
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkyl vs. aryl modifications: a comparative study on modular modifications of triphenylphosphonium mitochondrial vectors†
How Chee
Ong
 a,
João T. S.
Coimbra
b,
Germain
Kwek
a,
Maria J.
Ramos
b,
Bengang
Xing
a,
Pedro A.
Fernandes
*b and
Felipe
García
*a
a,
João T. S.
Coimbra
b,
Germain
Kwek
a,
Maria J.
Ramos
b,
Bengang
Xing
a,
Pedro A.
Fernandes
*b and
Felipe
García
*a
aSchool of Physical and Mathematical Sciences, Division of Chemistry and Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore. E-mail: fgarcia@ntu.edu.sg
bLAQV, REQUIMTE, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre s/n, 4169-007, Portugal. E-mail: pafernan@fc.up.pt
First published on 25th August 2021
Abstract
Triphenylphosphonium (TPP+) moieties are commonly conjugated to drug molecules to confer mitochondrial selectivity due to their positive charge and high lipophilicity. Although optimisation of lipophilicity can be achieved by modifying the length of the alkyl linkers between the TPP+ moiety and the drug molecule, it is not always possible. While methylation of the TPP+ moiety is a viable alternative to increase lipophilicity and mitochondrial accumulation, there are no studies comparing these two separate modular approaches. Thus, we have systematically designed, synthesised and tested a range of TPP+ molecules with varying alkyl chain lengths and degree of aryl methylation to compare the two modular methodologies for modulating lipophilicity. The ability of aryl/alkyl modified TPP+ to deliver cargo to the mitochondria was also evaluated by confocal imaging with a TPP+-conjugated fluorescein-based fluorophore. Furthermore, we have employed molecular dynamics simulations to understand the translocation of these molecules through biological membrane model systems. These results provide further insights into the thermodynamics of this process and the effect of alkyl and aryl modular modifications.
Introduction
The triphenylphosphonium (TPP+) moiety has drawn widespread interest as a molecular vector for selective mitochondrial delivery owing to its high lipophilicity, cationic nature, high stability in physiological conditions and its ease of conjugation to molecular cargo.1 The positive charge on the TPP+ moiety allows for the accumulation within the mitochondrial matrix (100–1000× for a monocation) due to the high mitochondrial membrane potential (Δψ, ca. 180 mV) in accordance with the Nernst equation.2–4 The TPP+ platform has been demonstrated to be highly versatile, and has been employed for the delivery of a wide range of cargo, such as spin-traps, antioxidants, prodrugs, protonophores, fluorophores, photodynamic therapy sensitisers as well as positron emitters into the mitochondria.1,5–14 With mitochondria dysfunction being inextricably linked to a myriad of diseases from neurodegenerative and cardiovascular diseases to cancer,15–19 the ability to deliver compounds to mitochondria is vital for the development of novel therapeutics. Thus, this work focuses on improving the efficacy of the TPP+ vector for enhanced mitochondrial delivery.An effective strategy for enhancing mitochondrial uptake has been focused around tuning the molecule's lipophilicity, which has been demonstrated to be highly relevant for TPP+-conjugated systems.20–24 The typical strategy for enhancing lipophilicity for TPP+-conjugated compounds is by selecting a molecular fragment (module) with a higher lipophilicity as the linker – usually a longer alkyl chain6,23,24 – between the TPP+ moiety and the cargo, which increases the rate of membrane permeation and hence the mitochondrial accumulation.25 However, chain length modulation has its limitations. It may be more challenging to modify linkers with a specific function, such as cleavable linkers used for drug release purposes, where modification may lead to alteration of function.14,26,27
Aryl methylation thus presents itself as an attractive alternative when linker modification is not synthetically viable or when it affects linker functionalities. Furthermore, it enables a modular approach for the synthesis of conjugated cargo with variable lipophilicity by varying the starting phosphine. It has also been demonstrated that aryl methylation of TPP+ can improve mitochondrial uptake and lipophilicity, in a similar manner to chain length modification.21,22,28,29 While judicious alkylation of the phenyl rings in TPP+ moieties has been shown to be an effective method to enhance lipophilicity in both mono-TPP+ and bis-TPP+, no systematic comparison between these two modular approaches has been reported in the literature.21 A comparative study between the two methodologies was thus needed to evaluate the mitochondrial delivery efficacies of the two series of phosphonium salts.
Herein we report the first study comparing the effects of alkyl vs. aryl modification on mitochondrial uptake for triphenylphosphonium moieties (Fig. 1). For this purpose, we synthesised and evaluated nine alkyltriarylphosphonium salts, with permutations of three different aryl groups (phenyl, p-tolyl and 3,5-dimethylphenyl) as well as three alkyl groups of different lengths (ethyl, pentyl and octyl), and assessed the effects of the additional groups on lipophilicity and bioactivity. Conjugation to a fluorescein-based dye followed by confocal microscopy was employed to ascertain the mitochondrial targeting effects of these species to compare the two methodologies. To further understand the thermodynamics of these cations transport across biological membranes, we have also performed umbrella sampling molecular dynamics simulations using a 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) membrane model system.
 | ||
| Fig. 1 (a) T*PP+ salts and (b) T*PP+–fluorescein conjugates studied in this work. | ||
Results and discussion
Synthesis and characterisation of alkyltriarylphosphonium salts
The choice of substituted triarylphosphines was based on our previously reported work. The selected alkylated aryl groups (p-tolyl and 3,5-dimethylphenyl) were chosen as they exhibited the largest increase in mitochondrial uptake among mono- and di-methylated phosphonium species.21,22 The length of alkyl chain was chosen such that any increase in aryl substitution can be matched by an identical increase in alkyl chain length based on its total atom count (i.e., structural isomerism). As such, an increase in aryl substitution can be matched by an equivalent increase in alkyl chain length (i.e.1b/2a, 1c/2b/3a, 2c/3b are isomers of each other). The synthesis and characterisation for compounds 1a–1c was previously reported,22 whereas compounds 2a–3c were obtained in good yields (75–99%) upon reacting triarylphosphines with the respective alkyl bromides in acetonitrile or toluene under reflux (Scheme 1).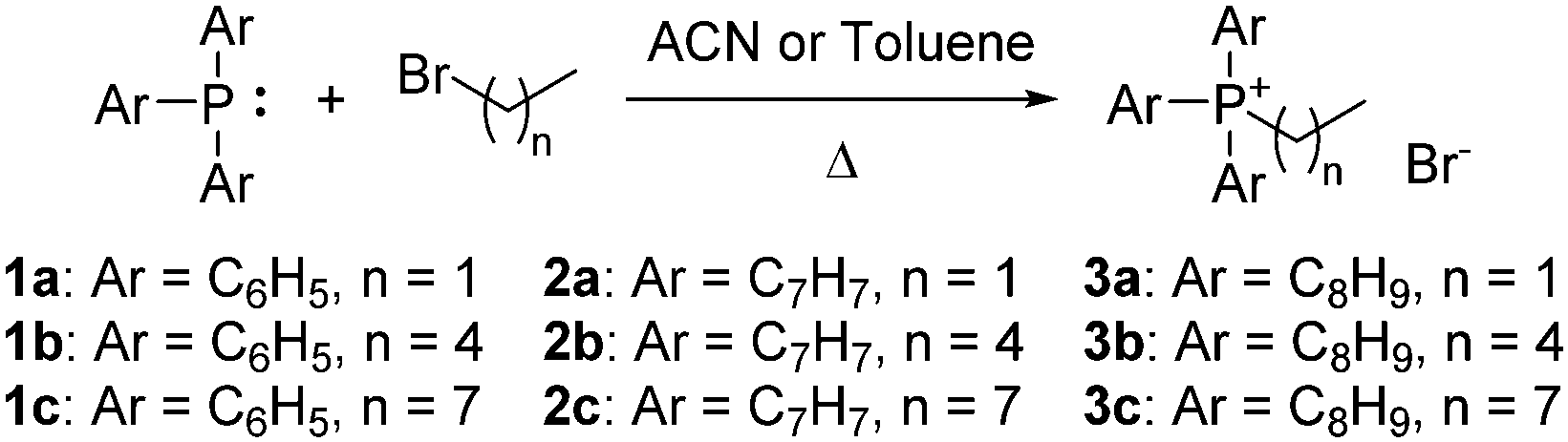 | ||
| Scheme 1 Synthesis of TPP+ cations 1a–3c. | ||
The compounds were characterised by 1H, 31P{1H} and 13C{1H} NMR spectroscopy, high-resolution mass spectrometry (HRMS), and single crystal X-ray structures were obtained for compounds 1b–3b.
The characterisation data was fully consistent with the proposed structures, indicating the successful synthesis of the phosphonium salts.21,22 All single-crystal structures for series a and b were obtained, with two previously unreported structures (see ESI†).22,30 Unfortunately, compounds 1c–3c resisted all attempts at crystallisation, presumably due to the poor packing of the long and flexible alkyl chains, forming waxy solids instead.31
Measurement of lipophilicity
The role of lipophilicity in drugs is well-documented and influences their biological properties greatly, and is frequently quantified by log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, where P is the partition coefficient between two immiscible phases. This is especially relevant for TPP+ compounds, as there are many studies in the literature correlating the accumulation, toxicity, or drug efficacies to lipophilicity.23,24,32,33 Due to the large impact that lipophilicity has on these systems, we quantified the lipophilicity of our compounds in water and 1-octanol and the values are presented in Table 1. The measurements were done using a shake-flask method as described in the literature.34 The logP values obtained correlate well with the increasing atom count, with larger molecules having larger lipophilicity. Steric parameters (molecular volume, and solvent accessible surface area) were found to have a good correlation with lipophilicity (r2 = 0.96), in line with our previous work (see Table 1 and ESI,† Fig. S7).21,22 An increase of the solvent accessible surface area (SASA) of the cations, has been linked with an increased (hydrophobic) surface area that is exposed to the solvents, favouring the partition to the 1-octanol phase.21,22 The molecular volume upon normalisation with respect to charge, has also been described as an important structural parameter for the prediction of mitochondrial accumulation of a series of TPP+ molecules.21 Alchemical free energy calculations focused on the van der Waals (vdW) contribution to the free energy of transfer between water and octanol, also showed a good correlation (r2 = 0.98) with the experimental logP results (see ESI,† Fig. S7). This is also in line with our previous works.
P, where P is the partition coefficient between two immiscible phases. This is especially relevant for TPP+ compounds, as there are many studies in the literature correlating the accumulation, toxicity, or drug efficacies to lipophilicity.23,24,32,33 Due to the large impact that lipophilicity has on these systems, we quantified the lipophilicity of our compounds in water and 1-octanol and the values are presented in Table 1. The measurements were done using a shake-flask method as described in the literature.34 The logP values obtained correlate well with the increasing atom count, with larger molecules having larger lipophilicity. Steric parameters (molecular volume, and solvent accessible surface area) were found to have a good correlation with lipophilicity (r2 = 0.96), in line with our previous work (see Table 1 and ESI,† Fig. S7).21,22 An increase of the solvent accessible surface area (SASA) of the cations, has been linked with an increased (hydrophobic) surface area that is exposed to the solvents, favouring the partition to the 1-octanol phase.21,22 The molecular volume upon normalisation with respect to charge, has also been described as an important structural parameter for the prediction of mitochondrial accumulation of a series of TPP+ molecules.21 Alchemical free energy calculations focused on the van der Waals (vdW) contribution to the free energy of transfer between water and octanol, also showed a good correlation (r2 = 0.98) with the experimental logP results (see ESI,† Fig. S7). This is also in line with our previous works.
| Compound | logP (±SD) |
IC50 (±95% CI)/μM | Volume (±SD)/nm3 | SASA (±SD)/nm2 | ΔGvdWaq→oct/kJ mol−1 | logPmem |
|---|---|---|---|---|---|---|
| 1a | −1.36 ± 0.09 | 16.15 ± 1.29 | 0.8912 ± 0.0264 | 5.381 ± 0.189 | −61.77 ± 0.57 | −0.73 ± 0.01 |
| 1b | −0.84 ± 0.01 | 1.64 ± 0.14 | 1.0481 ± 0.0313 | 6.255 ± 0.229 | −71.87 ± 0.68 | −0.47 ± 0.17 |
| 1c | 0.22 ± 0.02 | 0.34 ± 0.03 | 1.2053 ± 0.0359 | 7.137 ± 0.284 | −82.70 ± 0.71 | 0.45 ± 0.70 |
| 2a | −0.50 ± 0.05 | 3.83 ± 0.50 | 1.0502 ± 0.0323 | 6.296 ± 0.211 | −73.84 ± 0.54 | −0.54 ± 0.31 |
| 2b | 0.26 ± 0.02 | 0.45 ± 0.05 | 1.2087 ± 0.0365 | 7.184 ± 0.244 | −84.58 ± 0.53 | −0.38 ± 0.20 |
| 2c | 1.57 ± 0.02 | 0.30 ± 0.04 | 1.3646 ± 0.0420 | 8.056 ± 0.318 | −97.36 ± 0.83 | 1.69 ± 0.33 |
| 3a | 0.08 ± 0.02 | 0.80 ± 0.06 | 1.2063 ± 0.0350 | 7.130 ± 0.225 | −83.89 ± 0.69 | −0.28 ± 0.26 |
| 3b | 0.90 ± 0.01 | 0.36 ± 0.04 | 1.3633 ± 0.0387 | 7.983 ± 0.269 | −94.93 ± 0.81 | 1.10 ± 0.41 |
| 3c | 2.25 ± 0.05 | 0.32 ± 0.06 | 1.5112 ± 0.0485 | 8.756 ± 0.384 | −107.79 ± 0.87 | 2.30 ± 0.38 |
In vitro biological studies
To evaluate the cytotoxicity of 1a–3c, the 72 hour cell viability assay was conducted using HeLa cells by the resazurin reduction assay. In addition to compounds 1a–3c, methyltriphenylphosphonium (TPMP) iodide was also included as a comparison. The cytotoxic effects of TPP+ has been noted in the literature and has been linked to increased membrane uncoupling.24,35 However, these bioenergetic effects induced by TPP+ moieties are unlikely to be detrimental at relevant dosages.36 Notably, from previously published work, mitochondrial accumulation of similar species has been found to correlate with the IC50 values in TPP+ species.21 The absolute IC50 values (±95% confidence intervals) are presented in Table 1, and the dose–response curves are plotted against the experimental logP in Fig. 2.
