
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElemental sulphur in the synthesis of sulphur-containing polymers: reaction mechanisms and green prospects
Natalia P. Tarasova *,
Alexey A. Zanin,
Efrem G. Krivoborodov and
Yaroslav O. Mezhuev
*,
Alexey A. Zanin,
Efrem G. Krivoborodov and
Yaroslav O. Mezhuev
Dmitry Mendeleev University of Chemical Technology of Russia, Miusskaya Sq. 9, Moscow, 125047, Russia. E-mail: tarasnp@muctr.ru
First published on 1st March 2021
Abstract
The synthesis of polymers using elemental sulphur as a chemical agent has been studied in relation to the worldwide overproduction of cyclo-octasulphur. Herein, the mechanisms of the processes leading to the inclusion of elemental sulphur into macromolecules have been reviewed and the main methods for reduction of the reaction temperature required for the S8 ring opening have been shown. Approaches to the activation of cyclo-octasulphur in the synthesis and macromolecule cross-linking reactions were discussed in the context of finding the chemical agents and conditions that satisfy the principles of green chemistry.
Introduction
Although the properties of sulphur have been well known for a number of centuries, it remains the subject of a wide range of research.1 Currently, approximately two-thirds of the elemental sulphur produced originates from oil refineries and gas purification plants.2–7 As a result of the continuous development of the oil and gas industry, the overall global production of sulphur has exceeded its consumption for a number of years.8–11 According to the latest estimates, there is an annual sulphur production surplus of approximately seven million tonnes. Elemental sulphur is a manufacturing by-product, which is stored in large storage facilities and has a detrimental effect on the environment and human health.12–16 It should be stressed that the current trends in caustobiolith production and processing relating to the necessity of opening up new sulphur-rich oilfields as well as the constant tightening of regulations pertaining to the sulphur content in transportation fuels has resulted in the inevitable increase in the volume of surplus sulphur produced.17 Consequently, the challenge of using elemental sulphur in large-scale chemical engineering processes that produce sought-after materials containing the maximum number of bound sulphur atoms has become an important research topic.Sulphur-containing polymers form a complex group of macromolecular compounds with varied and often unique properties.18 This defines their wide range of applications in optical, optoelectronic, and photochemical materials as well as pharmaceutical preparations based on biopolymers, proton-conducting electrolytes, cathodes in lithium–sulphur batteries, and electromembrane processes.19–21 The inclusion of sulphur atoms allows the regulation of chemical and thermal stability, as well as the mechanical, electrical, and optical properties of these polymers.22–26 One of the possible ways of using surplus elemental sulphur is the development of processes that involve its direct conversion into sulphur-containing polymers.27,28 The key step of this group of processes is always the opening of the cyclo-octasulphur ring.2,29–31 The opening of the S8 ring can involve either a radical or ionic mechanism (Scheme 1). The latter mechanism allows the activation of sulphur via nucleophilic (Scheme 1B) or electrophilic catalysis (Scheme 1C).
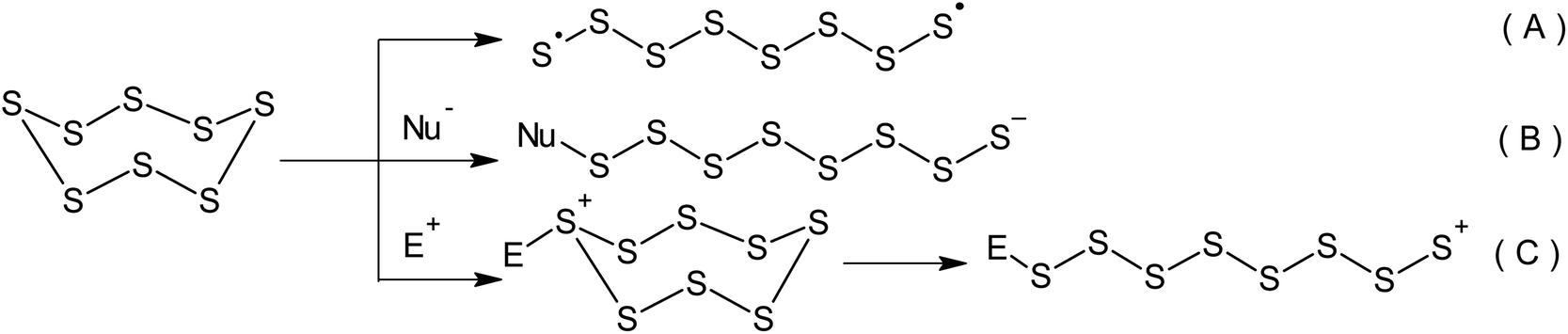 | ||
| Scheme 1 Radical (A), nucleophilic (B) and electrophilic (C) mechanisms of the opening the S8 ring. | ||
Although the radical processes used for the inclusion of elemental sulphur in polymers (Scheme 1A) remain a more popular and widespread field of research, these reactions require high temperatures and therefore, a high energy cost is required for the homolysis of S–S bonds. However, in the modern world the development of greener and more energy-efficient technologies in this research area is a promising way forward due to the constant increase in global energy consumption.32–36 Cutting the energy consumption during the synthesis of sulphur-containing polymers is aligned with the sixth principle of green chemistry,37–39 a concept published by Anastas and Warner in 1998.40,41 This concept is concerned with mitigating the negative impact of chemical technology on the environment and human health.42 Green chemistry is a widely recognised tool that can dramatically change the nature of the production and application of chemicals43 in order to achieve sustainability.44,45 Green chemistry principles are increasingly taken into consideration during the development of new technologies in the main areas of the chemical industry,46 such as pharmaceuticals,47–49 basic organic synthesis,50 petrochemistry, and the synthesis of polymeric materials.51–61
The synthesis of polymers using elemental sulphur not only allows the assimilation of elemental sulphur, but also the complete inclusion of the parent materials in non-toxic end products, often in one step and without the use of solvents, which aligns with green chemistry principles 1–5 and 8. However, significant energy consumption is a barrier to the green incorporation of sulphur into macromolecules. These high energy demands can be minimised by choosing suitable initiating and catalytic systems that enable the opening of the S8 ring under exceptionally mild conditions. Therefore, it is necessary to decrease the energy consumption by choosing suitable conditions and reagents, i.e. bring the synthesis of sulphur-containing polymers in line with the sixth principle of green chemistry via the rational implementation of the ninth principle of green chemistry.
Although there are a number of reviews dedicated to various aspects of the synthesis of sulphur-containing polymers and low-molecular-weight heterocyclic compounds,2,27,62–67 until present there have been no studies examining the inclusion of S8 into macromolecules from both the reaction mechanisms viewpoint and in the context of the opportunities for green development of this area of research. Therefore, the present review looks into the reaction mechanisms that lead to the inclusion of elemental sulphur into polymers and discusses the opportunities for the implementation of such processes under conditions that comply with the principles of green chemistry.
Homolytic and radical processes involved in opening of the S8 ring in polymer synthesis
Under normal conditions, sulphur is a solid crystalline substance, which is stable in the form of two allotropes: rhombic (α-sulphur) and monoclinic (β-sulphur) sulphur. Both allotropes are formed by non-planar cyclic octatomic crown-shaped S8 molecules with an S–S bond energy of 225.7 kJ mol−1. The difference between the two allotropes is the mutual orientation of the molecules in the crystal lattice.68–75The fundamental property of elemental sulphur is catenation,76 which is responsible for allotropism and polymorphism in simple substances,77 as well as cluster formation. The ability of sulphur to catenate is related to the energy gain (109 kJ mol−1) achieved when one S–S π-bond transforms into two σ-bonds. Over the past three decades, there have been a large number of studies published on the electronic structure of sulphur clusters. These studies use ab initio methods as well as density functional theory utilising varying degrees of complexity.72 Among the allotropic forms of sulphur, the S8 form is the most stable under normal conditions.78,79 This form exists as a highly symmetrical cyclic D4d (crown-shaped) structure. It has been reliably characterised using quantum chemistry calculations.80–82 The S8 conformation includes structures such as C2 (endo–exo ring), C2 (twisted ring), D2d (boat), C2h (chair), C2 (cluster), C1 (S7![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S), and C1 (S7Seq), as well as a few branched and unbranched forms. The crown-shaped S8 octatomic conformation (D4d) has been confirmed to be a global energy minimum using various levels of theory.83–85
S), and C1 (S7Seq), as well as a few branched and unbranched forms. The crown-shaped S8 octatomic conformation (D4d) has been confirmed to be a global energy minimum using various levels of theory.83–85
Elemental sulphur is mainly represented by the S8 ring, which is capable of forming linear chains at temperatures >432 K via the formation of biradical intermediates. Homopolymerisation of the S8 ring is an equilibrium process, which is shifted towards depolymerisation at room temperature,86 limiting the application of its corresponding macromolecules. Moreover, elemental sulphur is able to cross-link polymer chains and copolymerise with a number of compounds, which allow it to be incorporated into polymer products that are stable at ambient temperatures.
The ability of elemental sulphur to cross-link polymer chains at high temperature has been known since the nineteenth century, when, in 1839, Charles Nelson Goodyear discovered that heating uncured rubber with S8 produces a material with enhanced mechanical properties.87,88 Initially, the vulcanisation of rubber using elemental sulphur was performed at 413 K over 5 h. The vulcanisation mechanism of cis-poly(buta-1,3-diene) involves the homolytic opening of the S8 ring and the formation of oligosulphide biradicals that simultaneously remove allylic hydrogen atoms and link the resulting radicals formed in the main polymer chain, thus producing an irregular network (Scheme 2).89–92 The radical mechanism of the vulcanisation of rubber was confirmed by the possibility of increasing the speed of this process in the presence of a number of substances that are capable of thermal decomposition and forming radicals, such as dithiobenzothiazoles, benzothiazolesulfenamides, thiurams, and dithiocarbamates.93
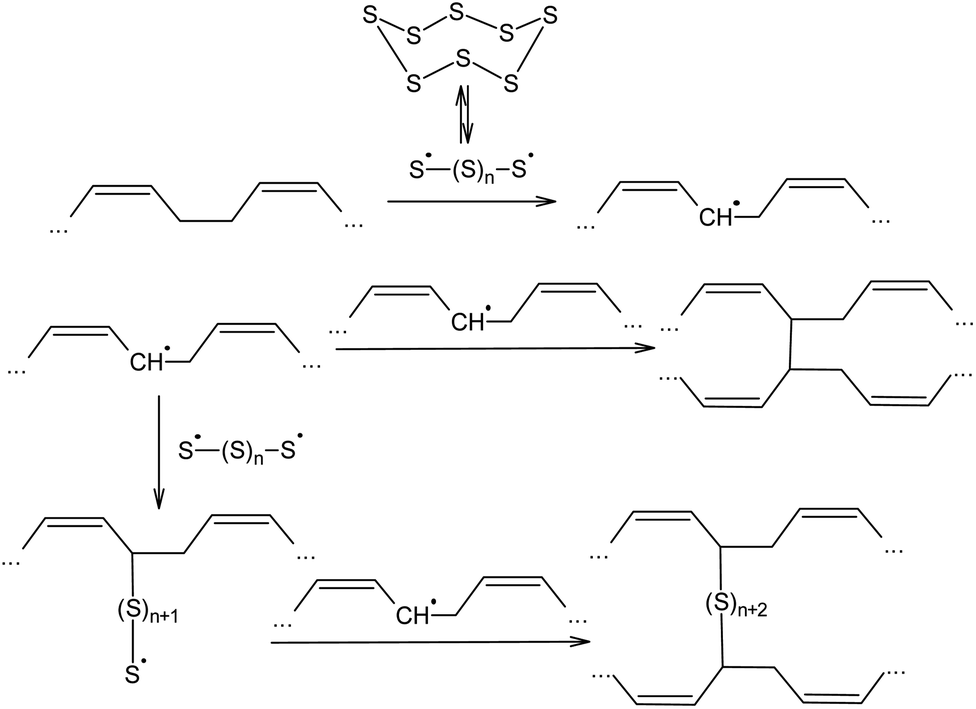 | ||
| Scheme 2 Mechanism of the vulcanisation of cis-poly(buta-1,3-diene). | ||
Another promising way of binding elemental sulphur is inverse vulcanisation, which is the copolymerisation of the S8 ring with unsaturated monomers, such as diisopropenylbenzene.4,24,26,94–99 Griebel et al.26 reported the synthesis of a thermoplastic copolymer as a result of the reaction between S8 and 1,3-diisopropenylbenzene performed at 458 K. The resulting polymer contains 50–80 wt% sulphur and had a refractive index of 1.8, making it suitable for possible use in IR-optics (Scheme 3).
