
Precursor polymorph determines the organic semiconductor structure formed upon annealing†
Lorenzo
Pandolfi
 a,
Andrea
Giunchi
a,
Arianna
Rivalta
a,
Simone
D'Agostino
b,
Raffaele Guido
Della Valle
a,
Marta
Mas-Torrent
c,
Massimilano
Lanzi
a,
Elisabetta
Venuti
*a and
Tommaso
Salzillo
*cd
a,
Andrea
Giunchi
a,
Arianna
Rivalta
a,
Simone
D'Agostino
b,
Raffaele Guido
Della Valle
a,
Marta
Mas-Torrent
c,
Massimilano
Lanzi
a,
Elisabetta
Venuti
*a and
Tommaso
Salzillo
*cd
aDipartimento di Chimica Industriale “Toso Montanari”, University of Bologna, Viale del Risorgimento 4, 40136, Bologna, Italy. E-mail: elisabetta.venuti@unibo.it
bDipartimento di Chimica ‘‘G. Ciamician’’, University of Bologna, Via F. Selmi 2, 40126, Bologna, Italy
cInstitut de Ciència de Materials de Barcelona, ICMAB-CSIC, Campus de la UAB, 08193, Bellaterra, Spain
dDepartment of Chemical and Biological Physics, Weizmann Institute of Science, Rehovot 76100, Israel. E-mail: tommaso.salzillo@weizmann.ac.il
First published on 18th June 2021
Abstract
Films of the chemical precursor tBoc-quinacridone obtained by the spin-coating and bar-assisted meniscus shearing methods were subjected to thermal deprotection to recover the organic semiconductor quinacridone in its crystalline form. We found that the final crystal structure of the semiconductor on the Si/SiO2 substrate is in fact determined by the chemical precursor starting structure, which is in turn induced by the deposition method. Indeed, the samples prepared by spin coating display the precursor structure known from the literature, which transforms into the β-quinacridone phase. The shearing technique instead yields highly homogeneous films composed of a novel tBoc-quinacridone polymorph, which acts as a trigger for the subsequent formation of a pure, well oriented α-quinacridone phase. Although this crystalline form is the least stable of the many quinacridone polymorphs, here it turns out to be selectively induced and stabilized. Finally, the organic field effect transistor charge mobility of the α-quinacridone films was measured.
1. Introduction
Organic semiconductors hold the promise to be complementary to the silicon-based electronics, coupling the low cost of both material and processability with potential for applications requiring flexible, stretchable and conformable devices.1,2The development of the new functional materials employed in this field has recently raised much attention to the subject of their polymorphism, which occurs quite commonly in systems where the molecules are kept together by weak dispersion forces.3 In fact, the crystal packing, i.e. the relative molecular orientations, has a significant effect on field effect mobilities, which depend both on the strength and the direction of the electronic coupling between neighbouring molecules.4–6 Thus, the capability to modify the crystal phase offers the chance to control the mobility of the charge carriers, and this may be achieved in thin films by selection of the growing conditions or by post-deposition processing.5,7–9
In the search of functional materials with low environmental impact, as recommended by the green policies,10 pigments belonging to the class of the indigoids11–13 were found to possess attractive features, displaying balanced, albeit not high, ambipolar field effect charge transport coupled with operational stability at ambient conditions.14 All elements of the class also exhibit extended polymorphism, with phases which are metastable in the processing conditions getting stabilized at surface growths or forming genuine thin film phases.11–13,15,16 Among other pigment candidates as semiconductors, quinacridone (QA) has the peculiarity of possessing a molecular structure appearing like that of OSC pentacene,17 where however the π conjugation on the molecular skeleton is broken by the substituents on the rings. Thus, similar to the case of indigo,15 the solid-state assembly is driven both by the very strong intermolecular H-bonds due to the substituents and by the π stacking of neighboring molecules. The interplay of interactions results for QA in the existence of four known polymorphs. These are characterized by quite different packing arrangements, for which different electronic and charge carrier properties can be foreseen, making this system a benchmark for the theoretical study between crystal structure and charge transport.18 On more practical grounds, QA has been investigated in view of its use as a potential very low cost and stable active material in electronic devices.19,20 Owing to the ability to form H-bonds and the lack of toxicity, it has also been suggested that its versatility could be extended to using it as a living cell interface for applications in bioelectronics.21 QA based organic field effect transistors (OFET) have been reported by Glowacki and co-workers,14 who demonstrated its ambipolar character with hole mobilities in the order of 0.1 cm2 V−1 s−1 for high vacuum evaporated films having tetratetracontane as a gate dielectric. A good reason to prefer vapor deposition methods is the scarce solubility of QA in water and in most organic solvents, a factor which prevents its easy processability by large area scalable techniques based on solution processing. However, a strategy to overcome such a drawback is via the formation of the “latent pigment”, following the methodology developed by Zambounis of CIBA Specialty Chemicals in 1994 and applied to pyrrolo-pyrrole pigments.22 The chemical functionalization has also been applied to QA,23 using the same synthetic route (Fig. 1).
