
Straight-chain ω-amino-α,β-unsaturated carbonyl compounds: versatile synthons for the synthesis of nitrogen-containing heterocycles via organocatalytic reactions
Shuhui
Li†
,
Xiaoxuan
Li†
,
Hui
Yao
 *,
Mengting
Tan
,
Dan
Xu
,
Nianyu
Huang
* and
Nengzhong
Wang
*
*,
Mengting
Tan
,
Dan
Xu
,
Nianyu
Huang
* and
Nengzhong
Wang
*
Hubei Key Laboratory of Natural Products Research and Development, Key Laboratory of Functional Yeast (China National Light Industry), College of Biological and Pharmaceutical Sciences, China Three Gorges University, Yichang 443002, P. R. China. E-mail: yaohui@ctgu.edu.cn; huangny@ctgu.edu.cn; wangnz@ctgu.edu.cn
First published on 30th November 2023
Abstract
Straight-chain ω-amino-α,β-unsaturated carbonyl compounds have been recognized as versatile organic synthons participating in various organocatalytic reactions for the creation of five- and six-membered saturated nitrogen-containing heterocycles, which are important subunits in a broad range of naturally occurring compounds, biologically active molecules, and drugs. Consequently, these organic synthons have attracted significant interest from synthetic and pharmaceutical chemists, and a series of methodologies and strategies have been efficiently developed. In this review, we elaborate and discuss the recent applications of such versatile synthons according to the following organocatalytic reaction modes: (1) covalent catalysis (secondary amine catalysis, phosphine catalysis, and N-heterocyclic carbene catalysis), (2) non-covalent catalysis (Brønsted acid catalysis), and (3) bifunctional catalysis (bifunctional tertiary amine/primary amine catalysis, bifunctional tertiary amine/squaramide catalysis, and bifunctional tertiary amine/thiourea catalysis).
1. Introduction
Five- and six-membered saturated nitrogen-containing heterocycles, such as pyrrolidines and piperidines, are common scaffolds in a wide range of naturally occurring compounds, biologically active molecules, and drug molecules, and have attracted considerable attention from the synthetic and pharmaceutical communities (Fig. 1).1 In order to obtain them quickly and easily, synthetic chemists have designed various versatile organic synthons and developed a series of efficient methods.2 Among the designed synthons, straight-chain ω-amino-α,β-unsaturated carbonyl compounds bearing both a nitrogen-centered nucleophile and a Michael acceptor could undergo intra- and intermolecular aza-Michael addition reactions for the synthesis of a C–N bond in nitrogen-containing heterocycles. The length of the carbon chain can be adjusted to generate five- or six-membered nitrogen-containing heterocycles. In addition, multi-heteroatom rings can also be constructed when one of the carbon atoms on the carbon chain is replaced with other heteroatoms, such as nitrogen, oxygen, and sulfur atoms. Therefore, straight-chain ω-amino-α,β-unsaturated carbonyl compounds, including α,β-unsaturated aldehydes/ketones/esters/thioesters/acids/amides, prove to be versatile organic synthons participating in diverse organic reactions.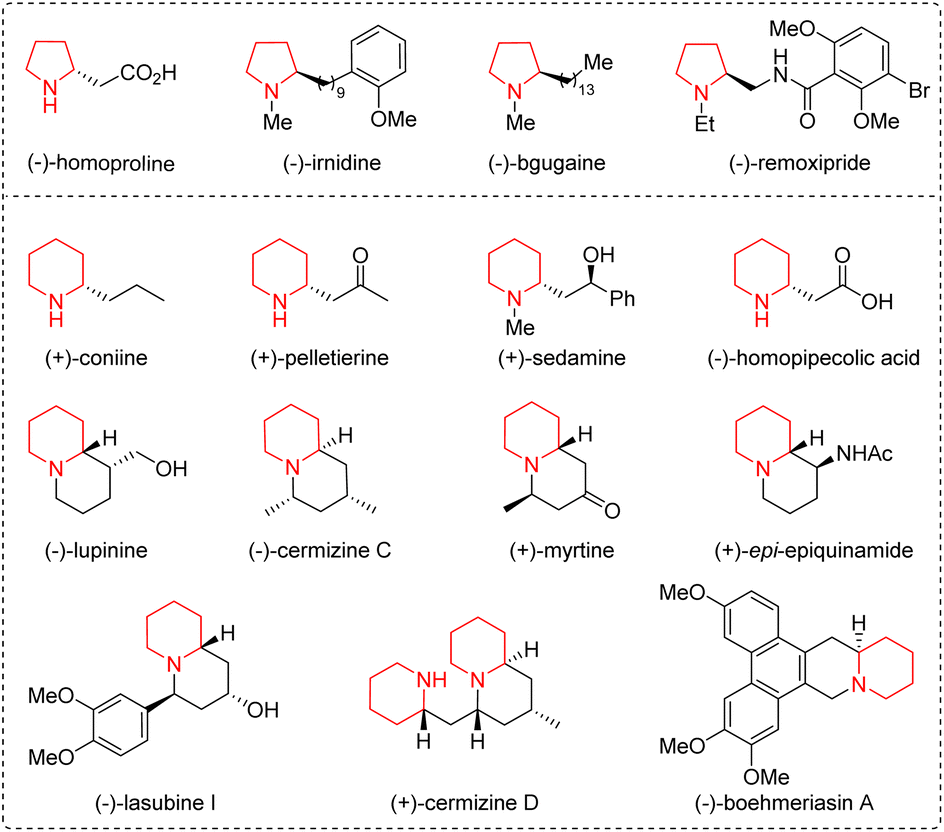 | ||
| Fig. 1 Representative natural products and drugs containing pyrrolidine/piperidine subunits. | ||
Organocatalysis, besides metal catalysis and biocatalysis, is now recognized as the third pillar of catalysis.3 In light of the advantages of organocatalysis, including readily available starting materials and catalysts, atom- and step-economy, environmental friendliness, and mild reaction conditions, in constructing ring structures, numerous novel methods have been developed. Among them, the organocatalytic aza-Michael addition reaction is one of the most common and extensively used strategies for the construction of the C–N bond in nitrogen-containing heterocycles.4
Since the pioneering work on the application of straight-chain ω-amino-α,β-unsaturated carbonyl compounds in the organocatalytic aza-Michael addition reaction by Fustero and co-workers in 2007,5 more than 40 original literature examples have been published. Herein, this review focuses on the recent advances in straight-chain ω-amino-α,β-unsaturated carbonyl compounds as versatile synthons for the preparation of five- and six-membered saturated nitrogen-containing heterocycles via organocatalytic reactions, including intramolecular aza-Michael addition reactions, cascade intermolecular aza-Michael addition/Michael addition reactions, cascade Mannich/aza-Michael addition reactions, and so on.
In the area of organocatalysis, several types of small organic molecules have been designed and classified based on the activation mode between the catalyst and substrate. As shown in Fig. 2, all the organic catalysts that have been described in this review are summarized. From a mechanistic viewpoint, secondary amines,6 phosphines,7 and N-heterocyclic carbenes8 are termed covalent catalysts because of the formation of a covalent bond between the catalyst and substrate. Brønsted acid catalysts9 rely on hydrogen-bonding interactions between the catalyst and substrate, and can be regarded as representative non-covalent catalysts.
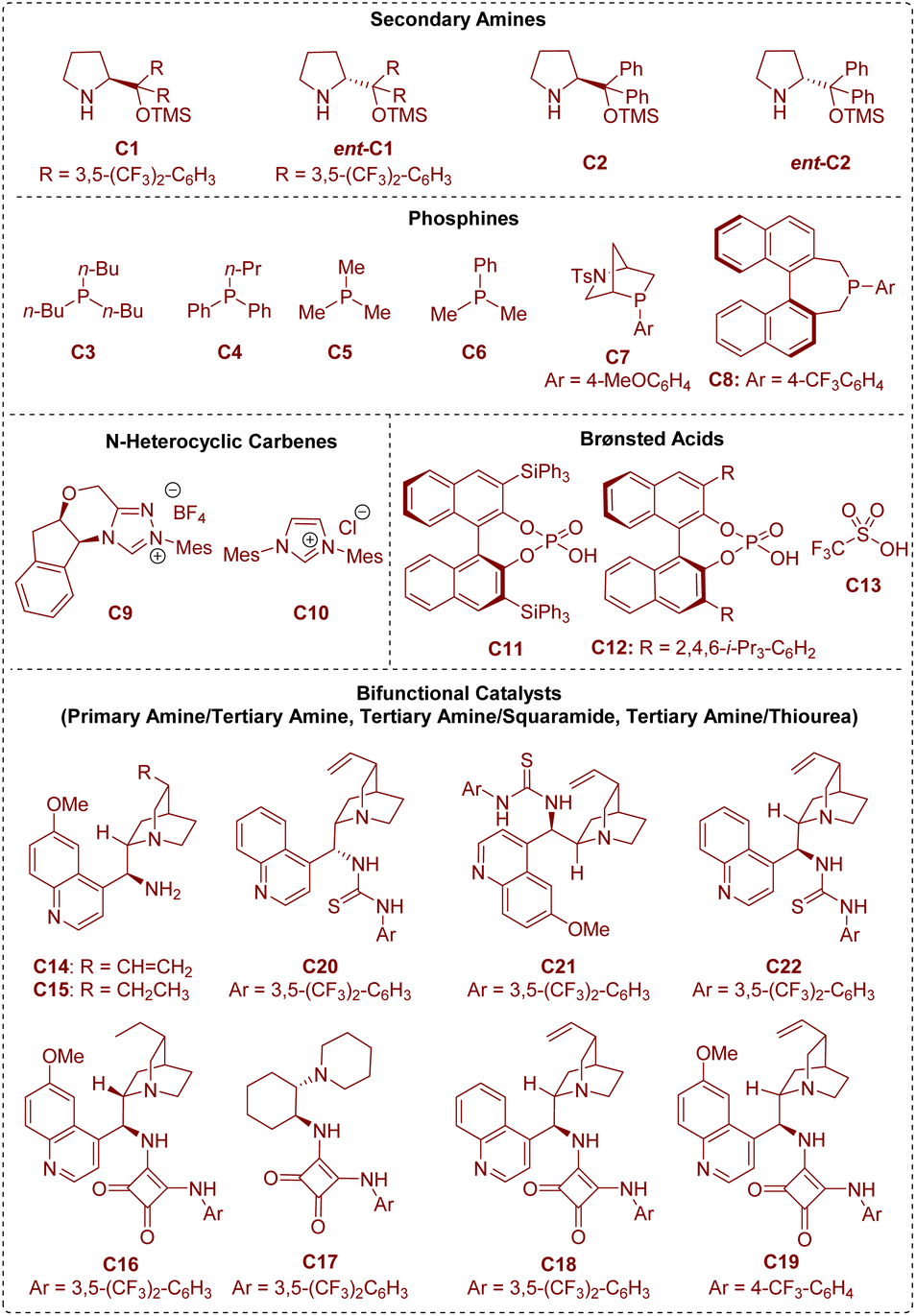 | ||
| Fig. 2 Organocatalysts utilized in the studies that have been described in this review. | ||
Besides, there is another kind of compound called a bifunctional catalyst based on covalent interactions/non-covalent interactions (bifunctional tertiary amines/primary amines10) or non-covalent interactions/non-covalent interactions (bifunctional tertiary amines/squaramides11 and bifunctional tertiary amines/thioureas12). According to the above organocatalytic reaction modes, this review will be described in the following three sections: (1) covalent catalysis (secondary amine catalysis, phosphine catalysis, and N-heterocyclic carbene catalysis), (2) non-covalent catalysis (Brønsted acid catalysis), and (3) bifunctional catalysis (bifunctional tertiary amine/primary amine catalysis, bifunctional tertiary amine/squaramide catalysis, and bifunctional tertiary amine/thiourea catalysis).
2. Covalent catalysis
2.1. Secondary amine catalysis
Secondary amine catalysis is ubiquitous and extremely important in organic synthesis. Since first reported by Knoevenagel in 1894,13 secondary amine catalysis has been widely used in the development of various synthetic methods for the creation of novel molecules and frequently used as a key step in the total synthesis of natural products and drug molecules.6 The catalytic reaction mechanism of secondary amine catalysis can be categorized into two modes: iminium ion activation and enamine activation. In this review, we will discuss the iminium ion activation mode derived from α,β-unsaturated aldehydes with diarylprolinol silyl ethers. In such reactions, the diarylprolinol silyl ethers not only effectively shield one of the enantiotropic faces of the α,β-unsaturated aldehydes but also ensure excellent chemoselectivity, producing 1,4-adducts as the only products.In 2007, Fustero and co-workers developed an enantioselective intramolecular aza-Michael reaction of straight-chain ω-amino-α,β-unsaturated aldehydes 1 in the presence of diarylprolinol silyl ether C1 and benzoic acid as a co-catalyst, producing several five- and six-membered saturated nitrogen-containing heterocycles 2 (10 examples) in moderate to good yields (30–80%) with excellent enantioselectivities (85–99% ee) (Scheme 1).5 This methodology was a general protocol to perform a highly enantioselective intramolecular aza-Michael reaction in an efficient manner. The authors proposed a possible mechanism for the catalytic reaction. Initially, the condensation of ω-amino-α,β-unsaturated aldehydes 1 with the catalyst C1 formed iminium ion TS-1. Subsequently, the intramolecular Michael addition of the nucleophile to the β-carbon atom generated enamine intermediate TS-2. The Si face of the iminium ion TS-1 was shielded by the chiral steric hindrance group in the catalyst, resulting in a nitrogen atom on the Re face. It is worth noting that the nitrogen atom attacked the β-carbon atom of the iminium ion TS-1 on the Si face when X = NCbz, O, or S because the priority of the substituents was reversed. Tautomerization of enamine intermediate TS-2 produced iminium ion TS-3, which was hydrolysed to give the desired products 2 and regenerated the catalyst C1. Reduction of aldehydes 3 with NaBH4 in MeOH afforded primary alcohols 2. The developed methodology was applied to the synthesis of three piperidine alkaloids, namely (+)-coniine (4), (+)-sedamine (5), and (+)-allosedamine (6). The Wittig reaction of piperidine 3a followed by hydrogenation of the double bond and Cbz gave (+)-coniine (4) in 65% yield. Furthermore, treatment of piperidine 3b with PhMgBr generated a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of secondary alcohols, which were separated by chromatography and the Boc group was reduced with LiAlH4 to forge (+)-sedamine (5) and (+)-allosedamine (6).
:2 mixture of secondary alcohols, which were separated by chromatography and the Boc group was reduced with LiAlH4 to forge (+)-sedamine (5) and (+)-allosedamine (6).
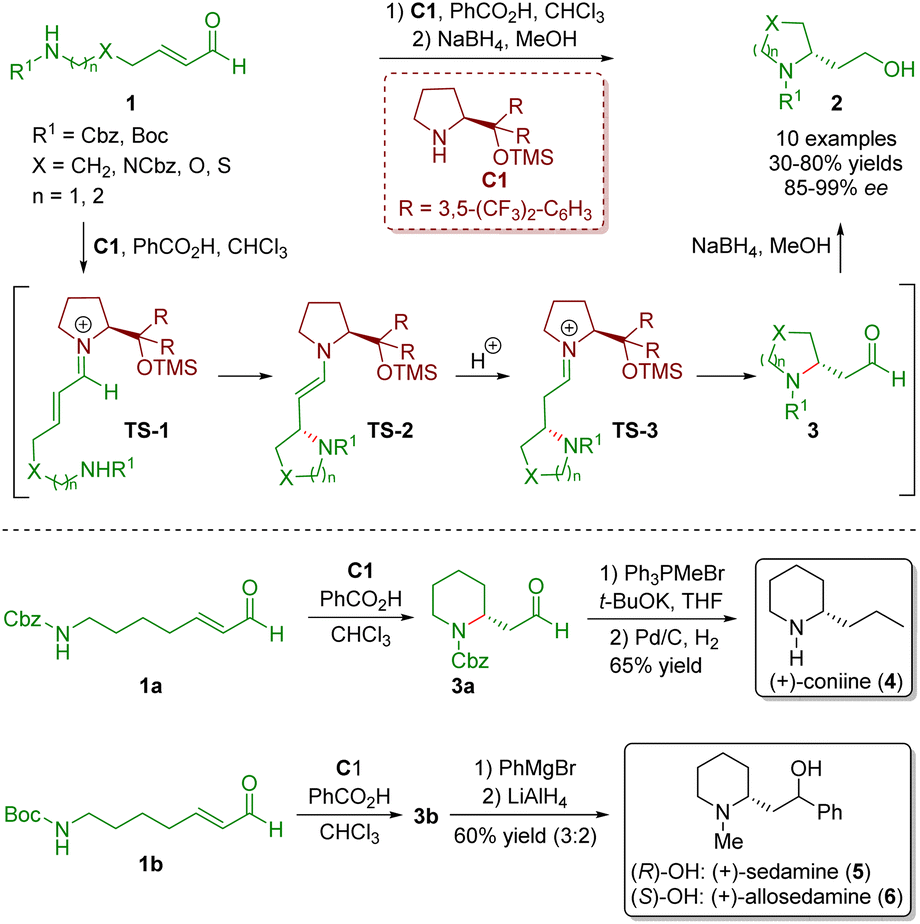 | ||
| Scheme 1 Diarylprolinol silyl ether-catalyzed intramolecular aza-Michael reaction and synthesis of three piperidine alkaloids (Fustero, 20075). | ||
In order to extend the developed novel strategy, they accomplished the asymmetric total synthesis of three quinolizidine alkaloids, namely (+)-myrtine (7), (+)-lupinine (9), and (+)-epi-epiquinamide (10), from the above common intermediate 3b in 5–9 steps (Scheme 2).14
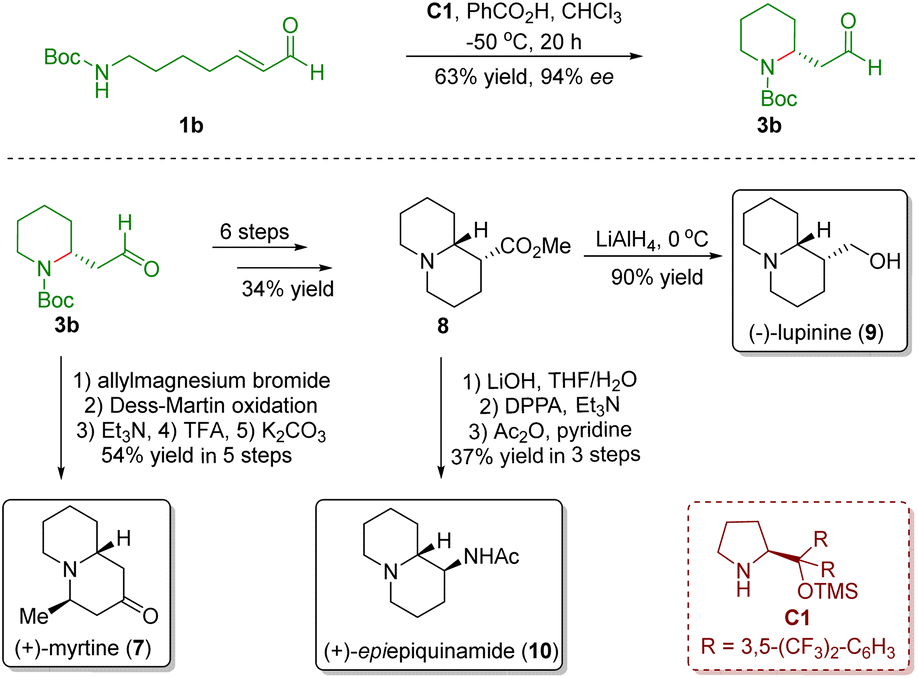 | ||
| Scheme 2 Enantioselective divergent synthesis of three quinolizidine alkaloids (+)-myrtine, (+)-lupinine, and (+)-epi-epiquinamide (Fustero and del Pozo, 201114). | ||
In 2008, Carter and co-workers demonstrated a strategy similar to that reported by Fustero and co-workers, which involved the use of MeOH/DCE as solvent and did not need acidic additives (Scheme 3).15 The authors also applied the developed methodology to the asymmetric synthesis of (−)-homoproline (11), (−)-homopipecolic acid (12), and (+)-pelletierine (13). The key intermediates pyrrolidine 3c and piperidine 3a were obtained via a diarylprolinol silyl ether-catalyzed intramolecular aza-Michael reaction of ω-amino-α,β-unsaturated aldehydes 1c and 1a, respectively. Pinnick oxidation of aldehydes 1c and 1a followed by Cbz-deprotection in the presence of Pd/C and H2 completed the synthesis of (−)-homoproline (11) and (−)-homopipecolic acid (12), respectively. On the other hand, Grignard addition of aldehyde 3a yielded secondary alcohol 14, which was converted into (+)-pelletierine (13) via Dess–Martin oxidation and then Cbz-deprotection.