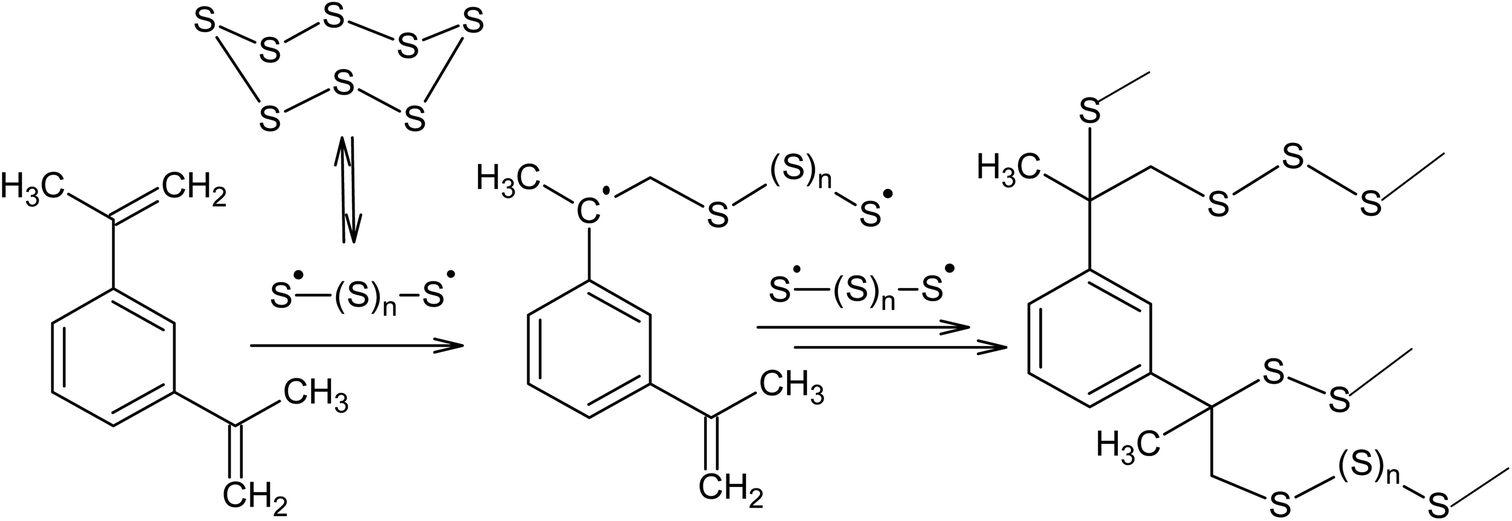 | ||
| Scheme 3 Reaction between S8 and 1,3-diisopropenylbenzene. | ||
The same approach was used to obtain a cross-linked polymer, which proved to be effective for the extraction of palladium and mercury salts from aqueous solutions, utilising the reaction between elemental sulphur and D-limonene at 443–453 K.100 Polymers with a high sulphur content were also obtained upon substituting D-limonene with perillyl alcohol, squalene,101 and botryococcene.102 In the latter case, the obtained polymer material can contain 50–90 wt% sulphur depending on the ratio of the initial reagents used, which has the potential to be used as a component in lithium–sulphur batteries. It should be noted that elemental sulphur can be found in a sample when the weight percentage was increased to >70 wt%. This was attributed to the linear oligosulphide fragments reaching a certain length, at which they become thermodynamically unstable and release elemental sulphur.
The inverse vulcanisation method allows not only a number of new polymer materials to be obtained, but also opens up a pathway toward the synthesis of nanocomposites. For example, it is possible to obtain nanocomposites as a result of the introduction of nanoparticles prepared in advance,103 but also via the formation of these particles during the curing process when sulphur reacts with unsaturated compounds.104 Both of these approaches were implemented by filling the high-sulphur network with lead sulfide.103,104 Both elemental sulphur and vulcanised cis-polyisoprene can be used as a source of sulphur atoms. This approach has been used to reinforce vulcanised polyisoprene rubber with cellulose nanofibres, which were modified with the residual unsaturated oleic acid.105
An increase in the curing temperature allows compounds containing triple carbon–carbon bonds to be involved in the reaction with S8. For example, 1,3-diethynylbenzene reacts with elemental sulphur at 473 K to form a macromolecular compound, which can be used as the cathode material in lithium–sulphur batteries.106 The requirement for a higher reaction temperature when reacting 1,3-diethynylbenzene reaction with sulphur compared to 1,3-diisopropenylbenzene corresponds to the formation of vinyl radicals, which are less stable than benzyl radicals.
Finally, the inverse vulcanisation reaction can be used to introduce certain groups into the polymer network that make electrochemical oxidative polymerization possible, such as 3,4-alkylenedioxythiophene.107 This approach is clearly advantageous when compared to the synthesis of similar materials via condensation with sulphur dichloride and their subsequent electrochemical oxidative copolymerization reaction.108
Polymerisation at 432 K in the presence of elemental sulphur is not only possible for alkenes and alkynes, but also for other compounds that can act as chain transfer agents in relation to oligosulphide radicals, such as N-phenylbenzoxazine (Scheme 4).109
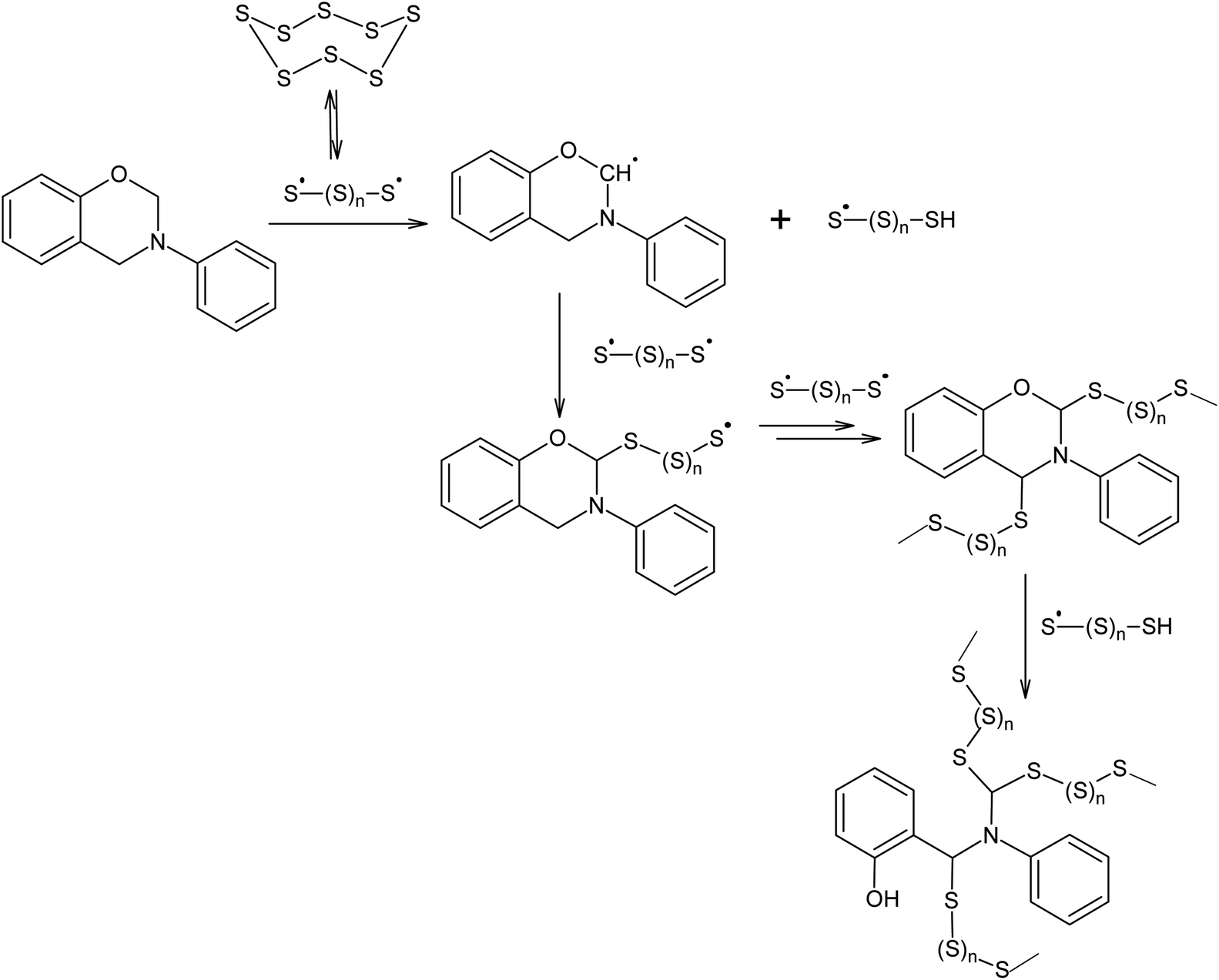 | ||
| Scheme 4 Benzoxazines used in the reaction with elemental sulphur. | ||
Aliphatic110 and aromatic111 dithiols can be used as chain transfer agents, which lead to the formation of their corresponding polymeric polysulfides (Scheme 5). This reaction usually takes place at 400 K using nucleophilic catalysis. As will be shown later, nucleophilic catalysis facilitates the opening of the S8 ring via an anionic mechanism and the homolysis of the S–S bonds in the linear chain of the sulphur atoms is responsible for the formation of radicals.