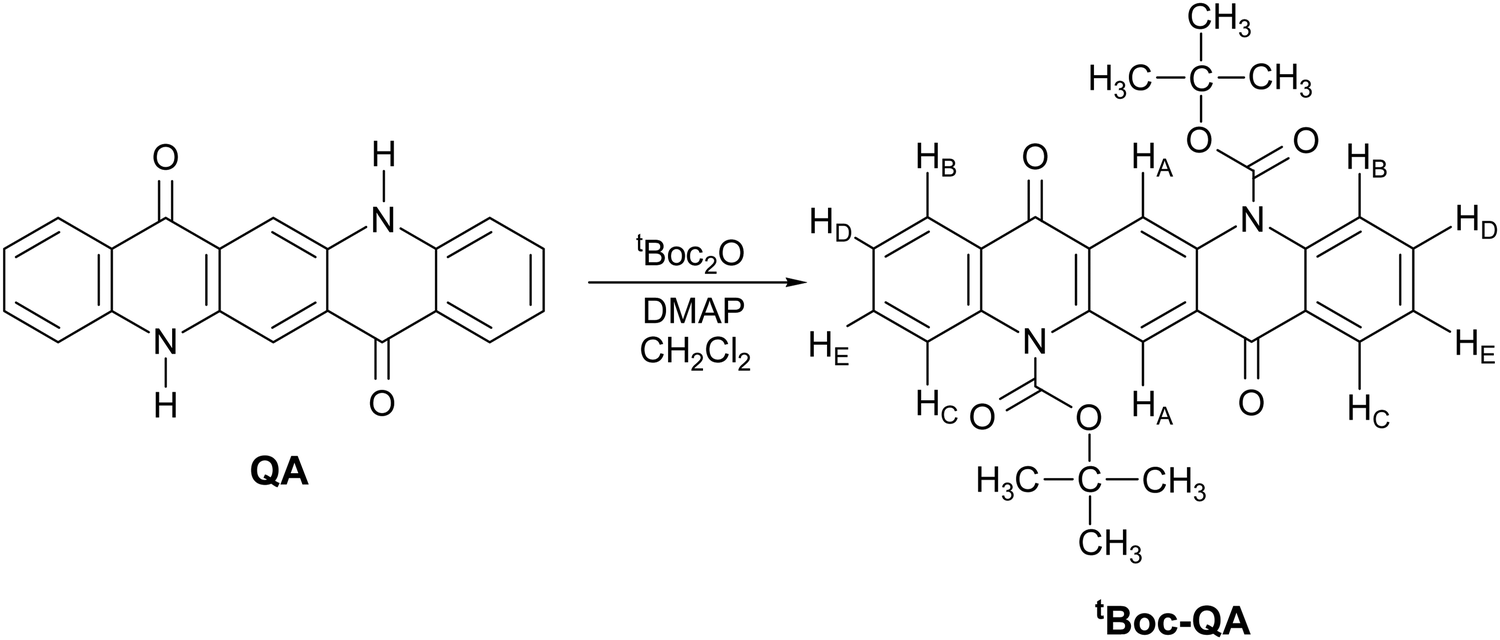 | ||
| Fig. 1 Synthetic route from quinacridone pigment (QA) to the latent pigment (tBoc-QA). | ||
The latent pigment is prepared by replacing the H atom of the NH group with the t-butoxycarbonyl (tBoc) group, a common “protective” agent in organic synthesis. This turns off the N–H⋯O intermolecular H bonds holding together neighboring molecules, thus highly increasing the solubility in all the common laboratory solvents with respect to the parent system. The pigment itself can be easily regenerated by the thermochemical cleavage of the tBoc group.
The aim of this work was to use the same strategy and prepare films of the QA pigment by large area solution printing techniques via the intermediate deposition of the t-butoxycarbonyl-quinacridone (tBoc-QA) derivative. The focus of the study was on the influence of the process on the polymorphic composition of the resulting QA film.
The outcomes of the classical spin-coating technique and of the roll-to-roll compatible bar-assisted meniscus shearing (BAMS) method24,25 were compared, with lattice phonon Raman spectroscopy and X-ray diffraction (XRD) providing an efficient polymorph screening. Notably, after deprotection, the films prepared by spin-coating were found to display the β form of QA, whereas those prepared by BAMS turned out to be entirely consisting of the metastable α form. The most stable γ phase instead was never observed. As the thermal cleavage treatment was identical in the two cases, the structural characterization of the precursor phase was undertaken, showing that the two deposition techniques produce the growth of two different polymorphs of tBoc-QA. This is an example of how the control of the crystalline form of the compound also implies the knowledge of the history of the solid phase that generated it. The implications of this finding for film fabrication processes are discussed, and the characteristics of OFETs based on the α quinacridone polymorph reported.
2. Experimental
Materials and synthesis
Quinacridone was purchased from Sigma-Aldrich, technical grade purity, and purified by sublimation at low pressure in nitrogen atmosphere. Reagent grade solvents bought from Sigma-Aldrich were used without further purification.The protection of the –NH groups of QA by tBoc groups was performed according to ref. 26 by reacting QA pigment powder with di-tert-butyl dicarbonate and 4-dimethylaminopyridine in methylene chloride at room temperature under Ar atmosphere. The progression of the reaction was followed by TLC and after 48 h no trace of the starting product was found. tBoc-QA was purified with column chromatography and recrystallized from ethyl acetate. The 1H-NMR spectrum of the obtained product shows five signals in the low-field region ascribable to the aromatic protons and a singlet at 1.77 ppm which is due to the methylic protons of the tBoc protecting group. The comparison between the integrated intensities of the aromatic protons and the aliphatic protons shows that all the aminic groups had been protected by tBoc. This result is also confirmed by the mass spectrum (MS), in which the progressive loss of tBu and tBoc groups is clearly observable (Fig. S1a and b, ESI†).
Pigment tBoc-QA may appear in two different polymorphic modifications, named here form I and II.
Single crystals of tBoc-QA form I were grown by evaporation of an acetone solution kept at room temperature. The solution was previously filtered and ultra-sonicated. We obtained hexagonal prismatic yellowish crystallites.
In order to grow mainly tBoc-QA form II single crystals, an acetone solution was filtered, ultra-sonicated and left to evaporate on a hot stage at 75 °C. In addition to that, we used a crystallite of form II from the drop cast sample as growth seed to perform another crystallization from solution, with the aim of growing large crystal, suitable for X-ray analysis.