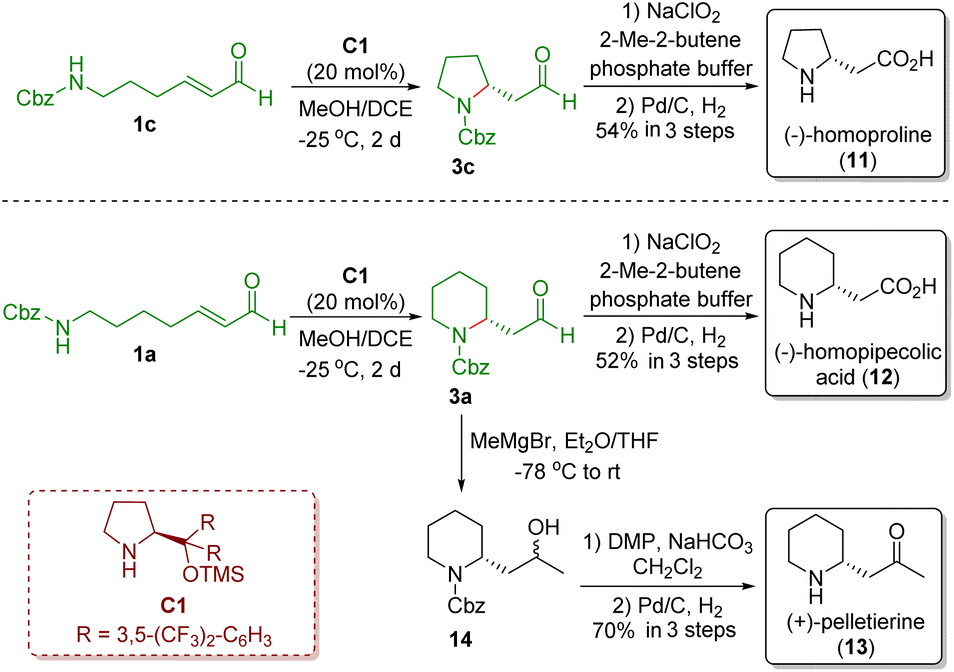 | ||
| Scheme 3 Asymmetric synthesis of (−)-homoproline, (−)-homopipecolic acid, and (+)-pelletierine (Carter, 200815). | ||
Based on the above protocol, Carter and co-workers also accomplished the asymmetric total synthesis of (+)-cermizine D (18) in 2012 (Scheme 4).16 First, the diarylprolinol silyl ether-catalyzed intramolecular aza-Michael reaction of ω-amino-α,β-unsaturated aldehyde 1b generated piperidine ent-3b in 85% yield with 96% ee. The Horner–Wadsworth–Emmons olefination of aldehyde ent-3b gave unsaturated sulfone 15 in 83% yield with a 4:1 E/Z ratio. Then, conjugate addition of Me2CuLi with 15 afforded sulfone 16 in 55% yield and 1:1.2 dr. Addition of sulfone 16 with aldehyde ent-3b in the presence of LDA generated hydroxy sulfone 17 in 93% yield and 1.5:1 dr. Finally, hydroxy sulfone 17 was transformed into (+)-cermizine D (18) via a five-step sequence including oxidation, reduction, desulfurization, Boc-deprotection, and cyclization.
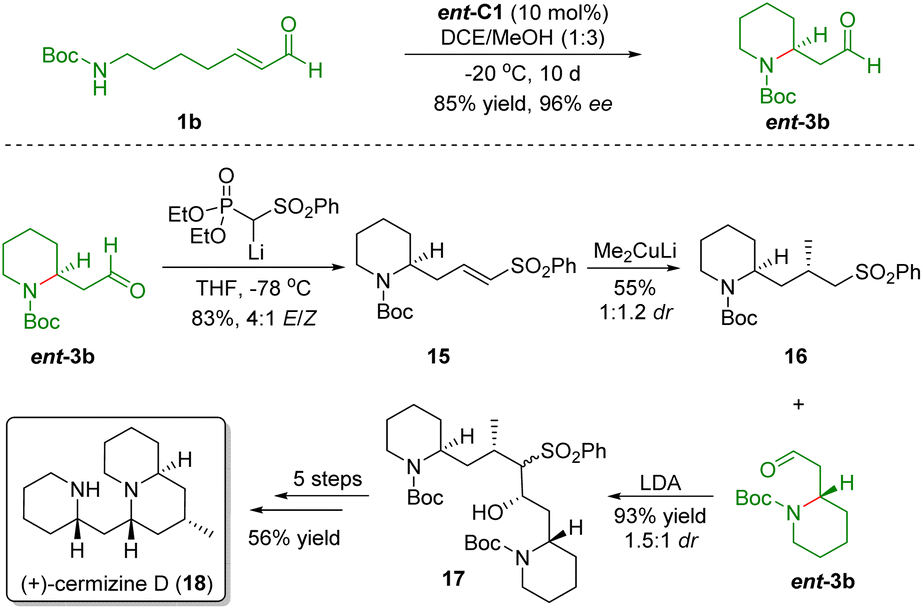 | ||
| Scheme 4 Asymmetric total synthesis of (+)-cermizine D (Carter, 201216). | ||
After completing the synthesis of (+)-cermizine D, Carter and co-workers showcased the formal synthesis of cermizine C (22) and senepodine G (23) using the same key reaction and the common intermediate 17 (Scheme 5).17 The poor diastereoselectivity of sulfone 16 reported in their previous work prompted them to develop a novel approach. In this work, the authors employed a chiral auxiliary to improve the diastereoselectivity from 1:1.2 to 10:1. Treatment of sulfone 16 with LDA followed by desulfurization afforded the known lactam intermediate 21 reported by Snider and co-workers.18 In this manner, the formal synthesis of cermizine C (22) and senepodine G (23) was accomplished.
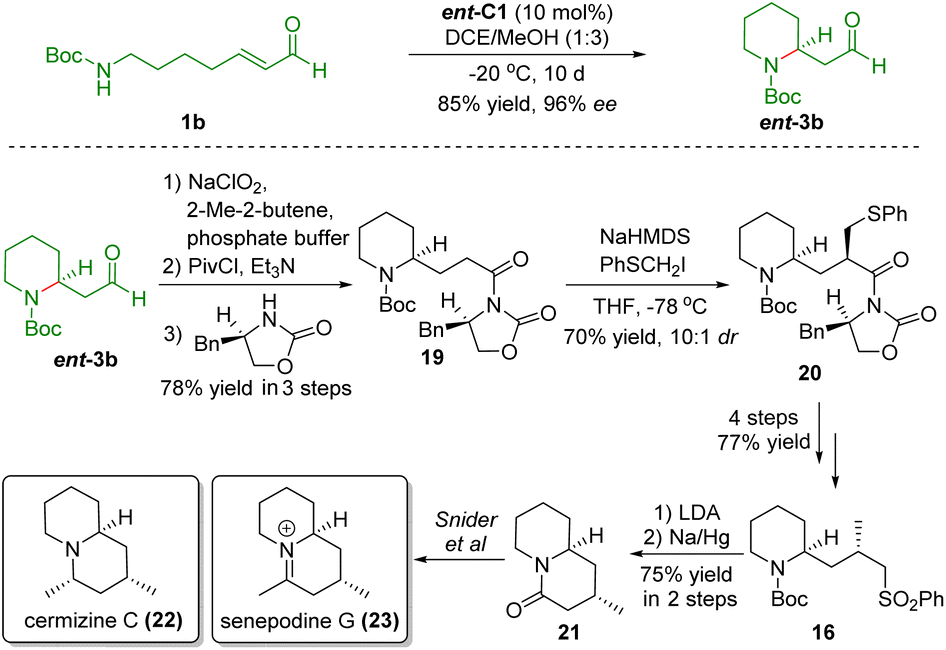 | ||
| Scheme 5 Formal synthesis of senepodine G and cermizine C (Carter, 201317). | ||
In 2011, Hong and co-workers described a diarylprolinol silyl ether-catalyzed intramolecular aza-Michael reaction of ω-amino-α,β-unsaturated aldehydes 1 for the stereoselective synthesis of both 2,6-cis- and 2,6-trans-piperidines 24 and 25 which was promoted by the gem-disubstituent effect (Scheme 6).19 Subsequently, the authors employed the developed methodology to complete the concise synthesis of (−)-epimyrtine (28) and (+)-myrtine (7). The key reaction was carried out in the presence of diarylprolinol silyl ethers C2 or ent-C2 to give the piperidines 24 or 25 in high yields (75–97%) with moderate to excellent stereoselectivities (1:1–>20:1). The Wittig reaction of aldehyde 24 with methyl(triphenylphosphoranylidene)acetate gave (E)-α,β-unsaturated ester 26, which was converted into (−)-epimyrtine (28) via a four-step sequence involving Ts-deprotection, LiAlH4-reduction, mesylation/intramolecular N-alkylation, and deprotection of 1,3-dithiane. On the other hand, Still–Gennari olefination of the inseparable mixture of 25 and 24 (4:1) afforded (Z)-α,β-unsaturated ester 27 by a separation process. The (Z)-α,β-unsaturated ester 27 was converted into (+)-myrtine (7) by following the procedures described above.
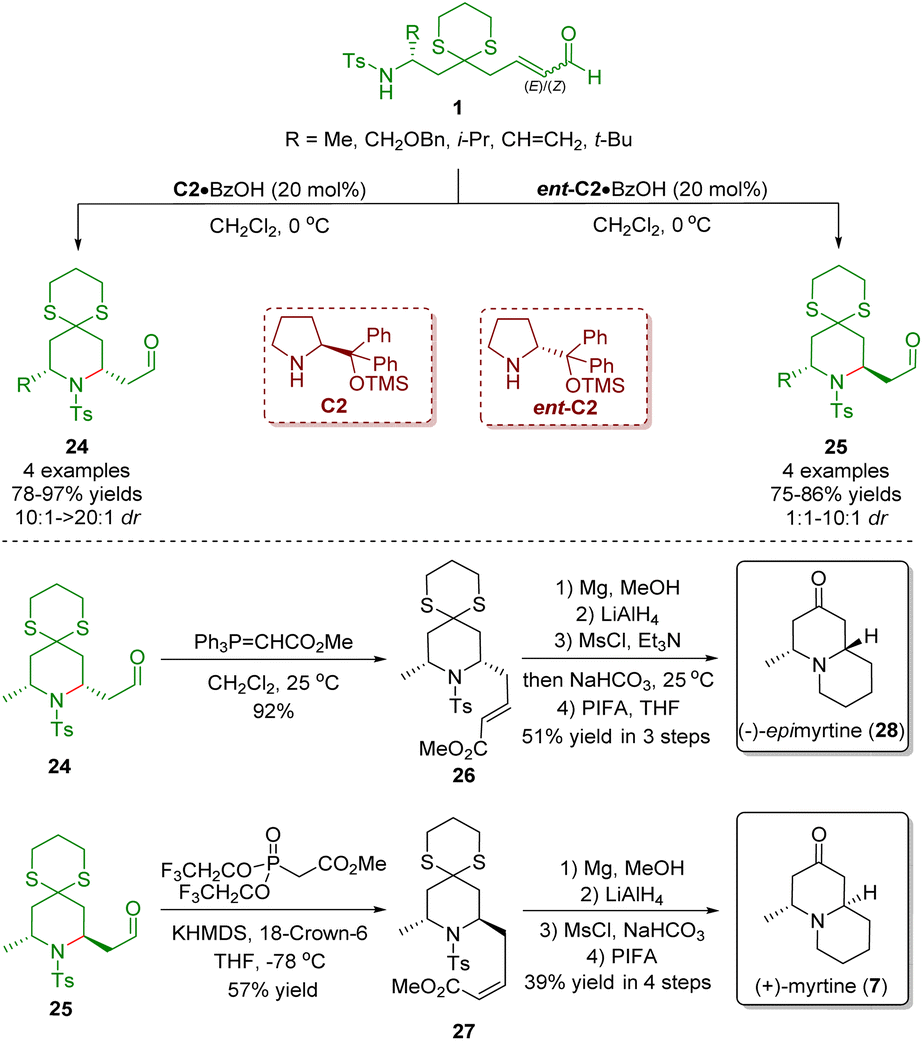 | ||
| Scheme 6 Asymmetric synthesis of (−)-epimyrtine and (+)-myrtine (Hong, 201119). | ||
In addition to intramolecular cyclization reactions, diaryl–prolinol silyl ether-catalyzed intermolecular annulation reactions have also been successfully developed. In particular, various multi-substituted heterocycles can be smoothly constructed via diarylprolinol silyl ether-catalyzed intermolecular reactions of ω-amino-α,β-unsaturated aldehydes 1 with different partners.
In 2008, Wang and co-workers developed an unprecedented highly enantio- and diastereoselective cascade aza-Michael/Michael reaction of trans-γ-Ts protected amino α,β-unsaturated ester 1d with α,β-unsaturated aldehydes 29 for establishing highly functionalized trisubstituted chiral pyrrolidines 30 in high yields (80–94%) with outstanding levels of enantioselectivities (96–>99% ee) and good to excellent diastereoselectivities (7:1–>30:1 dr) (Scheme 7).20
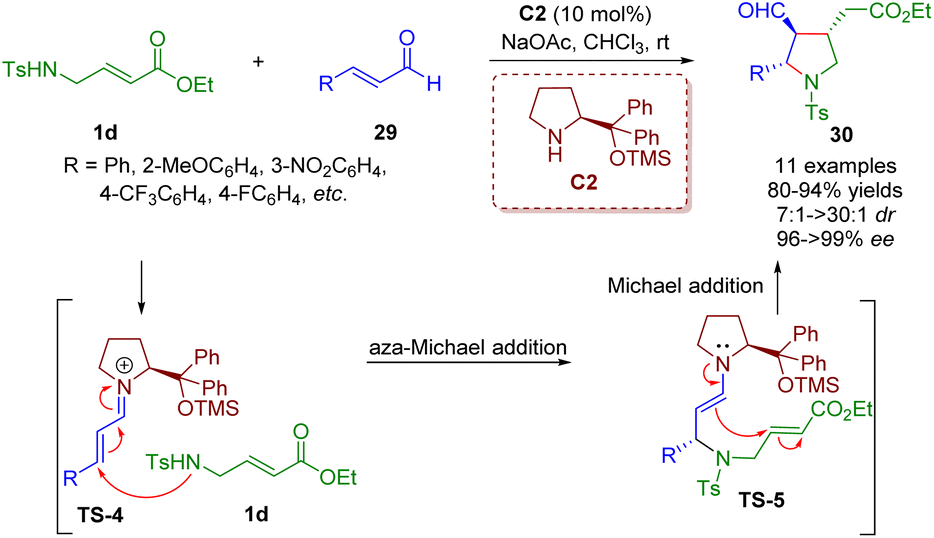 | ||
| Scheme 7 Diarylprolinol silyl ether-catalyzed enantio- and diastereoselective cascade aza-Michael/Michael reaction (Wang, 200820). | ||
By replacing γ-amino-substituted α,β-unsaturated ester 1d with δ-amino-substituted α,β-unsaturated ester 1e, Kanger and co-workers accomplished a diarylprolinol silyl ether-catalyzed cascade aza-Michael/Michael reaction for the asymmetric synthesis of 2,3,4-trisubstituted piperidines 31 (Scheme 8).21
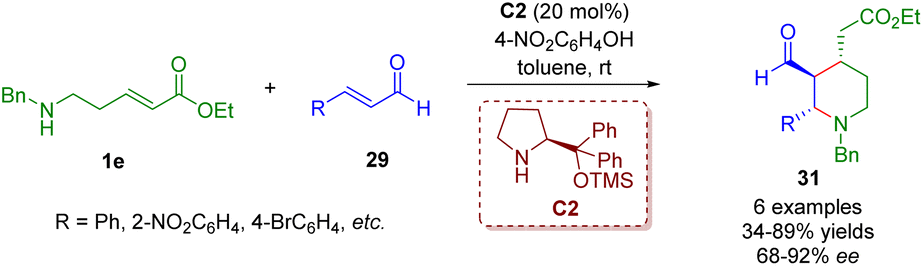 | ||
| Scheme 8 Diarylprolinol silyl ether-catalyzed enantioselective cascade aza-Michael/Michael reaction (Kanger, 201721). | ||
Soon after, Albrecht and co-workers reported the asymmetric synthesis of polysubstituted tetrahydro-1,2-oxazine derivatives 32 in high yields (up to 89%) with excellent stereocontrol (up to >20:1 dr, >99.5:0.5 er), relying on a diarylprolinol silyl ether-catalyzed cascade aza-Michael/Michael reaction of amino α,β-unsaturated esters/ketones 1 and α,β-unsaturated aldehydes 29 (Scheme 9).22
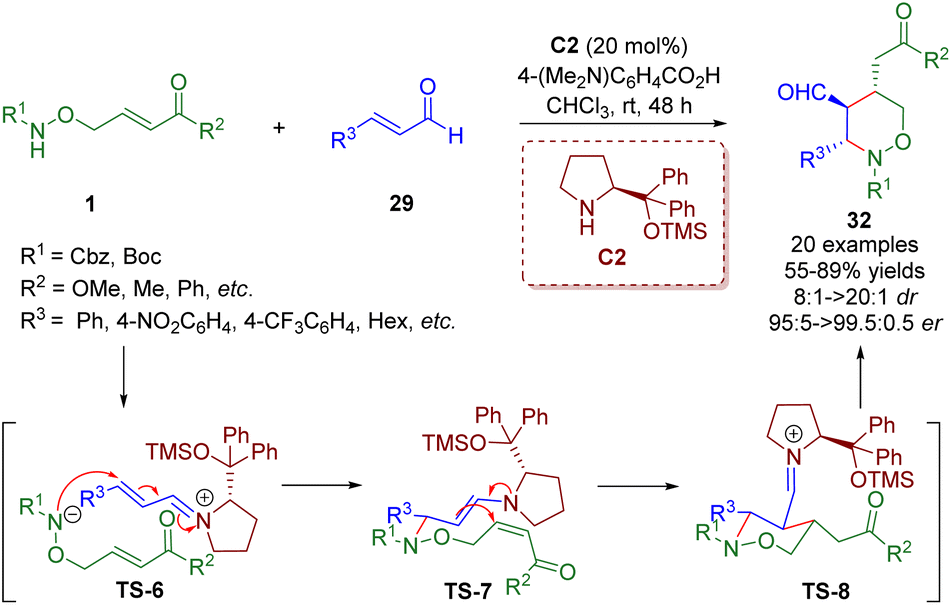 | ||
| Scheme 9 Diarylprolinol silyl ether-catalyzed enantioselective cascade aza-Michael–Michael reaction (Albrecht, 201722). | ||
2.2. Phosphine catalysis
In addition to the construction of five- and six-membered saturated nitrogen-containing heterocycles catalyzed by secondary amines, phosphine catalysis7 is also a practical and powerful tool for the creation of heterocyclic compounds, and has the advantages of readily available starting materials, environmental friendliness, and metal-free and mild reaction conditions.In 2018, Guo and co-workers reported a phosphine-catalyzed [5 + 1] annulation between δ-sulfonamido-substituted enones 1 and N-sulfonylimines 33 (Scheme 10).23 A series of 1,2,3,6-tetrahydropyridines (35 examples) with good to excellent yields (41–98%) were synthesized via simple and practical operation. The asymmetric version of the model [5 + 1] annulation reaction gave the chiral product in up to 73% ee. It is important to mention that δ-sulfonamido-substituted enones 1 acted as five-membered synthons. According to the results of control experiments, the authors proposed a plausible mechanism to explain the reaction. The reaction started with an intermolecular nucleophilic 1,4-conjugate addition between phosphine catalyst PBu3 and δ-sulfonamido-substituted enone 1f to afford intermediate TS-9. Then another nucleophilic addition of the intermediate TS-9 with N-sulfonylimine 33 led to intermediate TS-10. An intramolecular proton transfer of the intermediate TS-10 delivered intermediate TS-11, which was converted to intermediate TS-12via an elimination reaction. The intermediate TS-12 was transformed into the intermediate TS-13via the abstraction of a proton with the help of a 4-methyl-benzenesulfonamide anion. Subsequently, an intramolecular nucleophilic 1,4-conjugate addition of the intermediate TS-13 formed intermediate TS-14, which gave the desired product 34a concomitant with the phosphine catalyst PBu3 turning over.