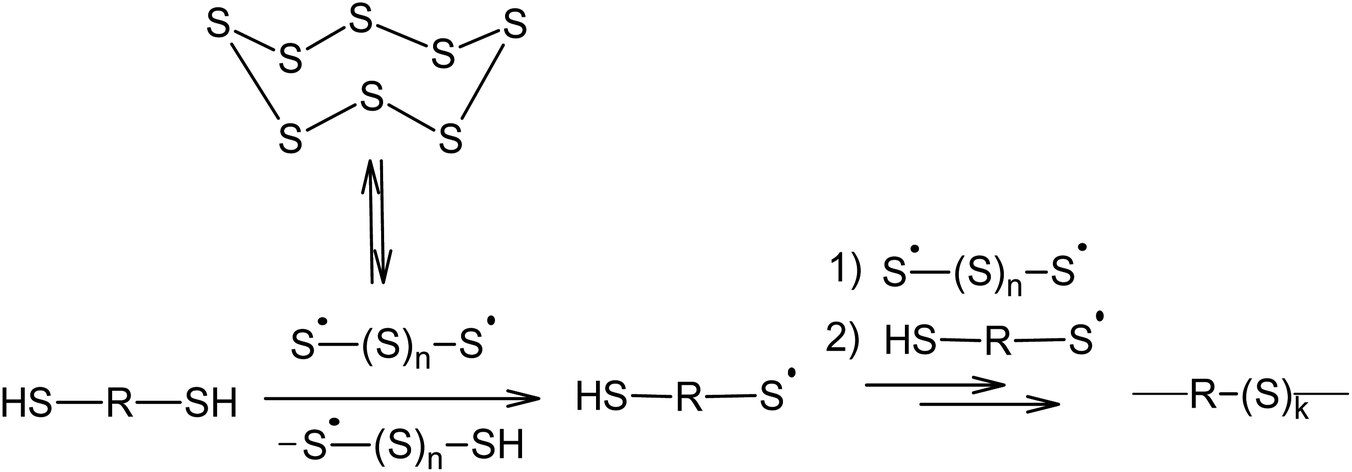 | ||
| Scheme 5 Dithiols used in the reaction with elemental sulphur. | ||
Therefore, the incorporation of elemental sulphur in macromolecules via a polyrecombination reaction is a general method in which the temperature allows for the formation of oligosulphide biradicals if the partner compound is capable of reacting with the resulting radicals or has sites that are active in radical substitution reactions.
The copolymerisation of elemental sulphur with unsaturated compounds in the absence of solvent allows high sulphur content polymer networks exhibiting a number of specific properties to be obtained. However, the inverse vulcanisation reaction requires high temperature, which does not allow this process to be fully classified as a green synthesis method. This is why a lot of effort has been dedicated to replacing the thermal activation of cyclo-octasulphur with an activation step initiated by different physical factors, although there has been relatively little success in this area reported to date. For example, UV irradiation at 254 nm and 20 W power causes free-radical grafting of linear polysulfide chains to the walls of carbon nanotubes as a result of their interaction with cyclo-octasulphur.112 Some studies113,114 have presented detailed investigations on the mechanism of bond breaking in the S8 ring induced by an impulse laser using ab initio molecular dynamics modelling. It has been shown that a bond in the S8 ring is easily broken immediately after an electron is transferred from the HOMO to LUMO, which was confirmed by the reduction in the starting sulphur polymerisation temperature upon photoinduction. For example, a photoinduced S8 ring opening process has been described at 403 K under irradiation of a 10 Hz impulse laser.115
It is likely that the most promising approaches to reduce the temperature required for the inclusion of elemental sulphur in polymeric materials will be related to the substitution of the radical process with ionic reactions. It is known that the vast majority of ionic reactions have a lower activation energy when compared to radical processes, which allows the possibility of achieving suitable velocities for S8 ring opening even at room temperature. The implementation of this possibility will simultaneously bring sulphur assimilation processes in line with the sixth and ninth principles of green chemistry as well as the undeniably green character of the majority of sulphur-containing polymers. The next chapter is dedicated to the ionic processes of S8 ring opening.
Nucleophilic and electrophilic activation of the S8 ring in polymer synthesis
Reactions between aromatic amines and sulphur have been known since the beginning of the twentieth century.116 Benzylamine and dimethylaniline react with S8 in the presence of lead oxide, even at 413 K;117 this temperature is lower than that used in sulphur homopolymerisation reactions. The EPR spectra obtained for the systems formed after dissolving elemental sulphur in an aliphatic amine only show one signal with a g-factor of 2.030, which suggests the formation of radicals that are localised on the sulphur atoms.118 The formation of radicals localised on sulphur atoms can be explained by the reversible homolysis of the S–S bonds in the oligosulphide fragments of the polythiobisamines and amino-polysulfides formed during the reaction118 and is probably not related to the formation of radical intermediates in the S8 ring opening reaction. It is very likely that in the presence of amines, the mechanism of S8 ring opening is anionic.119–121 The anionic opening of the S8 ring has been suggested for the Willgerodt–Kindler reaction,122,123 as well as for the transformation of benzothiadiazoles into benzopentathiepines124–126 under nucleophilic catalysis using DABCO (Scheme 6),126 although in the latter case, the reaction takes place at temperatures >433 K.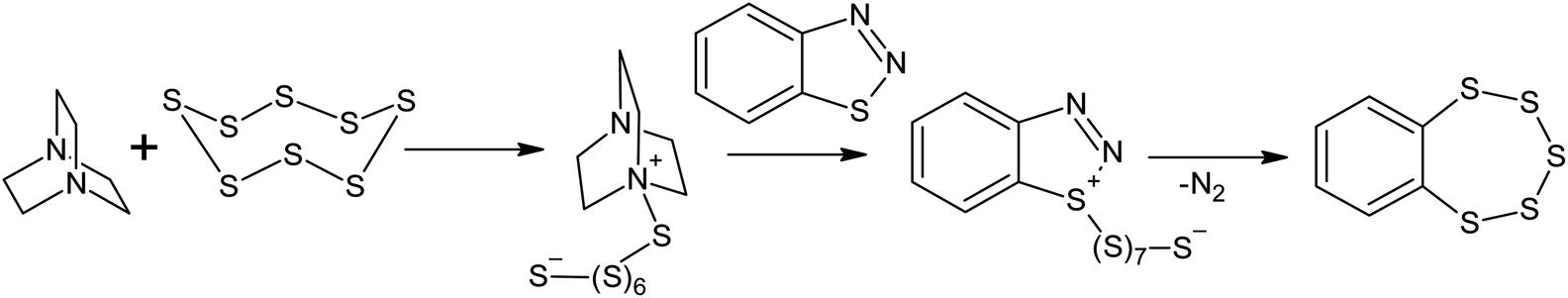 | ||
| Scheme 6 The transformation of benzothiadiazoles into benzopentathiepines in the presence of elemental sulphur. | ||
The use of alkynes makes it possible to intercept the polysulfide anions formed after the opening of the S8 ring, which opens up a new approach toward the synthesis of thioamides using amine catalysis (Scheme 7A).127 The latter reaction allows elemental sulphur to be incorporated into the multicomponent polycondensation of dienynes and diamines at temperatures as low as 373 K (Scheme 7B).128
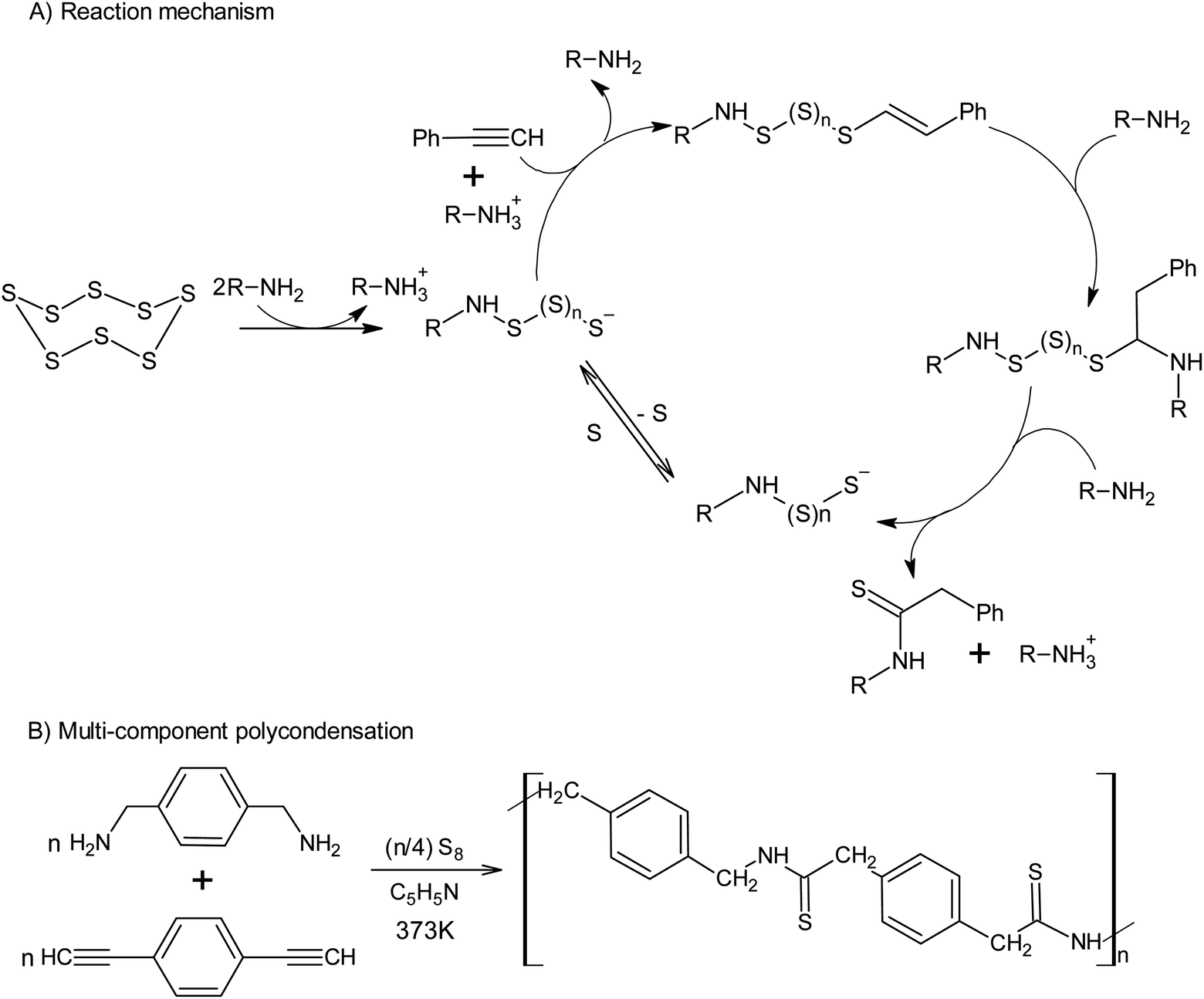 | ||
| Scheme 7 Synthesis of polythioamides using elemental sulphur. | ||