Raman and IR spectroscopy
Raman spectra of all the crystalline and thin film samples in the lattice phonon region (10–150 cm−1) and on the intramolecular range (150–2000 cm−1) were collected with a Horiba Jobin Yvon T64000 triple monochromator spectrometer interfaced with the optical stage of an Olympus BX40 microscope. The excitation wavelength was a Kr+ gas laser tuned at 647.1 nm with a nominal power of 1 W reduced by a neutral optical density filter to avoid sample damage.FTIR-ATR measurements in the wavenumber range 400–4000 cm−1 with a resolution of 2 cm−1 were carried out on a PerkinElmer Spectrum Two spectrophotometer, equipped with a Universal ATR accessory.
X-ray diffraction
Single-crystal data for all compounds were collected at RT on an Oxford XCalibur S CCD diffractometer equipped with a graphite monochromator (Mo-Kα radiation, λ = 0.71073 Å). All the tBoc-QA form II samples displayed crystal twinning. Thus, data were treated with the default configuration for twinned crystals of CrysAlisPro, and structure solution and refinement were performed on the HKLF4 file containing the non-overlapped reflections. The structures were solved by intrinsic phasing with SHELXT27 and refined on F2 by full-matrix least squares refinement with SHELXL28 implemented in the Olex28 software.29 All non-hydrogen atoms were refined anisotropically applying the rigid-body RIGU restraint.30 HCH atoms for all compounds were added in calculated positions and refined riding on their respective carbon atoms. Data collection and refinement details are listed in Table S1 (ESI†). The Mercury31 program was used to calculate intermolecular interactions and for molecular graphics. Crystal data can be obtained free of charge viahttp://www.ccdc.cam.ac.uk/conts/retrieving.html.†For phase identification and variable temperature X-ray powder diffraction experiments, diffractograms were recorded on a PANalytical X’Pert Pro automated diffractometer equipped with an X’Celerator detector in Bragg−Brentano geometry, using Cu-Kα radiation (λ = 1.5418 Å) without monochromator in the 2θ range between 5° and 40° (continuous scan mode, step size 0.0167°, counting time 19.685 s, Soller slit 0.04 rad, antiscatter slit 1/2, divergence slit 1/4, 40 mA·40 kV), and an Anton-Paar TTK 450 + LNC. The Mercury31 program was used for the calculation of the X-ray powder patterns on the basis of single-crystal data either retrieved from the Cambridge structural database (CSD): α-QA: QNACRD06, β-QA: QNACRD07, and γ-QA: QNACRD08, or collected in this work: tBoc-QA form I and form II, see ESI† (Fig. S2–S4).
Hot stage (HSM) and cross-polarized optical microscopy (POM)
Hot stage experiments were carried out using a Linkam TMS94 device connected to a Linkam LTS350 platinum plate and equipped with polarizing filters. Pictures were collected with the VisiCam Analyzer imaging software, from an Olympus BX41 stereomicroscope.Polarized optical microscopy (POM) images were taken with an Olympus BX51 microscope equipped with polarizer and analyzer at 90° in reflection mode.
Thermal analysis
Thermogravimetric analyses (TGA) were performed with a PerkinElmer TGA-7. Each sample, contained in a platinum crucible, was heated in a nitrogen flow (20 cm3 min−1) at a rate of 5 °C min−1, up to decomposition. Samples weights were in the range 5–10 mg.Differential scanning calorimetric (DSC) measurements were performed with a PerkinElmer DSC-7 equipped with a PII intracooler. Temperature and enthalpy calibrations were performed using high-purity standards (n-decane, benzene and indium). The heating of the aluminium open pans containing the samples (3–5 mg) was carried out at 5 °C min−1 in the temperature range of 40–300 °C.
Thin films and OFET fabrication
Spin-coated films were prepared with 50 μL of solutions of tBoc-QA (10 mg mL−1) in various solvents (namely, hexane, diethyl ether, ethanol and acetonitrile) deposited at 1500 rpm for about two minutes either on 10–10 mm2 glass microscope slides or on Si/SiO2 substrates from Si-Mat (thermally grown SiO2 with thickness of 200 nm).For the preparation of the films by the BAMS method, the solution concentration of the latent pigment tBoc-QA in chlorobenzene (CB, Sigma-Aldrich) was fixed at 20 mg mL−1, and the experimental setup used was the one reported in detail in previous works.24,25 The BAMS hot bed was kept at a constant temperature of 105 °C, and the best optimal speed (i.e. yielding highly homogeneous films) was found to be 1 mm s−1. Before depositing the layer of material, the gold electrodes were functionalized with pentafluorobenzothiol (PFBT, Sigma-Aldrich) by first exposing the substrates for 25 minutes to ozone and then immersing them for 15 min in a 2 μL mL−1 PFBT solution in isopropanol (IPA) solution.
Bottom gate/bottom contact (BGBC) OFETs were fabricated on p-doped Si/SiO2 wafers purchased from Si-Mat (SiO2 of thickness 200 nm, C = 17.25 nF cm−2) with pre-patterned interdigitated Cr/Au (5 nm/40 nm) electrodes with different channel lengths (L = 25 or 50 μm, W/L ratio = 100) fabricated by means of photolithography (Micro-Writer ML3 from Durham Magneto Optics Ltd.) with a lateral resolution of 5 μm.
The de-protection process needed to obtain the films of QA was performed by heating the tBoc-QA samples at 180–200 °C on a hot plate for 5 min. The color change observed in the organic material is a good indicator of the progress of the reaction.