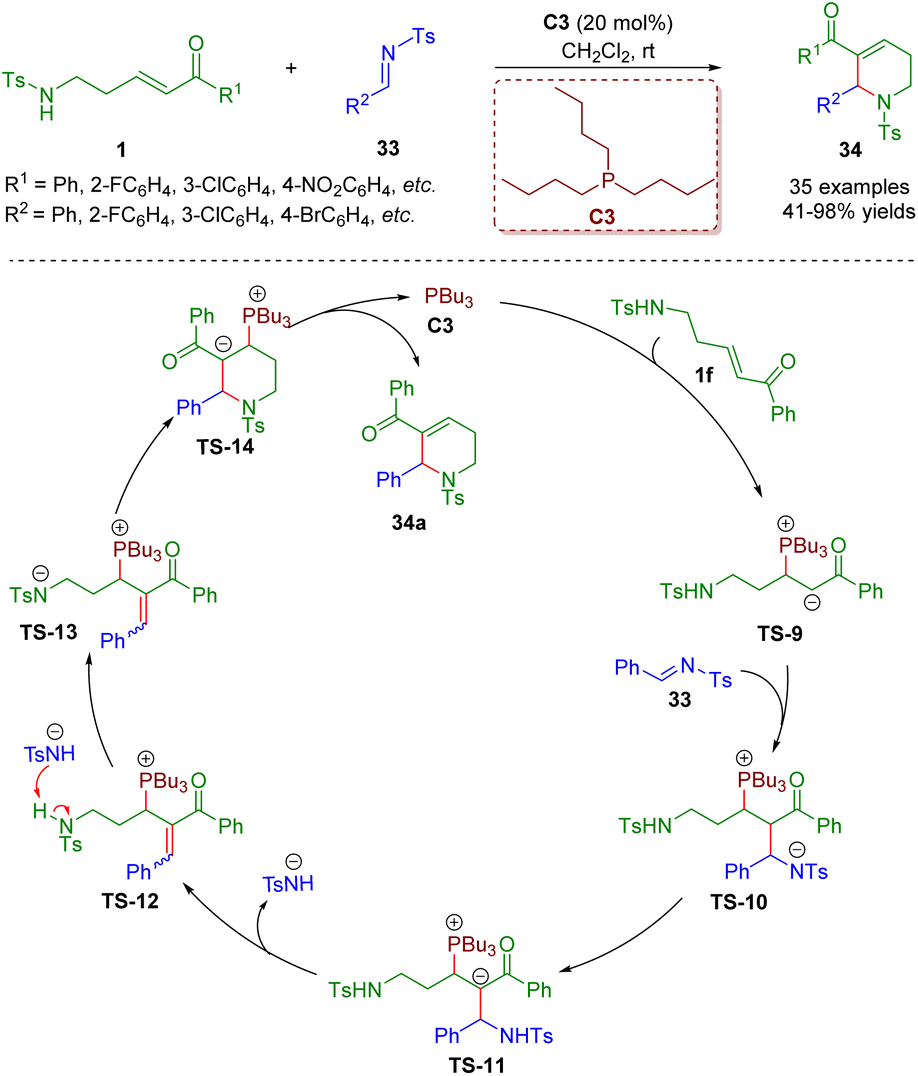 | ||
| Scheme 10 Phosphine-catalyzed [5 + 1] annulation between δ-sulfonamido-substituted enones and N-sulfonylimines (Guo, 201823). | ||
Different from the above creative work, δ-sulfonamido-substituted enones 1 acting as four-membered synthons were also achieved. Consequently, δ-sulfonamido-substituted enones 1 have been adopted as versatile building blocks. Recently, Guo, Zheng and co-workers developed a phosphine-catalyzed [4 + 2] annulation of δ-sulfonamido-substituted enones 1 and 1,1-dicyanoalkenes 35 for the synthesis of piperidine derivatives (36 examples) in moderate to excellent yields (37–95%) with good to excellent diastereoselectivities (4:1–>19:1) (Scheme 11).24 The authors presented a plausible reaction mechanism on the basis of experimental results and the theory of phosphine catalysis. The phosphine catalyst attacked δ-sulfonamido-substituted enones 1 to produce intermediate TS-15, which was converted to intermediate TS-16via a proton transfer. Then nucleophilic addition of TS-16 with 1,1-dicyanoalkenes 36 afforded intermediate TS-17. Finally, intramolecular nucleophilic substitution of intermediate TS-17 accomplished annulation to furnish product 36 and simultaneously liberated the phosphine catalyst PBu3.
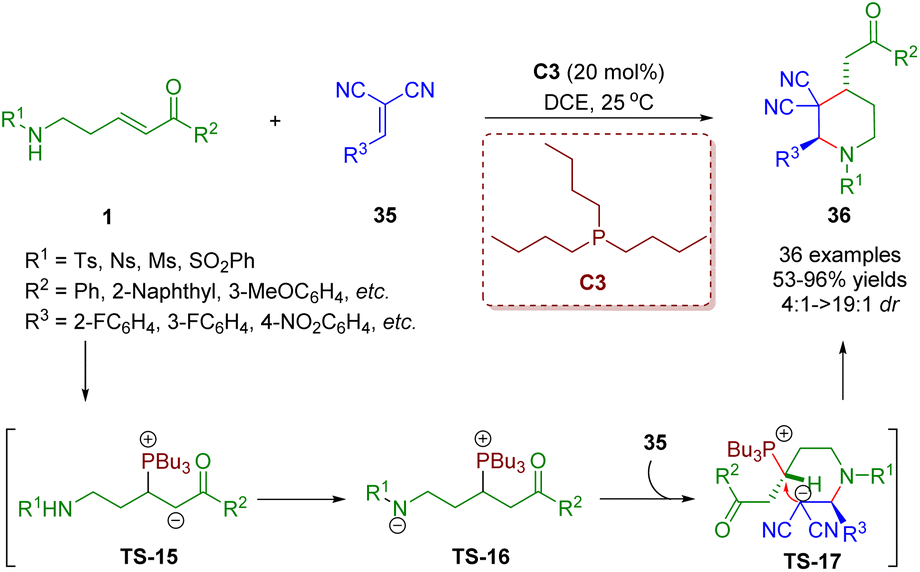 | ||
| Scheme 11 Phosphine-catalyzed [4 + 2] annulation between δ-sulfonamido-substituted enones and 1,1-dicyanoalkenes (Guo and Zheng, 202124). | ||
In 2019, Guo, Xiao and co-workers reported a phosphine-catalyzed [3 + 2] annulation of β-sulfonamido-substituted enones 1 and sulfamate-derived cyclic imines 37, giving a series of imidazoline derivatives (29 examples) in moderate to excellent yields (37–95%) with good to excellent diastereoselectivities (4:1–>20:1) (Scheme 12).25 In this reaction, β-sulfonamido-substituted enones acted as C–C–N synthons for annulations. To prove the practicability of the [3 + 2] annulation, a scale-up reaction was carried out, which smoothly worked under the mild reaction conditions. A possible mechanism was proposed on the basis of the results obtained. Similar to the above mechanism, the catalytic cycle was completed via four processes, including intermolecular nucleophilic addition, proton transfer, intermolecular nucleophilic addition, and intramolecular nucleophilic substitution.
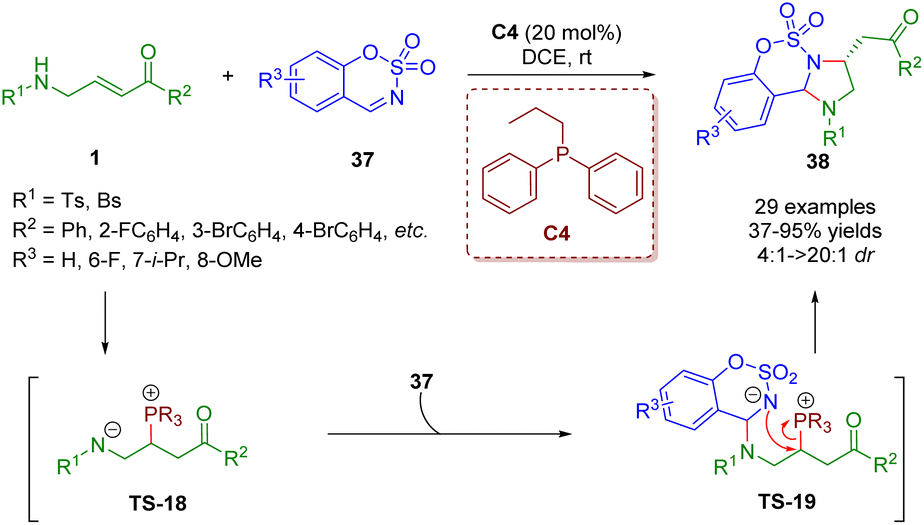 | ||
| Scheme 12 Phosphine-catalyzed [3 + 2] annulation of β-sulfonamido-substituted enones and sulfamate-derived cyclic imines (Guo and Xiao, 201925). | ||
In a similar fashion, Liu, Gao and co-workers showcased a phosphine-catalyzed [3 + 2] annulation of β-sulfonamido-substituted enones 1 with trans-α-cyano-α,β-unsaturated ketones 39 in 2021 for the creation of highly substituted pyrrolidines (31 examples) under mild conditions with high yields (66–86%) and moderate to good diastereoselectivities (4.5:1–14:1) (Scheme 13).26 In this reaction, β-sulfonamido-substituted enones served as C–C–N synthons for annulations. This strategy was realized by Pan and Mukhopadhyay via bifunctional squaramide catalysis in 2018.
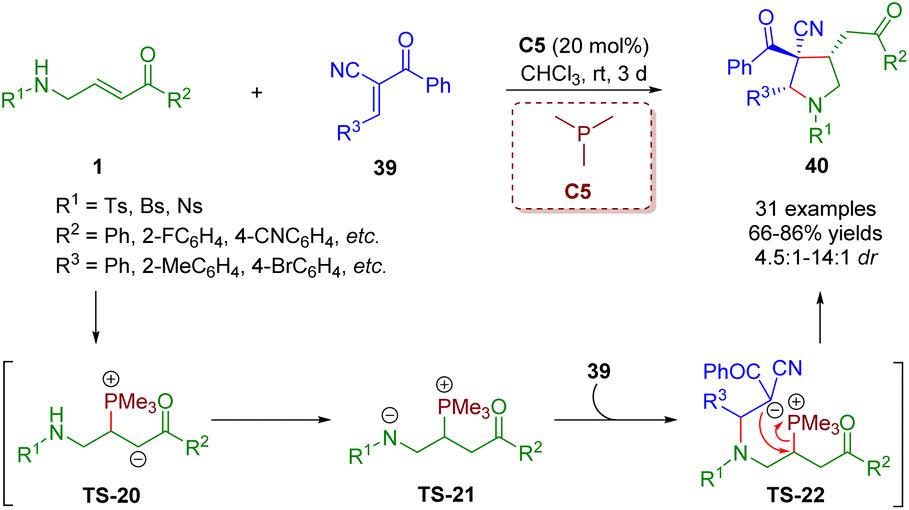 | ||
| Scheme 13 Phosphine-catalyzed [3 + 2] annulation of β-sulfonamido-substituted enones with trans-α-cyano-α,β-unsaturated ketones (Liu and Gao, 202126). | ||
In 2021, Lupton, Hooper and co-workers developed an unprecedented phosphine-catalyzed tandem isomerization/[3 + 2] annulation between β-sulfonamido-substituted α,β-unsaturated esters 1 and γ-substituted allenoates 41 (Scheme 14).27 A series of pyrrolines (24 examples) with moderate to good yields (27–77%) and outstanding levels of diastereoselectivities (all >20:1) were obtained.
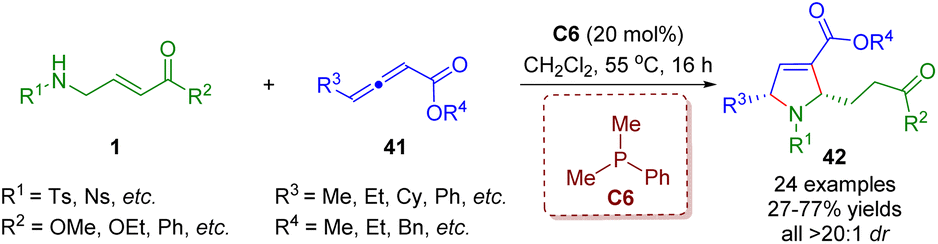 | ||
| Scheme 14 Phosphine-catalyzed tandem isomerization/[3 + 2] annulation sequence of β-sulfonamido-substituted α,β-unsaturated esters and γ-substituted allenoates (Lupton and Hooper, 202127). | ||
Soon after, a phosphine-catalyzed asymmetric tandem isomerization/[3 + 2] annulation sequence between β-sulfonamido-substituted α,β-unsaturated esters 1 and γ-substituted allenoates 41 was realized by Guo, Chen and co-workers (Scheme 15).28 A series of chiral pyrrolines 42 (24 examples) were obtained in high yields (21–87%) and excellent enantioselectivities (79–>99% ee). Bioactive molecule-based allenoates prepared from specific drugs containing acetic acid units also afforded good results. The mechanism involved two unlinked catalytic processes. The catalytic cycle A involved the redox isomerization of amino crotonates 1 to afford tosylimines 1a-3 followed by a (3 + 2) annulation with γ-substituted allenoates 41.
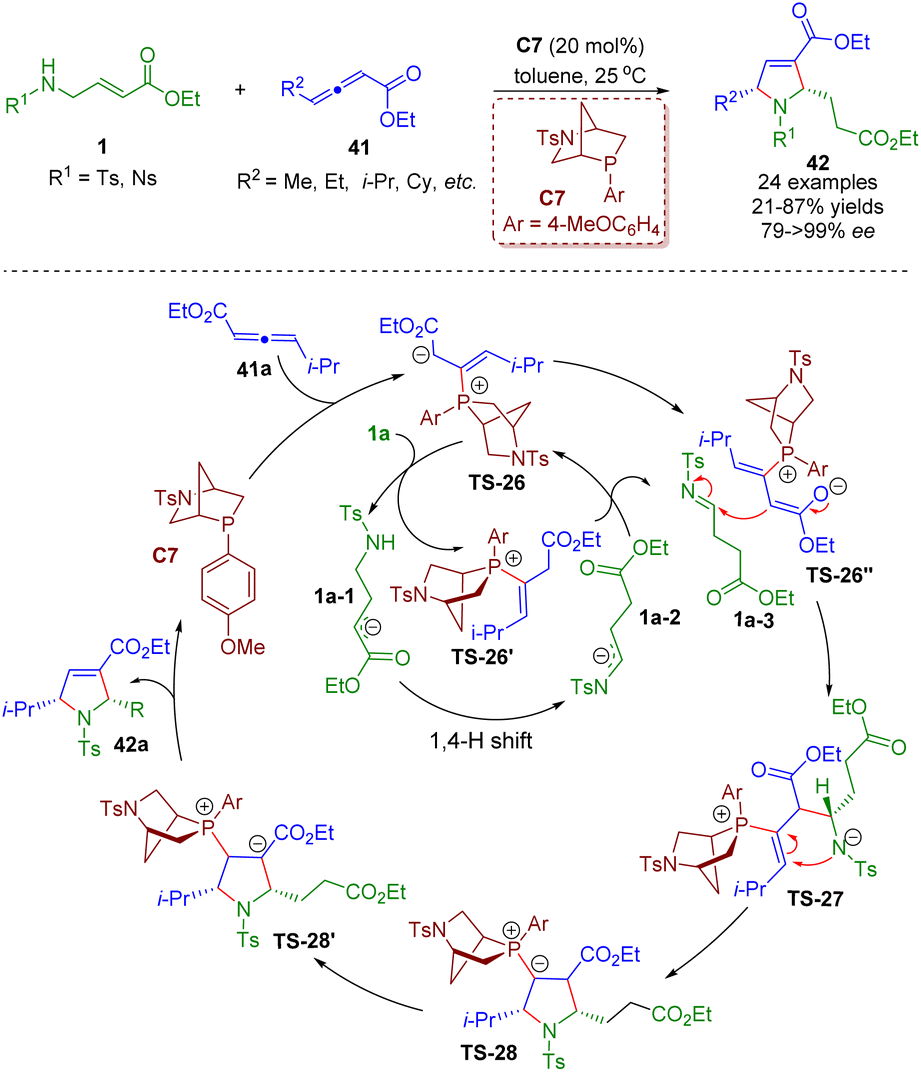 | ||
| Scheme 15 Phosphine-catalyzed asymmetric tandem isomerization/[3 + 2] annulation sequence of β-sulfonamido-substituted α,β-unsaturated esters (Guo and Chen, 202128). | ||
More recently, Lupton and co-workers reported a phosphine-catalyzed asymmetric [3 + 2] annulation between β-sulfonamido-substituted α,β-unsaturated esters 1 and allenoates 41, producing functionalized pyrrolidines 43 (18 examples) in good yields (55–91%) with excellent enantioselectivities (86:14–97:3 er) and moderate to good diastereoselectivities (2:1–5:1 dr) (Scheme 16).29 The reaction involved the umpolung γ-amination of the allenoates and β-umpolung intramolecular conjugate addition. On the basis of mechanistic studies, the authors proposed a plausible mechanism to explain the reaction.
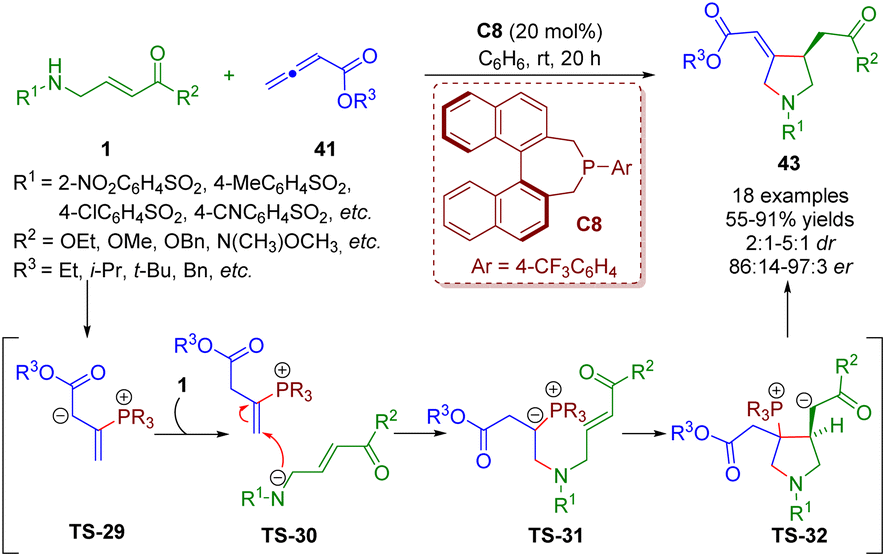 | ||
| Scheme 16 Phosphine-catalyzed asymmetric [3 + 2] annulation between β-sulfonamido-substituted α,β-unsaturated esters and allenoates (Lupton, 202229). | ||
2.3. N-Heterocyclic carbene catalysis
Sulfonamido-substituted α,β-unsaturated carbonyl compounds as versatile building blocks were also employed in N-heterocyclic carbene catalysis. In 2018, Ye and co-workers reported an N-heterocyclic carbene-catalyzed [3 + 2] and [4 + 2] annulation of γ-/δ-sulfonamido-substituted α,β-unsaturated ketones 1 with α-chloroaldehydes 44, producing the corresponding pyrrolidones (16 examples) and piperidones (5 examples) in good yields (21–81%) with exclusive trans-selectivities (all >20:1 dr) and excellent enantioselectivities (95–>99% ee) (Scheme 17).30 The authors provided a plausible mechanism for the NHC-catalyzed [3 + 2] and [4 + 2] annulations. First, N-heterocyclic carbene catalyst C9′ was generated from the precursor C9 in the presence of Cs2CO3. Then, NHC C9′ attacked α-chloroaldehyde 44a and afforded Breslow intermediate TS-33. The removal of hydrochloride gave azolium enolate TS-34 with the help of Cs2CO3. Azolium enolate TS-34 attacked α,β-unsaturated ketone 1avia the Si–Si face to give intermediate TS-35, which regenerated the catalyst and pyrrolidone derivative 45avia a lactamization reaction.