Nucleophilic amine catalysis allows the inverse vulcanisation temperature to be significantly reduced. For example, the inverse vulcanisation of styrene takes place in the presence of N-methylimidazole and 4-aminostyrene at temperatures as low as 383 K.129 The introduction of even small amounts of these activators provides for a significant increase in the velocity of the inverse vulcanisation of styrene. There is little doubt that the mechanism of activation of elemental sulphur by N-methylimidazole and 4-aminostyrene is generally the same because the kinetic curves obtained for the styrene inverse vulcanisation are practically identical in the presence of both accelerators.129 Moreover, the initial formation of oligosulphide anions does not produce an anionic mechanism for polymer chain growth because the velocity of the inverse vulcanisation increases for a series of monomers as follows: styrene < 3-aminostyrene ≪ 4-aminostyrene.130 Therefore, it is possible to assume that after the nucleophilic opening of the S8 ring, the linear chains made of sulphur atoms are subjected to reverse homolysis of the S–S bonds (Scheme 8).129 The interception of the radicals formed in this process by unsaturated compounds present in the reaction mixture leads to the development of an anionic inverse vulcanisation reaction. The change in the nature of the active centre may be a result of an increase in the velocity of the homolysis of the S–S bonds in the linear chains when compared to the initial S8 ring. The increased homolysis velocity of the S–S bonds in the linear sulphur chains corresponds to the EPR spectral data,118 as well as to the instability of polymeric sulphur at temperatures <432 K and the possibility of the inverse vulcanisation of 4-aminostyrene at temperatures as low as 333 K if S8 is substituted by the products of the inverse vulcanisation of styrene.129
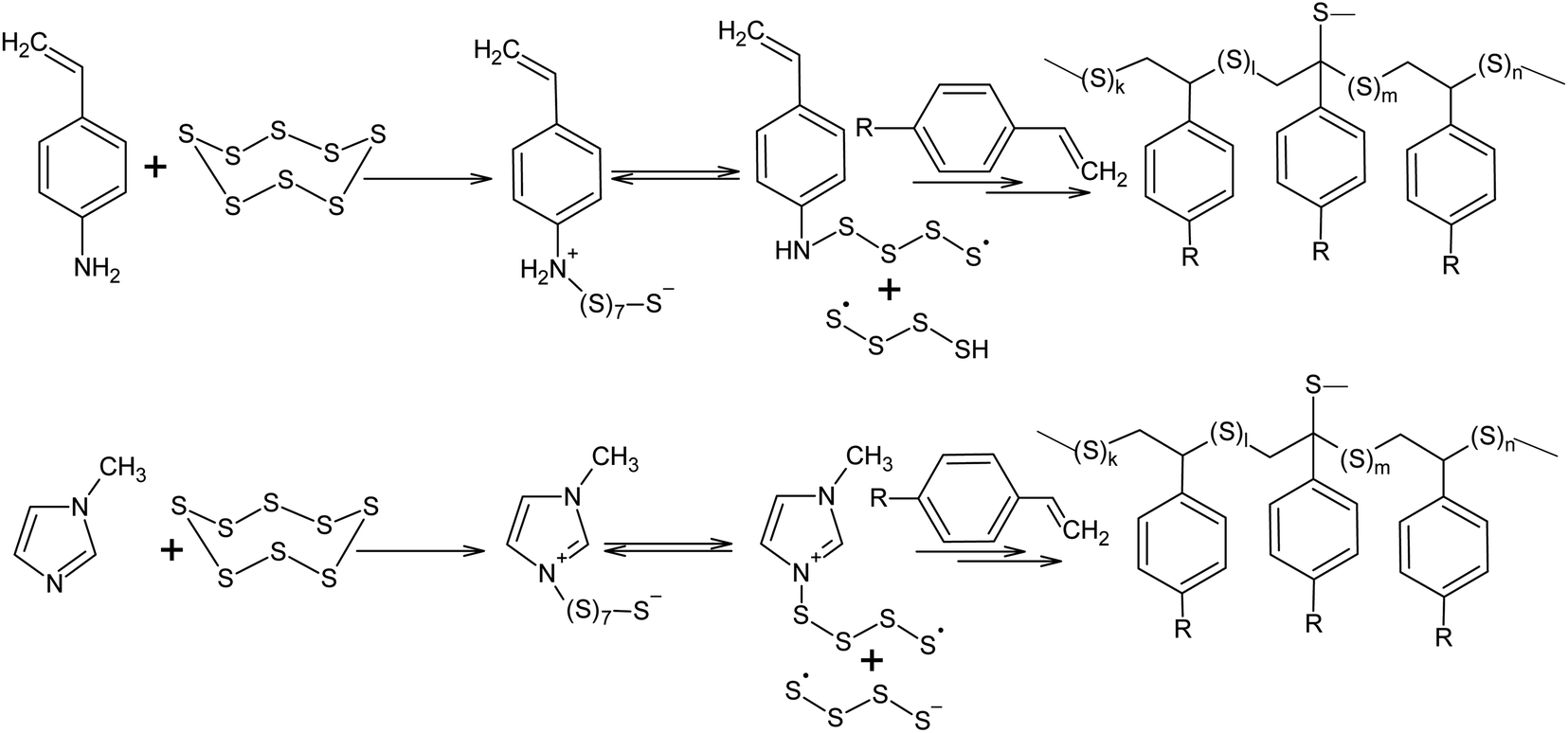 | ||
| Scheme 8 Acceleration of the inverse vulcanisation of styrene derivatives in the presence of nucleophilic activators (4-aminostyrene and N-methylimidazole). | ||
Even if there are no nucleophilic activators present, styrene is capable of participating in the inverse vulcanisation reaction with elemental sulphur at a temperatures as low as 403 K. However, 1,3-diisopropylbenzene is only active in the reaction when the temperature is >433 K. This points to the participation of benzyl protons in the reaction and explains the branching mechanism in the chains. It also corresponds with the formation of radicals as the active centres.131
However, it is possible that these nucleophilic activators act differently, in a way which has not been previously described in the literature. It is known that the addition of 4-aminostyrene to styrene significantly increases the velocity of the inverse vulcanisation of the latter. Moreover, the velocity of the inverse vulcanisation of 3-aminostyrene is only slightly higher than that of pure styrene.130 However, the nucleophilicity and basicity of the amino groups in 3-aminostyrene and 4-aminostyrene are similar, which contradicts the nucleophilic activation mechanism of elemental sulphur, as shown in Scheme 8 (first step). To eliminate this contradiction, we assume that the activation mechanism of the S8 ring is not related to nucleophilic assistance, but to single-electron transfer (Scheme 9). In addition, the mechanism shown in Scheme 9 explains the large number of free amino groups in the polymer after the interaction of sulphur and styrene in the presence of 4-aminostyrene ended. Although the mechanism of cyclo-octasulphur activation in the presence of 4-aminostyrene and N-methylimidazole is a matter for discussion, the radical nature of the inverse vulcanisation process itself has been firmly established.
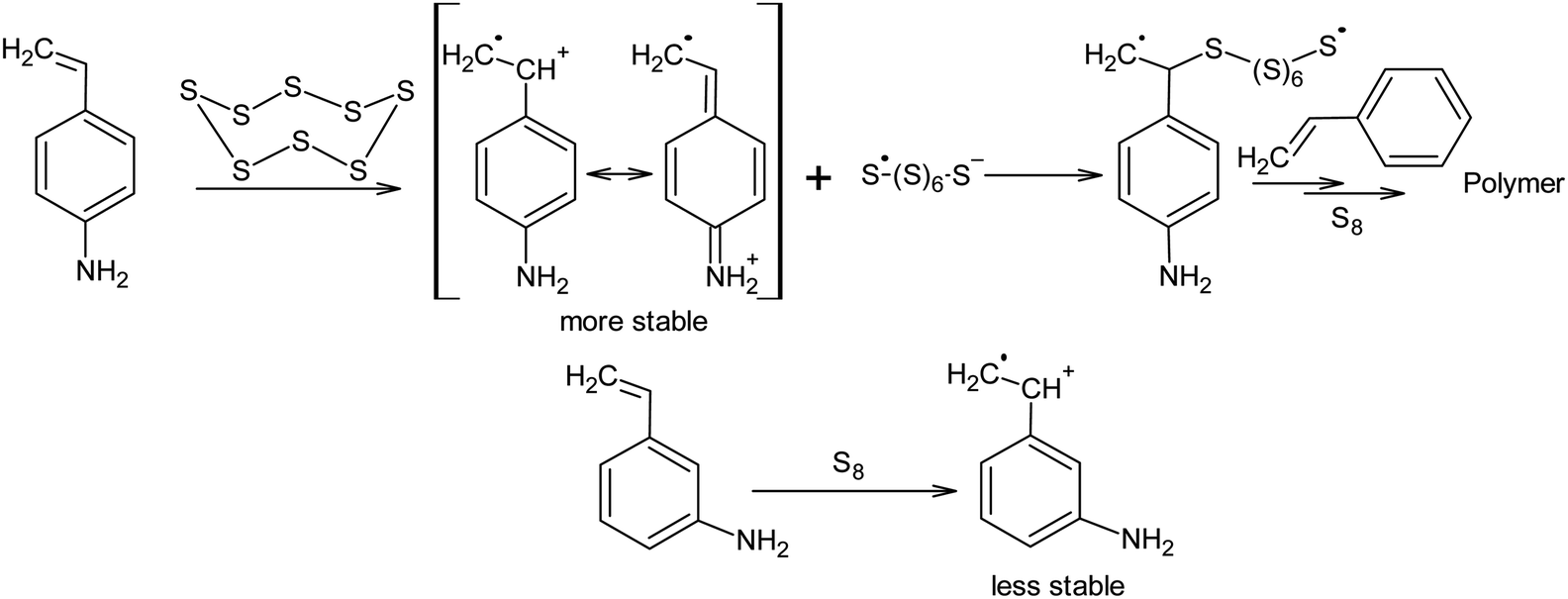 | ||
| Scheme 9 Cyclo-octasulphur activation as a result of single-electron reduction. | ||
Elemental sulphur can act as an electrophile in electrophilic aromatic substitution reactions. The interaction between S8 and m-phenylenediamine at 403 K has been reported to produce low-molecular oligomers with irregular structures that can be used in the further curing and modification of epoxy resins. It was assumed that, in the cases described above, the reaction begins with anionic S8 ring opening, although due to the excessive electron density in the aromatic system of m-phenylenediamine, a regrouping of the N-substituted oligosulphide into its corresponding C-derivative is possible (Scheme 10).132
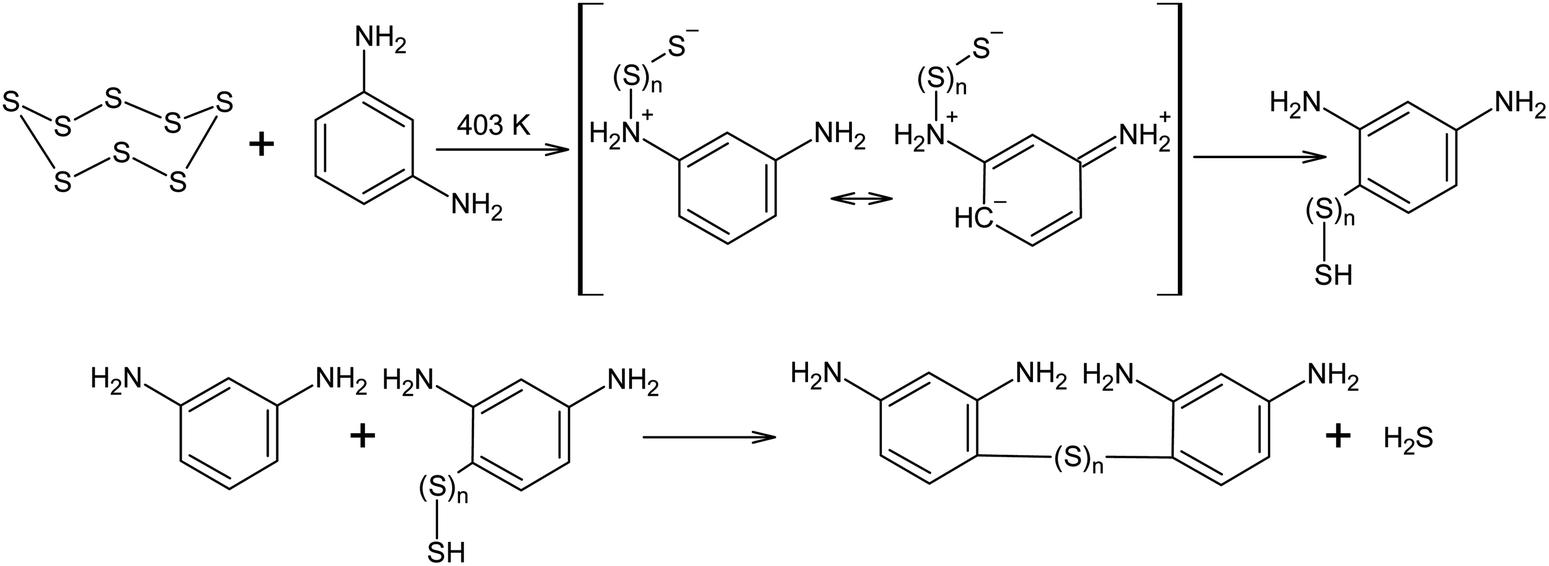 | ||
| Scheme 10 The mechanism of the electrophilic aromatic substitution using elemental sulphur. | ||
Using amines and other nitrogen-containing compounds for the nucleophilic activation of sulphur allows the temperature required for inclusion of sulphur in the polymers to be reduced by a few tens of degrees. The possibility of using the products of vulcanisation as a source of sulphur in inverse vulcanisation reactions at temperatures as low as 330 K opens up new opportunities for the recycling of sulphur-containing elastomers. Therefore, elemental sulphur and amine-based systems are of significant interest in the context of using elemental sulphur as a component in macromolecules.
It should be noted that elemental sulphur can be activated by sulphides under exceptionally mild conditions with the further formation of inorganic polysulfides.133,134 For example, Na2S6 is obtained when sodium sulphide reacts with elemental sulphur at temperatures as low as 323 K.135 Oligomerisation of elemental sulphur is possible in aqueous solutions of sodium sulphide, even at room temperature. The possibility of S8 ring opening during the interaction with sulphide anions at room temperature points to the low activation energy of the reaction, as shown in Scheme 11, and other similar reactions.
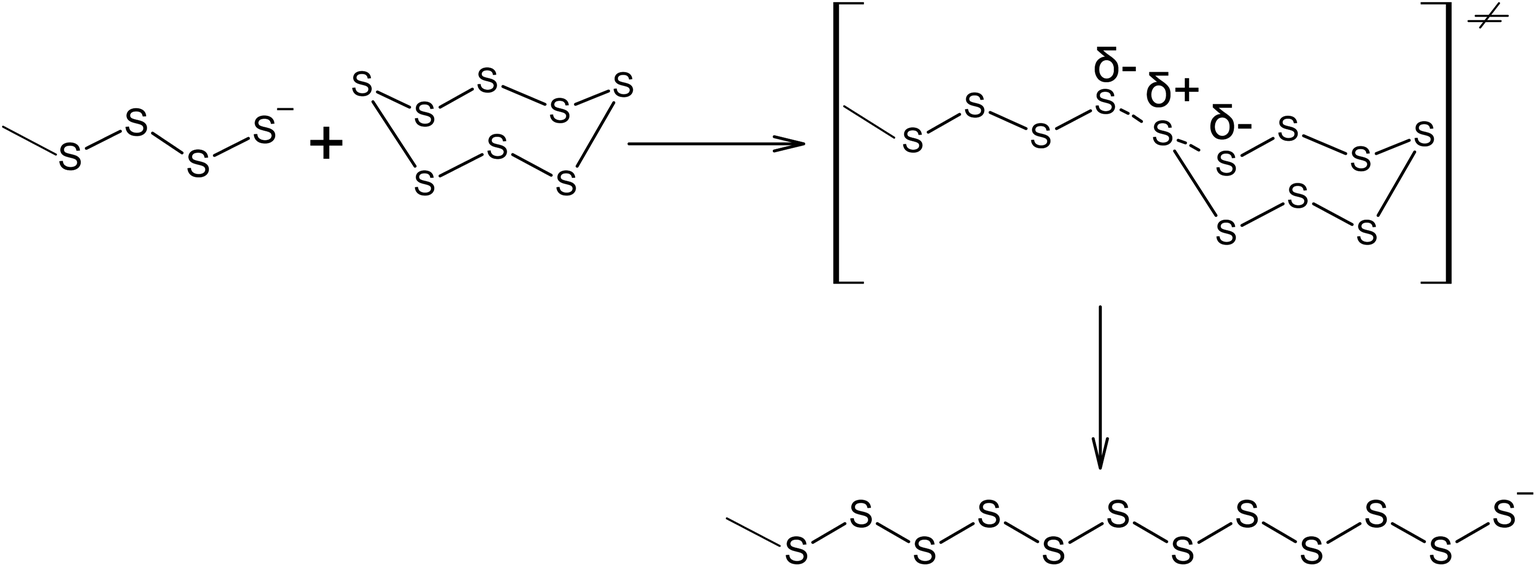 | ||
| Scheme 11 Cyclo-octasulphur ring opening under nucleophilic attack by an oligosulphide anion. | ||
As an example of the nucleophilic activation of sulphur, the interaction of diallyl disulphide derivatives with cyclo-octasulphur at 393 K, leading to the formation of diallyl polysulfides containing between 3 to 22 sulphur atoms (Scheme 12),136 can be considered.
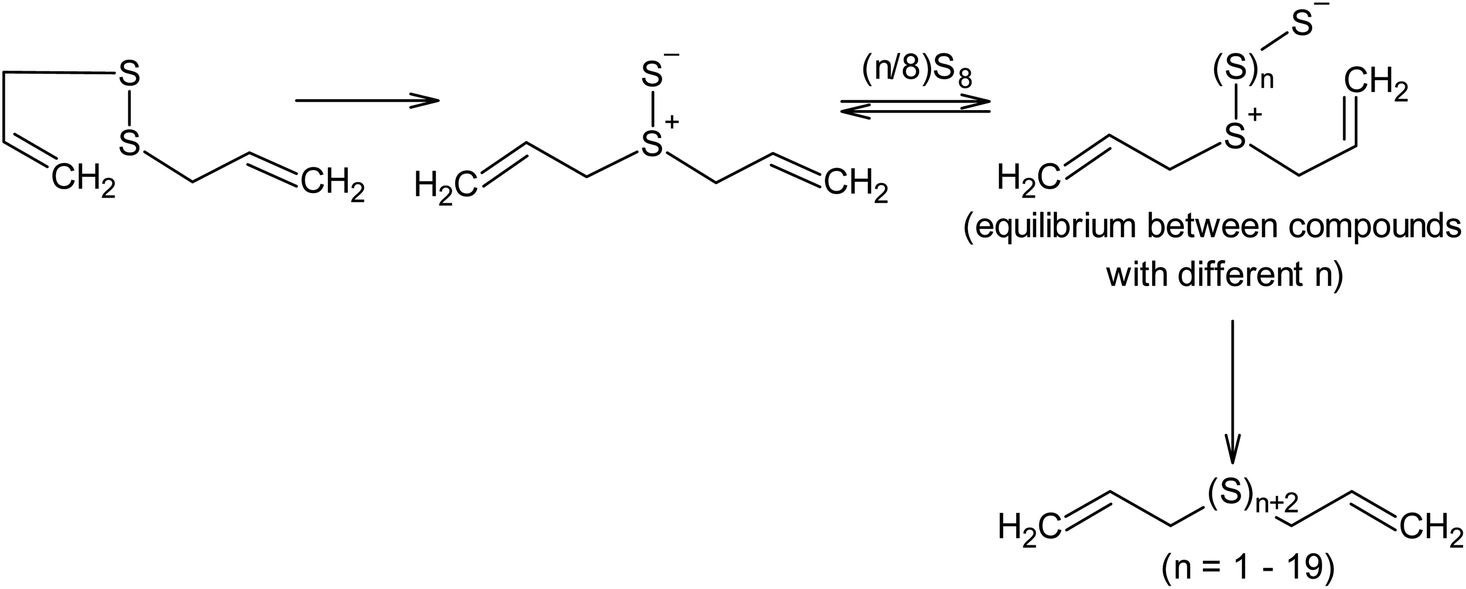 | ||
| Scheme 12 Mechanism of cyclo-octasulphur activation in the presence of diallyl disulphide. | ||
The strategy for the nucleophilic activation of the S8 ring by a sulphide anion opens up a pathway for the synthesis of linear macromolecules via the copolymerisation of thiirane derivatives using elemental sulphur.137 The activity of propylene sulphide and cyclo-octasulphur in the anionic copolymerisation is approximately the same. However, the activity of alkyl derivatives of thiirane in the copolymerisation process increases in the following order: 1,1-dimethylthiirane < methylthiirane < thiirane,138 which corresponds to the anionic nature of the active centres. The copolymerisation of elemental sulphur with methylthiirane was conducted in the presence of sodium thiophenoxide as an initiator in benzene upon the addition of dibenzo-18-crown-6 ether at 80 °C (Scheme 13A). The radical copolymerisation of styrene and elemental sulphur resulting from inverse vulcanisation leads to the formation of a branched copolymer. However, the transition to anionic initiation opens the way toward the synthesis of linear copolymers of styrene and elemental sulphur (Scheme 13B). The formation of a linear copolymer of elemental sulphur and styrene was observed when the reaction was initiated using potassium thiophenoxide at 95 °C.139 1,2,3-Trithiolanes are capable of homopolymerisation and copolymerisation with S8 via a living anionic mechanism, which forms macromolecules with an average molecular weight of up to 105 (Scheme 13C).140 The formation of macromolecules with an average molecular weight of up to 105 was also observed in the copolymerisation of thiiranes used in wire coatings with elemental sulphur.141
 | ||
| Scheme 13 The inclusion of elemental sulphur in polymers using anionic copolymerisation with: (A) methylthiirane, (B) phenylthiirane, (C) norbornene. | ||
The low-temperature synthesis of copolymers using elemental sulphur and styrene 1,3-butadiene or isoprene is possible in THF upon initiation by metallic sodium.142 At the optimized reaction temperature (233 K), the hydrocarbon co-monomers oxidise sodium faster than elemental sulphur, which actively intercepts the carbanions formed during the reaction. Although using sodium as a component in the reaction system provides for the low-temperature bonding of elemental sulphur and the formation of polymeric products, the conditions used in this reaction clearly do not conform to the principles of green chemistry.
Recently, the authors of this review demonstrated the possibility of the nucleophilic activation of elemental sulphur using ionic liquids containing dimethyl phosphate, such as 1,3-dimethylimidazolium dimethyl phosphate and tri-n-butyl(methyl)phosphonium dimethyl phosphate, even at room temperature (Scheme 14).143–145
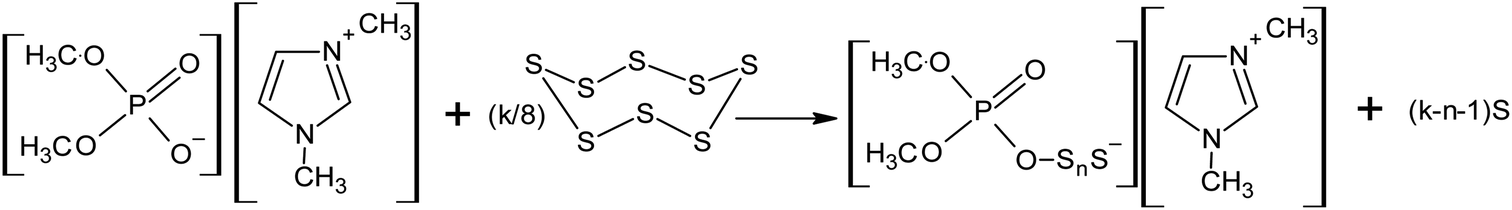 | ||
| Scheme 14 The interaction of 1,3-dimethylimidazolium dimethyl phosphate with elemental sulphur. | ||
Anionic copolymerisation of elemental sulphur with thiiranes opens up new possibilities for the synthesis of linear polymers with a high sulphur content and controlled structures, while achieving complete atom economy. Although the copolymerisation of sulphur and thiiranes takes place under mild conditions, this reaction is still not possible at room temperature. At the same time, the search continues for new initiating and catalytic systems and media that will allow the process temperature to be reduced. The fundamental possibility of sulphur oligomerisation, even at room temperature, in water and ionic liquids containing dimethyl phosphate has already been demonstrated. Therefore, catalysis using suitable nucleophiles allows the temperature required for a direct inclusion of elemental sulphur in polymers to be significantly reduced and ultimately conditions that would approximate the requirements of green chemistry to be achieved.
Unlike the nucleophilic activation of elemental sulphur, the processes that use electrophilic catalysis in the S8 ring opening reaction used toward the synthesis of high molecular compounds have been studied to a much lesser degree. The difficulties associated with the use of Lewis acids in the catalysis of cationic polymerisation and copolymerisation of sulphur are likely related to the high velocity of the degradation of the linear sulphur chains under such conditions. At the same time, the immobilisation of electrophiles on solid surfaces opens up a completely new opportunity for the utilisation of the S8 ring in anionic oligomerisation or polymerisation. For example, it is possible to open the S8 ring in the presence of silica gel, which binds the electrophilic centres of Lewis acids (Scheme 15). The effectiveness of zinc chloride,146,147 titanium(IV) chloride,148–150 iron(III) chloride,151 and aluminium chloride152,153 in the presence of silica gel has been demonstrated, which widens the options for regulating the properties of their corresponding sorbents.
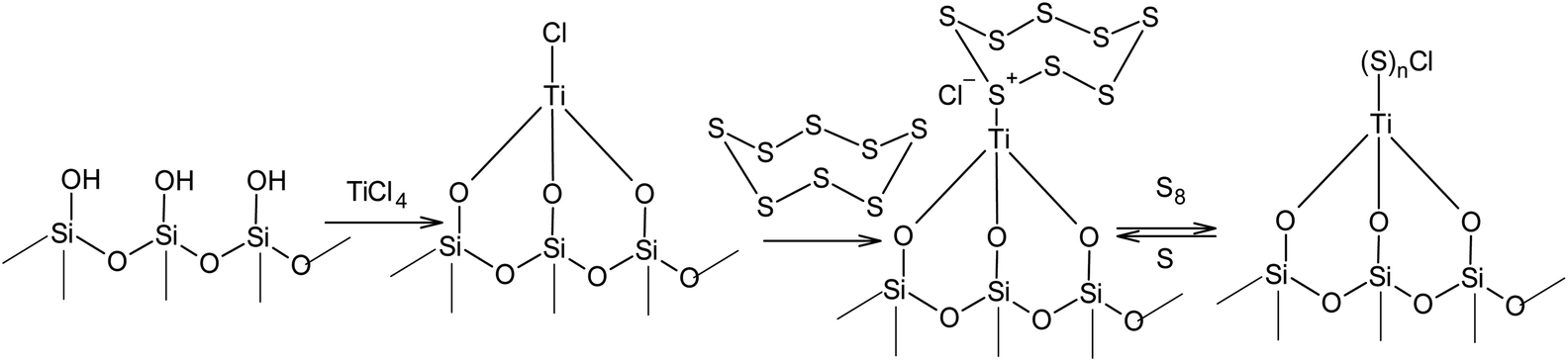 | ||
| Scheme 15 Electrophilic activation of sulphur in the presence of Lewis acids immobilised on the surface of silica gel. | ||
Another example of the possible electrophilic activation of the S8 ring is the catalysis of the vulcanisation of diene rubber in the presence of zinc oxide and 2-mercaptobenzothiazole (Scheme 16). In this case, a polysulfide formation step catalysed by Zn2+ occurs after the formation of the zinc salt and this ultimately leads to an increase in velocity of diene rubber cross-linking reaction. Possible mechanisms for vulcanisation and the impact of vulcanisation accelerators have been detailed in other publications.154,155
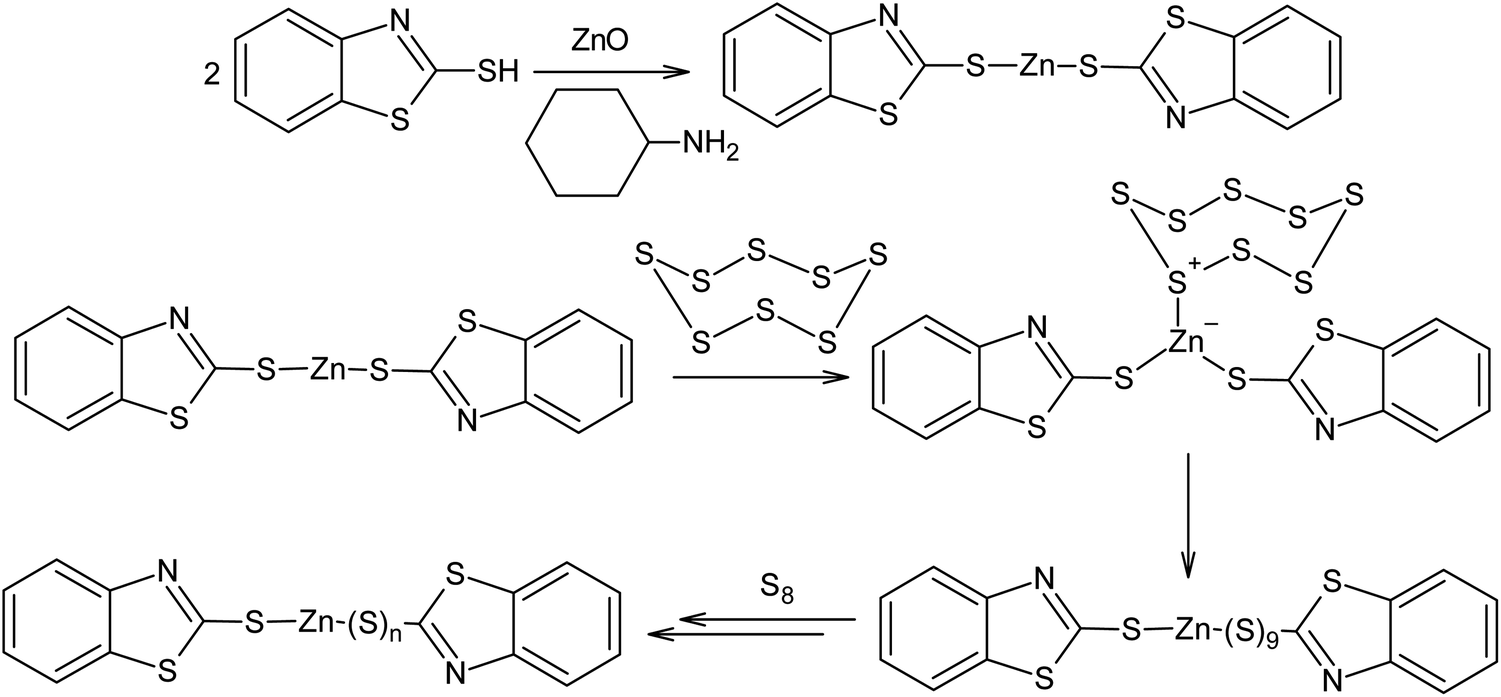 | ||
| Scheme 16 Possible mechanism for the activation of elemental sulphur during the vulcanisation of rubber using zinc oxide and 2-mercaptobenzothiazole. | ||
Conclusions
With the continuing growth of the petrochemical industry's output and constant tightening of the regulations pertaining to the purification of oil products obtained from sulphur and sulphur-containing compounds, the production of elemental sulphur is running at a significant surplus. Thus, there is a need to develop technologies for the disposal of elemental sulphur, including those for its inclusion in polymers. The synthesis of sulphur-containing polymers from elemental sulphur using traditional methods requires high temperatures. However, the nucleophilic and electrophilic activation of cyclo-octasulphur allows the energy consumption to be significantly reduced. At the same time, sulphur-containing polymers have good mechanical properties, are active in a number of electrochemical processes, and have promising applications as sorbents. This leads to the expectation that elemental sulphur will be considered as a valuable resource. It has been demonstrated that a decrease in the temperature required for the S8 ring opening reaction is a key objective and can be reached by selecting suitable ionic catalytic and initiating systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The reported study was funded by Dmitry Mendeleev University of Chemical Technology.Notes and references
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC.
- Mineral Commodity Summaries, U.S. Department of the Interior, Washington, DC, USA, 2020, p. 160 Search PubMed.
- J. A. Smith, X. Wu, N. G. Berry and T. Hasell, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1777–1781 CrossRef CAS.
- J. G. Wagenfeld, Waste Manag., 2019, 95, 78–89 CrossRef.
- A. Davarpanah, B. Mirshekari, T. Jafari Behbahani and M. Hemmati, J. Pet. Explor. Prod. Technol., 2018, 8, 743–751 CrossRef CAS.
- R. Khatun, M. Reza, M. Moniruzzaman and Z. Yaakob, Renewable Sustainable Energy Rev., 2017, 76, 608–619 CrossRef CAS.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y. E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS.
- F. Crescenzi, A. Crisary, E. D'Angeli and A. Nardella, Environ. Sci. Technol., 2006, 40, 6782–6786 CrossRef CAS.
- T. B. Nguyen, Adv. Synth. Catal., 2017, 359, 1066–1130 CrossRef CAS.
- P. Harrisson, Global Sulphur Market Outlook, 2016 Search PubMed.
- G. Kutney, Sulfur: History, Technology, Applications & Industry, Chem. Tec. Publishing, Toronto, 2013 Search PubMed.
- T. A. Rappold and K. S. Lackner, J. Energy, 2010, 35, 1368–1380 CrossRef CAS.
- P. Gosselin, S. E. Hrudey, M. A. Naeth, A. Plourde, R. Therrien and G. Van Der Kraak, Environmental and Health Impacts of Canada's Oil Sands Industry, Royal Society of Canada, Ottawa, 2010 Search PubMed.
- J. A. Smith, R. Mulhall, S. Goodman, G. Fleming, H. Allison, R. Raval and T. Hasell, ACS Omega, 2020, 5, 5229–5234 CrossRef CAS.
- Official website of the Astrakhan region of the Russian Federation URL: https://www.astrobl.ru.
- J. Lim, J. Pyun and K. Char, Angew. Chem., Int. Ed., 2015, 54, 3249–3258 CrossRef CAS.
- A. Hoefling and P. Theato, Nachr. Chem., 2016, 64, 9–12 CrossRef CAS.
- A. Kultys, in Encyclopedia of Polymer Science and Technology: Sulfur-containing Polymers, ed. H. Mark, Wiley, NY, 2010, p. 67 Search PubMed.
- A. Kausar, S. Zulfiqar and M. I. Sarwar, Polym. Rev., 2016, 54, 185–267 CrossRef.
- Y. Zhang, D. Wu and Z. Weng, Org. Chem. Front., 2017, 4, 2226–2229 RSC.
- F. Garbassi and R. Po, in Encyclopedia of Polymer Science and Technology: Engineering Thermoplastics, Overview, Wiley, NJ, 2003, p. 383 Search PubMed.
- M. Polk, in Encyclopedia of Polymer Science and Technology: High Performance Fibers, ed. J. Kroschwitz and H. Mark, Wiley, NJ, 2003, p. 214 Search PubMed.
- A. Kultys and A. Puszka, Pol. J. Chem. Technol., 2013, 15, 65–70 CAS.
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y.-E. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229–232 CrossRef CAS.
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K. J. Kim, J. van der Laan, N. A. Nguyen, E. L. Dereniak, M. E. Mackay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26, 3014–3018 CrossRef CAS.
- J. J. Griebel, R. S. Glass, K. Char and J. Pyun, Prog. Polym. Sci., 2016, 58, 90–125 CrossRef CAS.
- A. Kausar, S. Zulfiqar and M. I. Sarwar, Polym. Rev., 2016, 54, 185–267 CrossRef.
- V. F. Kozhevnikov, W. B. Payne and J. K. Olson, J. Chem. Phys., 2004, 121, 7379–7386 CrossRef CAS.
- R. W. Gomez, J. L. Perez, M. V. Marquina, R. Ridaura and M. L. Marquina, Rev. Mex. Fis., 2007, 53, 30–33 CAS.
- R. Steudel, S. Passlack-Stephan and G. Holdt, Z. Anorg. Allg. Chem., 1984, 517, 7–42 CrossRef CAS.
- A. Makarov, T. Mitrova, F. Veselov, A. Galkina and V. Kulagin, Therm. Eng., 2017, 10, 5–16 Search PubMed.
- A. Makarov, L. Grigoriev and T. Mitrova, World Energy Markets Evolution and its Consequences For Russia, Russian Federation, Moscow, 2015 Search PubMed.
- L. Ossowska, D. Janiszewska, N. Bartkowiak-Bakun and G. Kwiatkowsk, Eur. Res. Stud., 2020, 3, 185–198 CrossRef.
- K. Karpov, Stud. Russ. Econ. Dev., 2019, 30, 38–43 CrossRef.
- G. Sun, C. Yuan and M. Hafeez, Environ. Sci. Pollut. Res., 2020, 27, 7105–7118 CrossRef CAS.
- M. I. Hoffert, K. Caldeira, G. Benford, R. David, D. R. Criswell, C. Christopher Green, H. Herzog, A. K. Jain, H. S. Kheshgi, K. S. Lackner, J. S. Lewis, H. D. Lightfoot, W. Manheimer, J. C. Mankins, M. E. Mauel, L. J. Perkins, M. E. Schlesinger, T. Volk and T. Wigley, Science, 2002, 5595, 981–987 CrossRef.
- A. Ivankovic, A. Dronjic, A. Martinovic Bevanda and S. Talic, Int. J. Sustainable Green Energy, 2017, 3, 39–48 CrossRef.
- S. D. Dhage and K. K. Khisodiya, Int. Res. J. Pharm., 2013, 4, 1–4 CrossRef.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- P. T. Anastas and T. C. Williamson, Green Chemistry: Frontiers in Chemical Synthesis and Processes, Oxford University Press, Oxford, 1998 Search PubMed.
- I. Beletskaya and L. Kustov, Russ. Chem. Rev., 2010, 79, 441–461 CrossRef CAS.
- M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807–810 CrossRef CAS.
- M. Eissen, J. O. Metzger, E. Schmidt and U. Schneidewind, Angew. Chem., Int. Ed., 2002, 41, 414–436 CrossRef CAS.
- T. L. Chen, H. Kim, S. Y. Pan, P. C. Tseng, Y. P. Lin and P. C. Chiang, Sci. Total Environ., 2020, 716, 136998–137014 CrossRef CAS.
- N. Tarasova, A. Makarova, S. Vavailov, S. Varlamova and M. Schukina, Herald Russ. Acad. Sci., 2013, 12, 1068–1075 Search PubMed.
- R. K. Sindhu, A. Verma, D. Sharma, S. Gupta and S. Arora, Arch. Med. Pharm. Sci. Res., 2017, 1, 39–44 Search PubMed.
- S. Sharma, J. Das, W. M. Braje, A. K. Dash and S. Handa, ChemSusChem, 2020, 13, 2859–2873 CrossRef CAS.
- A. Dogan, C. C. Eylem and N. E. B. Akduman, Microchem. J., 2020, 157, 104895 CrossRef CAS.
- I. I. Moiseev, Russ. Chem. Rev., 2013, 82, 616–623 CrossRef.
- I. I. Moiseev, Kinet. Catal., 2011, 3, 337–347 CrossRef.
- R. Höfer and M. Selig, Polymer Science: A Comprehensive Reference, 2012, vol. 19, pp. 5–14 Search PubMed.
- E. Engel and J. L. Scott, Green Chem., 2020, 22, 3693–3715 RSC.
- H. Pinkowska, Polymers, 2006, 51, 836–842 CAS.
- S. B. Aziz, O. G. Abdullah, A. M. Hussein and H. M. Ahmed, Polymers, 2017, 9, 626–641 CrossRef.
- S. Kobayashi, Struct. Chem., 2017, 28, 461–474 CrossRef CAS.
- Y. Yoshimura, M. Nojiri, M. Arimoto, K. Ishimoto, Y. Aso, H. Ohara and S. Kobayashi, Polymers, 2016, 90, 342–350 CrossRef CAS.
- S. Sen and J. Puskas, Molecules, 2015, 20, 9358–9379 CrossRef CAS.
- S. Iliescu and G. Ilia, Curr. Green Chem., 2014, 1, 40–50 CrossRef CAS.
- V. K. Thakur, M. K. Thakur, P. Raghavan and M. R. Kessler, ACS Sustainable Chem. Eng., 2014, 2, 1072–1092 CrossRef CAS.
- M. A. Dube and S. Salehpour, Macromol. React. Eng., 2014, 8, 7–28 CrossRef CAS.
- E. J. Goethals, J. Macromol. Sci., Polym. Rev., 1968, 2, 73–144 CrossRef CAS.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef.
- Y. Zhang, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2019, 10, 4078–4096 RSC.
- N. P. Tarasova and A. A. Zanin, Pure Appl. Chem., 2019, 91, 671–686 CrossRef CAS.
- S. Liu, G.-J. Deng and H. Huang, Synlett, 2021, 32(02), 142–158 CrossRef CAS.
- T. B. Nguyen, Adv. Synth. Catal., 2020, 362, 3448–3482 CrossRef CAS.
- B. Eckert, R. Okazaki, R. Steudel, N. Takeda, N. Tokitoh and M. W. Wong, in Elemental Sulfur and Sulfur-Rich Compounds II, ed. R. Steudel, Springer-Verlag Berlin Heidelberg, Berlin, 2003, p. 240 Search PubMed.
- C. Nims, B. Cron, M. Wetherington, J. Macalady and J. Cosmidis, Sci. Rep., 2019, 9, 1–12 CrossRef CAS.
- R. Steudel, Top. Curr. Chem., 2003, 230, 1–134 CrossRef CAS.
- M. W. Wong, Top. Curr. Chem., 2003, 231, 1–30 CrossRef CAS.
- R. Steudel and B. Eckert, J. Phys. Chem., 2004, 121, 6573–6574 CrossRef CAS.
- G. O. Sofekun, E. Evoy, K. L. Lesage, N. Chou and R. A. Marriott, J. Rheol., 2018, 62, 469–476 CrossRef CAS.
- C. Meyer and N. Kharasch, in Elemental Sulfur: Chemistry and Physics, ed. C. Meyer, B. Meyer and N. Kharasch, Interscience Publishers, Oakland, 1965, p. 390 Search PubMed.
- B. A. Trofimov, L. M. Sinegovskaya and N. K. Gusarova, J. Sulfur Chem., 2009, 30, 518–554 CrossRef CAS.
- K. S. Andrikopoulos, A. G. Kalampounias, O. Falagara and S. N. Yannopoulos, J. Chem. Phys., 2013, 139, 124501 CrossRef CAS.
- T. Rauchfuss, Nat. Chem., 2011, 3, 648 CrossRef CAS.
- S. Diez, A. Hoefling, P. Theato and W. Pauer, Polymers, 2017, 9, 59–74 CrossRef.
- M. Mikuriya, K. Taniguchi, Y. Koyama, H. Watanabe, D. Yoshioka, R. Mitsuhashi and E. Asato, X-Ray Struct. Anal. Online, 2020, 36, 1–2 CrossRef CAS.
- D. Plasienka, P. Cifra and R. Martonak, J. Chem. Phys., 2015, 142, 154502 CrossRef.
- K. Raghavachari, C. M. Rohlfing and J. S. Binkley, J. Chem. Phys., 1990, 93, 5862–5873 CrossRef CAS.
- D. S. Warren and B. M. Gimarc, J. Phys. Chem., 1993, 97, 4031–4035 CrossRef CAS.
- J. Cioslowski, A. Szarecka and D. Moncrieff, J. Phys. Chem., 2001, 105, 501–505 CrossRef CAS.
- R. Ludwig, J. Behler, B. Klink and F. Weinhold, Angew. Chem., Int. Ed., 2002, 41, 3199–3202 CrossRef CAS.
- R. O. Jones and P. Ballone, J. Chem. Phys., 2003, 118, 9257–9265 CrossRef CAS.
- A. V. Tobolsky and A. Eisenberg, J. Am. Chem. Soc., 1959, 81, 780–782 CrossRef CAS.
- A. Ansarifar, Academic Journal of Polymer science, 2018, 1, 555568 CrossRef.
- J. E. Mark and B. Erman, Science and Technology of Rubber, Academic Press, San Diego, 2005 Search PubMed.
- T. Komatsu, Int. Polym. Sci. Technol., 2009, 36, 49–55 CrossRef.
- E. H. Farmer, J. Soc. Chem. Ind., 1947, 66, 86–92 CrossRef CAS.
- L. Bateman, C. G. Moore and M. Porter, J. Chem. Soc., 1958, 2866–2879 RSC.
- A. Ansarifar, K. Noulta, S. H. Sheikh, G. W. Weaver and K. G. Upul Wijayantha, Tire Technol. Int., 2018, 104–108 Search PubMed.
- P. Ghosh, S. Katare, P. Patkar, J. M. Caruthers, V. Venkatasubramanian and K. A. Walker, Rubber Chem. Technol., 2003, 76, 592–693 CrossRef CAS.
- Y. Wei, L. Xiang, Z. Xu, H. Sun, Y. C. Zheng, L. Peng, Z. Liu, C. Gao and M. Gao, Polym. Chem., 2015, 6, 973–982 RSC.
- J. J. Griebel, N. A. Nguyen, A. V. Astashkin, R. S. Glass, M. E. Mackay, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 1258–1261 CrossRef CAS.
- Y. Zou, X. Su and J. Jiang, J. Am. Chem. Soc., 2013, 135, 18377–18384 CrossRef CAS.
- S. Zhuo, Y. Huang, C. Liu, H. Wang and B. Zhang, Chem. Commun., 2014, 50, 11208–11210 RSC.
- J. J. Griebel, G. Li, R. S. Glass, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 173–177 CrossRef CAS.
- M. K. Salman, B. Karabay, C. Lutfiye and A. Cihaner, J. Appl. Polym. Sci., 2016, 133, 43655–43665 CrossRef.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalke, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS.
- D. J. Parker, S. T. Chong and T. Hassel, RSC Adv., 2018, 8, 27892–27899 RSC.
- S. Oishi, K. Oi, J. Kuwabara, R. Omoba, Y. Aihara, T. Fukuda, T. Toshikazu, J.-C. Choi, M. Watanabe and T. Kanbara, ACS Appl. Polym. Mater., 2019, 1, 1195–1202 CrossRef CAS.
- J. C. Bear, W. J. Peveler, P. D. McNaughter, I. P. Parkin, P. O'Brien and C. W. Dunnill, Chem. Commun., 2015, 51, 10467–10470 RSC.
- E. T. Kim, W. J. Chung, J. Lim, P. Johe, R. S. Glass, J. Pyun and K. Char, Polym. Chem., 2014, 5, 3617–3623 RSC.
- G. I. Sabakhova, R. T. Akhmetova, A. A. Yusupova and L. R. Baraeva, Vestn. Kazan. Tekhnol. Univ., 2013, 16, 48–50 CAS.
- Z. Sun, M. Xiao, S. Wang, D. Han, S. Song, G. Chen and Y. Meng, J. Mater. Chem., 2014, 2, 9280–9286 RSC.
- P. T. Dirlam, A. G. Simmonds, R. C. Shallcross, K. J. Arrington, W. J. Chung, J. J. Griebel and J. Pyun, ACS Macro Lett., 2015, 4, 111–114 CrossRef CAS.
- S. H. Hosseini, M. Malekdar and S. Naghdi, Polym. J., 2010, 42, 640–647 CrossRef CAS.
- H.-K. Lin and Y.-L. Liu, Macromol. Rapid Commun., 2018, 39, 1700832 CrossRef.
- B. K. Bordoloi and E. M. Pearce, J. Polym. Sci., Part A: Polym. Chem., 1978, 16, 3293–3300 CAS.
- A. Bhargav, M. E. Bell, Y. Cui and Y. Z. Fu, ACS Appl. Energy Mater., 2018, 1, 5859–5864 CrossRef.
- J. Mao, Y. Wang, J. Zhu and J. Yu, Appl. Surf. Sci., 2018, 447, 235–243 CrossRef CAS.
- F. Shimojo, K. Hoshino and Y. Zempo, J. Phys.: Condens. Matter, 1998, 10, 177–182 CrossRef.
- S. Munejiri, F. Shimojo and K. Hoshino, Comput. Phys. Commun., 2001, 142, 364–367 CrossRef.
- Y. Sakaguchi and K. Tamura, MRS Proc., 2006, 918, 135–141 CAS.
- H. H. Hodgson and A. G. Dix, J. Chem. Soc., Trans., 1914, 105, 952–956 RSC.
- M. L. Moore and T. B. Johnson, J. Am. Chem. Soc., 1935, 57, 1287–1289 CrossRef CAS.
- W. G. Hodgson, S. A. Buckler and G. Peters, J. Am. Chem. Soc., 1963, 85, 543–546 CrossRef CAS.
- E. K. Badeeva, E. V. Platova and E. S. Batyeva, Russ. J. Gen. Chem., 2014, 84, 1648–1650 CrossRef CAS.
- E. K. Badeeva, D. B. Krivolapov, A. T. Gubaidullin, I. A. Litvinov, E. S. Batyeva and O. G. Sinyashin, Mendeleev Commun., 2005, 15, 22–23 CrossRef.
- T. Kimura, T. Namauo, K. Amano, N. Takahashi, Y. Takaguchi, T. Hoshi and N. Kobayashi, J. Porphyrins Phthalocyanines, 2011, 15, 547–554 CrossRef CAS.
- B. Eftekhari-Sis, S. V. Khajeh and O. Buyukgungor, Synlett, 2013, 24, 977–980 CrossRef CAS.
- X. Li, Q. Pan, R. Hu, X. Wang, Z. Yang and S. Han, Asian J. Org. Chem., 2016, 5, 1353–1358 CrossRef CAS.
- E. Ramli, T. B. Rauchfuss and C. L. Stern, J. Am. Chem. Soc., 1990, 112, 4043–4044 CrossRef CAS.
- S. Dev, E. Ramli, T. B. Rauchfuss and C. L. Stern, J. Am. Chem. Soc., 1990, 112, 6385–6386 CrossRef CAS.
- B. L. Chenard and T. J. Miller, J. Org. Chem., 1984, 49, 1221–1222 CrossRef CAS.
- T. B. Nguyen, M. Q. Tran, L. Ermolenko and A. Al-Mourabit, Org. Lett., 2014, 16, 310–313 CrossRef CAS.
- W. Li, X. Wu, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, Macromolecules, 2015, 48, 7747–7754 CrossRef CAS.
- Y. Zhang, N. G. Pavlopoulos, T. S. Kleine, M. Karayilan, R. S. Glass, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 7–12 CrossRef CAS.
- Y. Zhang, T. Kleine, K. Carothers, D. Phan, R. S. Glass, M. Mackay, K. Char and J. Pyun, Polym. Chem., 2018, 9, 2290–2294 RSC.
- Y. Zhang, J. J. Griebel, P. T. Dirlam, N. A. Nguyen, R. S. Glass, M. E. Mackay, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 107–116 CrossRef CAS.
- M. Karayilan, T. S. Kleine, K. J. Carothers, J. J. Griebel, K. M. Frederick, D. A. Loy, R. S. Glass, M. E. Mackay, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2020, 58, 35–41 CAS.
- N. Hartler, J. Libert and A. Teder, Ind. Eng. Chem. Process Des. Dev., 1967, 6, 398–406 CrossRef CAS.
- W. E. Kleinjan, A. Keizer and A. J. H. Janssen, Colloids Surf., B, 2005, 43, 228–237 CrossRef CAS.
- H. Gan, D. Miao, Q. Pan, R. Hu, X. Li and S. Han, Chem.–Asian J., 2016, 11, 1770–1774 CrossRef CAS.
- K. Wang, M. Groom, R. Sheridan, S. Zhang and E. Block, J. Sulfur Chem., 2012, 34, 55–66 CrossRef.
- S. Penczek, R. Slazak and A. Duda, Nature, 1978, 273, 738–739 CrossRef CAS.
- A. Duda and S. Penczek, Macromolecules, 1982, 15, 36–40 CrossRef CAS.
- J. Wreczycki, D. M. Bielinski, M. Kozanecki, P. Maczugowska and G. Mloston, Materials, 2020, 13, 2597–2616 CrossRef CAS.
- T. Baran, A. Duda and S. Penczek, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 1085–1095 CrossRef CAS.
- S. Penczek and A. Duda, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 59, 47–62 CrossRef.
- J.-M. Catala, J.-M. Pujol and J. Brossas, Macromol. Chem. Phys., 1987, 188, 2517–2522 CrossRef CAS.
- N. P. Tarasova, A. A. Zanin, P. S. Sobolev and E. G. Krivoborodov, Dokl. Chem., 2017, 473, 78–79 CrossRef CAS.
- N. P. Tarasova, Y. O. Mezhuev, A. A. Zanin and E. G. Krivoborodov, Dokl. Chem., 2019, 484, 8–11 CrossRef CAS.
- N. Tarasova, E. Krivoborodov, A. Egorova, A. Z. L. Glukhov, I. Toropygin and Y. Mezhuev, Pure Appl. Chem., 2020, 92, 1297–1304 CrossRef CAS.
- G. I. Sabakhova, R. T. Akhmetova, A. A. Yusupova and L. R. Baraeva, Vestn. Kazan. Tekhnol. Univ., 2013, 16, 48–50 CAS.
- A. A. Yusupova, L. R. Baraeva, R. T. Akhmetova, A. I. Khatsrinov and A. A. Bobryshev, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 570, 012103 CAS.
- A. A. Ysupova, A. G. Shamov, R. T. Ahmetova, V. A. Pervushin and A. I. Khatsrinov, J. Quantum Chem., 2011, 111, 2575–2578 CrossRef CAS.
- A. A. Yusupova, A. I. Khatsrinov and R. T. Akhmetova, Inorg. Mater., 2018, 54, 809–814 CrossRef CAS.
- A. A. Yusupova, A. A. Bobryshev and A. A. Treschev, Solid State Phenom., 2018, 284, 1114–1118 Search PubMed.
- L. R. Baraeva, A. A. Yusupova, R. T. Akhmetova, A. I. Khatsrinov and Z. V. Mezhevich, Russ. J. Phys. Chem. A, 2019, 93, 1106–1110 CrossRef CAS.
- A. A. Yusupova, A. I. Khatsrinov and L. N. Shafigullin, Mater. Today: Proc., 2019, 19, 1932–1936 CAS.
- A. A. Yusupova, A. I. Khatsrinov and L. N. Shafigullin, Solid State Phenom., 2020, 299, 181–187 Search PubMed.
- A. M. Joseph, B. George, K. N. Madhusoosaban and R. Alex, Rubber Sci., 2015, 28, 82–121 Search PubMed.
- S. Mostoni, P. Milana, B. Di Credico, M. D'Arienzo and R. Scotti, Catalysts, 2019, 9, 664–686 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |