chemistry 2025 |