The α-QA film electrical characteristics were assessed under ambient conditions using an Agilent B1500A semiconductor parameter analyzer coupled with a Karl SÜSS probe station. Transfer characteristics were measured in the linear regime, with the drain voltage VD fixed at −5 V, while the gate voltage VG was scanned between +10 V and −60 V backward and forward. Output characteristics were measured under constant VG from 0 to −60 V with a step of 10 V, while VD was scanned between +2 V and −40 V, also backward and forward. Mobility values, threshold voltages and on/off current ratios were extracted in the linear regime using the classic MOSFETs equation:
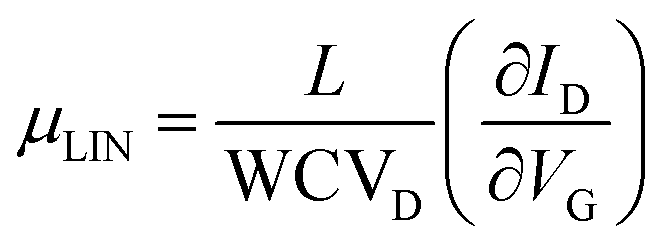
3. Results and discussion
The synthesized tBoc-QA was repeatedly purified by recrystallization in ethyl acetate (EtOAc), obtaining platelet-like yellow crystals (Fig. S5, ESI†). Single-crystal X-ray diffraction (SC-XRD) analysis yielded a monoclinic structure belonging to P21/n space group with Z = 2 molecules per unit cell, and crystallographic parameters (Table S1, ESI†) in agreement with those of the previously reported tBoc-QA structure, hereafter denoted as form I.32The regeneration of the parent QA in the bulk state was easily achieved by thermochemical treatment, as shown by the TGA analysis of Fig. S6, ESI.† The thermochemical process starts at 150 °C and ends at 230 °C with a loss of about 38% of the initial weight due to the decomposition of the two tBoc functionalities which evolve into the release of CO2 and isobutene.33 Hot stage microscopy images (Fig. S5, ESI†) and XRD (Fig. 2) reveal that in the bulk, and for the indicated temperature range, the transformation proceeds through a crystal-to-crystal process with the formation of the β-QA polymorph as the only product.34,35
 | ||
| Fig. 2 Comparison of the experimental powder XRD patterns with those calculated on the basis of the single-crystal data for compounds.34 Top: thermal deprotection of a polycrystalline sample of tBoc-QA leads to formation of β-QA. | ||
Characterization of films prepared by spin-coating
The preparation of homogeneous films of QA from solution requires achieving homogeneous films of its precursor first, and thus a number of different organic solvents were tested for the tBoc-QA spin-coating deposition, as well as different spinning conditions. Notwithstanding the excellent tBoc-QA solubility in all most solvents, the best film morphology and homogeneity was realized with ethanol solutions, as shown in the optical image of Fig. 3a. Based on literature data33 and on the tBoc-QA DSC and TGA thermograms of Fig. S6 (ESI†), the film deprotection process was performed as reported in the “Experimental” section, and the complete removal of the tBoc group was assessed by spectroscopic and XRD measurements prior to and after the thermochemical deprotection process, as reported in the following. FTIR spectroscopy is an effective and well-established method to verify that a complete deprotection of the latent pigment has taken place. The intensity decrease of the strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching signal, detected around 1750 cm−1, and the appearance of the characteristic signal of the N–H stretching mode between 3000 cm−1 and 3200 cm−1, which confirms the activation of H-bonding sites (Fig. S7, ESI†), are the markers for the reaction progress.36 We also employed Raman microscopy over the range 10–4000 cm−1 to probe both the removal of the tBoc groups and the formation of the QA crystalline structure on the same area of the film. With different selection rules, infrared and Raman spectroscopies provide similar information over the wavenumber range where molecular vibrations are detected and can both be used to monitor the chemical reaction. In addition, the Raman spectra in the terahertz range probe the so-called lattice vibrations (or lattice phonons), originating from the weak intermolecular force field. These yield a unique spectral pattern, which is representative of the crystal structure and can be used to identify the phases present in the films.
O stretching signal, detected around 1750 cm−1, and the appearance of the characteristic signal of the N–H stretching mode between 3000 cm−1 and 3200 cm−1, which confirms the activation of H-bonding sites (Fig. S7, ESI†), are the markers for the reaction progress.36 We also employed Raman microscopy over the range 10–4000 cm−1 to probe both the removal of the tBoc groups and the formation of the QA crystalline structure on the same area of the film. With different selection rules, infrared and Raman spectroscopies provide similar information over the wavenumber range where molecular vibrations are detected and can both be used to monitor the chemical reaction. In addition, the Raman spectra in the terahertz range probe the so-called lattice vibrations (or lattice phonons), originating from the weak intermolecular force field. These yield a unique spectral pattern, which is representative of the crystal structure and can be used to identify the phases present in the films.
 | ||
| Fig. 3 Optical images of the Raman characterized films as prepared (a) and after the thermochemical cleavage (b); Raman spectra in the lattice phonon range for spin-coated film of tBoc-QA before and after de-protection process compared with QA α and β polymorphs spectra from literature16 (c); 1400–1700 cm−1 intramolecular range for tBoc-QA spin-coated film before (yellow trace, assigment in-plane NRH bending modes) and after de-protection (red trace, assignment in-plane C–H bendings and ring C–C stretchings) (d). | ||
In Fig. 3 we report Raman spectra, which include the lattice phonon modes (Fig. 3c) and the range of the molecular vibrations (Fig. 3d), of the tBoc-QA spin-coated films spin-cast from an ethanol solution before and after the deprotection process, along with the optical images of the sample.
In Fig. 3d the intramolecular vibrational range 1400–1800 cm−1 is reported, which represents a diagnostic region sensitive to the tBoc cleavage process. The strongest bands (1567 and 1598 cm−1) detected here for QA belong to in-plane total symmetric NRH bending modes which are not present in the derivative. In fact, all the in plane bending modes in the region contain a contribution of this group, as confirmed by DFT simulations (see ESI†). Accordingly, the strongest bands of the derivative owe their intensity to total symmetric in-plane C–H bendings and ring C–C stretchings, with the mode at 1622 cm−1 mostly describing a CO bending.