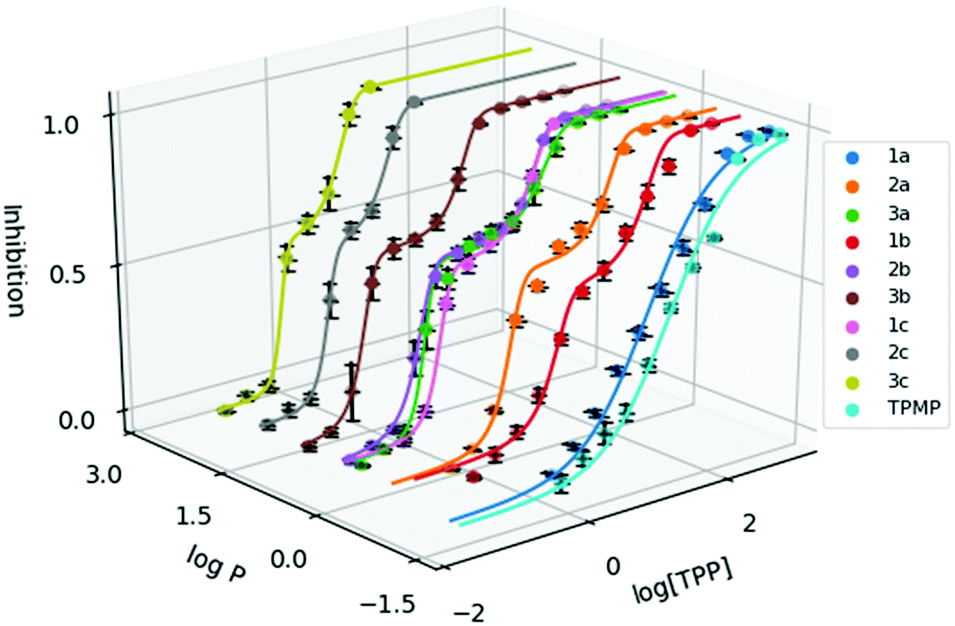 | ||
| Fig. 2 Dose response curve of the methylated TPP+ salts (1a–3c) with TPMP. Error bars for logP are shown for standard deviation and inhibition as standard error. [TPP] is shown in μM. | ||
IC50 was observed to decrease with both increased alkyl chain length, as well as increasing aryl methylation. With an increase of the alkyl chain length from 1a–1c, the IC50 decreased from 16.15 to 1.64 to 0.34 μM. On the other hand, with an increase of the aryl methylation from 1a–3a, the IC50 dropped from 16.15 to 3.83 to 0.80 μM. The two modular approaches appear to be compatible, with compound 2b having a lower IC50 compared to 1b and 2a. However, the effects appear to diminish with the more lipophilic compounds. For example, 3c, with the longest alkyl chain and the most substituted aryl ring, had an IC50 of 0.32 ± 0.06 μM, which was similar to both 2c (0.30 ± 0.04 μM) and 3b (0.32 ± 0.02 μM). This indicates that the scope of the compounds studied approaches the upper limit for the lipophilicity-linked cytotoxicity observed in the literature, with negligible changes in IC50 over an order of magnitude of logP.24
Interestingly, while a sigmoidal model provided a good description for 1a and TPMP, a double-sigmoidal model was a more accurate model for the remaining compounds, which – to the best of our knowledge – has not been previously reported for TPP+ species. While there are multiple explanations in the literature for this phenomenon, ranging from cell population heterogeneity, differing mode of action at different concentrations, to cell-cycle phase specificity, detailed mechanistic studies are necessary to determine the cause of this phenomenon.37,38
The effects of compounds 1a–3c on the mitochondrial membrane potential (MMP) was also monitored. After treatment of HeLa cells with 2.5 μM of each of the nine compounds for 4 hours, the MMP was evaluated using the JC-1 assay (see ESI† for detailed procedures). The experimental results are shown in Fig. 3. The treatment with 1a–3c resulted in significant impact on the MMP with a large decrease in the MMP for the highly lipophilic salts (2c, 3b, 3c), similar to FCCP treatment, despite high cell viability (see ESI†). On the other hand, salts with low lipophilicity (1a, 1b, 2a) showed membrane hyperpolarisation, possibly due to the inhibition of ATPase or specific protein complexes.39,40 The results from the MMP assay correlated well with logP, with an evident sigmoidal relationship between MMP and logP (see ESI†). Interestingly, clustering of 1c, 2b and 3a (isomers) in the plot was observed as well. These observations indicate a possible competition between multiple interactions affecting the MMP (ATPase inhibition, proton leak, etc.), but additional studies will be required to determine the exact mechanism.
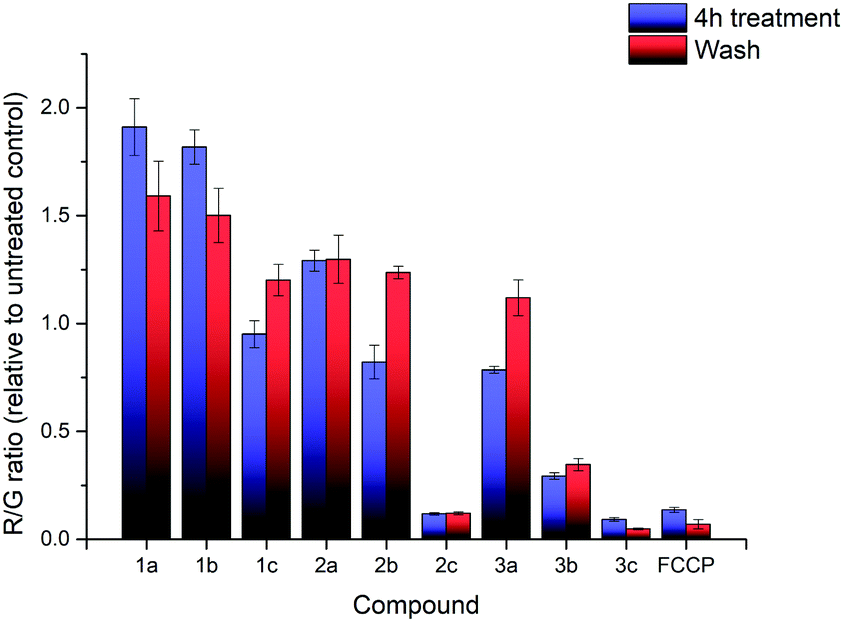 | ||
| Fig. 3 R/G ratio after treatment with compounds 1a–3c and FCCP, before washing (blue) and after washing (red). Error bars refer to the standard deviations. | ||
Additional investigation on the reversibility of this effect was carried out by replacing the medium with fresh DMEM. After an additional 4 hours of incubation, the JC-1 assay revealed that the R/G ratio of most compounds, with the exception of the high lipophilicity derivatives, converged towards 1.2× the untreated control, showing a reduction in the magnitude of both the hyperpolarising and depolarising effects previously observed. This was attributed to a low residual amount of TPP+ still present after washing. Cell viability of all nine salts remained relatively similar compared to the previous data, indicating that further cell death was not significant.
In summary, the MMP was affected from incubation with all TPP+ salts, which were known to induce proton leaks and affect mitochondrial respiration.41,42 The effects appear to be correlated with the lipophilicity of the nine salts, with no significant difference between alkyl chain extension and aryl methylation. Removal of TPP+ from the cell medium results in a slow restoration of the MMP, with no further cell death observed.
Computational studies
To understand the thermodynamics of the cations transport across biological membranes, we have also performed umbrella sampling molecular dynamics simulations. The full protocol details are provided in the ESI† accompanying this paper (see ESI†). We explored the thermodynamics of the cations transport using a hydrated POPC bilayer system as our model membrane. This model is much closer to biological membranes than biphasic 1-octanol:water systems, accounts for ion–membrane specific interactions,43–46 and thus can be used to examine the differences between the cation–membrane interactions as a result of structural changes in the TPP+ moiety (Fig. 4). In our study, the membrane model is divided into four regions based on the different density compositions, namely, bulk water (region IV); most of the charged phosphate and choline density (region III), the region that contains both hydrophobic tails and a portion of the polar headgroup density, ending where the lipid tail density intercepted the choline density (region II); and a central zone that contains only the hydrophobic lipid tails (region I). The translocation free energy profile of all nine triphenylphosphonium cations (1a–3c), as well as detailed parameters describing local free energy maxima (ΔGmax), minima (ΔGmin) and free energy barrier at the centre of the bilayer (ΔGB), are depicted in Fig. 4 and Table S3 (ESI†).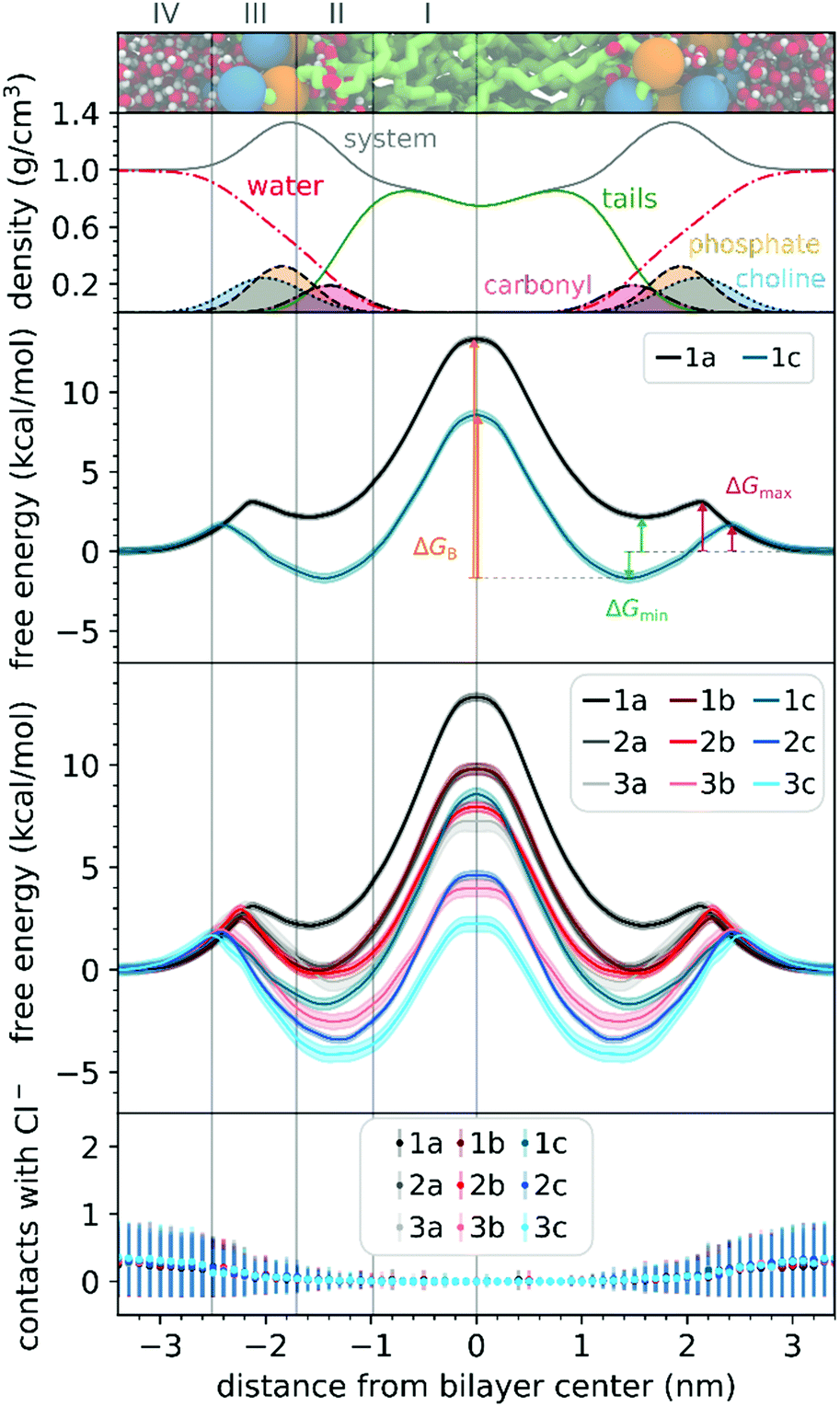 | ||
| Fig. 4 (top 2 panels) Hydrated bilayer model system and the partial density profiles for the different functional groups or molecules in the system. (third panel) Free energy profiles for the translocation of selected cations and illustration of ΔGmin, ΔGmax and ΔGB. (fourth panel) Free energy profiles for the translocation of all the studied cations. (bottom panel) Average number of contacts of any of the studied cations with Cl− counter-ions in the simulation cell (considering a distance threshold of 0.6 nm). Membrane regions (from left to right): region IV (bulk water); region III (charged phosphate and choline groups); region II (hydrophobic tails and a portion of the polar headgroup); and region I (hydrophobic lipid tails). | ||
The free energy profile can be qualitatively understood in 3 phases during translocation. As the TPP+ ion approaches the surface of the membrane, an increase in free energy (ΔGmax, region III) is observed. There is a subsequent decrease in free energy (ΔGmin, region II), and finally, we observe a free energy maximum in region I (ΔGB). The potential energy profiles of TPP+ ions with lipid bilayers have been previously written down as the sum of four terms, including Born, image and dipole energy contributions, and a neutral energy term.45,46 The free energy profile diagrams obtained are highly consistent with models previously reported in the literature.47,48
The free energy profile diagrams obtained further emphasise the importance of lipophilicity in TPP+ systems; since an increase in lipophilicity can be correlated with a respective decrease in free energy at all three critical points, ΔGmax, ΔGmin and ΔGB. Notably, the maximum values of the free energy profiles relative to water (defined at 0.0 kcal mol−1) of isomeric compounds are comparable, with 1b/2a at 9.80–9.85 kcal mol−1, 1c/2b/3a at 7.28–8.61 kcal mol−1, and 2c/3b at 3.99–4.63 kcal mol−1 (see ESI,† Table S3). We also observed that among the four compounds with the lowest IC50 (between 0.30 and 0.36 μM), 1c, 2c, 3c and 3b, showed the smallest free energy barrier for penetrating the high-density region of the bilayer (ΔGmax), and except for 1c, these compounds had low ΔGB values (see Fig. 4 and ESI,† Table S3).
To allow the full translocation free energy profile to be considered, the membrane partition coefficient (logPmem) was computed from the free energy profile diagrams by calculating the standard binding free energy of the cations to the membrane (Table 1, see ESI† for more details). We observed that the four compounds with the lowest IC50 (between 0.30 and 0.36 μM) – 2c, 3c, 1c and 3b, were the only ones to show a positive logPmem (between 0.45 and 2.30). The logPmem obtained were also found to be well-correlated with experimental logP, and consistent with previously reported translocation rate constant trends in black lipid membranes,49 (r2 = 0.89, Fig. S8, ESI†), with a mean unsigned difference of 0.29 ± 0.23. However, these differences were to be expected as the experimental results were obtained in water and 1-octanol.
The logPmem for the different isomers were then compared in terms of aryl substitution vs. alkyl chain length modules. For the 1b/2a isomers, very similar membrane partition (−0.47 ± 0.17 and −0.54 ± 0.31, respectively) were obtained. As for the other isomers, i.e., 1c/2b/3a and 2c/3b, the simulations showed that 1c and 2c had a higher membrane partition when compared with their respective isomers (ranging from 0.6 to 0.8). This suggests that a longer alkyl chain length module could have an increased tendency to associate with lipid membranes despite having similar lipophilicities.
To examine the possible different tendencies to form ion-pairs for 1a–3c, which may affect translocation profiles, an ionic concentration of ca. 0.15 M of NaCl was used throughout our studies. By following the number of contacts between the cations and the Cl− ions present in the system, we did not detect the formation of ion pairs as the cations translocated the hydrated bilayer system (see Fig. 4). Still, in a separate simulation, we have constrained the interatomic distance between one Cl− ion and the 1a cation, to investigate the effect of the formation of an ion pair in the translocation free energy profile of 1a (see ESI,† Fig. S9). We observed that ion pairing should not be favourable for the translocation of this cation, which is in agreement with existing literature for TPMP using DFT-based continuum model calculations by Hartley, Murphy et al., which have shown that the energetic penalty for TPMP to be transferred from water to the non-aqueous hexane phase as an ion-pair was larger than for the TPMP cation alone.50 Although the same partition was obtained with or without constraining interatomic distances, the free energy barrier of 1a without constraints were lower by ca. 2 kcal mol−1.
Mitochondrial uptake and localisation of TPP+-conjugated fluorescein
To quantify the difference in mitochondrial delivery between the various TPP+ vectors, a fluorescein-based dye was conjugated to the triarylphosphonium salts modules with differing chain length units. The compounds synthesised are shown in Fig. 1, with modifications from previously reported molecule (MitoFluo) by Antonenko et al.51,52 To control regioselectivity, as well as to restrict the protonophoric activity observed in mitoFluo, the methyl ester of fluorescein was used. The compounds 4a/4b/4c consist of a triphenylphosphonium linked to the fluorescent moiety with an increasing chain length, while compounds 4a/4d/4e consist of the fluorescent dye linked with a 5-carbon chain to different alkylated triarylphosphonium groups.The synthesis of TPP+-conjugated fluorescein was achieved in three steps: esterification of fluorescein, synthesis of ω-bromoalkyltriarylphosphonium bromide and Williamson ether synthesis for the conjugation of TPP+ to the dye (Scheme 2). 4a–4e were characterised by 1H, 13C{1H} and 31P{1H} NMR and HRMS. The experimental excitation/emission fluorescence spectra obtained for the five conjugated dyes were virtually identical in both intensities and wavelengths (see ESI,† Fig. S6). logP values for the TPP+–fluorescein conjugates were also quantified experimentally and presented in Table 2. The relative increase in logP values were highly consistent with the differences in the unconjugated TPP+ salts.
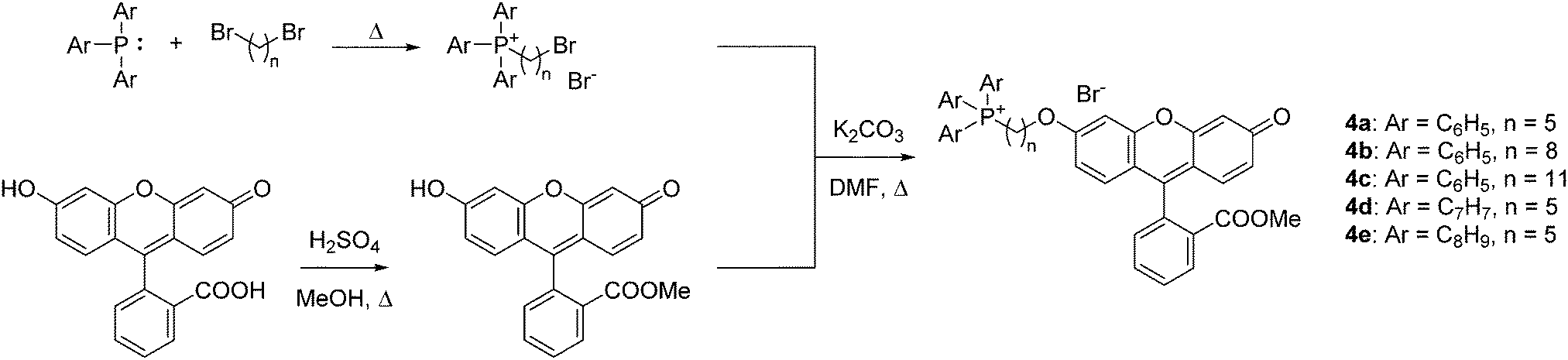 | ||
| Scheme 2 Synthesis of TPP+–fluorescein conjugates 4a–4e. | ||
| Compound | logP (±SD) |
Fluo/MT (±SD) | Pearson's coefficient | M1 | M2 |
|---|---|---|---|---|---|
| 4a | 0.42 ± 0.04 | 0.065 ± 0.053 | 0.568 ± 0.108 | 0.954 ± 0.023 | 0.874 ± 0.013 |
| 4b | 1.25 ± 0.05 | 0.521 ± 0.042 | 0.884 ± 0.006 | 0.956 ± 0.013 | 0.967 ± 0.007 |
| 4c | 2.72 ± 0.07 | 1.009 ± 0.091 | 0.896 ± 0.006 | 0.973 ± 0.004 | 0.974 ± 0.003 |
| 4d | 1.43 ± 0.01 | 0.361 ± 0.058 | 0.856 ± 0.021 | 0.956 ± 0.012 | 0.965 ± 0.004 |
| 4e | 2.37 ± 0.01 | 0.757 ± 0.058 | 0.877 ± 0.018 | 0.954 ± 0.007 | 0.967 ± 0.006 |
To measure the mitochondrial uptake and subcellular localisation of these conjugated dyes, HeLa cells were treated with compounds 4a–4e (100 nM) together with MitoTracker DeepRed FM (25 nM) in DMEM for 30 minutes, washed with 3 × 200 μL of PBS, and the medium was replaced with DMEM. The samples were then examined under a confocal microscope. The relative mitochondrial uptake among the cells was quantified using the ratio of fluorescence intensities between the 4a–4e and the MitoTracker (Fluo/Mt). Additional image analyses (n = 5) were performed to show colocalisation with MitoTracker Deep Red, and the data is presented in Fig. 5 and Table 2.
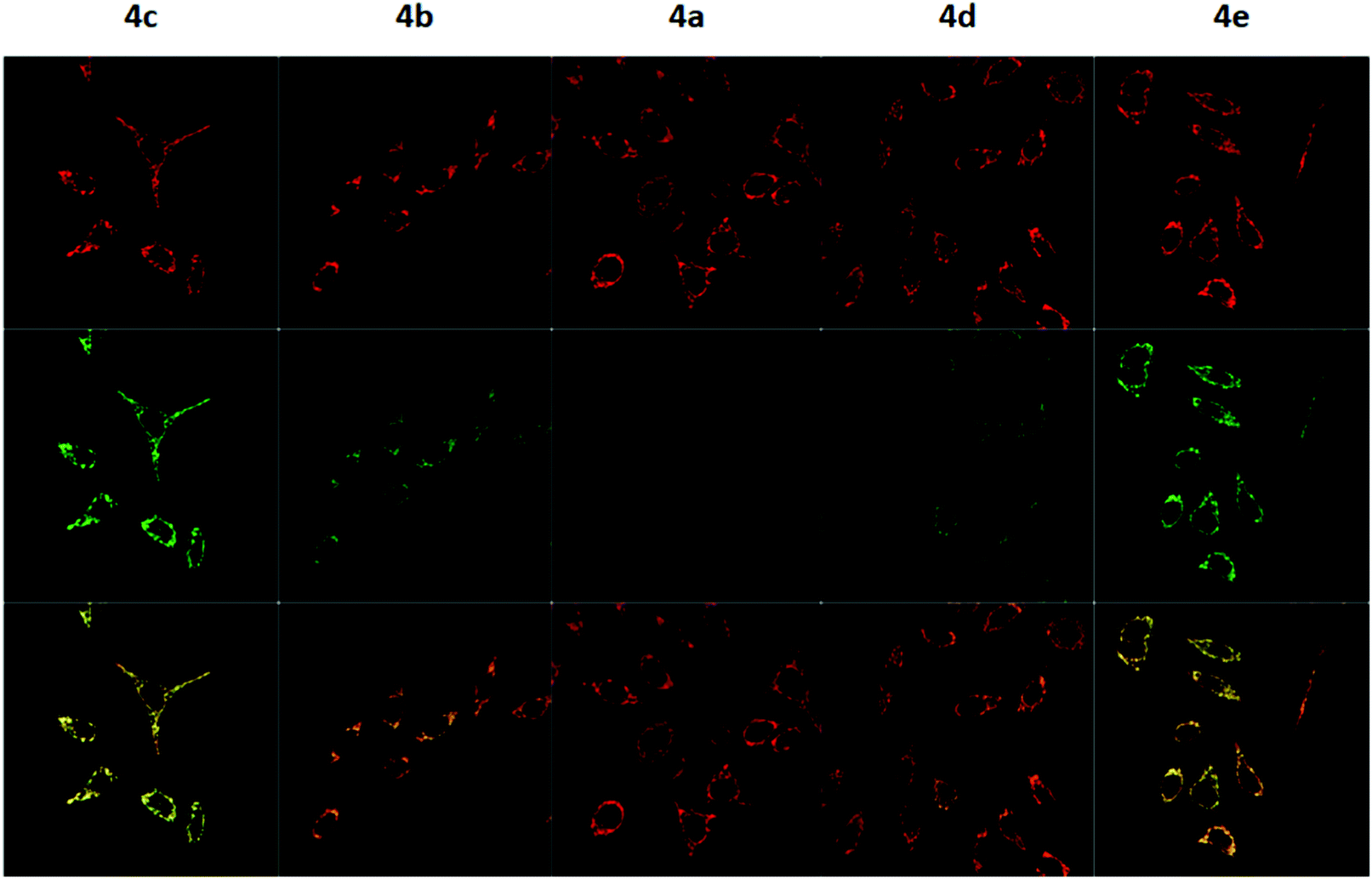 | ||
| Fig. 5 Confocal fluorescence microscopy images of HeLa cells treated with 4a–4e upon excitation with 488 nm and 644 nm lasers. The first row shows the fluorescence signal from MitoTracker Deep Red, the second row shows the fluorescence signal from 4a–4e, and the third row shows the overlay of the two channels. | ||
The images obtained revealed a colocalisation of the TPP+ conjugated dyes and MitoTracker Deep Red, indicating a high selectivity of compounds 4a–4e for the mitochondria. The Pearson's correlation coefficient for compounds 4b–4e were very high, ranging from 0.856 to 0.896. Although compound 4a has a relatively low r-value of 0.568, the Van Steensel's curve supports that like the other four dyes, 4a localises in the mitochondria, with the maximum in the cross-correlation function at dx = 0 for all compounds (see ESI†). The high Mander's coefficient, M1 and M2, which ranges from 0.954 to 0.974 for compounds 4b–4e indicates the bulk of the fluorescence signal overlaps between the MitoTracker and fluorescein channels, further supporting a high degree of mitochondrial selectivity.
Evidently, the fluorescent intensity from the fluorescein dyes were correlated with lipophilicity. As the chain length was extended from 5 to 11 carbons in compounds 4a/4b/4c, the Fluo/MT ratio increased 16 times from 0.065 to 1.009. Similarly, an increase in aryl groups methylation results in an increase in mitochondrial accumulation as the Fluo/MT ratio for compounds 4a/4d/4e increased 12 times from 0.065 to 0.757. Both methods were found to increase mitochondrial uptake drastically, indicating the viability of both methodologies for mitochondrial delivery, consistent with other work in the literature. Fluo/Mt was found to correlate well with logP values of the TPP+–fluorescein conjugate (r2 = 0.94). The slightly increased performance of 4c as compared to 4e was hence attributed to the higher lipophilicity (2.72 ± 0.07 for 4c and 2.37 ± 0.01 for 4e).
Conclusions
In conclusion, we have reported the first evaluation of aryl methylation as a complementary modular strategy to the traditional methods of chain length extension for enhancing mitochondrial targeting abilities. Both strategies result in lowered membrane permeation energy and greater mitochondrial accumulation with isomeric compounds displaying analogous translocation profiles, cytotoxicity, and mitochondrial accumulation. Most notably, both approaches are fully compatible, and can be simultaneously applied for the greatest increase in mitochondrial targeting. This work demonstrates that aryl methylation is both comparable and complementary with traditional methods.Our work underscores the modular nature and complementary nature of both approaches. We hope our work will catalyse the synthesis of a wide range of fine-tuned mitochondrial delivery vectors based on a broader range of phosphonium salts and other non-conventional delivery vectors.
Author contributions
H. C. O. and G. K. designed and carried out the experiments. J. T. S. C., M. J. R., P. A. F. designed and carried out the theoretical studies. X. B. G. provided the necessary instruments and equipment for biological studies. F. G., M. J. R., and P. A. F. obtained the necessary funding, F. G. and P. A. F. provided supervision for the project. All authors contributed to the writing and revising the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. G. would like to thank A*STAR AME IRG (A1783c0003 and A2083c0050), NTU for a start-up grant (M4080552), MOE Tier 1 grants (RG 11/15 and RG 113/16) and H. S. S. for financial support. H. C. O. would like to thank NTU for NPGS scholarship J. T. S. C., M. J. R. and P. A. F. acknowledge financial support from the Associate Laboratory for Green Chemistry Unit – LAQV, which is financed by national funds through FCT/MCTES – Portuguese Foundation for Science and Technology within the scope of the project UIDB/50006/2020. J. T. S. C. has received financial support from the scientific employment stimulus – individual call of 2018 (CEECIND/01374/2018).Notes and references
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- M. P. Murphy, Biochim. Biophys. Acta, 2008, 1777, 1028–1031 CrossRef CAS PubMed.
- M. P. Murphy and R. A. Smith, Annu. Rev. Pharmacol. Toxicol., 2007, 47, 629–656 CrossRef CAS PubMed.
- L. B. Chen, Annu. Rev. Cell Biol., 1988, 4, 155–181 CrossRef CAS PubMed.
- G. A. Korshunova, A. V. Shishkina and M. V. Skulachev, Biochemistry, 2017, 82, 760–777 CAS.
- F. Sodano, B. Rolando, F. Spyrakis, M. Failla, L. Lazzarato, E. Gazzano, C. Riganti, R. Fruttero, A. Gasco and S. Sortino, ChemMedChem, 2018, 13, 1238–1245 CrossRef CAS PubMed.
- A. Logan, V. R. Pell, K. J. Shaffer, C. Evans, N. J. Stanley, E. L. Robb, T. A. Prime, E. T. Chouchani, H. M. Cocheme, I. M. Fearnley, S. Vidoni, A. M. James, C. M. Porteous, L. Partridge, T. Krieg, R. A. Smith and M. P. Murphy, Cell Metab., 2016, 23, 379–385 CrossRef CAS PubMed.
- Y. Zheng, X. Ji, B. Yu, K. Ji, D. Gallo, E. Csizmadia, M. Zhu, M. R. Choudhury, L. K. C. De La Cruz, V. Chittavong, Z. Pan, Z. Yuan, L. E. Otterbein and B. Wang, Nat. Chem., 2018, 10, 787–794 CrossRef CAS PubMed.
- G. F. Kelso, C. M. Porteous, C. V. Coulter, G. Hughes, W. K. Porteous, E. C. Ledgerwood, R. A. Smith and M. P. Murphy, J. Biol. Chem., 2001, 276, 4588–4596 CrossRef CAS PubMed.
- H. S. Jung, J. H. Lee, K. Kim, S. Koo, P. Verwilst, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2017, 139, 9972–9978 CrossRef CAS PubMed.
- C. J. Zhang, Q. Hu, G. Feng, R. Zhang, Y. Yuan, X. Lu and B. Liu, Chem. Sci., 2015, 6, 4580–4586 RSC.
- S. E. Brown, M. F. Ross, A. Sanjuan-Pla, A. R. Manas, R. A. Smith and M. P. Murphy, Free Radical Biol. Med., 2007, 42, 1766–1780 CrossRef CAS PubMed.
- M. P. Murphy, K. S. Echtay, F. H. Blaikie, J. Asin-Cayuela, H. M. Cocheme, K. Green, J. A. Buckingham, E. R. Taylor, F. Hurrell, G. Hughes, S. Miwa, C. E. Cooper, D. A. Svistunenko, R. A. Smith and M. D. Brand, J. Biol. Chem., 2003, 278, 48534–48545 CrossRef CAS PubMed.
- S. Chalmers, S. T. Caldwell, C. Quin, T. A. Prime, A. M. James, A. G. Cairns, M. P. Murphy, J. G. McCarron and R. C. Hartley, J. Am. Chem. Soc., 2012, 134, 758–761 CrossRef CAS PubMed.
- V. Gogvadze, S. Orrenius and B. Zhivotovsky, Trends Cell Biol., 2008, 18, 165–173 CrossRef CAS PubMed.
- F. Di Lisa, N. Kaludercic, A. Carpi, R. Menabo and M. Giorgio, Pharmacol. Rep., 2009, 61, 123–130 CrossRef CAS PubMed.
- N. A. Haelterman, W. H. Yoon, H. Sandoval, M. Jaiswal, J. M. Shulman and H. J. Bellen, Annu. Rev. Neurosci., 2014, 37, 137–159 CrossRef CAS PubMed.
- F. Celsi, P. Pizzo, M. Brini, S. Leo, C. Fotino, P. Pinton and R. Rizzuto, Biochim. Biophys. Acta, 2009, 1787, 335–344 CrossRef CAS PubMed.
- P. H. Reddy, Neuromol. Med., 2008, 10, 291–315 CrossRef CAS PubMed.
- G. Park and F. P. Gabbai, Angew. Chem., Int. Ed., 2020, 59, 5298–5302 CrossRef CAS PubMed.
- H. C. Ong, Z. Hu, J. T. S. Coimbra, M. J. Ramos, O. L. Kon, B. Xing, E. K. L. Yeow, P. A. Fernandes and F. Garcia, Inorg. Chem., 2019, 58, 8293–8299 CrossRef CAS PubMed.
- Z. Hu, Y. Sim, O. L. Kon, W. H. Ng, A. J. Ribeiro, M. J. Ramos, P. A. Fernandes, R. Ganguly, B. Xing, F. Garcia and E. K. Yeow, Bioconjugate Chem., 2017, 28, 590–599 CrossRef CAS PubMed.
- J. Asin-Cayuela, A. R. Manas, A. M. James, R. A. Smith and M. P. Murphy, FEBS Lett., 2004, 571, 9–16 CrossRef CAS PubMed.
- L. S. Khailova, P. A. Nazarov, N. V. Sumbatyan, G. A. Korshunova, T. I. Rokitskaya, V. I. Dedukhova, Y. N. Antonenko and V. P. Skulachev, Biochemistry, 2015, 80, 1589–1597 CAS.
- M. F. Ross, T. A. Prime, I. Abakumova, A. M. James, C. M. Porteous, R. A. Smith and M. P. Murphy, Biochem. J., 2008, 411, 633–645 CrossRef CAS PubMed.
- J. Lu, F. Jiang, A. Lu and G. Zhang, Int. J. Mol. Sci., 2016, 17, 561 CrossRef PubMed.
- J. Ripcke, K. Zarse, M. Ristow and M. Birringer, ChemBioChem, 2009, 10, 1689–1696 CrossRef CAS PubMed.
- A. Haslop, L. Wells, A. Gee, C. Plisson and N. Long, Mol. Pharmaceutics, 2014, 11, 3818–3822 CrossRef CAS PubMed.
- A. J. Smith, B. E. Osborne, G. P. Keeling, P. J. Blower, R. Southworth and N. J. Long, Dalton Trans., 2020, 49, 1097–1106 RSC.
- E. W. Czerwinski, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o2393–o2394 CrossRef CAS.
- G. A. Gainanova, G. I. Vagapova, V. V. Syakaev, A. R. Ibragimova, F. G. Valeeva, E. V. Tudriy, I. V. Galkina, O. N. Kataeva, L. Y. Zakharova, S. K. Latypov and A. I. Konovalov, J. Colloid Interface Sci., 2012, 367, 327–336 CrossRef CAS PubMed.
- N. S. Chandel, BMC Biol., 2014, 12, 34 CrossRef PubMed.
- M. F. Ross, T. Da Ros, F. H. Blaikie, T. A. Prime, C. M. Porteous, I. I. Severina, V. P. Skulachev, H. G. Kjaergaard, R. A. Smith and M. P. Murphy, Biochem. J., 2006, 400, 199–208 CrossRef CAS PubMed.
- A. Andres, M. Roses, C. Rafols, E. Bosch, S. Espinosa, V. Segarra and J. M. Huerta, Eur. J. Pharm. Sci., 2015, 76, 181–191 CrossRef CAS PubMed.
- J. Trnka, M. Elkalaf and M. Andel, PLoS One, 2015, 10, e0121837 CrossRef PubMed.
- C. Reily, T. Mitchell, B. K. Chacko, G. Benavides, M. P. Murphy and V. Darley-Usmar, Redox Biol., 2013, 1, 86–93 CrossRef CAS PubMed.
- L. M. Levasseur, H. K. Slocum, Y. M. Rustum and W. R. Greco, Cancer Res., 1998, 58, 5749–5761 CAS.
- S. N. Gardner, Cancer Res., 2000, 60, 1417–1425 CAS.
- M. Forkink, G. R. Manjeri, D. C. Liemburg-Apers, E. Nibbeling, M. Blanchard, A. Wojtala, J. A. M. Smeitink, M. R. Wieckowski, P. H. G. M. Willems and W. J. H. Koopman, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1247–1256 CrossRef CAS PubMed.
- D. G. Nicholls and S. L. Budd, Physiol. Rev., 2000, 80, 315–360 CrossRef CAS PubMed.
- C. Reily, T. Mitchell, B. K. Chacko, G. A. Benavides, M. P. Murphy and V. M. Darley-Usmar, Redox Biol., 2013, 1, 86–93 CrossRef CAS PubMed.
- C. A. Kulkarni, B. D. Fink, B. E. Gibbs, P. R. Chheda, M. Wu, W. I. Sivitz and R. J. Kerns, J. Med. Chem., 2021, 64, 662–676 CrossRef CAS PubMed.
- B. H. Honig, W. L. Hubbell and R. F. Flewelling, Annu. Rev. Biophys. Bioeng., 1986, 15, 163–193 CrossRef CAS PubMed.
- R. F. Flewelling and W. L. Hubbell, Biophys. J., 1986, 49, 531–540 CrossRef CAS PubMed.
- R. F. Flewelling and W. L. Hubbell, Biophys. J., 1986, 49, 541–552 CrossRef CAS PubMed.
- B. Silver, The physical chemistry of membranes: an introduction to the structure and dynamics of biological membranes, Springer Science & Business Media, 2012 Search PubMed.
- A. Ono, S. Miyauchi, M. Demura, T. Asakura and N. Kamo, Biochemistry, 1994, 33, 4312–4318 CrossRef CAS PubMed.
- B. Ketterer, B. Neumcke and P. Lauger, J. Membr. Biol., 1971, 5, 225–245 CrossRef CAS PubMed.
- T. I. Rokitskaya, V. B. Luzhkov, G. A. Korshunova, V. N. Tashlitsky and Y. N. Antonenko, Phys. Chem. Chem. Phys., 2019, 21, 23355–23363 RSC.
- E. L. Robb, J. M. Gawel, D. Aksentijevic, H. M. Cocheme, T. S. Stewart, M. M. Shchepinova, H. Qiang, T. A. Prime, T. P. Bright, A. M. James, M. J. Shattock, H. M. Senn, R. C. Hartley and M. P. Murphy, Free Radical Biol. Med., 2015, 89, 883–894 CrossRef CAS PubMed.
- S. S. Denisov, E. A. Kotova, E. Y. Plotnikov, A. A. Tikhonov, D. B. Zorov, G. A. Korshunova and Y. N. Antonenko, Chem. Commun., 2014, 50, 15366–15369 RSC.
- Y. N. Antonenko, S. S. Denisov, D. N. Silachev, L. S. Khailova, S. S. Jankauskas, T. I. Rokitskaya, T. I. Danilina, E. A. Kotova, G. A. Korshunova, E. Y. Plotnikov and D. B. Zorov, Biochim. Biophys. Acta, 2016, 1860, 2463–2473 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2078443 and 2078444. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cb00099c |
| This journal is © The Royal Society of Chemistry 2021 |