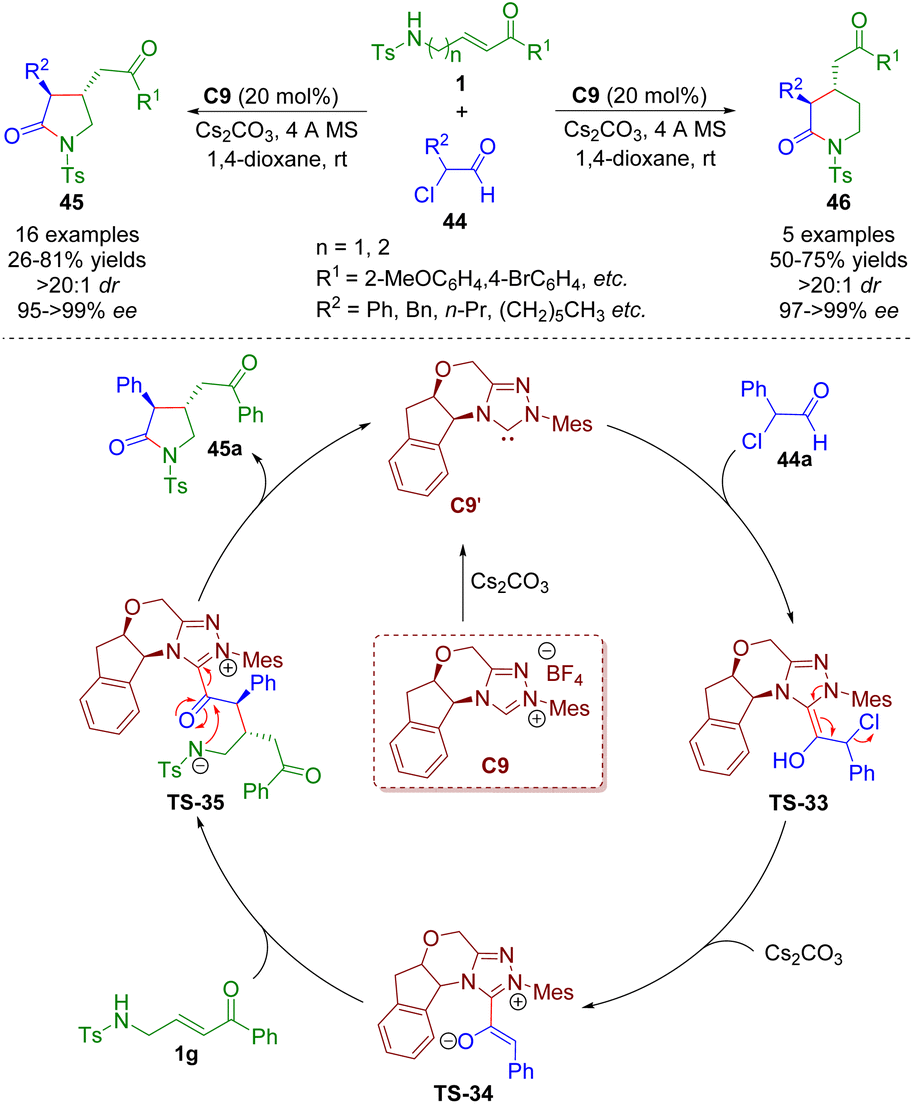 | ||
| Scheme 17 N-Heterocyclic carbene-catalyzed [3 + 2] and [4 + 2] annulation of γ-/δ-sulfonamido-substituted α,β-unsaturated ketones with α-chloroaldehydes (Ye, 201830). | ||
In 2021, Ye, Zhang and co-workers described N-heterocyclic carbene-catalyzed intramolecular aza-Michael addition of sulfonamido-substituted α,β-unsaturated carboxylic acids 1 for the establishment of pyrrolidones 47 (18 examples) and piperidones 48 (8 examples) in good yields (50–95%) (Scheme 18).31 The plausible catalytic mechanism is similar to that described above.
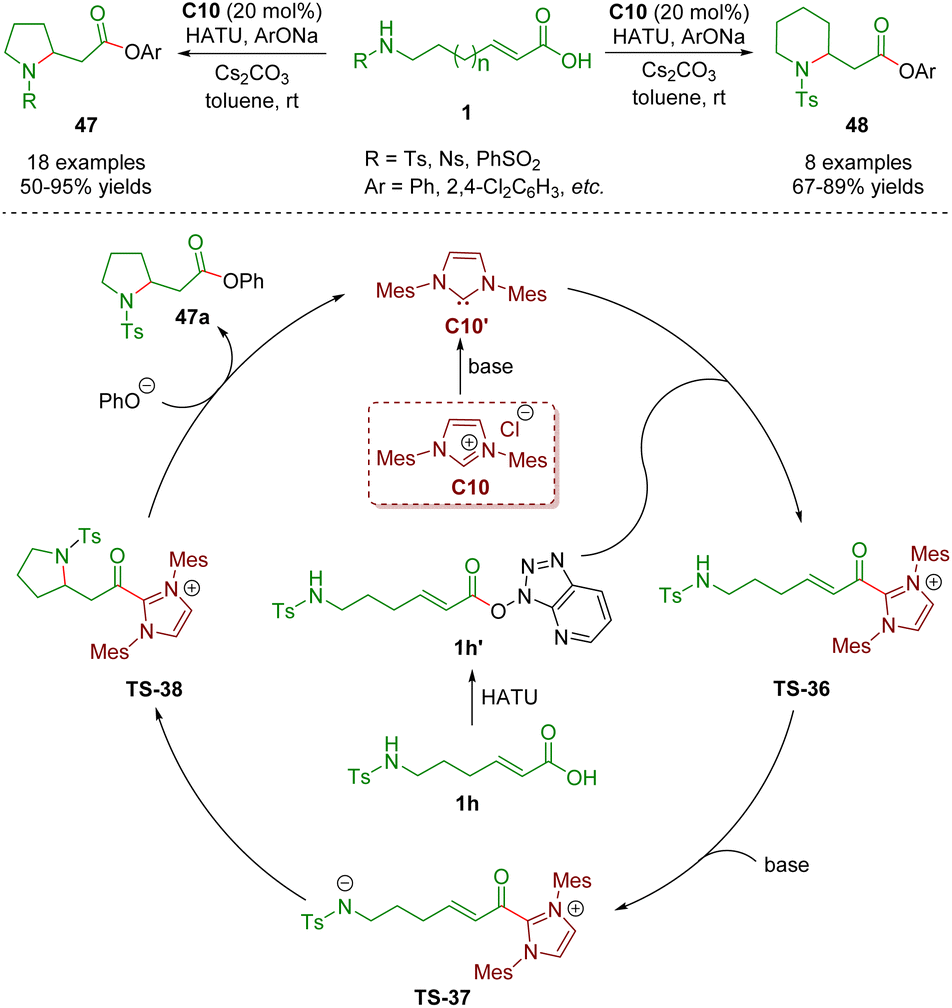 | ||
| Scheme 18 N-Heterocyclic carbene-catalyzed intramolecular aza-Michael addition of sulfonamido-substituted α,β-unsaturated carboxylic acids (Ye and Zhang, 202131). | ||
3. Non-covalent catalysis
3.1. Chiral phosphoric acid catalysis
Chiral phosphoric acid catalysis has been recognized as a significant organocatalysis9 since the seminal contributions on BINOL-derived phosphoric acid-catalyzed asymmetric Mannich reactions by the research groups of Akiyama and Terada in 2004.32 Chiral phosphoric acid catalysis has also been found to be used in the synthesis of homo-/multi-substituted and spiro-fused pyrrolidines via an intramolecular aza-Michael addition reaction, a cascade intramolecular aza-Michael addition/intramolecular Michael addition reaction or a cascade condensation/isomerization/intramolecular Michael addition.In 2013, Yu and co-workers reported the first chiral phosphoric acid-catalyzed enantioselective intramolecular aza-Michael addition of amino-substituted α,β-unsaturated ketones 1 (Scheme 19).33 A series of 2-substituted pyrrolidines 49 (12 examples) were efficiently synthesized in good to excellent yields (84–97%) and enantioselectivities (91:9–98:2 er).
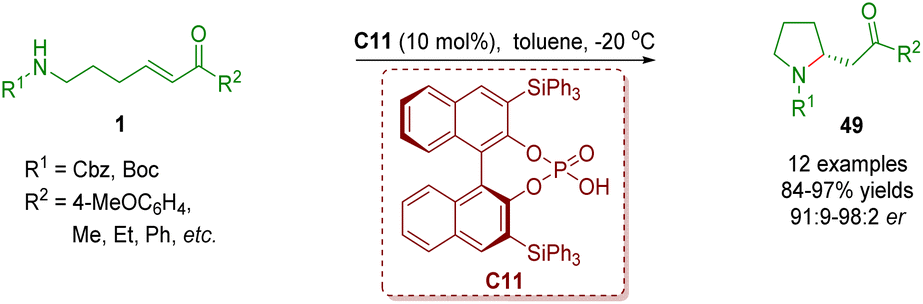 | ||
| Scheme 19 Chiral phosphoric acid-catalyzed enantioselective intramolecular aza-Michael addition of amino-substituted α,β-unsaturated esters (Yu, 201333). | ||
Recently, Clark, Ermanis and co-workers achieved the total synthesis of (−)-irnidine (53) and (−)-bgugaine (54), which possess DNA binding and antibacterial properties, from common β-homoproline intermediates 52 (Scheme 20).34 The intermediates 52 were obtained by a “clip-cycle” strategy involving cross metathesis in the presence of a second-generation Hoveyda–Grubbs catalyst and then a chiral phosphoric acid-catalyzed enantioselective intramolecular aza-Michael addition reaction. The developed methodology was suitable for the construction of 2,2- and 3,3-disubstituted pyrrolidines and spiropyrrolidines 52, and was applied to the synthesis of N-methylpyrrolidine alkaloids (−)-irnidine (53) and (−)-bgugaine (54).
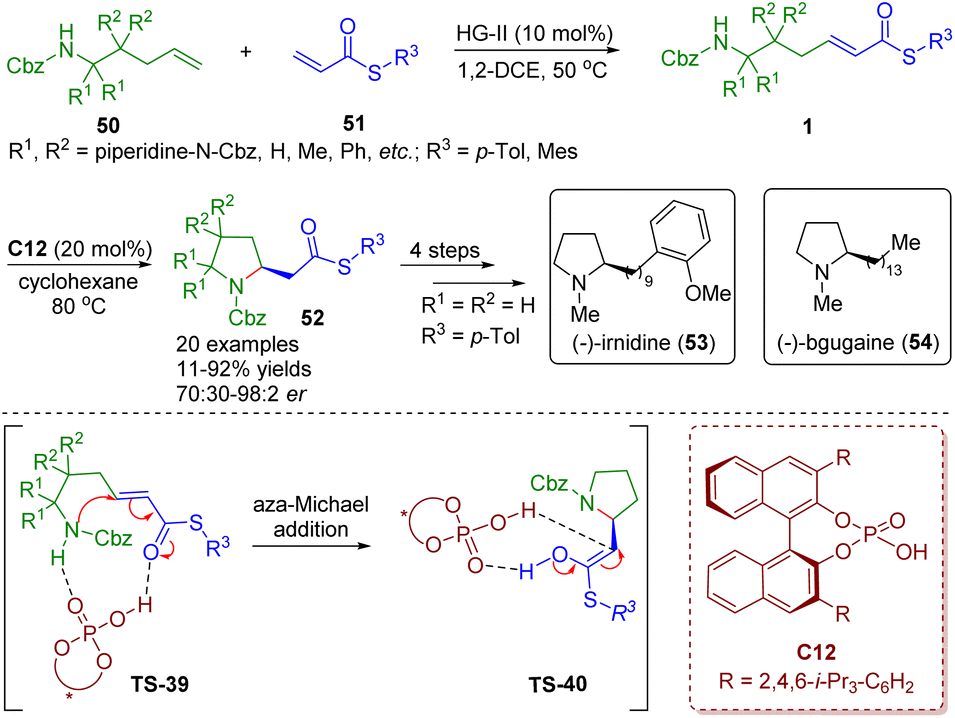 | ||
| Scheme 20 Chiral phosphoric acid-catalyzed enantioselective aza-Michael addition of amino-substituted α,β-unsaturated thioesters (Clark and Ermanis, 202034). | ||
About one month later, del Pozo and co-workers reported a similar strategy involving cross metathesis/chiral phosphoric acid-catalyzed enantioselective intramolecular aza-Michael addition/intramolecular Michael addition for the synthesis of poly-substituted pyrrolizidinones 57 (14 examples) in moderate to good yields (35–81%) and excellent enantioselectivities (95–99% ee) (Scheme 21).35
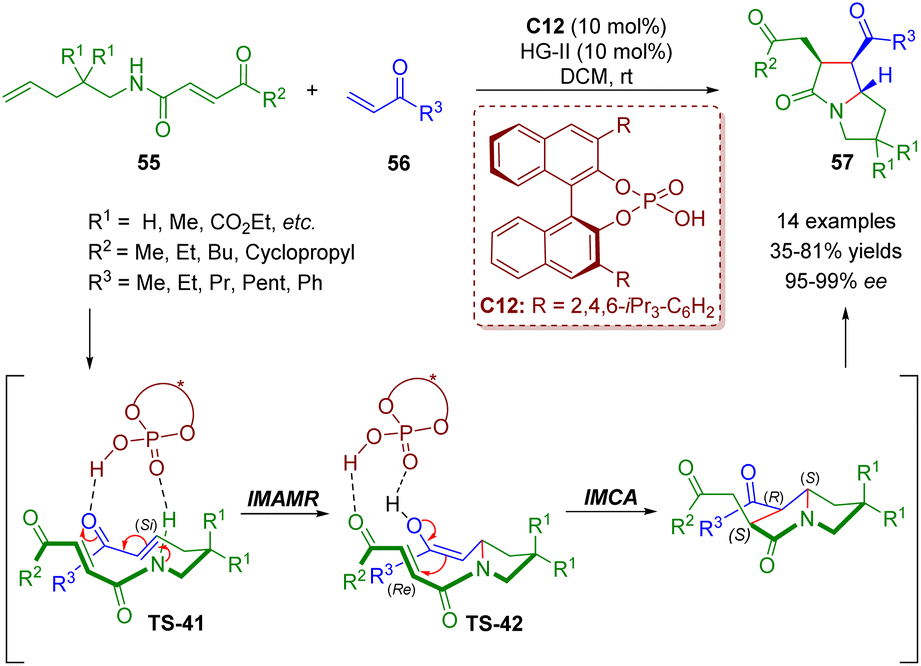 | ||
| Scheme 21 Triple-tandem strategy to access chiral pyrrolizidinone derivatives (del Pozo, 202035). | ||
More recently, Ghorai and Panda reported a chiral phosphoric acid-catalyzed enantioselectively modified Heyns rearrangement for the synthesis of a wide range of 1-azaspiro cyclobutanone-containing spiropyrrolidine derivatives 59 (18 examples) in good yields (52–81%) with excellent stereoselectivities (up to >99% ee, >20:1 dr) (Scheme 22).36 First, the condensation of δ-amino-α,β-unsaturated ketones 1 with the catalyst C11 formed iminium ion TS-43, which is subsequently isomerized to enamine TS-44. Finally, the enamine intermediate TS-44 underwent intramolecular Michael addition to generate spiropyrrolidine derivatives 59 with the help of the catalyst C11.
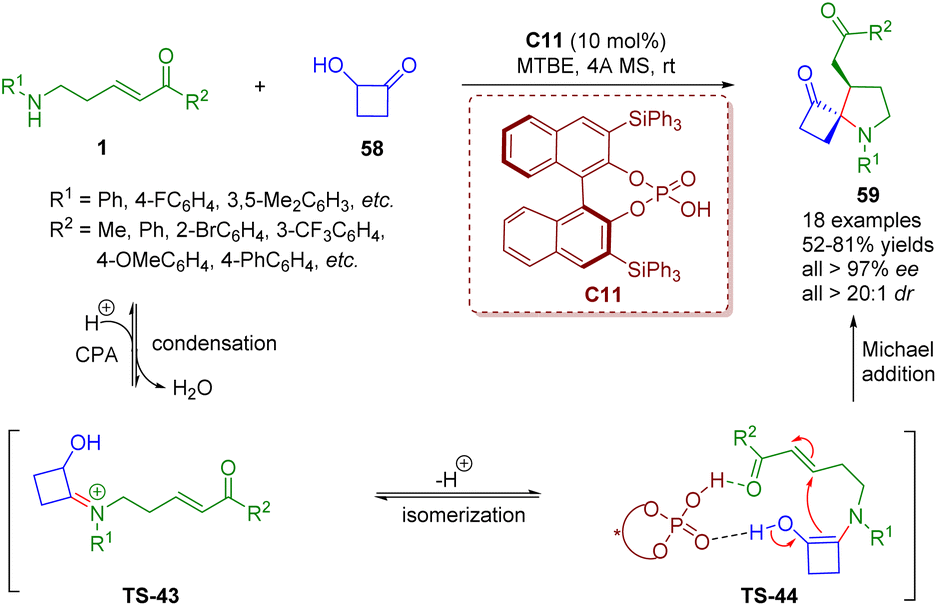 | ||
| Scheme 22 Chiral phosphoric acid-catalyzed enantioselective modified Heyns rearrangement of amino-substituted α,β-unsaturated esters (Ghorai and Panda, 202336). | ||
3.2. Other Brønsted acid catalyses
More recently, the Liu group developed a Brønsted acid-catalyzed cascade process, including hemiaminalization/dehydration/intramolecular Michael addition, hemiaminalization/oxa-intramolecular Michael addition, hemiaminalization/dehydration/double Michael addition, and hemiaminalization/dehydration/Michael addition/Friedel–Crafts cyclization of amino-α,β-unsaturated ketones 1 with different partners 60–63 for the synthesis of a series of functionalized six-membered heterocycles (Scheme 23).37 Furthermore, δ-sulfonamido-substituted α,β-unsaturated ketone 1f underwent a dimerization reaction to give highly functionalized piperidine derivative 68 in 58% yield when there was no aldehyde in the reaction solution. Interestingly, N,N-acetal 69 was obtained via a three-component reaction of β-sulfonamido-substituted α,β-unsaturated ketone 1g, aldehyde 60a, and p-toluenesulfonamide.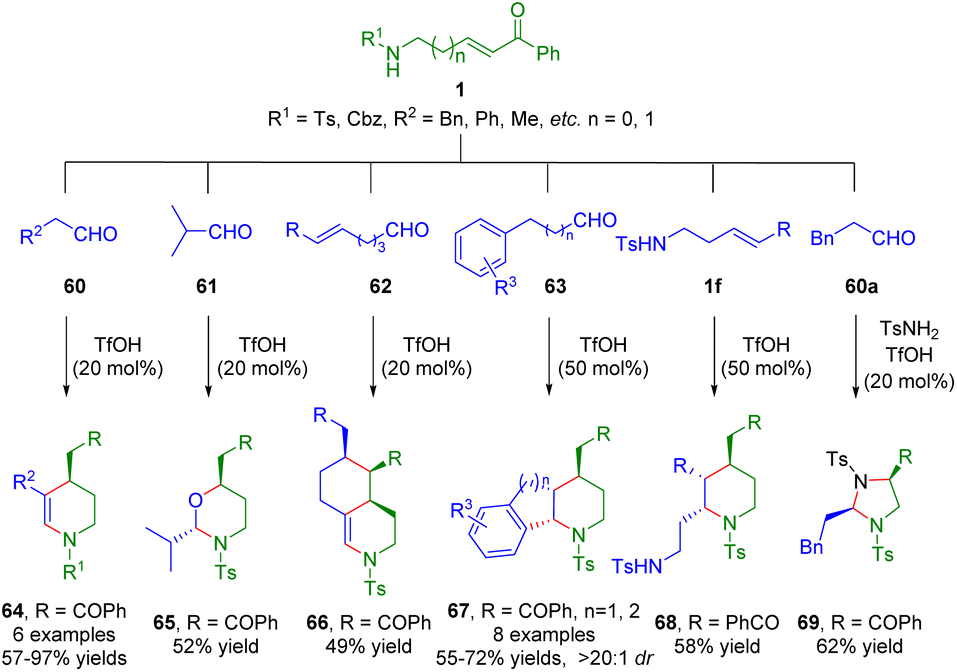 | ||
| Scheme 23 Brønsted acid-catalyzed cascade process of amino α,β-unsaturated ketones (Liu, 202337). | ||
4. Bifunctional catalysis
4.1. Bifunctional tertiary amine/primary amine catalysis
As described in section 2.1, chiral secondary amines, such as diarylprolinol silyl ethers, are undoubtedly the important catalysts for condensation with α,β-unsaturated aldehydes to form iminium ions. However, it is difficult for them to generate congested iminium ions with α,β-unsaturated ketones due to the bulky steric hindrance. Primary amines, especially cinchona alkaloid-derived primary amines, could overcome the inherent difficulties of secondary amines. This part will summarize and discuss cinchona alkaloid-derived primary amine-catalyzed enantioselective intramolecular aza-Michael addition of amino α,β-unsaturated ketones 1, producing a series of functionalized five- and six-membered nitrogen-containing heterocycles 70 (Scheme 24).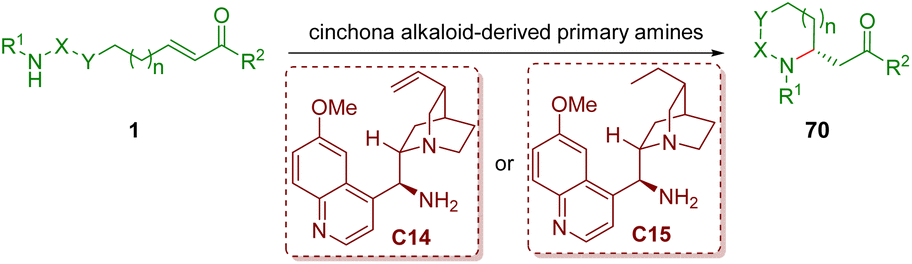 | ||
| Scheme 24 Cinchona alkaloid-derived primary amine-catalyzed enantioselective intramolecular aza-Michael addition of amino α,β-unsaturated ketones. | ||
In 2011, Fan, Chen, and co-workers first reported a general method for performing enantioselective intramolecular aza-Michael addition of amino α,β-unsaturated ketones 1 (Scheme 25).38 The reaction was carried out in the presence of cinchona alkaloid-derived primary amine C14 as a catalyst and CF3CO2H as a co-catalyst to produce five- and six-membered heterocycles 71 in high yields (75–97%) and good to excellent enantioselectivities (78–99% ee). According to the absolute configuration of the heterocycles 71, the authors proposed a mechanistic model for the observed enantioselectivity in the reaction. The iminium ions TS-45 and TS-45′ were generated via the condensation of amino α,β-unsaturated ketones 1 with the catalyst C14. The iminium ion TS-45′ was the energetically favored transition state, resulting in the corresponding products 71 with the R absolute configuration. It should be noted that catalyst C14 showed a bifunctional catalysis mode of hydrogen-bonding interaction between the protonated tertiary amine and the carbamate carbonyl oxygen.
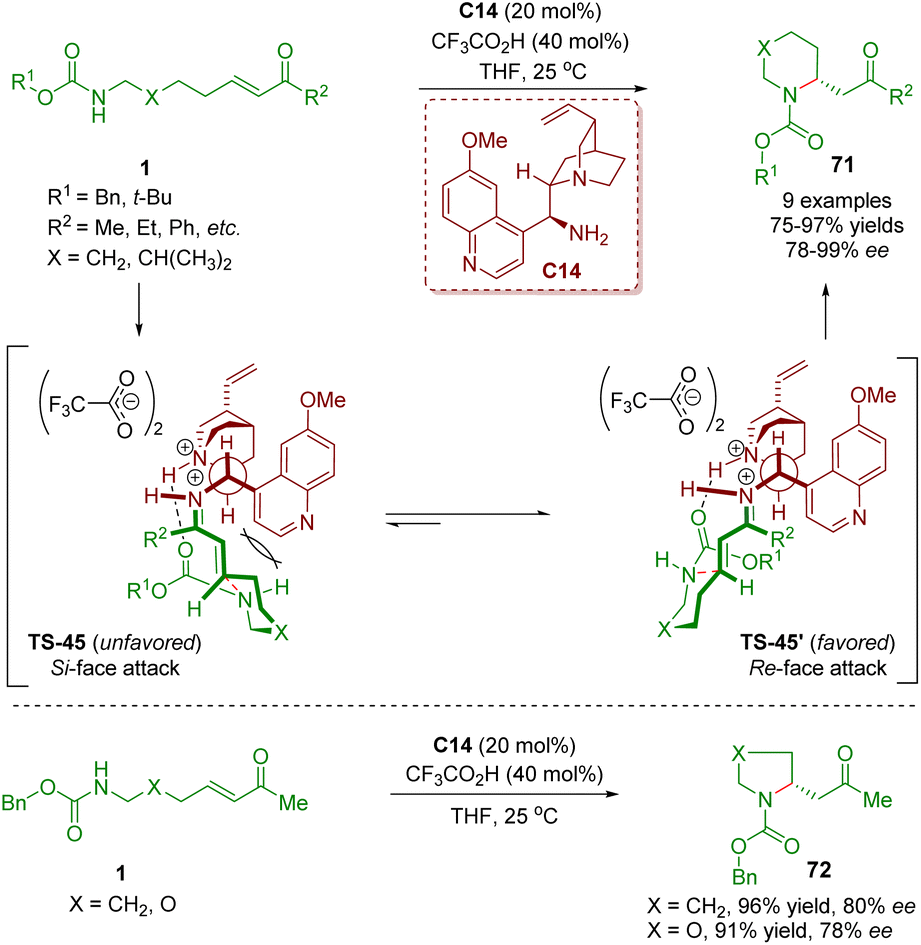 | ||
| Scheme 25 Cinchona alkaloid-derived primary amine-catalyzed enantioselective intramolecular aza-Michael addition (Fan, 201138). | ||
Almost simultaneously, Sánchez-Roselló, Fustero and co-workers reported the synthesis of 2-substituted piperidines 73, which was similar to the above-described method (Scheme 26).39 The reaction was catalyzed by cinchona alkaloid-derived primary amine C15 and CF3CF2CO2H as a co-catalyst to give the desired products 73 in good yields (58–97%) and excellent enantioselectivities (82–98% ee). Furthermore, the use of microwave irradiation can not only improve the efficiency of the process but also be compatible with yields and enantiomeric excesses.
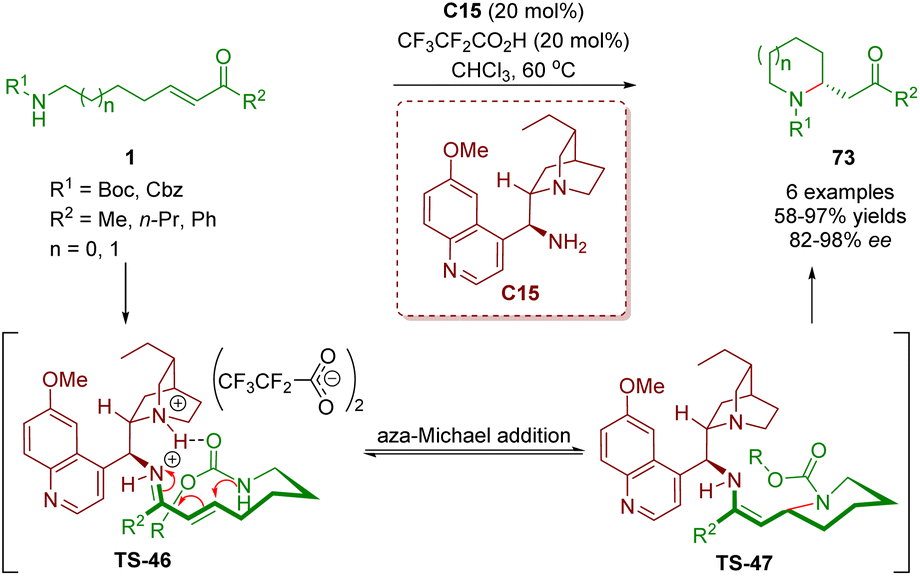 | ||
| Scheme 26 Cinchona alkaloid-derived primary amine-catalyzed enantioselective intramolecular aza-Michael addition (Sánchez-Roselló and Fustero, 201139). | ||
In 2018, del Pozo, Fustero and co-workers reported another example of the synthesis of six-membered nitrogen-containing heterocycles 74 in moderate to excellent yields (62–99%) and good to excellent enantioselectivities (76–97% ee) via a cinchona alkaloid-derived primary amine-catalyzed enantioselective intramolecular aza-Michael addition of amino α,β-unsaturated ketones 1 (Scheme 27).40
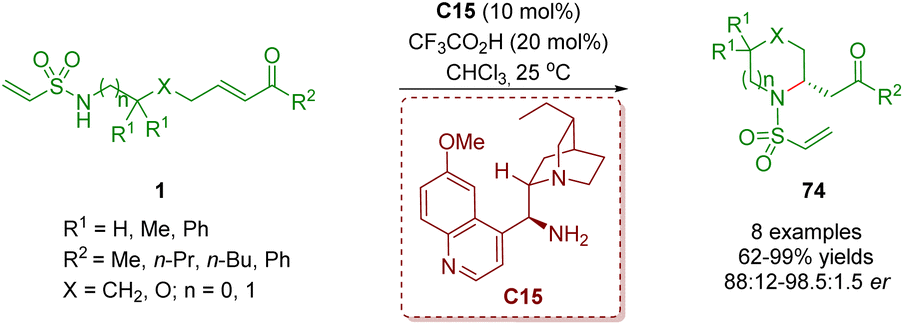 | ||
| Scheme 27 Cinchona alkaloid-derived primary amine-catalyzed enantioselective intramolecular aza-Michael addition (del Pozo and Fustero, 201840). | ||
About one month later, the same group reported an organocatalytic enantioselective desymmetrization of amino α,β-unsaturated ketones 1 in the presence of cinchona alkaloid-derived primary amine C15 and TFA as a co-catalyst to give 2,5- and 2,6-disubstituted piperidines 75 in good yields (35–87%) with low diastereoselectivities (1.5:1–5:1 dr) and moderate to good enantioselectivities (Scheme 28).41
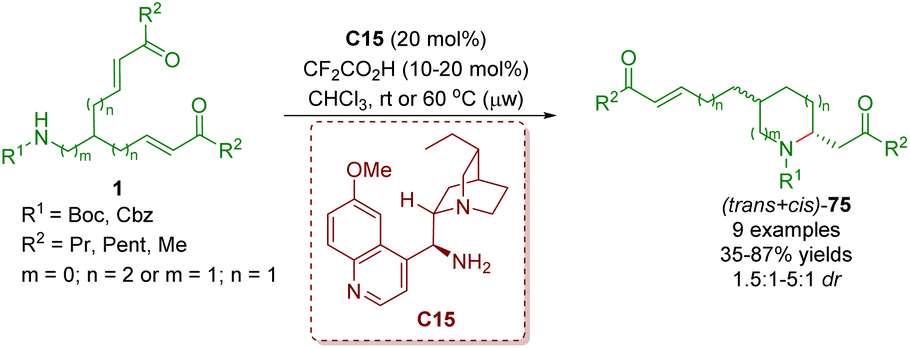 | ||
| Scheme 28 Cinchona alkaloid-derived primary amine-catalyzed enantioselective desymmetrization (del Pozo, 201841). | ||
In 2020, another example of organocatalytic enantioselective desymmetrization of amino α,β-unsaturated ketones 1 was developed by the same group (Scheme 29).42 The reaction took place in the presence of catalyst C15 and TFA as a co-catalyst to provide 2,5,5-trisubstituted piperidines 76 bearing one all-carbon quaternary stereogenic center in good to excellent yields (67–99%) with good diastereoselectivities (4:1–12:1 dr) and excellent enantioselectivities (76–99% ee).
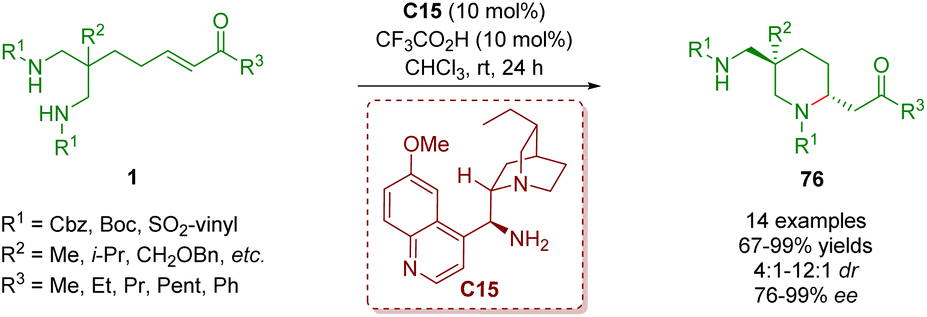 | ||
| Scheme 29 Cinchona alkaloid-derived primary amine-catalyzed enantioselective desymmetrization (del Pozo, 202042). | ||
In 2012, Yu and co-workers achieved the asymmetric synthesis of phenanthroquinolizidine alkaloids (−)-cryptopleurine (78a) and (−)-boehmeriasin A (78b) featuring a cinchona alkaloid-derived primary amine-catalyzed enantioselective intramolecular aza-Michael addition of amino-substituted α,β-unsaturated ketones 1 (Scheme 30).43 Piperidines 77 were smoothly obtained in good yields (84% and 86%) and enantioselectivities (93:7 and 95:5 er). Piperidines 77 were converted into (−)-cryptopleurine (78a) and (−)-boehmeriasin A (78b) in a five-step sequence.
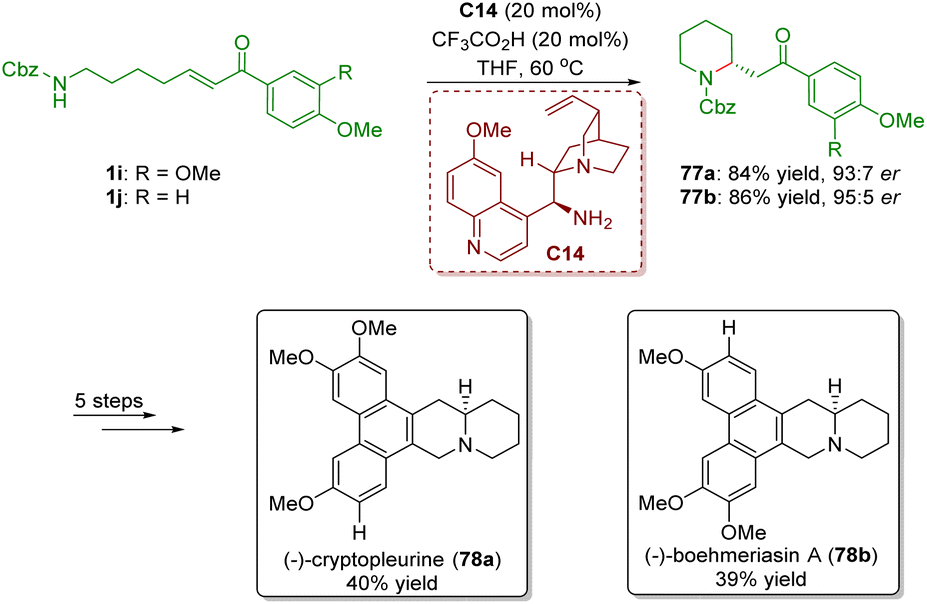 | ||
| Scheme 30 Asymmetric synthesis of phenanthroquinolizidine alkaloids (−)-cryptopleurine and (−)-boehmeriasin A (Yu, 201243). | ||
In 2014, Yu and co-workers reported a highly enantioselective intramolecular aza-Michael addition to give chiral 3-substituted 1,2-oxazinanes 79 in moderate to excellent yields (55–99%) and enantioselectivities (86:14–98:2 er) (Scheme 31).44 These reactions were carried out in the presence of cinchona alkaloid-derived primary amine C14 as a catalyst and pentafluoropropionic acid (PFP) as a co-catalyst.
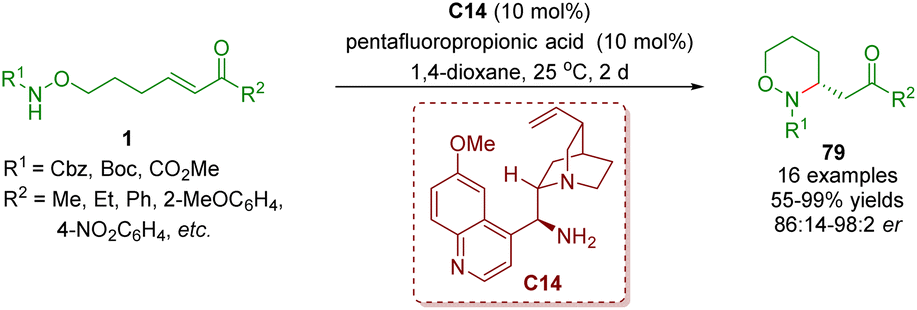 | ||
| Scheme 31 Cinchona alkaloid-derived primary amine-catalyzed enantioselective intramolecular aza-Michael addition (Yu, 201444). | ||
In 2017, Yang and co-workers developed an enantioselective intramolecular aza-Michael addition to afford 2-substituted pyrrolidines and piperidines 80 in moderate to excellent yields (55–99%) and enantioselectivities (92–97.5% ee) (Scheme 32).45 The reaction was catalyzed by cinchona alkaloid-derived primary amine C14 and diphenyl phosphate (DPP) as a co-catalyst.