The analysis of the lattice phonon region of the Fig. 3, with its intense peaks, reveals the high degree of crystallinity of the tBoc-QA spin-coated samples. The spectral features recorded over this energy range result to be independent of the solvent used for the fabrication (Fig. S8, ESI†) and do correspond to those found for the freshly synthesized and re-crystalized tBoc-QA, confirming that form I is present in these films.32 The corresponding Raman spectra at a first approximation can be interpreted in the framework of the so-called rigid body approach, which considers separately vibrational modes arising from the intermolecular and intramolecular force fields.37 In a centrosymmetric structure with Z = 2, we can predict at low frequencies the occurrence of six Raman active intermolecular modes, classified according to the irreducible representations of the factor symmetry group in 3Ag + 3Bg. Each of these modes describes a pure concerted vibration of the whole molecule around its inertia axes. In the spectrum of Fig. 3c, tBoc-QA displays at least six easily identifiable peaks below 100 cm−1. Additional less intense scatterings might be indeed hidden in the envelope of the strong broad bands that make up the spectrum and could be solved by deconvolution. This is not surprising, as the tBoc substituent endows the parent pigment molecule with highly flexible groups, activating intramolecular modes at low wavenumbers and making in fact the rigid body approximation not fully effective for a detailed spectral analysis. However, what is important here is that the unique phonon pattern of tBoc-QA first allows for the identification of the compound crystal state, and then for the in situ detection of its cleavage reaction, which proceeds with the formation of the QA crystalline form. The previously reported spectral and structural characterization of several QA polymorphs16,34 allows in turn for the identification of the one occurring in the film. Based on the presence of the broad bands at 111 cm−1 and at 78 cm−1, as well as of the features below 50 cm−1,16 the pattern of Fig. 3c is recognized as that of the powder of the QA β-form, given for comparison in the same Figure. The presence of β-QA is further confirmed by the pair of bands at 215 and 230 cm−1,16 normally used to discriminate this polymorph from γ-QA, which is the structure most commonly occurring in the bulk. The similarity of the spectrum to that of a powder hints to the absence of a texture in the film.
The β-QA polymorph assignment given by Raman on the spin-coating samples is confirmed by XRD measurements and agrees with the transformation taking place for the tBoc-QA form I polycrystalline sample (see Fig. 2). As for the films, the most recent literature reports that those grown by direct QA sublimation also belong to β-QA phase.14,20 Previously, the only growth of the γ-QA phase was observed by sublimation at high temperature (≈600 K). This phase was then assumed to be the most stable in these conditions, whereas β-QA would grow at much lower temperatures, around 500 K.32,38,39 In some instances, small changes in the sublimation conditions have been found to induce the concomitant growth of β-QA and α-QA forms,20 similarly to what is known for systems such as of pentacene.40 Finally, the films obtained via the precursor by Yanagisawa at al.23 contained β-QA.
Characterization of films prepared by the BAMS technique
The fabrication of films by the BAMS technique24 was undertaken to obtain more homogeneous and reproducible samples, compatible with the requirements of large area processing. As previously reported, the BAMS technique is a variant of the traditional blade coating method, which consists in pouring the solution of the material to deposit between a substrate placed on a hot plate and a bar located ∼500 μm apart. This allows for the formation of a confined meniscus, which is subsequently displaced when the substrate itself is moved horizontally at a constant speed. In recent years, this method has proved to induce high crystallinity in thin-films at a speed compatible with roll-to-roll processes using as active materials OSCs and OSCs blended with insulating polymers, and even for the more challenging formation of charge transfer complexes.41The tBoc-QA films prepared by this simple and yet effective solution processing technique feature a high homogeneous deposition, with a very good degree of coverage of the whole substrate area. This can be seen in Fig. 4a, which shows the optical microscopy image of a thin-film fabricated on a Si/SiO2 substrate having interdigitated gold-prepatterned contacts, prepared for OFET measurements. The images recorded in cross-polarized optical microscopy (POM) highlight the morphology and the polycrystalline texture of the sample (Fig. 4b). It is worth noting that such characteristics are retained after the annealing process at 200 °C (see Fig. 4c and d), confirming that the thermal cleavage is accompanied by a crystal-to-crystal transformation, in which the formation of the new lattice of QA follows the disruption of that of tBoc-QA.
 | ||
| Fig. 4 Optical (a and c) and POM (b and d) images of tBoc-QA thin films fabricated by BAMS technique before (a and b) and after (c and d) the thermal treatment which leads to QA. | ||
The analysis of such a QA lattice reveals the presence of the α form, as demonstrated in Fig. 5. Fig. 5a shows the comparison of the Raman lattice phonon spectra of the film formed by thermal annealing with that of the α-QA single crystal from the literature.16 The assignment is unmistakable, as the spectrum of the Z = 1 triclinic α form is characterized by the same broad and structureless band centred at 97 cm−1.16 The β form found in the spin-coated samples is absent and cannot be found even by the Raman mapping of extended areas of the films. The XRD data of Fig. 5c, where the patterns of the various QA polymorphs are compared to that of the films, clearly confirm that the metastable α-QA is the only structure occurring in the BAMS films. The stabilization of metastable forms in growth processes that take place at the interfaces is a common phenomenon. What is surprising in this case is that contrary to what happens in samples obtained by spin-coating, there is complete selectivity towards this structure, to the detriment of the more widely encountered β form. The reasons for the formation of the different QA phases must be traced back to the crystalline phase of the precursor itself in the two deposition methods. In Fig. 6 the tBoc-QA Raman spectra of films obtained by spin coating and BAMS are compared. The spectra differ in the lattice phonon region (Fig. 6a), which is indicative of different crystal structures. As mentioned before, the 1400–1800 cm−1 interval selected in Fig. 6b is specific for intramolecular modes sensitive to the cleavage process and is thus relevant to chemical diagnostics. The lack of significant differences between the spectra over this range is an indication that the two processing methods do not modify the chemical nature of tBoc-QA. Such a test is fully justified by the BAMS higher deposition temperature, which could have promoted the precursor partial cleavage, with the restoration of a fraction of amine groups. In agreement with the Raman findings, the X-ray diffraction pattern of these samples (Fig. 5c) displays peaks which do not correspond to reflections of the known tBoc-QA form I. Thus, a new tBoc-QA polymorph, hereafter named form II, must be responsible for the formation of the α-QA films after the thermal treatment. Its range of thermodynamic stability is expected to fall at temperatures higher than those of existence of form I. Further, it should be noted that the BAMS technique in these conditions tends to give rise to kinetic and metastable polymorphs.42–44
 | ||
| Fig. 5 (a) Raman characterization in the lattice phonon and (b) in the intramolecular range used for the compound identification of the films fabricated by BAMS technique. (c) XRD patterns of the same BAMS films as prepared and after thermochemical cleavage of tBoc groups compared with the different QA and tBoc-QA polymorphs. | ||
 | ||
| Fig. 6 (a) Lattice phonon and (b) intramolecular Raman spectra of spin cast and BAMS tBoc-QA films; (c) diffraction patterns of BAMS films and the two tBoc-QA polymorphs. Inset: Orientation of the two tBoc-QA molecules with respect of the (20−2) and (200) Miller planes in form II. HCH atoms omitted for clarity. | ||
To isolate and characterize the new phase, tBoc-QA solutions in acetone were drop cast at increasing temperatures and recovered after solvent evaporation at ambient conditions. At a solution and substrate T = 75 °C, the block shaped crystallites obtained at ambient T were accompanied by the formation of needle shaped specimens, as shown in the images of Fig. S9 (ESI†). Accordingly, two distinct lattice phonon patterns were identified by Raman, one related to tBoc-QA form I, the other to the phase presented in the BAMS processed films. Raman spectra recorded in polarized light for the two cases (Fig. S9, ESI†) assist the analysis of each of the patterns in more detail and confirm their belonging to two different structures, proving the existence of a second crystal phase. Crystallites of this phase singled out by probing their Raman spectrum could finally be structurally characterized by SC-XRD (Table S1, ESI†).
Like form I, tBoc-QA form II is found to have a monoclinic unit cell with space group P21/c and the parameters reported in Table S1 (ESI†). Also, the ESI,† details the main structural differences between form I and form II.
As expected, the peak positions of the XRD spectrum collected on the BAMS tBoc-QA film correspond to those of form II, as displayed in detail in Fig. 6c. In particular, two sets of Miller planes progressions can be identified. The first one coincides with the (2n00) series of tBoc-QA P21/c form II, an indication of a growth with the (100) plane lying on the surface. The second, much weaker, coincides with the series (2n0–2n) of the same structure. The molecular orientations within these planes are shown in the Fig. 6c, where also the arrangement of the quinacridone core face-to-face stackings with respect to the interface is easily recognizable in either case. Information about the presence of two predominant arrangements of the precursor molecule on the surface is also supplied by the Raman spectra of the films reported in Fig. S10 (ESI†). Basically, only two lattice phonon patterns are found in the Raman mapping of large areas of a film kept in a fixed position with respect to the laboratory reference frame. Each pattern is characterized by its own motif of relative peak intensities. This suggests the presence of two (main) different ways in which the crystalline domains are oriented with respect to the impinging polarized excitation laser beam, which agrees with the observation of two different series of peaks in the diffractograms.
The knowledge of how the crystal lattice of the latent pigment is oriented in the film may help the modeling of the arrangement of the QA molecules with respect to the substrate after deprotection. In fact, since the thermochemical cleavage is found to proceed through a crystal-to-crystal transformation, the removal of the gaseous products must take place without a large disruption of the reactant lattice, possibly with small movements of the molecular cores to adjust in the product lattice.
OFET performance of α-QA films
Previous works and a patent report QA based OFETs manufactured with high vacuum deposited thin films both on Si/SiO2 wafers and on tetratetracontane, where the H-bonded molecules were found to form π-stacks aligned parallel to the gate dielectric substrate and the best charge mobilities were achieved.17,45,46 On the other hand, QA relevance for applications could be greatly increased if crystalline films for OFETs were prepared by printing, overcoming the disadvantages of the lack of solubility.β-QA OFETs fabricated by the spin-coating technique via the formation of the latent pigment as a chemical precursor were characterized by Yanagisawa and co-workers.23 With an understanding of how to access it in thin films, in the present work we evaluate the electrical performances of the α-polymorph based OFETs prepared by the BAMS technique.
OFETs were manufactured in a bottom gate/bottom contact configuration on Si/SiO2 substrates with pre-patterned gold interdigitated electrodes and channel dimensions of 25 and 50 microns, keeping constant W/L = 100. Fig. 7a and b show their transfer curves (drain current IDvs. gate voltage VG) in the linear regime and the output characteristic curves (drain current IDvs. drain voltage VD), respectively, measured at room temperature under ambient conditions.
 | ||
| Fig. 7 (a) Transfer characteristic curves in linear regime in forward (full line) and backward (dotted line) sweeps and (b) corresponding output electrical characteristics of the QA thin film transistor prepared (L = 25 μm, W/L = 100) by depositing by BAMS tBoc-QA followed by cleavage of tBoc groups. | ||
The curves display typical FET characteristics with current modulation in negative source-gate voltage, which is in accordance with hole transport (i.e., p-type behavior) occurring in the QA based devices. Some hysteresis is visible between the backward and forwards sweeps. From the linear regime, we determined the average linear mobility on at least 20 devices, giving a value of μh,av = (1.1 ± 0.2) × 10−4 cm2 V−1 s−1 with a threshold voltage of VTH,av = −7.9 ± 3.7 V and on–off ratio of 300. The performances are not influenced by the different channel lengths suggesting low contact resistance, which is also in agreement with the linearity of the output curves at low drain voltages. The mobility and on–off ratio found here are both only one order of magnitude higher than those reported for the spin-coated β-QA films,23 meaning that semiconducting properties of QA are preserved in the α-QA crystal form, but without much improvement. As not even β-QA vacuum deposited films23 on Si/SiO2 displayed mobilities as high as those measured on tetratetracontane,14 the way OSC/substrate interactions drive the assembly at the interface of either crystal modification must remain the main factor determining the electrical properties of this compound.
4. Conclusions
The strategy of preparing films of crystalline QA via a chemical precursor with higher solubility, coupled to the deposition of the precursor by means of the BAMS method, has proved to be successful in improving sample homogeneity and reproducibility. Unexpectedly, however, the films thus prepared are entirely formed by the α quinacridone polymorph, at variance from what is commonly observed in the spin coating depositions, which contain the β form of the pigment. The formation of α-QA turns out to be induced by the presence of a different polymorph of the precursor itself, whose growth is most likely triggered by the solution shearing crystallisation process combined with higher deposition temperatures. The α-QA films display OFET hole mobilities comparable to those measured in ref. 23 for the β form prepared by either the same chemical precursor method or high vacuum deposition on Si/SiO2. Much better mobilities were recorded for QA by combining ultra-high vacuum fabrication with different substrates and electrode materials,14 but a direct comparison with these experiments is not meaningful.The main piece of information we want to convey here is the when chemical precursor methods are employed the precursor polymorph may determine the resulting OSC polymorph.
Hence, this method may constitute a means for accessing and test different structures, in addition to all those already used in crystal engineering to exploit the versatile and multifaced quality of polymorphism.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. S. acknowledges the H2020-MSCA-COFUND-2014 Programme (P-SPHERE, Grant agreement 665919). This work was supported by the MIUR-PRIN project “From natural to artificial light-harvesting systems: unveiling fundamental processes towards a bio-inspired materials design” (HARVEST) protocol 201795SBA3. This work was funded by DGI (Spain) with the project GENESIS PID2019-111682RB-I00, the Generalitat de Catalunya (2017-SGR-918), and the Spanish Ministry of Economy and Competitiveness, through the “Severo Ochoa” Programme for Centers of Excellence in R&D (FUNFUTURE CEX2019-000917-S). The authors thank Dr Raphael Pfattner (ICMAB-CSIC) for fruitful discussions.References
- Y. H. Lee, M. Jang, M. Y. Lee, O. Y. Kweon and J. H. Oh, Chem, 2017, 3, 724–763 CAS.
- V. Raghuwanshi, D. Bharti, A. K. Mahato, A. K. Shringi, I. Varun and S. P. Tiwari, ACS Appl. Electron. Mater., 2020, 2, 529–536 CrossRef CAS.
- J. Bernstein, Polymorphism in Molecular Crystals, New York, 2002 Search PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Bredas, Top. Curr. Chem., 2007, 107, 926 CAS.
- H. Chung and Y. Diao, J. Mater. Chem. C, 2016, 4, 3915–3933 RSC.
- M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833–4856 CrossRef CAS PubMed.
- G. E. Purdum, N. Yao, A. Woll, T. Gessner, R. T. Weitz and Y. L. Loo, Adv. Funct. Mater., 2016, 26, 2357–2364 CrossRef CAS.
- T. Salzillo, N. Montes, R. Pfattner and M. Mas-torrent, J. Mater. Chem. C, 2020, 8, 15361–15367 RSC.
- R. Pfattner, S. T. Bromley, C. Rovira and M. Mas-torrent, Adv. Funct. Mater., 2016, 26, 2256–2275 CrossRef CAS.
- M. Irimia-Vladu, Y. Kanbur, F. Camaioni, M. E. Coppola, C. Yumusak, C. V. Irimia, A. Vlad, A. Operamolla, G. M. Farinola, G. P. Suranna and N. González-Benitez, et al. , Chem. Mater., 2019, 31, 6315–6346 CrossRef CAS PubMed.
- T. Salzillo, S. D’Agostino, A. Rivalta, A. Giunchi, A. Brillante, R. G. Della Valle, N. Bedoya-Martínez, E. Zojer, F. Grepioni and E. Venuti, J. Phys. Chem. C, 2018, 122, 18422–18431 CrossRef CAS.
- A. Rivalta, A. Giunchi, L. Pandolfi, T. Salzillo, S. D’agostino, O. Werzer, B. Schrode, N. Demitri, M. Mas-Torrent, A. Brillante, R. G. Della Valle and E. Venuti, Dyes Pigm., 2020, 172, 107847 CrossRef.
- L. Pandolfi, A. Rivalta, T. Salzillo, A. Giunchi, S. D’Agostino, R. G. Della Valle, A. Brillante and E. Venuti, J. Phys. Chem. C, 2020, 124, 17702–17710 CrossRef CAS.
- E. D. Głowacki, M. Irimia-Vladu, M. Kaltenbrunner, J. Gsiorowski, M. S. White, U. Monkowius, G. Romanazzi, G. P. Suranna, P. Mastrorilli, T. Sekitani, S. Bauer, T. Someya, L. Torsi and N. S. Sariciftci, Adv. Mater., 2013, 25, 1563–1569 CrossRef PubMed.
- A. Rivalta, C. Albonetti, D. Biancone, M. Della Ciana, S. d’Agostino, L. Biniek, M. Brinkmann, A. Giunchi, T. Salzillo, A. Brillante, R. G. Della Valle and E. Venuti, Surf. Interfaces, 2021, 24, 101058 CrossRef.
- T. Salzillo, A. Rivalta, N. Castagnetti, S. D’Agostino, M. Masino, F. Grepioni, E. Venuti, A. Brillante and A. Girlando, CrystEngComm, 2019, 21, 3702–3708 RSC.
- E. Daniel Głowacki, L. Leonat, M. Irimia-Vladu, R. Schwödiauer, M. Ullah, H. Sitter, S. Bauer and N. Serdar Sariciftci, Appl. Phys. Lett., 2012, 101, 1–5 CrossRef.
- C. Winkler, A. Jeindl, F. Mayer, O. T. Hofmann, R. Tonner and E. Zojer, Chem. Mater., 2019, 31, 7054–7069 CrossRef CAS.
- T. L. Chen, J. J. A. Chen, L. Catane and B. Ma, Org. Electron., 2011, 12, 1126–1131 CrossRef CAS.
- D. Berg, C. Nielinger, W. Mader and M. Sokolowski, Synth. Met., 2009, 159, 2599–2602 CrossRef CAS.
- M. Sytnyk, M. Jakešová, M. Litviňuková, O. Mashkov, D. Kriegner, J. Stangl, J. Nebesářová, F. W. Fecher, W. Schöfberger, N. S. Sariciftci, R. Schindl, W. Heiss and E. D. Głowacki, Nat. Commun., 2017, 8, 91 CrossRef.
- Y. Imura, Y. Yamashita, T. Senju and J. Mizuguchi, J. Imaging Soc. Jpn., 2005, 44, 138–142 CAS.
- H. Yanagisawa, J. Mizuguchi, S. Aramaki and Y. Sakai, Jpn. J. Appl. Phys., 2008, 47, 4728–4731 CrossRef CAS.
- F. G. Pozo, S. Fabiano, R. Pfattner, S. Georgakopoulos, C. Rovira, X. Crispin, M. Berggren and M. Mas-torrent, Adv. Funct. Mater., 2016, 26, 2379–2386 CrossRef.
- I. Temiño, F. G. Del Pozo, M. R. Ajayakumar, S. Galindo, J. Puigdollers and M. Mas-Torrent, Adv. Mater. Technol., 2016, 1, 1600090 CrossRef.
- M. Sytnyk, E. D. Głowacki, S. Yakunin, G. Voss, W. Schöfberger, D. Kriegner, J. Stangl, R. Trotta, C. Gollner, S. Tollabimazraehno, G. Romanazzi, Z. Bozkurt, M. Havlicek, N. S. Sariciftci and W. Heiss, J. Am. Chem. Soc., 2014, 136, 16522–16532 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Thorn, B. Dittrich and G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2012, 68, 448–451 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- J. Mizuguchi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, o474–o475 CrossRef CAS.
- E. D. Głowacki, G. Voss, K. Demirak, M. Havlicek, N. Sünger, A. C. Okur, U. Monkowius, J. Gasiorowski, L. Leonat and N. S. Sariciftci, Chem. Commun., 2013, 49, 6063–6065 RSC.
- E. F. Paulus, F. J. J. Leusen and M. U. Schmidt, CrystEngComm, 2007, 9, 131–143 RSC.
- T. E. Gorelik, C. Czech, S. M. Hammer and M. U. Schmidt, CrystEngComm, 2016, 18, 529–535 RSC.
- I. Maqueira-Albo, G. Ernesto Bonacchini, G. Dell’Erba, G. Pace, M. Sassi, M. Rooney, R. Resel, L. Beverina and M. Caironi, J. Mater. Chem. C, 2017, 5, 11522–11531 RSC.
- T. Salzillo, A. Giunchi, M. Masino, N. Bedoya-Martĺnez, R. G. Della Valle, A. Brillante, A. Girlando and E. Venuti, Cryst. Growth Des., 2018, 18, 4869–4873 CrossRef CAS.
- F. H. Chung and R. W. Scott, J. Appl. Crystallogr., 1971, 4, 506–511 CrossRef CAS.
- J. Kalinowski, W. Stampor, P. Di Marco and V. Fattori, Chem. Phys., 1994, 182, 341–352 CrossRef CAS.
- R. G. Della Valle, E. Venuti, A. Brillante and A. Girlando, J. Chem. Phys., 2003, 118, 807–815 CrossRef CAS.
- T. Salzillo, A. Campos and M. Mas-Torrent, J. Mater. Chem. C, 2019, 7, 10257–10263 RSC.
- S. Riera-Galindo, A. Tamayo and M. Mas-Torrent, ACS Omega, 2018, 3, 2329–2339 CrossRef CAS.
- S. Galindo, A. Tamayo, F. Leonardi and M. Mas-Torrent, Adv. Funct. Mater., 2017, 27, 1700526 CrossRef.
- T. Salzillo, A. Campos, A. Babuji, R. Santiago, S. T. Bromley, C. Ocal, E. Barrena, R. Jouclas, C. Ruzie, G. Schweicher, Y. H. Geerts and M. Mas-Torrent, Adv. Funct. Mater., 2020, 2006115, 1–9 Search PubMed.
- M. Irimia-Vladu, E. D. Gåowacki, P. A. Troshin, G. Schwabegger, L. Leonat, D. K. Susarova, O. Krystal, M. Ullah, Y. Kanbur, M. A. Bodea, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Adv. Mater., 2012, 24, 375–380 CrossRef CAS.
- E. D. Głowacki, L. Leonat, G. Voss, M. A. Bodea, Z. Bozkurt, A. M. Ramil, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, AIP Adv., 2011, 1, 042132 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2067646 and 2067647. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc01313k |
| This journal is © The Royal Society of Chemistry 2021 |