
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge transport in liquid crystal network of terthiophene-siloxane block molecules†
Hirotoshi
Sakaino
ab,
Stefan C. J.
Meskers
 c,
E. W.
Meijer
a and
Ghislaine
Vantomme
*a
c,
E. W.
Meijer
a and
Ghislaine
Vantomme
*a
aInstitute for Complex Molecular Systems and Laboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: g.vantomme@tue.nl
bElectronic & Imaging Materials Research Laboratories, Toray Industries, Inc., 3-1-2 Sonoyama, Otsu, Shiga 520-0842, Japan
cInstitute for Complex Molecular Systems and Molecular Materials and Nanosystems, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands
First published on 1st November 2022
Abstract
In their thermotropic liquid-crystalline state, molecular semiconductors can show charge transport with high carrier mobility. Polymerization of the corresponding mesogens into a cross-linked network often deteriorates the charge transport. Here, we report that mesogens consisting of a terthiophene core and discrete oligodimethylsiloxane side-chains terminated by acrylate units can be photopolymerized in the columnar phase with retention of nanoscale order and charge transport capabilities. We argue that the strong tendency for microphase segregation protects the semiconducting block from reacting with free radicals during polymerization. This work provides new insights into the design of electroactive materials with charge transport properties.
Organic electronics are actively pursued for application in stretchable conductors, semiconductors, and E-skin.1,2 In comparison with traditional crystalline semiconductors, polymeric and molecular charge transporting materials are more flexible.3–5 Yet this advantageous mechanical property is often accompanied by less favorable charge transport characteristics. The large number of conformations and intermolecular arrangements that molecules can adopt promotes flexibility and as a result this structural disorder tends to localize and induce trapping sites for charge carriers. To avoid this, one would like to decouple the mechanical from the electronic functionality of the material. Liquid-crystalline networks (LCNs) made of mesogens with π-conjugated, charge transporting cores and flexible polymerizable end-groups offer such an opportunity. These LC materials have been exploited to make an organic field-effect transistor (OFET),6–8 as spontaneous molecular order at the nanoscale promotes charge carrier mobility.9,10 While the photopolymerized end-groups provide structural integrity and mechanical stiffness. However, it remains a challenge to make such LCNs for charge transport11 because the polymerization step can change the molecular packing.12–15 An additional difficulty is that intermediates in the polymerization reaction, e.g. free radicals, may attack the π-electron system of the mesogens leading to deep traps that impair charge transport.
Recently, materials combining the molecular directionality of liquid crystals (LCs) with the phase segregation of block copolymers have been explored. Early examples are phase-segregated LCs,16,17 while more recent examples are based on discrete length block co-oligomers with one block consisting of discrete oligodimethylsiloxane (oDMS).18–23 These materials assemble into well-defined phase-segregated structures with sub-10 nm feature sizes. The discrete nature of both the siloxanes and the complementary block promotes phase separation.24 Hence, we envision using this design to accomplish nanophase segregation in LCNs such that the charge transport functionality carried by the semiconducting moiety is separated in space from the photopolymerizable functionality buried in the siloxane part. Therefore, chemical reactions of reactive species involved in the photopolymerization process on the delicate, unsaturated π-electron system of the semiconducting block may be prevented. Furthermore, after photopolymerization, the phase separation may prevent charge carriers injected into the semiconducting block from getting trapped at potential trap sites posed by unreacted polymerizable groups or remnants of photoinitiator. In addition, the flexible, liquid-like nature of the siloxane units may allow for enough mechanical flexibility such that the molecular packing of the semiconducting block is not impacted by strain that may have built up during the photopolymerization.
Here, we study the network formation and charge transport in phase-segregated LC RM-TTPSi3 (Fig. 1). The mesogen is composed of a terthiophene central core decorated with short siloxane tails and terminated with polymerizable acrylate units. The morphological and electronic properties of the material before and after photopolymerization show that the charge transport functionality is maintained going from the unpolymerized LC state to the LCN state.
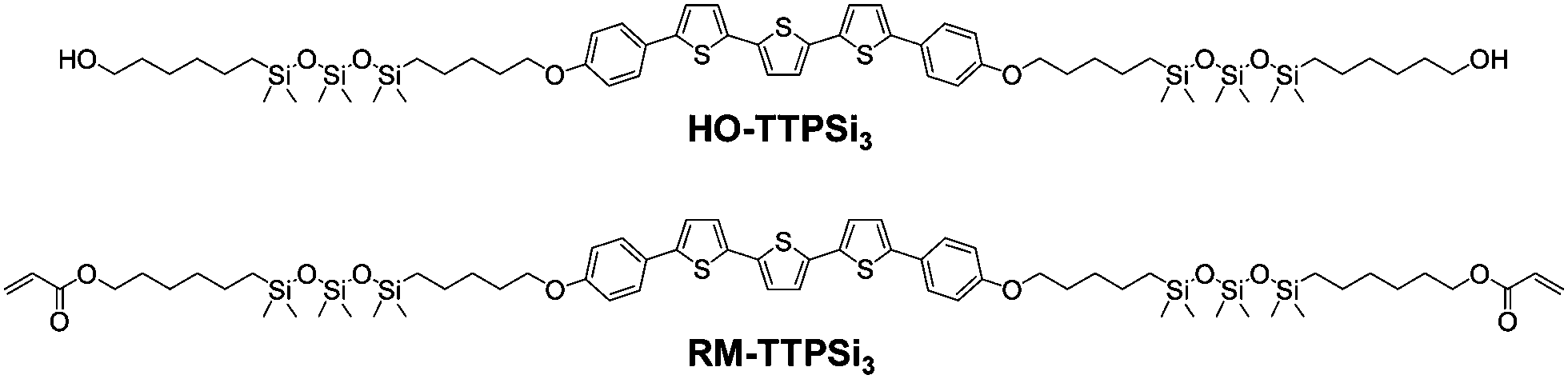 | ||
| Fig. 1 Chemical structures of terthiophene-siloxane mesogens HO-TTPSi3 and RM-TTPSi3. | ||
The reactive terthiophene-siloxane triblock mesogen (RM-TTPSi3, Fig. 1) was synthesized in four steps by coupling of the symmetric olefin-terminated terthiophene and asymmetric alcohol-functionalized oDMS-monohydride (Fig. S1, ESI†). The terthiophene central block was linked to the alcohol-functionalized oDMS-monohydride via a platinum-catalyzed hydrosilylation reaction. And the triblock molecule HO-TTPSi3 obtained was derivatized with acryloyl end-groups. The reactive mesogen RM-TTPSi3 was a waxy orange solid at room temperature and was characterized by NMR and MALDI-TOF mass spectrometry (Fig. S2–S13, ESI†). The thermal properties of HO-TTPSi3 and RM-TTPSi3 were studied using differential scanning calorimetry (DSC) and polarized optical microscopy (POM) (Fig. 2 and Fig. S14–S19, ESI†). In the cooling run of the DSC thermogram, HO-TTPSi3 and RM-TTPSi3 showed two major exothermic transitions between 67 °C and 144 °C. Going through these transitions as monitored by POM, HO-TTPSi3 and RM-TTPSi3 changed from an isotropic melt to a birefringent viscous liquid characteristic of ordered LC phases, which phases were determined in more detail by small-angle X-ray scattering (SAXS) as discussed below.
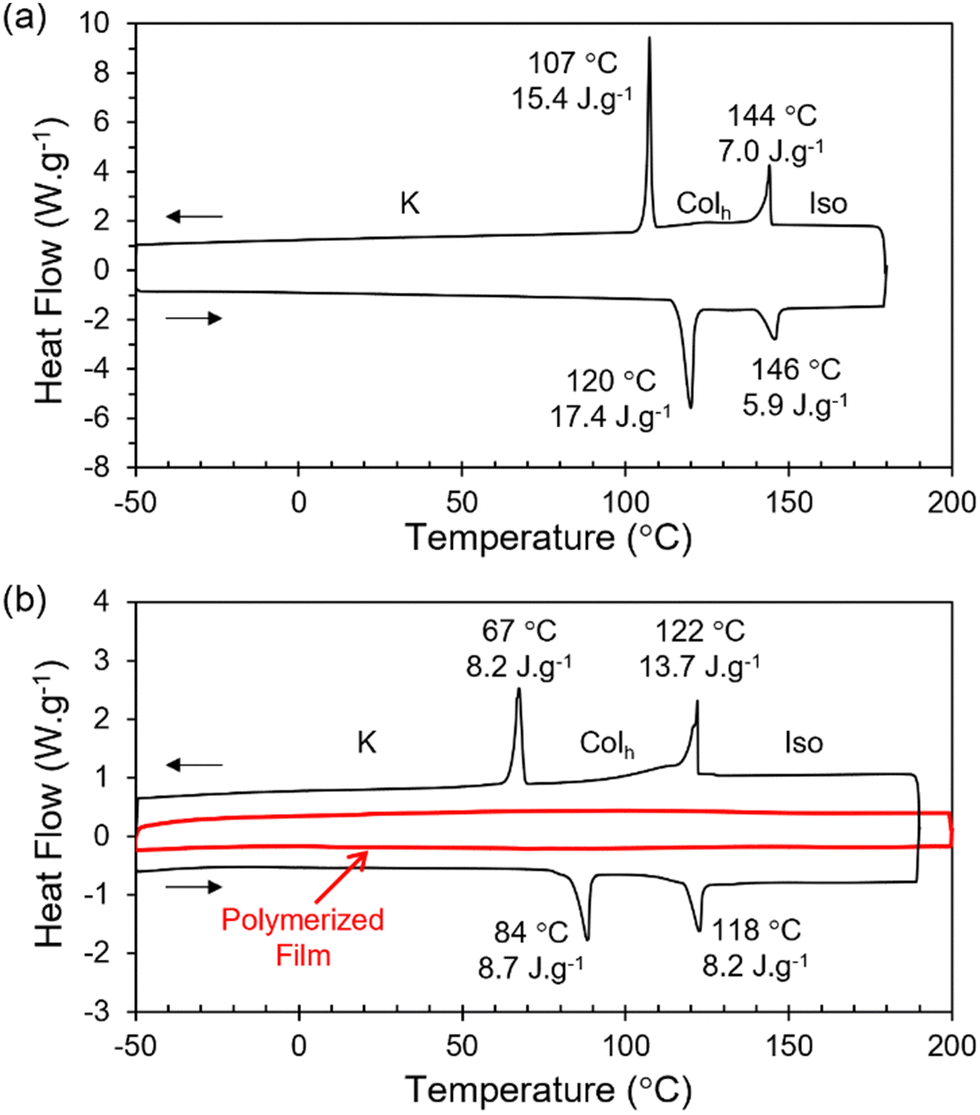 | ||
| Fig. 2 DSC traces (second heating and cooling run) of (a) HO-TTPSi3 and (b) RM-TTPSi3 before (black trace) and after (red trace) photopolymerization at 25 °C. A temperature ramp of 10 K min−1 was used. Exothermic heat flows have a positive value. | ||
To analyze the conformation of the terthiophene units in the LC state, UV experiments and further POM observations were performed. In dichloromethane solution, RM-TTPSi3 exhibits a maximum absorption at 412 nm corresponding to the π–π* absorption band of the terthiophene units (Fig. S20, ESI†). On spin-cast thin films, the maximum absorption of RM-TTPSi3 red-shifts giving three maxima at 417, 438 and 470 nm, indicating the formation of J-type aggregated structures of the terthiophenes in the columnar morphology.6 Variable-temperature SAXS measurements were conducted to confirm the morphological properties of HO-TTPSi3 and RM-TTPSi3. Two distinct morphologies with domain spacings sub-10 nm were characterized: a hexagonal columnar (Colh) phase at 120 °C and a crystalline (K) phase of lamellar structure at 20 °C for HO-TTPSi3 (Fig. 3a) and a Colh phase at 100 °C and a K phase at 20 °C for RM-TTPSi3 (Fig. 3b). Moreover, a broad scattering reflection around 9 nm−1 is visible in the transmission scattering profile representative for the amorphous siloxane halo. It indicates that well-defined nanoscale morphologies are induced by the self-aggregation of the oDMS moieties in the solid state. In addition, the appearance of sharp scattering peaks at 20 °C in the wide-angle region (q > 7 nm−1) suggests the presence of π-stacking between the terthiophene aromatic moieties.
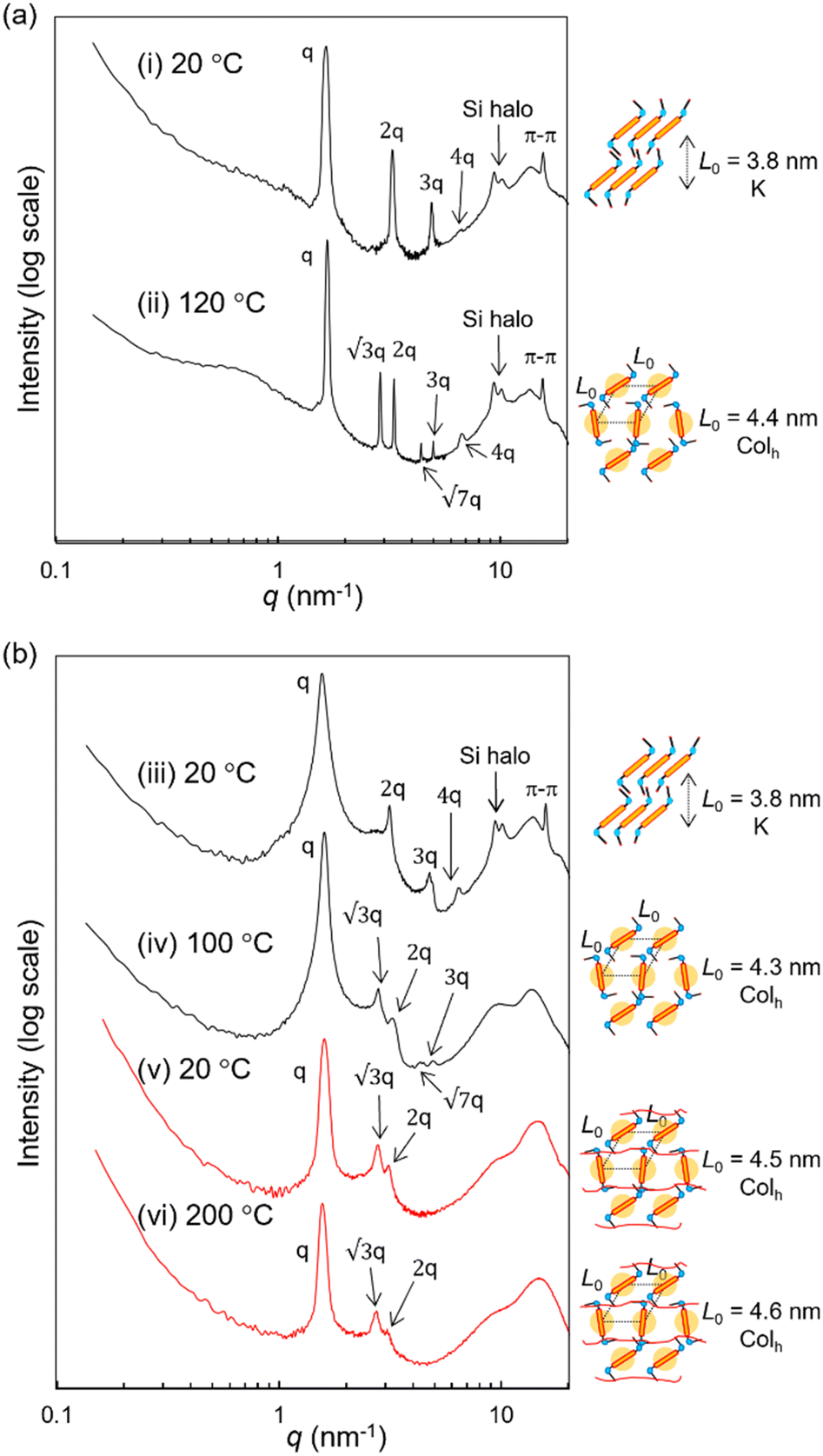 | ||
| Fig. 3 1D transmission scattering profiles of (a) HO-TTPSi3 and (b) RM-TTPSi3 and schematic representations of the molecular packing. The black and red lines indicate a bulk and a film after photo-polymerization at 90 °C, respectively. The SAXS measurements were carried out at (i) 20 °C, (ii) 120 °C, (iii) 20 °C, (iv) 100 °C, (v) 20 °C and (vi) 200 °C. | ||
We prepared LCN films by photopolymerization of a mixture of RM-TTPSi3 and radical photo-initiator (∼2 wt%) at 90 °C. The films obtained were glassy, birefringent and did not show any phase transitions in the DSC thermogram even when scanning up to 200 °C (Fig. 2b and Fig. S21, ESI†). After polymerization, the band of aliphatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching disappeared in FT-IR spectroscopy, indicating complete photopolymerization (Fig. S22, ESI†). Furthermore, in SAXS, the polymerized film exhibited a Colh phase showing that the LC structure is retained after photo-initiated cross-linking (Fig. 3b). SAXS measurements at 200 °C also showed that the domain spacing L0 of the polymerized film did not change with changes in temperature, due to the cross-linking density of the polymer network. These results indicate that the three-dimensional structure of the LC phase is largely preserved in the polymer network formed after photopolymerization.
C stretching disappeared in FT-IR spectroscopy, indicating complete photopolymerization (Fig. S22, ESI†). Furthermore, in SAXS, the polymerized film exhibited a Colh phase showing that the LC structure is retained after photo-initiated cross-linking (Fig. 3b). SAXS measurements at 200 °C also showed that the domain spacing L0 of the polymerized film did not change with changes in temperature, due to the cross-linking density of the polymer network. These results indicate that the three-dimensional structure of the LC phase is largely preserved in the polymer network formed after photopolymerization.
We then explored the transport of charge carriers through the bulk of the material by time-of-flight (TOF) mobility measurements in LC cells with two transparent ITO electrodes separated by 20 mm. Transient photocurrent traces for HO-TTPSi3 and polymerized RM-TTPSi3 in the Colh phase are shown in Fig. 4. After generating charge carriers with a short pulse of UV light at t = 0 μs, the photocurrent shows transport of charges through the material towards the oppositely charged electrode. Both electron and hole transport are observed. Upon arrival of a charge carrier at the electrode opposite to the side of illumination, its contribution to the overall photocurrent ends. The slow decay of the photocurrent over time observed for the bulk sample implies a broad distribution of transit times for individual carriers, i.e. dispersive transport. Nevertheless, at high temperatures (T = 130 °C), a characteristic time for transport can be obtained from the kink in the photocurrent when plotted using logarithmic axes.
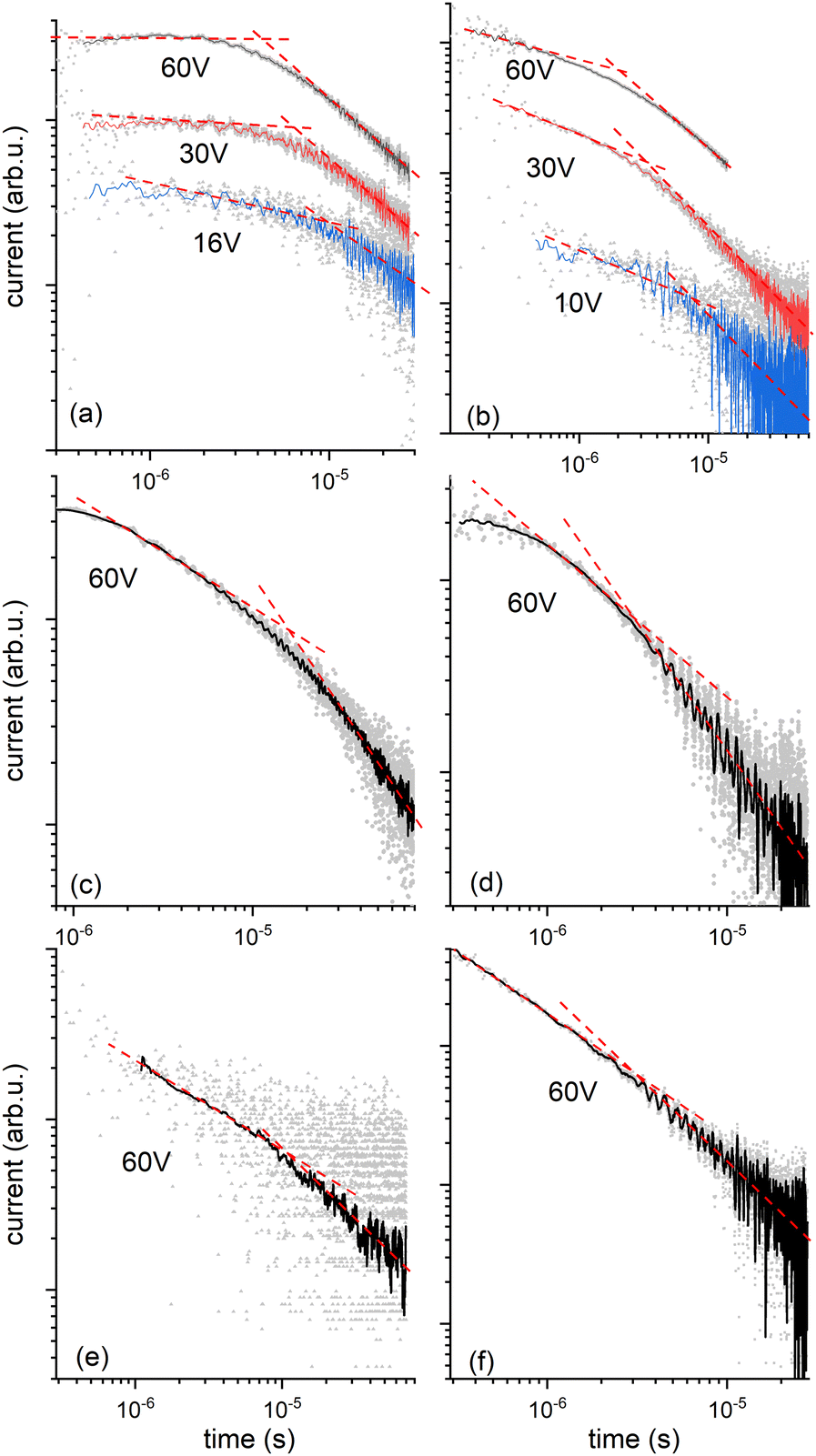 | ||
| Fig. 4 Transient photocurrent curves in time-of-flight mobility measurements. (a) Hole current in HO-TTPSi3 in the Colh phase at 130 °C, for various applied bias voltages. The sample thickness is 20 μm in all cases shown here. (b) Electron current of HO-TTPSi3 in the Colh phase at 130 °C. (c) Hole current of RM-TTPSi3 polymerized at 90 °C. (d) Electron current of RM-TTPSi3 polymerized at 90 °C. (e) Hole current of RM-TTPSi3 polymerized at 50 °C. (f) Electron current of RM-TTPSi3 polymerized at 50 °C. (c–f) Measurements conditions: 140 °C and applied bias 60V. | ||
For compound HO-TTPSi3, almost nondispersive transport of holes is observed in the Colh phase (T = 130 °C, Fig. 4a and Table 1) with a mobility of 2 × 10−2 cm2 V−1 s−1. This value for hole mobility in an LC phase is comparable to mobilities reported for alkyl-functionalized terthiophenes in their LC state.25–29 Preliminary measurements of hole transport at room temperature in the crystalline phase indicate that the charge transport characteristics of the Colh phase are largely maintained in the crystalline phase (Table 1). Transport of electrons is clearly dispersive, yet average mobilities for electrons at high temperatures are similar to those for holes (μelectron = 3 × 10−2 cm2 V−1 s−1, T = 130 °C, Fig. 4b and Table 1). In the crystalline phase near room temperature, transport of electrons is highly dispersive and no characteristic arrival time for the electrons could be obtained from the data. At temperatures above the clearing point, charge transport is impaired, with mobilities for positive charges at least an order of magnitude lower than in the organized phases showing that the disorder in the isotropic phase affects the charge carrier transport.
| Material | T pol [°C] | T meas [°C] | μ hole [cm2 V−1 s−1] | μ electron [cm2 V−1 s−1] |
|---|---|---|---|---|
| a Temperature at which photopolymerization was performed. b Temperature of the photocurrent transient measurement. c Polymerized. | ||||
| HO-TTPSi3 | — | 130 | 2 × 10−2 | 3 × 10−2 |
| HO-TTPSi3 | — | 30 | 2 × 10−2 | Dispersive |
RM-TTPSi3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
90 | 140 | 4 × 10−3 | 2 × 10−2 |
|
RM-TTPSi3c |
90 | 100 | 1 × 10−3 | 1 × 10−2 |
|
RM-TTPSi3c |
50 | 140 | 5 × 10−3 | 2 × 10−2 |
The reactive mesogen RM-TTPSi3 in its non-polymerized state was also investigated. The photocurrents observed were highly dispersive and no clear estimates of transit times could be obtained, suggesting that the acrylate end-groups may interfere with the transport of charges. In addition the broader peaks in the SAXS measurements of RM-TTPSi3, compared to HO-TTPSi3 (Fig. 3) indicate a lower degree of order in the Colh phase of RM-TTPSi3, which may also contribute to poorer transport characteristics.
The charge transport after photopolymerization of RM-TTPSi3 was then measured at high temperature (T = 140 °C, see Fig. 4c, d and Table 1). Remarkably, the thin film LCN retains its ambipolar charge transport capability. In comparison with the unpolymerized HO-TTPSi3 material, the transport of holes is now strongly dispersive showing only featureless photocurrent transients at low applied voltages. Nevertheless, at the highest field strengths used (E = 3 × 104 V cm−1), arrival times could be estimated for both electrons and holes yielding mobilities μhole = 4 × 10−3 cm2 V−1 s−1 and μelectron = 2 × 10−2 cm2 V−1 s−1. Although these mobilities are slightly lower than those for the unpolymerized material, the charge transport capabilities are maintained upon polymerization. These results suggest that the molecular packing of the mesogens is preserved during polymerization and fixed by the polymer network, showing the advantage of the phase-segregated design of the monomer compared to a classical design with alkyl spacers.12–15 The role of the siloxane spacer units is then to isolate the terthiophene cores from the acrylate units, which contributes to minimizing radical damage to the terthiophene cores during polymerization. Nevertheless, the hole mobility is lower than the electron mobility, suggesting that the hole transport process is influenced by hole traps in the bulk state. Moreover, the results were similar for a thin film LCN polymerized at 50 °C in the crystalline phase, confirming the charge transport characteristics of the Colh phase are largely maintained in the crystalline phase (Fig. 4e, f and Table 1).
The present work is a step forward in the efforts to make nanoscale ordered materials with charge transport functionality and exciting applications in organic electronics.
The authors would like to thank Prof. Dirk J. Broer for fruitful discussions, and Brigitte A. G. Lamers for the SAXS measurements. The authors are thankful for the financial support from Toray Industries Inc. and from the Royal Netherlands Academy of Arts and Sciences and the Dutch Ministry of Education, Culture and Science (Gravity program 024.001.035) and the Netherlands Organization for Scientific Research (NWO-TOP PUNT Grant: 10018944, NWO-VENI Grant 722.017.003).
Conflicts of interest
There are no conflicts to declare.References
- J. C. Yang, J. Mun, S. Y. Kwon, S. Park, Z. Bao and S. Park, Adv. Mater., 2019, 31(48), 1904765 CrossRef PubMed.
- P. Friederich, A. Fediai, S. Kaiser, M. Konrad, N. Jung and W. Wenzel, Adv. Mater., 2019, 31, 1808256 CrossRef PubMed.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66 CrossRef.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832 CrossRef.
- A. Pron, P. Gawrys, M. Zagorska, D. Djuradoa and R. Demadrillea, Chem. Soc. Rev., 2010, 39, 2577 RSC.
- A. J. J. M. van Breemen, P. T. Herwig, C. H. T. Chlon, J. Sweelssen, H. F. M. Schoo, S. Setayesh, W. M. Hardeman, C. A. Martin, D. M. de Leeuw, J. J. P. Valeton, C. W. M. Bastiaansen, D. J. Broer, A. R. Popa-Merticaru and S. C. J. Meskers, J. Am. Chem. Soc., 2006, 128(7), 2336 CrossRef CAS.
- B.-H. Huisman, J. J. P. Valeton, W. Nijssen, J. Lub and W. Ten Hoeve, Adv. Mater., 2003, 15, 2002 CrossRef CAS.
- H. Iino, T. Usui and J.-I. Hanna, Nat. Commun., 2015, 6828 CrossRef CAS PubMed.
- J. L. Bredas, J. P. Calbert, D. A. Da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(9), 5804 CrossRef CAS.
- M. O’Neill and S. M. Kelly, Adv. Mater., 2011, 23, 566 CrossRef PubMed.
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- I. McCulloch, M. Coelle, K. Genevicius, R. Hamilton, M. Heckmeier, M. Heeney, T. Kreouzis, M. Shkunov and W. Zhang, Jpn. J. Appl. Phys., 2008, 47(1), 488 CrossRef CAS.
- R. J. Baldwin, T. Kreouzis, M. Shkunov, M. Heeney, W. Zhang and I. McCulloch, J. Appl. Phys., 2007, 101, 023713 CrossRef.
- I. Bleyl, C. Erdelen, K.-H. Etzbach, W. Paulus, H.-W. Schmidt, K. Siemensmeyer and D. Haarer, Mol. Cryst. Liq. Cryst., 1997, 299, 149 CrossRef CAS.
- I. McCulloch, W. Zhang, M. Heeney, C. Bailey, M. Giles, D. Graham, M. Shkunov, D. Sparrowe and S. Tierney, J. Mater. Chem., 2003, 13, 2436–2444 RSC.
- T. Kato, Science, 2002, 295, 2414 CrossRef CAS PubMed.
- C. Tschierske, J. Mater. Chem., 1998, 8(7), 1485 RSC.
- J. Newton, H. Coles, P. Hodge and J. Hannington, J. Mater. Chem., 1994, 4(6), 869 RSC.
- G. Johansson, V. Percec, G. Ungar and K. Smith, Chem. Mater., 1997, 9(1), 164 CrossRef.
- K. Nickmans, J. N. Murphy, B. de Waal, P. Leclère, J. Doise, R. Gronheid, D. J. Broer and A. P. H. J. Schenning, Adv. Mater., 2016, 28, 10068 CrossRef PubMed.
- B. Oschmann, J. Lawrence, M. W. Schulze, J. M. Ren, A. Anastasaki, Y. Luo, M. D. Nothling, C. W. Pester, K. T. Delaney, L. A. Connal, A. J. McGrath, P. G. Clark, C. M. Bates and C. J. Hawker, ACS Macro Lett., 2017, 6(7), 668 CrossRef PubMed.
- W. Zhang, W. Yang, H. Pan, X. Lyu, A. Xiao, D. Liu, Y. Liu, Z. Shen, H. Yang and X.-H. Fan, Soft Matter, 2022, 18, 3430 RSC.
- H. Sakaino, B. A. G. Lamers, S. C. J. Meskers, E. W. Meijer and G. Vantomme, J. Polym. Sci., 2021, 59(12), 1131 CrossRef CAS.
- H. Sakaino, D. J. Broer, S. C. J. Meskers, E. W. Meijer and G. Vantomme, Angew. Chem., Int. Ed., 2022, 61, e202200839 CrossRef CAS PubMed.
- M. Funahashi and J.-I. Hanna, Adv. Mater., 2005, 17, 594 CrossRef CAS.
- M. Funahashi, F. Zhang and N. Tamaoki, Adv. Mater., 2007, 19, 353 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed.
- J.-I. Hanna, A. Ohno and H. Iino, Thin Solid Films, 2014, 554, 58 CrossRef CAS.
- F. Zhanga, M. Funahashia and N. Tamaoki, Org. Electron., 2009, 10, 73 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04911b |
| This journal is © The Royal Society of Chemistry 2022 |