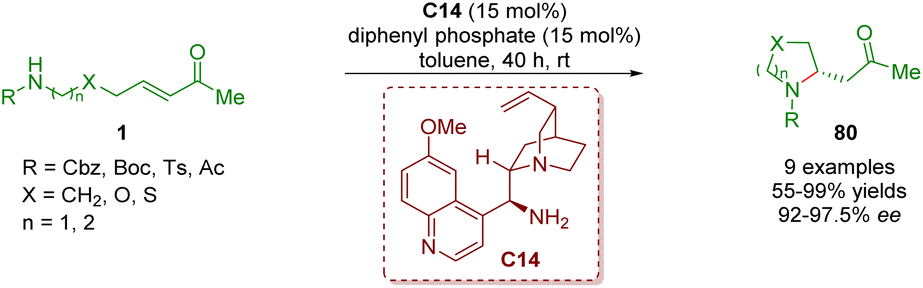 | ||
| Scheme 32 Cinchona alkaloid-derived primary amine-catalyzed enantioselective intramolecular aza-Michael addition (Yang, 201745). | ||
4.2. Bifunctional tertiary amine/squaramide catalysis
Bifunctional tertiary amines/squaramides are an important branch of organocatalysts and a powerful tool for the asymmetric construction of complex structures via multiple hydrogen-bonding interactions between catalysts and substrates.In 2015, Du and Zhao developed a bifunctional tertiary amine/squaramide-catalyzed asymmetric cascade intermolecular aza-Michael/intramolecular Michael addition of γ-sulfonamido-substituted α,β-unsaturated esters/ketones 1 and 3-ylidene-oxindoles 81 (Scheme 33).46 A series of spiro[pyrrolidine-3,3′-oxindole]s 82 (40 examples) were obtained in high yields (72–99%) with moderate to excellent diastereoselectivities (73:27–>99:1 dr) and enantioselectivities (67–>99% ee). The authors proposed a catalytic reaction mechanism based on the previous research experience and the absolute configuration of the products. First, 3-ylideneoxindole 81a was activated and oriented by the bifunctional tertiary amine/squaramide C16via double hydrogen-bonding interactions. On the other hand, substrate 1g was deprotonated by the tertiary amine of the catalyst C16 to enhance its nucleophilicity. The nitrogen anion of 1g attacked the 3-ylideneoxindole 81a on the Si face via intermolecular aza-Michael addition to deliver transition state TS-49. Subsequently, intramolecular Michael addition of TS-50 from the Si face afforded transition state TS-51, which underwent protonation to form the desired product 82a and regenerate the catalyst C16.
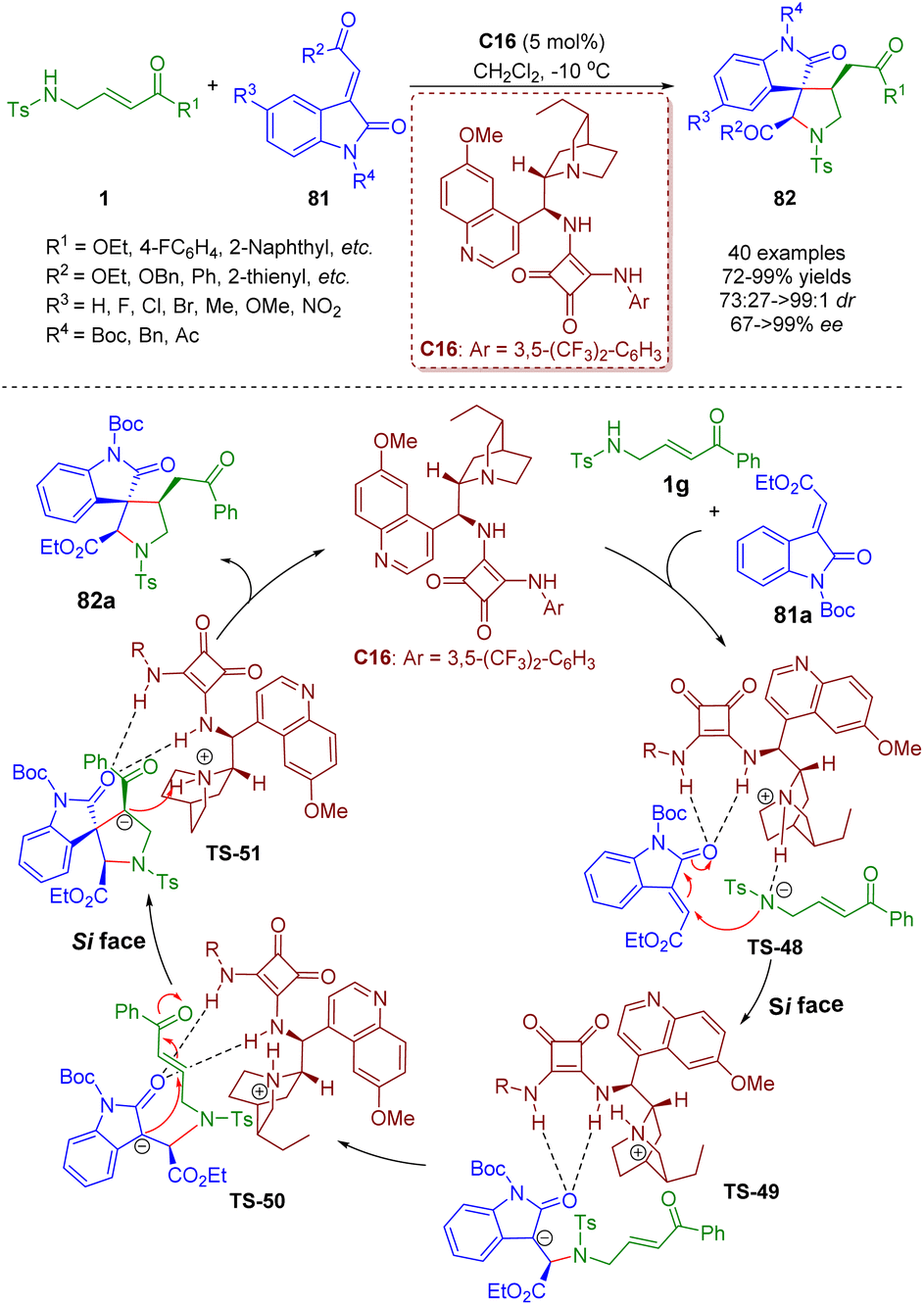 | ||
| Scheme 33 Bifunctional squaramide-catalyzed asymmetric cascade intermolecular aza-Michael/intramolecular Michael addition (Du, 201546). | ||
Twenty days later, the same group reported a bifunctional tertiary amine/squaramide-catalyzed asymmetric cascade inter-molecular aza-Michael/intramolecular Michael addition between γ-sulfonamido-substituted α,β-unsaturated esters/ketones 1 and nitroalkenes 83 (Scheme 34).47 This organocatalytic cascade reaction can afford various highly functionalized chiral trisubstituted pyrrolidines 84 (21 examples) in moderate to excellent yields (43–99%) with moderate to good diastereoselectivities (53:47–91:9 dr) and good to excellent enantioselectivities (56–>99% ee) under mild conditions.
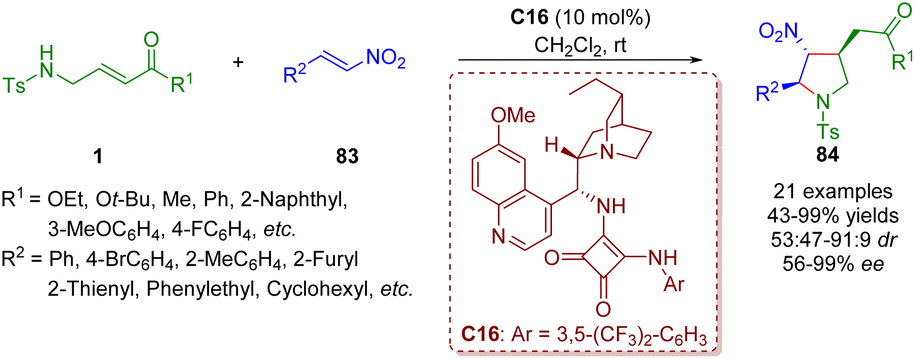 | ||
| Scheme 34 Bifunctional squaramide-catalyzed asymmetric cascade intermolecular aza-Michael/intramolecular Michael addition (Du, 201547). | ||
In 2016, a bifunctional tertiary amine/squaramide-catalyzed cascade intermolecular aza-Michael/intramolecular Michael addition of γ-sulfonamido-substituted α,β-unsaturated ketones 1 and pyrazolones 85 was developed by the Du group (Scheme 35).48 A series of spiropyrrolidine-pyrazolones 86 were synthesized in good to excellent yields (49–98%) with excellent diastereoselectivities (all >20:1 dr) and high to excellent enantioselectivities (71–98% ee).
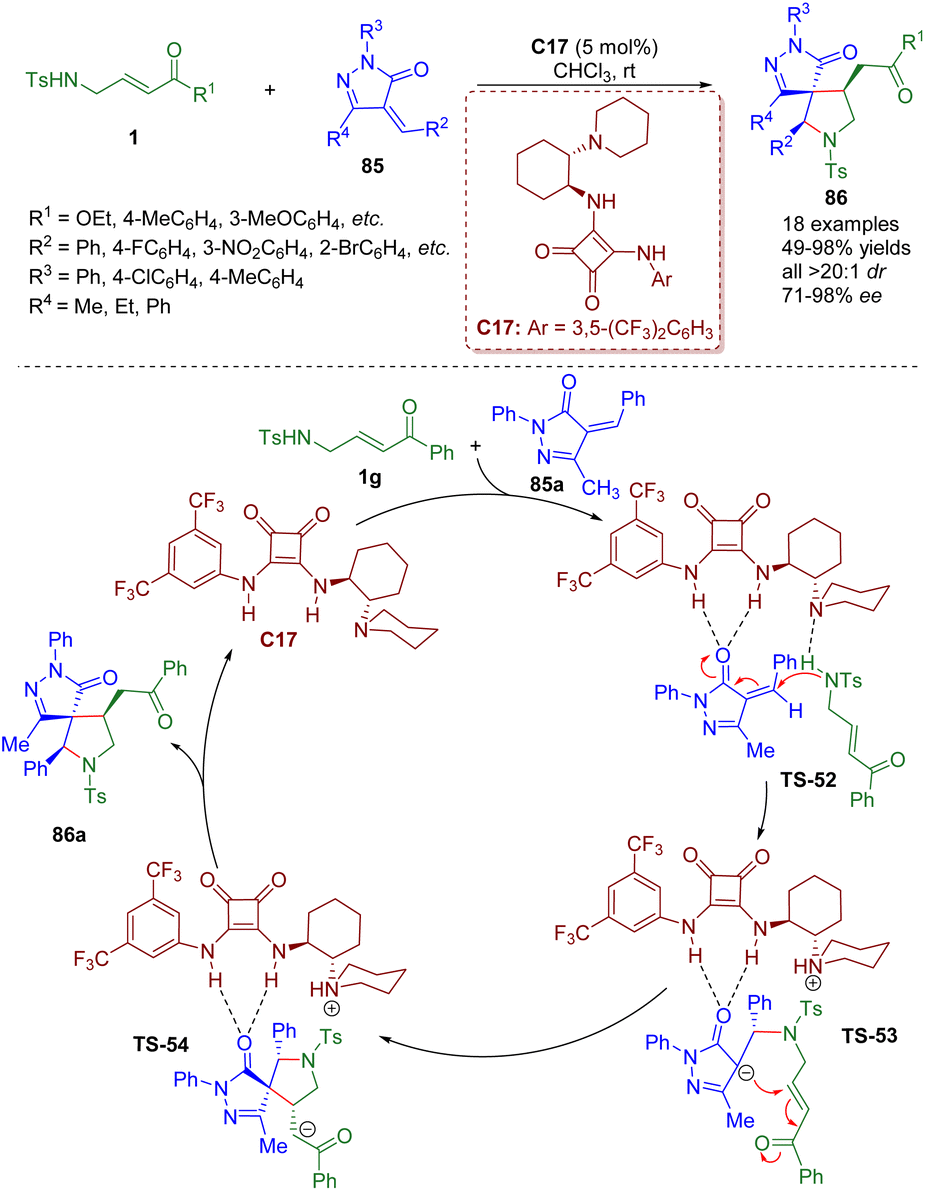 | ||
| Scheme 35 Bifunctional squaramide-catalyzed asymmetric cascade intermolecular aza-Michael/intramolecular Michael addition (Du, 201648). | ||
In 2018, Pan and Mukhopadhyay developed a formal [3 + 2] annulation of γ-sulfonamido-substituted α,β-unsaturated ketones 1 and N-tosyl imines 33 by asymmetric bifunctional squaramide catalysis (Scheme 36).49 This methodology can afford 2,4-disubstituted imidazolidines 87 in good to high yields (60–82%) with high diastereoselectivities (10:1–>20:1 dr) and enantioselectivities (73–92% ee) under mild and simple reaction conditions.
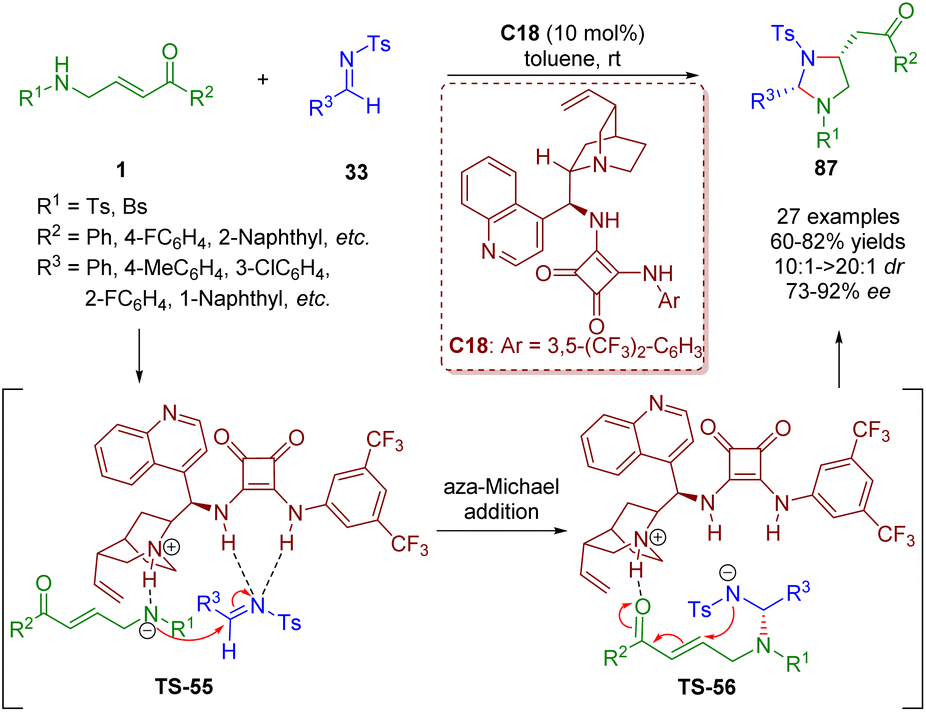 | ||
| Scheme 36 Bifunctional squaramide-catalyzed [3 + 2] annulation (Pan, 201849). | ||
In the same year, Pan and Mukhopadhyay reported an asymmetric bifunctional squaramide-catalyzed formal [3 + 2] annulation of γ-sulfonamido-substituted α,β-unsaturated ketones 1 and trans-α-cyano-α,β-unsaturated ketones 39 for the synthesis of highly substituted pyrrolidines 88 bearing an all-carbon quaternary stereocenter (Scheme 37).50
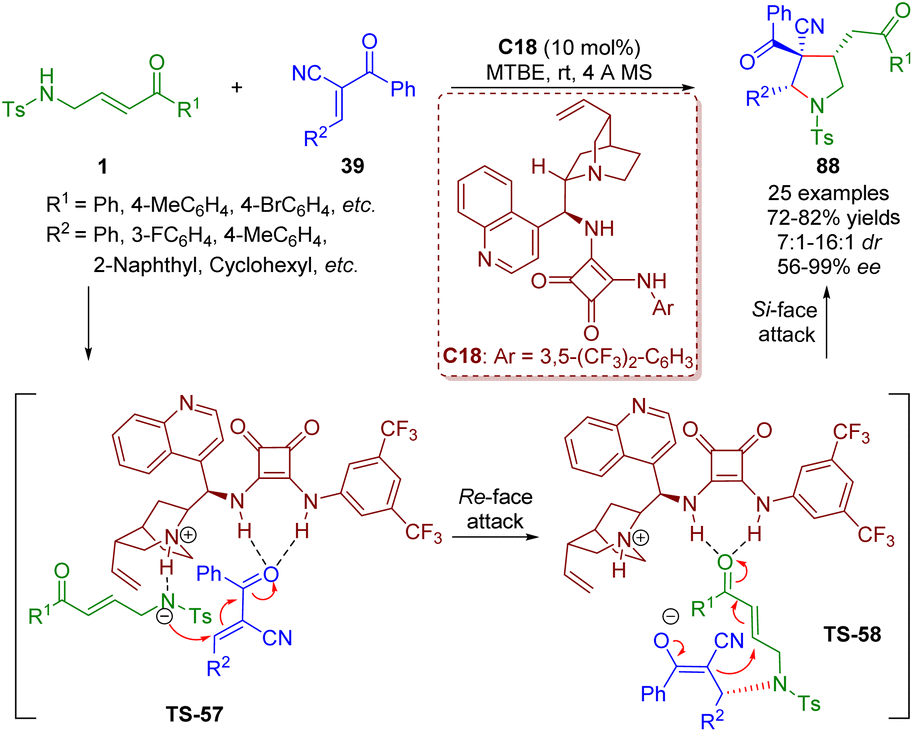 | ||
| Scheme 37 Bifunctional squaramide-catalyzed [3 + 2] annulation (Pan, 201850). | ||
In a similar fashion, the first asymmetric bifunctional squaramide-catalyzed formal [3 + 2] annulation of γ-sulfonamido-substituted α,β-unsaturated ketones 1 with simple alkyl aldehydes 89 was reported by Pan and Mukhopadhyay in 2019 (Scheme 38).51 Oxazolidines 90 were obtained in good yields (68–76%) with high diastereoselectivities (1:1–>20:1 dr) and enantioselectivities (86–99% ee).
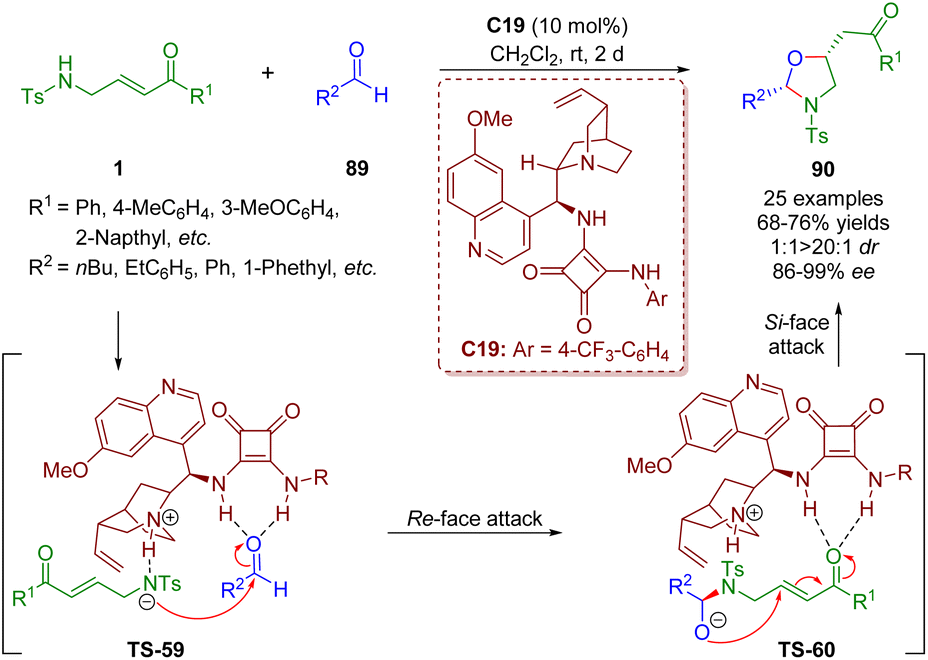 | ||
| Scheme 38 Bifunctional squaramide-catalyzed [3 + 2] annulation (Pan, 201951). | ||
More recently, the Kim group reported a bifunctional squaramide-catalyzed [3 + 2] annulation of γ-sulfonamido-α,β-unsaturated ketones 1 with imines 37, producing a series of imidazolidines 38 (23 examples) in good yields (53–99%) with moderate to excellent diastereoselectivities (4:1–>30:1 dr) and enantioselectivities (42–95% ee) under mild reaction conditions (Scheme 39).52 This strategy was realized by Guo and coworkers via phosphine catalysis in 2019.
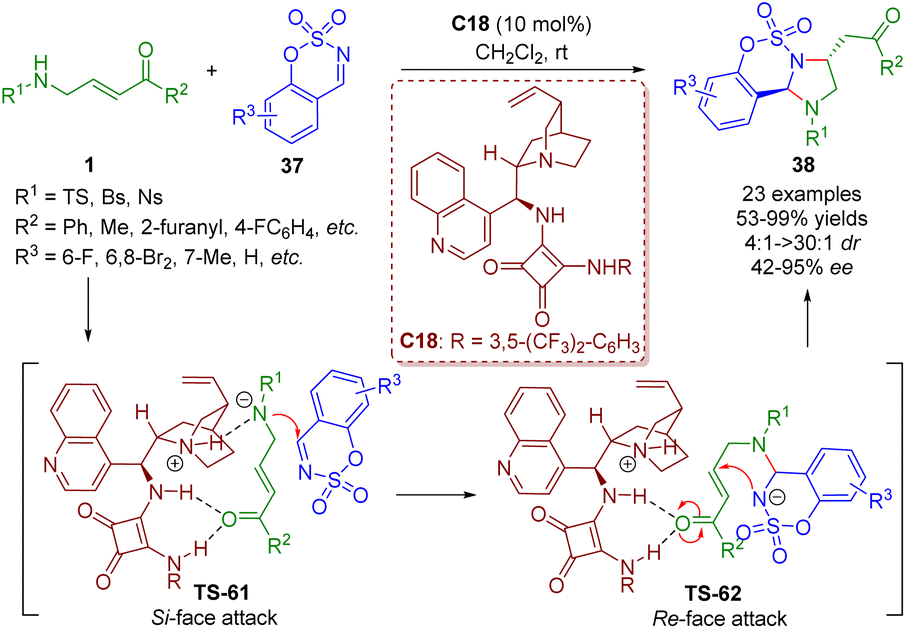 | ||
| Scheme 39 Bifunctional squaramide-catalyzed [3 + 2]-annulation (Kim, 202352). | ||
Soon after, by replacing γ-sulfonamido-substituted α,β-unsaturated ketones 1 with δ-sulfonamido-substituted α,β-unsaturated ketones 1, Kim and co-workers obtained functionalized hexahydropyrimidines 91 in the presence of the bifunctional squaramide catalyst C18 (Scheme 40).53
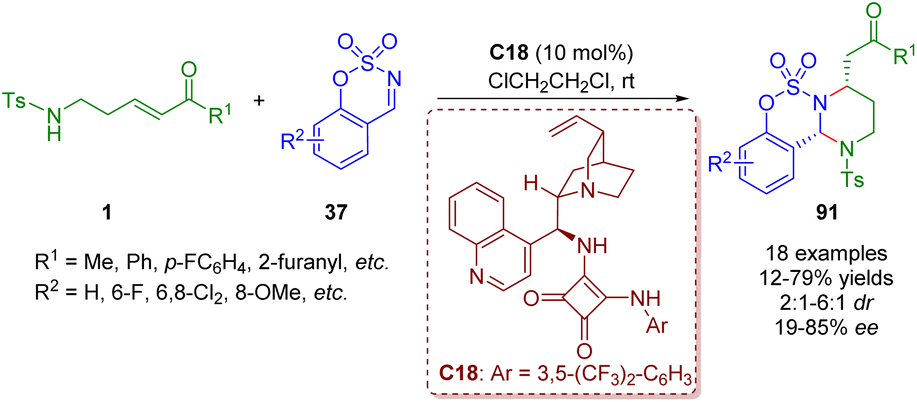 | ||
| Scheme 40 Bifunctional squaramide-catalyzed [3 + 2]-annulation of δ-sulfonamido-α,β-unsaturated ketones with cyclic N-sulfimines (Kim, 202353). | ||
4.3. Bifunctional tertiary amine/thiourea catalysis
Bifunctional tertiary amines/thioureas, the same as bifunctional tertiary amines/squaramides, have emerged as powerful multiple hydrogen-bonding catalysts for generating complex molecular structures.In 2017, Kanger and co-workers accomplished a bifunctional thiourea-catalyzed cascade aza-Michael/Michael reaction of δ-amino-substituted α,β-unsaturated esters 1e and trans-β-nitrostyrene 83 for the asymmetric synthesis of 2,3,4-trisubstituted piperidines 92 and 93 as a mixture (1:1–1.4:1) (Scheme 41).54
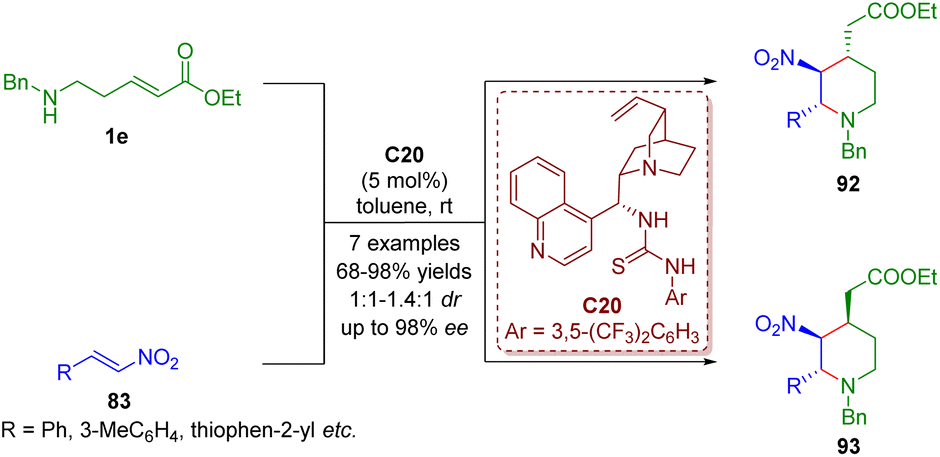 | ||
| Scheme 41 Bifunctional thiourea-catalyzed cascade aza-Michael–Michael reaction (Kanger, 201754). | ||
In 2017, Takayama and co-workers accomplished the asymmetric total synthesis of (−)-lasubine (98), relying on a bifunctional thiourea-catalyzed enantioselective intramolecular aza-Michael addition of amino-substituted α,β-unsaturated ketones 1 (Scheme 42).55 The key reaction of the synthesis is the treatment of N-Bus perhydropyridine 94a (95% ee) with TFA followed by cyclization with K2CO3, which produced quinolizidinone 96 in 67% yield with 74% ee. On the other hand, deprotection of the Ns group in 94b in the presence of K2CO3/PMPSH afforded quinolizidinone 96 in 58% yield with 69% ee. It is noteworthy that the decrease of the enantiomeric excess in the above two different courses was probably caused by the partial racemization via retro aza-Michael addition under the strongly acidic/basic conditions.
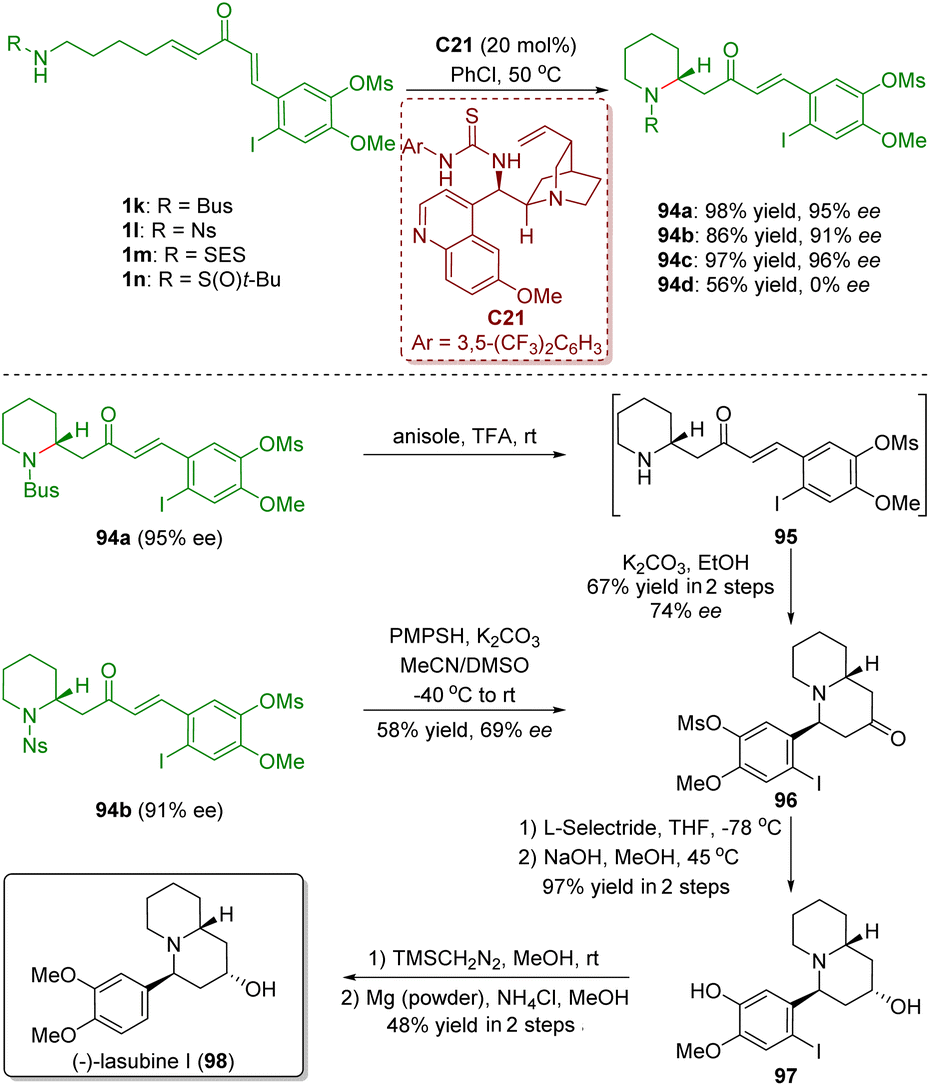 | ||
| Scheme 42 Total synthesis of (−)-lasubine (Takayama, 201755). | ||
Based on the diarylprolinol silyl ether-catalyzed cascade aza-Michael/Michael reaction of δ-amino α,β-unsaturated esters/ketones 1 and α,β-unsaturated aldehydes 29 (Scheme 9),22 a bifunctional tertiary amine/thiourea-catalyzed cascade aza-Michael/Michael reaction of δ-amino-substituted α,β-unsaturated esters/ketones 1 and trans-β-nitrostyrene 83 was reported by Albrecht and co-workers in 2018 (Scheme 43).56 This methodology can produce tetrahydro-1,2-oxazines 99 in high yields (66–97%) and excellent diastereoselectivities (18:1–>20:1 dr) and enantioselectivities (88:12–99.5:0.5 er).
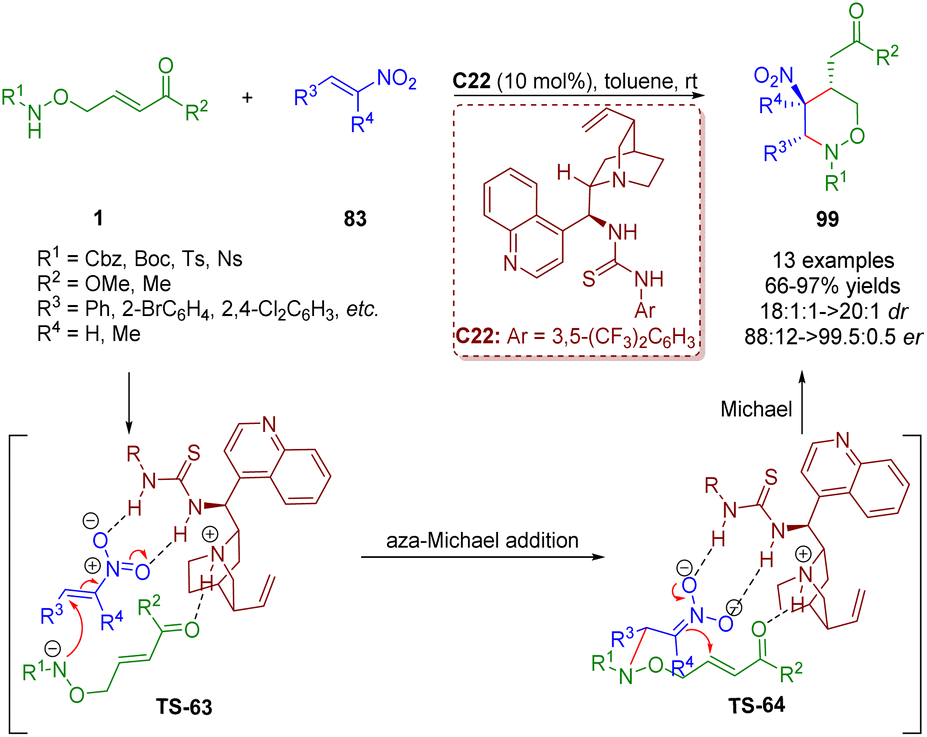 | ||
| Scheme 43 Bifunctional thiourea-catalyzed cascade aza-Michael/Michael reaction (Albrecht, 201856). | ||
5. Conclusions and outlook
In summary, straight-chain ω-amino-α,β-unsaturated carbonyl compounds prove to be versatile building blocks participating in various organocatalytic reactions, including intramolecular aza-Michael addition reactions, cascade intermolecular aza-Michael addition/Michael addition reactions, cascade Mannich/aza-Michael addition reactions, and so on, for the synthesis of five- and six-membered saturated nitrogen-containing heterocycles. This review discussed the recent applications of such organic synthons according to the following reaction modes: (1) covalent catalysis (secondary amine catalysis, phosphine catalysis, and N-heterocyclic carbene catalysis), (2) non-covalent catalysis (Brønsted acid catalysis), and (3) bifunctional catalysis (bifunctional tertiary amine/primary amine catalysis, bifunctional tertiary amine/squaramide catalysis, and bifunctional tertiary amine/thiourea catalysis).Despite the remarkable developments in the area of nitrogen-containing heterocycle synthesis using straight-chain ω-amino-α,β-unsaturated carbonyl compounds as versatile synthons over the past seventeen years, there are still some limitations and challenges for this strategy, such as the construction of medium-sized nitrogen-containing heterocycles and the applications of photo-/electro-catalyzed annulation reactions. We hope and believe that the current achievements will inspire synthetic and pharmaceutical chemists to develop novel reaction modes and catalytic systems, leading to a new chapter in the synthesis of nitrogen-containing heterocycles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support provided by the National Natural Science Foundation of China (22207063), the Higher Education Discipline Innovation Project (D20015), the Natural Science Foundation of Hubei Province (2022CFB838), the Hubei Provincial Department of Education (D20221204 and Q20221212), and the Natural Science Foundation of Yichang City (A23-2-002).References
- Selected reviews on natural products and drugs containing pyrrolidine/piperidine subunits: (a) Z. Amara, J. Caron and D. Joseph, Recent contributions from the asymmetric aza-Michael reaction to alkaloids total synthesis, Nat. Prod. Rep., 2013, 30, 1211–1225 RSC; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (c) N. Wang, X. Xiao, C.-X. Liu, H. Yao, N. Huang and K. Zou, Recent advances in the total synthesis of Aspidosperma and Kopsia alkaloids using tetracyclic pyridocarbazoles as versatile building blocks, Adv. Synth. Catal., 2022, 364, 2479–2501 CrossRef CAS; (d) G. Li, M. Lou and X. Qi, A brief overview of classical natural product drug synthesis and bioactivity, Org. Chem. Front., 2022, 9, 517–571 RSC; (e) X. Zhang and J. Xu, Five-membered carbocycle construction in the synthesis of Daphniphyllum alkaloids: recent strategic and methodological advances, Org. Chem. Front., 2022, 9, 6708–6716 RSC. For selected examples, see: (f) F.-F. Xu, J.-Q. Chen, D.-Y. Shao and P.-Q. Huang, Catalytic enantioselective reductive alkynylation of amides enables one-pot syntheses of pyrrolidine, piperidine and indolizidine alkaloids, Nat. Commun., 2023, 14, 6251 CrossRef CAS PubMed; (g) K.-L. Ji, S.-F. He, D.-D. Xu, W.-X. He, J.-F. Zheng and P.-Q. Huang, Concise total synthesis of (−)-quinocarcin enabled by catalytic enantioselective reductive 1,3-dipolar cycloaddition of secondary amides, Angew. Chem., Int. Ed., 2023, 62, e202302832 CrossRef CAS PubMed; (h) W. Zhou, S. Xi, H. Chen, D. Jiang, J. Yang, S. Liu, L. He, H. Qiu, Y. Lan and M. Zhang, A bridged backbone strategy enables collective synthesis of strychnan alkaloids, Nat. Chem., 2023, 15, 1074–1082 CrossRef CAS PubMed.
- For selected reviews, see: (a) M.-Y. Han, J.-Y. Jia and W. Wang, Recent advances in organocatalytic asymmetric synthesis of polysubstituted pyrrolidines, Tetrahedron Lett., 2014, 55, 784–794 CrossRef CAS; (b) M. Sánchez-Roselló, M. Escolano, D. Gaviña and C. del Pozo, Two Decades of progress in the asymmetric intramolecular aza–Michael reaction, Chem. Rec., 2022, 22, e202100161 CrossRef PubMed; (c) N. A. Frolov and A. N. Vereshchagin, Piperidine derivatives: recent advances in synthesis and pharmacological applications, Int. J. Mol. Sci., 2023, 24, 2937 CrossRef CAS PubMed; (d) K. T. Ashitha, M. S. Ajay Krishna, D. Basavaraja and S. B. Somappa, Recent advances in the transition metal-free synthesis of heterocycles from α,β-unsaturated ketones, Org. Chem. Front., 2022, 9, 5306–5357 RSC.
- Selected reviews on organocatalysis: (a) D. W. C. MacMillan, The advent and development of organocatalysis, Nature, 2008, 455, 304–308 CrossRef CAS PubMed; (b) C. M. Marson, Multicomponent and sequential organocatalytic reactions: diversity with atom-economy and enantiocontrol, Chem. Soc. Rev., 2012, 41, 7712–7722 RSC; (c) J. Alemán and S. Cabrera, Applications of asymmetric organocatalysis in medicinal chemistry, Chem. Soc. Rev., 2013, 42, 774–793 RSC; (d) G. Zhan, W. Du and Y.-C. Chen, Switchable divergent asymmetric synthesis via organocatalysis, Chem. Soc. Rev., 2017, 46, 1675–1692 RSC; (e) Y.-B. Wang and B. Tan, Construction of axially chiral compounds via asymmetric organocatalysis, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed; (f) N. Wang, Z. Wu, J. Wang, N. Ullah and Y. Lu, Recent applications of asymmetric organocatalytic annulation reactions in natural product synthesis, Chem. Soc. Rev., 2021, 50, 9766–9793 RSC; (g) Y. Hussain, Tamanna, M. Sharma, A. Kumar and P. Chauhan, Recent development in asymmetric organocatalytic domino reactions involving 1,6-addition as a key step, Org. Chem. Front., 2022, 9, 572–592 RSC; (h) R. Ballini, A. Palmieri and M. Petrini, Catalysts’ evolution in the asymmetric conjugate addition of nitroalkanes to electron-poor alkenes, Org. Chem. Front., 2022, 9, 6077–6113 RSC.
- Selected reviews on the organocatalytic aza-Michael addition reactions: (a) J. Wang, P. Li, P. Y. Choy, A. S. C. Chan and F. Y. Kwong, Advances and applications in organocatalytic asymmetric aza–Michael addition, ChemCatChem, 2012, 4, 917–925 CrossRef CAS; (b) M. Sánchez-Roselló, J. L. Aceña, A. Simón-Fuentes and C. del Pozo, A general overview of the organocatalytic intramolecular aza-Michael reaction, Chem. Soc. Rev., 2014, 43, 7430–7453 RSC; (c) M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Recent advances in the asymmetric synthesis of pharmacology-relevant nitrogen heterocycles via stereoselective aza-Michael reactions, Org. Biomol. Chem., 2019, 17, 3670–3708 RSC; (d) Y.-X. Song and D.-M. Du, Recent advances in catalytic asymmetric aza–Michael addition triggered cascade reactions, Adv. Synth. Catal., 2021, 363, 4667–4694 CrossRef CAS.
- S. Fustero, D. Jiménez, J. Moscardó, S. Catalán and C. del Pozo, Enantioselective organocatalytic intramolecular aza-Michael reaction: a concise synthesis of (+)-sedamine, (+)-allosedamine, and (+)-coniine, Org. Lett., 2007, 9, 5283–5286 CrossRef CAS PubMed.
- Selected reviews on secondary amine catalysis: (a) A. Erkkilä, I. Majander and P. M. Pihko, Iminium catalysis, Chem. Rev., 2007, 107, 5416–5470 CrossRef PubMed; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Asymmetric enamine catalysis, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed; (c) V. Marcos and J. Alemán, Old tricks, new dogs: organocatalytic dienamine activation of α,β-unsaturated aldehydes, Chem. Soc. Rev., 2016, 45, 6812–6832 RSC; (d) L. Klier, F. Tur, P. H. Poulsen and K. A. Jørgensen, Asymmetric cycloaddition reactions catalysed by diarylprolinol silyl ethers, Chem. Soc. Rev., 2017, 46, 1080–1102 RSC; (e) P. Chauhan, S. Mahajan and D. Enders, Achieving molecular complexity via stereoselective multiple domino reactions promoted by a secondary amine organocatalyst, Acc. Chem. Res., 2017, 50, 2809–2821 CrossRef CAS PubMed; (f) G. J. Reyes-Rodríguez, N. M. Rezayee, A. Vidal-Albalat and K. A. Jørgensen, Prevalence of diarylprolinol silyl ethers as catalysts in total synthesis and patents, Chem. Rev., 2019, 119, 4221–4260 CrossRef PubMed.
- Selected reviews on phosphine catalysis: (a) L.-W. Ye, J. Zhou and Y. Tang, Phosphine-triggered synthesis of functionalized cyclic compounds, Chem. Soc. Rev., 2008, 37, 1140–1152 RSC; (b) Y.-N. Gao and M. Shi, Phosphine-mediated enantioselective synthesis of carbocycles and heterocycles, Chin. Chem. Lett., 2017, 28, 493–502 CrossRef CAS; (c) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Phosphine organocatalysis, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed; (d) H. Ni, W.-L. Chan and Y. Lu, Phosphine-catalyzed asymmetric organic reactions, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed; (e) E.-Q. Li and Y. Huang, Recent advances in phosphine catalysis involving γ-substituted allenoates, Chem. Commun., 2020, 56, 680–694 RSC; (f) Y. Huang, J. Liao, W. Wang, H. Liu and H. Guo, Synthesis of heterocyclic compounds through nucleophilic phosphine catalysis, Chem. Commun., 2020, 56, 15235–15281 RSC.
- Selected reviews on N-heterocyclic carbene catalysis: (a) D. Enders, O. Niemeier and A. Henseler, Organocatalysis by N-heterocyclic carbenes, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (b) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An overview of N-heterocyclic carbenes, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (c) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Organocatalytic reactions enabled by N-heterocyclic carbenes, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed; (d) K. J. R. Murauski, A. A. Jaworski and K. A. Scheidt, A continuing challenge: N-heterocyclic carbene-catalyzed syntheses of γ-butyrolactones, Chem. Soc. Rev., 2018, 47, 1773–1782 RSC; (e) S. Mondal, S. R. Yetra, S. Mukherjee and A. T. Biju, NHC-catalyzed generation of α,β-unsaturated acylazoliums for the enantioselective synthesis of heterocycles and carbocycles, Acc. Chem. Res., 2019, 52, 425–436 CrossRef CAS PubMed; (f) L. Dai and S. Ye, Recent advances in N-heterocyclic carbene-catalyzed radical reactions, Chin. Chem. Lett., 2021, 32, 660–667 CrossRef CAS; (g) Y. Wang, Y. Liu, S. Zhao, Y. Long and X. Wu, Catalyst-controlled stereoselective carbon–heteroatom bond formations by N-heterocyclic carbene (NHC) organocatalysis, Org. Chem. Front., 2023, 10, 4437–4458 RSC.
- Selected reviews on Brønsted acid catalysis: (a) J. Yu, F. Shi and L.-Z. Gong, Brønsted-acid-catalyzed asymmetric multicomponent reactions, Acc. Chem. Res., 2011, 44, 1156–1171 CrossRef CAS PubMed; (b) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: history and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed; (c) X. Li and Q. Song, Recent advances in asymmetric reactions catalyzed by chiral phosphoric acids, Chin. Chem. Lett., 2018, 29, 1181–1192 CrossRef CAS; (d) Z.-L. Xia, Q.-F. Xu-Xu, C. Zheng and S.-L. You, Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions, Chem. Soc. Rev., 2020, 49, 286–300 RSC; (e) Y.-C. Zhang, F. Jiang and F. Shi, Organocatalytic asymmetric synthesis of indole-based chiral heterocycles: strategies, reactions, and outreach, Acc. Chem. Res., 2019, 53, 425–446 CrossRef PubMed.
- Selected reviews on bifunctional primary amine/tertiary amine catalysis: (a) L. Jiang and Y.-C. Chen, Recent advances in asymmetric catalysis with cinchona alkaloid-based primary amines, Catal. Sci. Technol., 2011, 1, 354–365 RSC; (b) J. Duan and P. Li, Asymmetric organocatalysis mediated by primary amines derived from cinchona alkaloids: recent advances, Catal. Sci. Technol., 2014, 4, 311–320 RSC.
- Selected reviews on bifunctional tertiary amine/squaramide catalysis: (a) P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Bifunctional amine–squaramides: powerful hydrogen–bonding organocatalysts for asymmetric domino/cascade reactions, Adv. Synth. Catal., 2015, 357, 253–281 CrossRef CAS; (b) B.-L. Zhao, J.-H. Li and D.-M. Du, Squaramide–catalyzed asymmetric reactions, Chem. Rec., 2017, 17, 994–1018 CrossRef CAS PubMed; (c) X.-Q. Hou and D.-M. Du, Recent advances in squaramide–catalyzed asymmetric mannich reactions, Adv. Synth. Catal., 2020, 362, 4487–4512 CrossRef CAS.
- Selected reviews on bifunctional tertiary amine/thiourea catalysis: (a) X. Fang and C.-J. Wang, Recent advances in asymmetric organocatalysis mediated by bifunctional amine–thioureas bearing multiple hydrogen-bonding donors, Chem. Commun., 2015, 51, 1185–1197 RSC; (b) S. Gandhi, V. Sivadas and B. Baire, Thiourea–tertiary amine promoted cascade catalysis: a tool for complexity generation, Eur. J. Org. Chem., 2020, 220–234 Search PubMed.
- E. Knoevenagel, Ueber eine darstellungsweise der glutarsäure, Ber. Dtsch. Chem. Ges., 1894, 27, 2345–2346 CrossRef.
- S. Fustero, J. Moscardó, M. Sánchez-Roselló, S. Flores, M. Guerola and C. del Pozo, Organocatalytic enantioselective synthesis of quinolizidine alkaloids (+)-myrtine, (−)-lupinine, and (+)-epiepiquinamide, Tetrahedron, 2011, 67, 7412–7417 CrossRef CAS.
- E. C. Carlson, L. K. Rathbone, H. Yang, N. D. Collett and R. G. Carter, Improved protocol for asymmetric, intramolecular heteroatom Michael addition using organocatalysis: enantioselective syntheses of homoproline, pelletierine, and homopipecolic acid, J. Org. Chem., 2008, 73, 5155–5158 CrossRef CAS PubMed.
- N. Veerasamy, E. C. Carlson and R. G. Carter, Expedient enantioselective synthesis of cermizine D, Org. Lett., 2012, 14, 1596–1599 CrossRef CAS PubMed.
- N. Veerasamy, E. C. Carlson, N. D. Collett, M. Saha and R. G. Carter, Enantioselective approach to quinolizidines: total synthesis of cermizine D and formal syntheses of senepodine G and cermizine C, J. Org. Chem., 2013, 78, 4779–4800 CrossRef CAS PubMed.
- B. B. Snider and J. F. Grabowski, Total synthesis of (−)-senepodine G and (−)-cermizine C, J. Org. Chem., 2007, 72, 1039–1042 CrossRef CAS PubMed.
- Y. Ying, H. Kim and J. Hong, Stereoselective synthesis of 2,6-cis- and 2,6-trans-piperidines through organocatalytic aza-Michael reactions: a facile synthesis of (+)-myrtine and (−)-epimyrtine, Org. Lett., 2011, 13, 796–799 CrossRef CAS PubMed.
- H. Li, L. Zu, H. Xie, J. Wang and W. Wang, Highly enantio- and diastereoselective organocatalytic cascade aza-Michael-Michael reactions: a direct method for the synthesis of trisubstituted chiral pyrrolidines, Chem. Commun., 2008, 5636–5638 RSC.
- K. Kriis, T. Melnik, K. Lips, I. Juhanson, S. Kaabel, I. Järving and T. Kanger, Asymmetric Synthesis of 2,3,4-Trisubstituted Piperidines, Synthesis, 2017, 604–614 CAS.
- P. Drelich, M. Moczulski and Ł. Albrecht, d0a3 Synthon equivalents for the stereocontrolled synthesis of functionalized 1,4-amino alcohol precursors, Org. Lett., 2017, 19, 3143–3146 CrossRef CAS PubMed.
- L. Zhou, C. Yuan, Y. Zeng, H. Liu, C. Wang, X. Gao, Q. Wang, C. Zhang and H. Guo, Phosphine-catalyzed [5 + 1] annulation of δ-sulfonamido-substituted enones with N-sulfonylimines: a facile synthesis of tetrahydropyridines, Chem. Sci., 2018, 9, 1831–1835 RSC.
- M. Liu, L. Zhou, W. Shi, Y. Hu, J. Liao, Z. Duan, W. Wang, Y. Wu, B. Zheng and H. Guo, Phosphine-catalyzed (4 + 2) annulation of δ-sulfonamido-substituted enones with 1,1-dicyanoalkenes: synthesis of piperidine derivatives, Org. Lett., 2021, 23, 7703–7707 CrossRef CAS PubMed.
- W. Shi, L. Zhou, B. Mao, Q. Wang, C. Wang, C. Zhang, X. Li, Y. Xiao and H. Guo, Phosphine-catalyzed [3 + 2] annulation of β-sulfonamido-substituted enones with sulfamate-derived cyclic imines, J. Org. Chem., 2019, 84, 679–686 CrossRef CAS PubMed.
- Z. Gao, L. Xie, L. Ji, X. Ma, X. Li, H. Liu and H. Guo, Phosphine-catalyzed [3 + 2] annulation of β-sulfonamido-substituted enones with trans-α-cyano-α, β-unsaturated ketones for the synthesis of highly substituted pyrrolidines, RSC Adv., 2021, 11, 40136–40139 RSC.
- J. T. Maddigan-Wyatt, M. T. Blyth, J. Ametovski, M. L. Coote, J. F. Hooper and D. W. Lupton, Redox isomerization/(3 + 2) allenoate annulation by auto-tandem phosphine catalysis, Chem. – Eur. J., 2021, 27, 16232–16236 CrossRef CAS PubMed.
- L. Zhou, X. Zhang, Q. Wang, M. Liu, W. Wang, Y. Wu, L. Chen and H. Guo, Phosphine-catalyzed asymmetric tandem isomerization/annulation of allyl amines with allenoates: enantioselective annulation of a saturated C-N bond, Org. Lett., 2021, 23, 9173–9178 CrossRef CAS PubMed.
- J. T. Maddigan-Wyatt, J. Cao, J. Ametovski, J. F. Hooper and D. W. Lupton, Enantioselective synthesis of pyrrolidines by a phosphine-catalyzed γ-umpolung/β-umpolung cascade, Org. Lett., 2022, 24, 2847–2852 CrossRef CAS PubMed.
- Z.-F. Zhang, C.-L. Zhang, Z.-Y. Song, Z.-H. Gao and S. Ye, N-Heterocyclic carbene-catalyzed annulation of α-chloroaldehydes with γ-/δ-amino-α,β-unsaturated ketones: enantioselective synthesis of pyrrolidones and piperidones, Chem. – Eur. J., 2018, 24, 8302–8305 CrossRef CAS PubMed.
- Z.-F. Zhang, Z.-H. Gao, C.-L. Zhang and S. Ye, N-heterocyclic carbene-catalyzed intramolecular aza-Michael addition of alkyl amines to α,β-unsaturated carboxylic acid: synthesis of pyrrolidines and piperidines, Tetrahedron, 2021, 94, 132337 CrossRef CAS.
- (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Enantioselective mannich-type reaction catalyzed by a chiral Brønsted acid, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed; (b) D. Uraguchi and M. Terada, Chiral Brønsted acid-catalyzed direct mannich reactions via electrophilic activation, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
- H. Liu, C. Zeng, J. Guo, M. Zhang and S. Yu, Enantioselective synthesis of 2-substituted pyrrolidines via domino cross metathesis/intramolecular aza-Michael addition, RSC Adv., 2013, 3, 1666–1668 RSC.
- C. J. Maddocks, K. Ermanis and P. A. Clarke, Asymmetric “clip-cycle” synthesis of pyrrolidines and spiropyrrolidines, Org. Lett., 2020, 22, 8116–8121 CrossRef CAS PubMed.
- M. Escolano, J. T. Fernández, F. Rabasa-Alcañiz, M. Sánchez-Roselló and C. del Pozo, Enantioselective synthesis of pyrrolizidinone scaffolds through multiple-relay catalysis, Org. Lett., 2020, 22, 9433–9438 CrossRef CAS PubMed.
- S. Panda and P. Ghorai, Expedient access to enantioenriched 1-azaspiro cyclobutanones enabled by modified heyns rearrangement, Angew. Chem., Int. Ed., 2023, e202306179 CAS.
- Y.-R. Ma, X.-J. Lv, Q. Dong, Y.-C. Ming and Y.-K. Liu, Brønsted-acid-catalyzed in situ formation of acyclic tertiary enamides and its application to the preparation of diverse nitrogen-containing heterocyclic compounds, Org. Lett., 2023, 25, 5929–5934 CrossRef CAS PubMed.
- J.-D. Liu, Y.-C. Chen, G.-B. Zhang, Z.-Q. Li, P. Chen, J.-Y. Du, Y.-Q. Tu and C.-A. Fan, Asymmetric organocatalytic intramolecular aza–Michael addition of enone carbamates: catalytic enantioselective access to functionalized 2-substituted piperidines, Adv. Synth. Catal., 2011, 353, 2721–2730 CrossRef CAS.
- S. Fustero, C. del Pozo, C. Mulet, R. Lazaro and M. Sánchez-Roselló, Microwave-assisted organocatalytic enantioselective intramolecular aza-Michael reaction with α,β-unsaturated ketones, Chem. – Eur. J., 2011, 17, 14267–14272 CrossRef CAS PubMed.
- C. Mulet, M. Escolano, S. Llopis, S. Sanz, C. R. de Arellano, M. Sánchez-Roselló, S. Fustero and C. del Pozo, Dual role of vinyl sulfonamides as N-nucleophiles and michael acceptors in the enantioselective synthesis of bicyclic δ-sultams, Adv. Synth. Catal., 2018, 360, 2885–2893 CrossRef CAS.
- M. Guerola, M. Escolano, G. Alzuet-Piña, E. Gómez-Bengoa, C. R. de Arellano, M. Sánchez-Roselló and C. del Pozo, Synthesis of substituted piperidines by enantioselective desymmetrizing intramolecular aza-Michael reactions, Org. Biomol. Chem., 2018, 16, 4650–4658 RSC.
- M. Escolano, M. Guerola, J. Torres, D. Gaviña, G. Alzuet-Piña, M. Sánchez-Roselló and C. del Pozo, Organocatalytic enantioselective synthesis of 2,5,5-trisubstituted piperidines bearing a quaternary stereocenter. Vinyl sulfonamide as a new amine protecting group, Chem. Commun., 2020, 56, 1425–1428 RSC.
- C. Zeng, H. Liu, M. Zhang, J. Guo, S. Jiang and S. Yu, Enantioselective synthesis of cryptopleurine and boehmeriasin a via organocatalytic intramolecular aza-Michael addition, Synlett, 2012, 2251–2254 CAS.
- S. Cheng and S. Yu, Enantioselective synthesis of 3-substituted 1,2-oxazinanes via organocatalytic intramolecular aza-Michael addition, Org. Biomol. Chem., 2014, 12, 8607–8610 RSC.
- X.-D. Zhai, Z.-D. Yang, Z. Luo and H.-T. Xu, Asymmetric catalyzed intramolecular aza-Michael reaction mediated by quinine-derived primary amines, Chin. Chem. Lett., 2017, 28, 1793–1797 CrossRef CAS.
- B.-L. Zhao and D.-M. Du, Organocatalytic enantioselective cascade aza-Michael/Michael addition sequence for asymmetric synthesis of chiral spiro[pyrrolidine-3,3′-oxindole]s, Asian J. Org. Chem., 2015, 4, 1120–1126 CrossRef CAS.
- B.-L. Zhao, Y. Lin, H.-H. Yan and D.-M. Du, Squaramide-catalysed asymmetric cascade aza-Michael/Michael addition reaction for the synthesis of chiral trisubstituted pyrrolidines, Org. Biomol. Chem., 2015, 13, 11351–11361 RSC.
- J.-H. Li, H. Wen, L. Liu and D.-M. Du, Diastereo- and enantioselective synthesis of spiro-pyrrolidine-pyrazolones by squaramide-catalyzed cascade aza-Michael/Michael reactions, Eur. J. Org. Chem., 2016, 2492–2499 CrossRef CAS.
- S. Mukhopadhyay and S. C. Pan, Organocatalytic asymmetric synthesis of 2,4-disubstituted imidazolidines via domino addition-aza-Michael reaction, Chem. Commun., 2018, 54, 964–967 RSC.
- S. Mukhopadhyay and S. C. Pan, Organocatalytic asymmetric synthesis of highly substituted pyrrolidines bearing a stereogenic quaternary centre at the 3-position, Org. Biomol. Chem., 2018, 16, 9349–9353 RSC.
- S. Mukhopadhyay and S. C. Pan, Organocatalytic asymmetric synthesis of 2,5-disubstituted oxazolidines, Adv. Synth. Catal., 2019, 361, 1028–1032 CrossRef CAS.
- Y. Kim, S. Y. Kim and S.-G. Kim, Organocatalytic asymmetric [3 + 2]-annulations of γ-sulfonamido/γ-hydroxy-α,β-unsaturated ketones with cyclic N-sulfimines: synthesis of chiral polyheterotricyclic imidazolidines and oxazolidines, J. Org. Chem., 2023, 88, 1113–1127 CrossRef CAS PubMed.
- S. Y. Kim, Y. Kim and S.-G. Kim, Organocatalytic enantioselective synthesis of 1,3-oxazinanes and hexahydropyrimidines via, [4 + 2]–annulation of cyclic N–sulfimines, Asian J. Org. Chem., 2023, 12, e202300022 CrossRef.
- K. Kriis, T. Melnik, K. Lips, I. Juhanson, S. Kaabel, I. Järving and T. Kanger, Asymmetric synthesis of 2,3,4-trisubstituted piperidines, Synthesis, 2017, 604–614 CAS.
- T. Hirama, T. Umemura, N. Kogure, M. Kitajima and H. Takayama, Synthetic study of biphenylquinolizidine alkaloids I. Asymmetric total synthesis of lasubine I featuring organocatalyzed asymmetric intramolecular aza-Michael addition, Tetrahedron Lett., 2017, 58, 223–226 CrossRef CAS.
- M. Moczulski, P. Drelich and Ł. Albrecht, Bifunctional catalysis in the stereocontrolled synthesis of tetrahydro-1,2-oxazines, Org. Biomol. Chem., 2018, 16, 376–379 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |