
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine-mediated formal [3 + 2] annulation for synthesis of furocoumarin from oxime esters†
Quyen T. Pham a,
Phong Q. Le*b,
Ha V. Danga,
Hiep Q. Hac,
Huong T. D. Nguyend,
Thanh Truong*c and
Tri Minh Le*a
a,
Phong Q. Le*b,
Ha V. Danga,
Hiep Q. Hac,
Huong T. D. Nguyend,
Thanh Truong*c and
Tri Minh Le*a
aSchool of Medicine, VNU-HCM, Linh Trung Ward, Thu Duc District, Ho Chi Minh City, Vietnam. E-mail: leminhtri@ump.edu.vn
bSchool of Biotechnology, International University, VNU-HCM, Quarter 6, Linh Trung Ward, Thu Duc District, Ho Chi Minh City, Vietnam. E-mail: lqphong@hcmiu.edu.vn
cDepartment of Chemical Engineering, HCMC University of Technology, VNU-HCM, 268 Ly Thuong Kiet, District 10, Ho Chi Minh City, Vietnam. E-mail: tvthanh@hcmut.edu.vn
dDepartment of Chemistry, HCMC University of Science, VNU-HCM, 227 Nguyen Van Cu Street, District 5, Ho Chi Minh City, Vietnam
First published on 16th December 2020
Abstract
A novel synthesis of furocoumarins was developed by a reaction between oxime esters and 4-hydroxycoumarins. The reaction was proposed to undergo radical mechanism mediated by iodine, a cheap and common laboratory reagent. Mechanistic studies showed the key for the successful transformation was the presence of α-iodoimine intermediate which facilitated the ring-closing step. The developed conditions produced good functional group tolerance with a wide range of high-profile furocoumarin product. The potential for this strategy to be applied in other syntheses of heterocyclic compounds is highly achievable.
1. Introduction
Heterocyclic compounds play crucial roles in the pharmaceutical industry because of their presence in substantial amount of drugs.1 In addition to offering protein binding functional groups, heterocycles also affect favorably drugs' solubility and their pharmacokinetic properties. Among the important heterocyclic compounds, furocoumarins have been commonly targeted due to their remarkable potential biological activities (Fig. 1).2,3 They have been demonstrating anti-bacteria, anti-viruses (including HIV), anti-cancer, and inhibitory hyperplasia activities.4 Furthermore, furocoumarins also showed potential applications in photosensitive medications, pesticides and molecular biology.5 In this report, we described the first metal-free formal [3 + 2] annulation between two versatile synthetic building blocks, oxime esters and 4-hydroxylcoumarine, to synthesize these important heterocyclic compounds.Approaches for furocoumarin synthesis varied but extensively were integrated with 4-hydroxycoumarin starting materials. These 4-hydroxycoumarin substrates are known as adaptable synthons in the synthesis of countless heterocycles,6 such as pyrazolinones,7 1,2-benzisoxazones,8 quinoline.9 For the generation of furocoumarin, 4-hydroxycoumarin would frequently be under the catalysis of metal salts, for instance, Ce(IV),10 Ru(II),11 Cu(II),12 multicopper oxidase13 or it could be in a multi-components reaction, mostly with benzaldehyde and isocyanide.14 The disadvantages of these methods are the employment of toxic metals and increasing complexity of the reaction that would be using multiple chemicals in a single procedure. Therefore, a simpler, non-metal catalyzed protocol to generate these valuable furocoumarins is much needed. With that standpoint, we found out that oxime esters are well-suitable reagents in developing a new synthetic methodology which is metal-free, rapid and from universal starting materials.
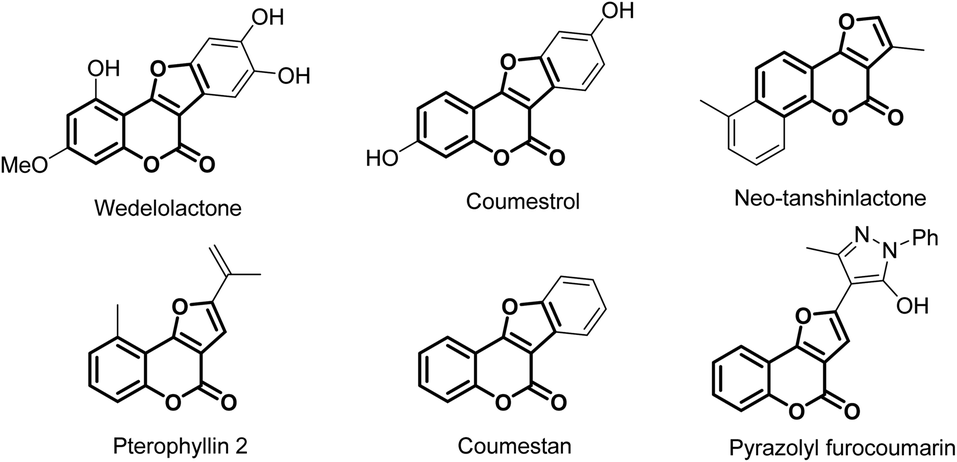 | ||
| Fig. 1 Selected examples of furocoumarin derivatives. | ||
The oxime ester compounds are widely employed in organic synthesis because they are readily accessable from oximes and acid chlorides or anhydrides.15 The N–O σ bond of oxime esters with an average energy of ∼57 kcal mol−1 causes this bond quite unstable. And because of the susceptible cleavage of the N–O bond by active metals, the oxime esters are frequently utilized in transition metal catalyzed transformation.16 However, there are still elegant studies which can convert these hydroxy amine derivatives into higher value products in a transition metal-free fashion. For example, in 2016, Huang and coworkers reported the metal-free synthesis of polysubstituted pyridines from ketoximes and acroleins (Scheme 1a).17 In this dedicated research, iodine and trimethylamine were used to facilitate the [3 + 3] reaction to form pyridine products with high chemoselectivity and wide tolerance of functional groups. The reaction was suggested to undergo a radical pathway initiated by single electron transfer from iodine to oxime substrates. Huang later found out that NH4I when incorporated with either Et3N or Na2S2O4 could also be used to generate imine radical intermediates in the reactions with 1,3-diketone and provided diversified polysubstituted pyridines (Scheme 1a and b).18,19 Besides, Gao developed I2-catalyzed condensation of ketoxime acetates with Et3N20a or aldehydes20b as the C1 source through formal [3 + 2 + 1] annulation to give structurally symmetrical pyridines or 2,4-diarylpyridines (Scheme 1c). In another research based on the transamidation of isatins, Gao and coworkers20c otherwise have demonstrated an efficient quinoline-4-carboxamide formation using the same substrate ketoxime acetates (Scheme 1d). Herein we envisioned that the oxime esters will practically achieve the formation of other heterocycles when cooperated with different organic moieties and with specifically 4-hydroxycoumarin to potentially form furocoumarin products.
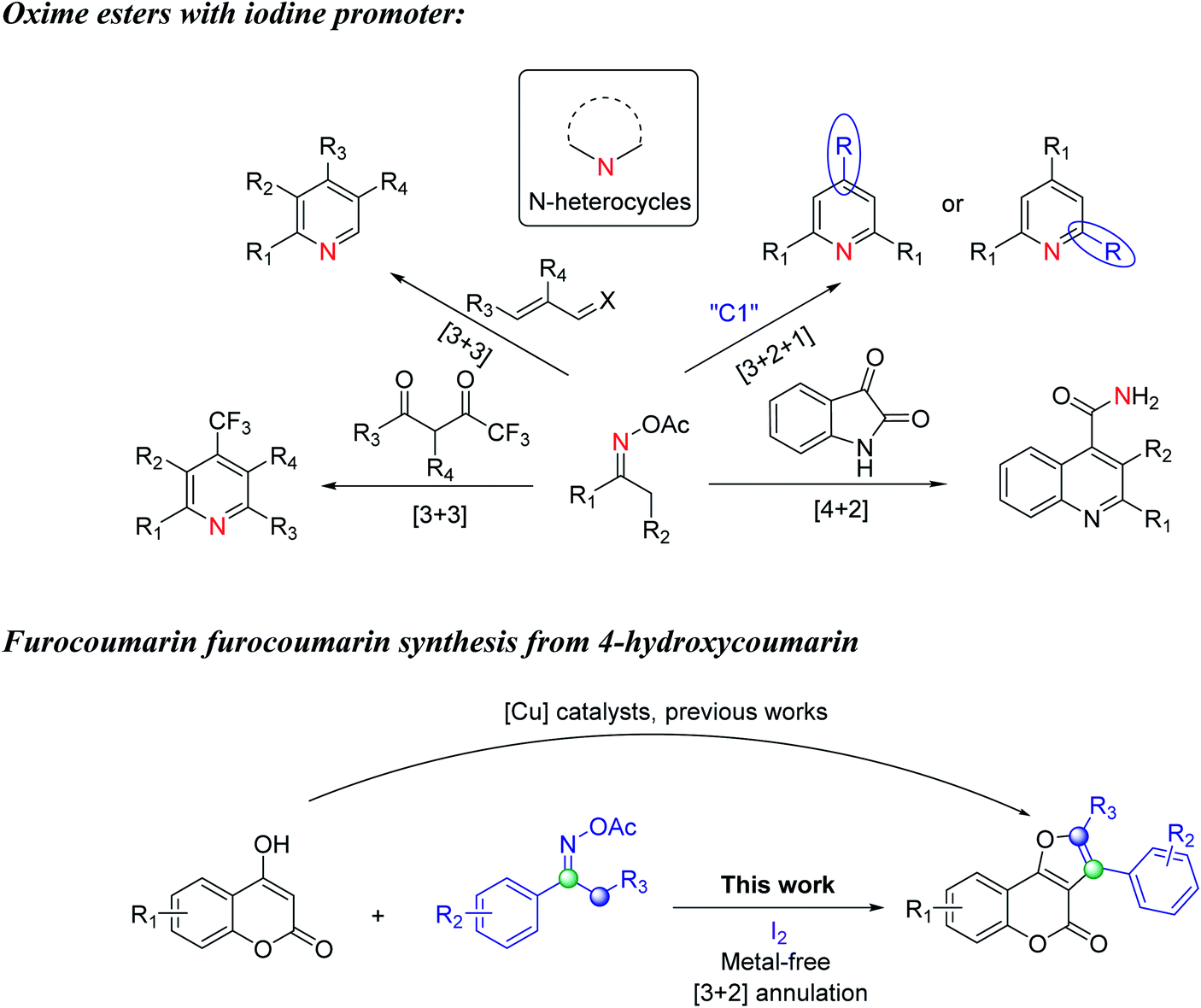 | ||
| Scheme 1 Previous works of oxime esters with iodine and furocoumarin synthesis from 4-hydroxycoumarin. | ||
2. Experimental
In a general procedure, 4-hydroxycoumarin (0.1 mmol, 16.2 mg), iodine (0.05 mmol, 12.7 mg) and mesitylene (2.0 mL) were added into the 8 mL vial containing a stirring bar. The reaction mixture was tightly sealed and heated at 140 °C in 12 hours. After completion, the reaction mixture was cooled down to room temperature and diphenylether (0.1 mmol, 17 mg) was added as internal standard. Subsequently, the resulting mixture was extracted with DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (3:1) and the organic layer was collected and dried by using anhydrous Na2SO4 and then recrystallized in ethylacetate to afford desired product.
:H2O (3:1) and the organic layer was collected and dried by using anhydrous Na2SO4 and then recrystallized in ethylacetate to afford desired product.
3. Results and discussion
Our investigation of the heterocycles synthesis started with the reaction of O-acetyl ketoxime 1 and 4-hydroxycoumarin 2a (Table 1). To our surprise, furocoumarin 3a was obtained in a small amount with DMSO as solvent at 120 °C without the need of any additives, giving very first signal that our reaction may follow different pathways rather than previous proposed mechanism (Table 1, entry 1). The structure of furocoumarin 3a was precisely confirmed by X-ray crystallography on its single crystal (Fig. 2, CCDC number is 2026801). Furthermore, other solvents, ranging from non-polar to polar, generated products with different results (Table 1, entries 1–8). Generally, solvents such as DMF DMSO which are known to consume a big portion of the iodine radical via radical-mediated reactions provided the desired product in low yields.21 Ethereal solvents were also tested and only moderate yields were achieved presumably due to the α-hydrogen abstraction of these solvents with radicals.22 Non-polar aromatic solvents which are quite inert to radical reagents afforded higher efficiency and optimal results, 58%, were obtained in mesitylene. Other sources of iodine were also tested, with inorganic salts KI and NH4I gave comparably low yield of product formation, 26% and 30% yield, respectively (entries 9 and 10), while I2O5 provided only trace amount of the product (entry 11). In absence of I2, no fucocoumarin was observed, which showed the crucial role of the iodine promoter in this reaction (entry 12).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp. (°C) | Yieldb (%) |
| a Reaction condition: 1a (0.15 mmol), 2a (0.3 mmol), I2 promoter (50 mol%) in solvent (1.5 mL) at T °C under Ar for 2 h.b GC yield.c The reaction was run under O2.d Open air conditions. | ||||
| 1 | I2 | DMSO | 120 | 8 |
| 2 | I2 | DMF | 120 | 14 |
| 3 | I2 | 1,4-Dioxane | 120 | 21 |
| 4 | I2 | Dibutyl ether | 120 | 26 |
| 5 | I2 | PhCl | 120 | 39 |
| 6 | I2 | Toluene | 120 | 32 |
| 7 | I2 | Xylene | 120 | 53 |
| 8 | I2 | Mesitylene | 120 | 58 |
| 9 | KI | Mesitylene | 120 | 26 |
| 10 | NH4I | Mesitylene | 120 | 30 |
| 11 | I2O5 | Mesitylene | 120 | Trace |
| 12 | — | Mesitylene | 120 | ND |
| 13 | I2 | Mesitylene | 80 | 44 |
| 14 | I2 | Mesitylene | 140 | 87 |
| 15c | I2 | Mesitylene | 140 | 67 |
| 16d | I2 | Mesitylene | 140 | 46 |
| 17 | I2 | Mesitylene | 160 | 86 |
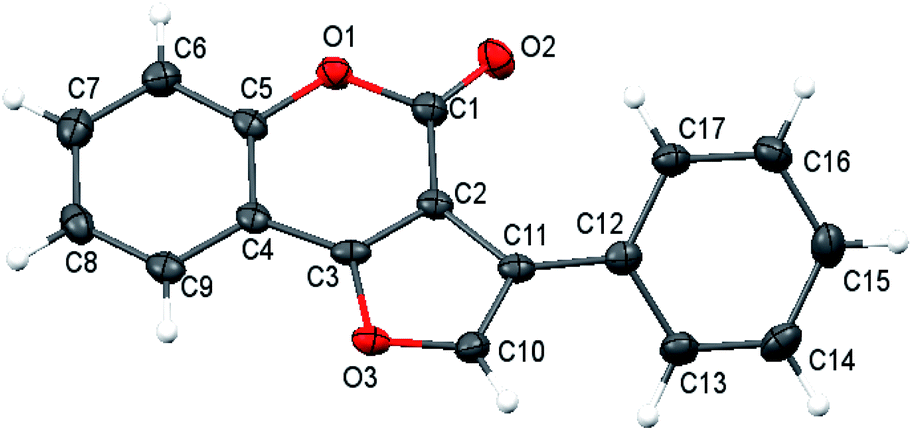 | ||
| Fig. 2 Crystal structure of 3a. | ||
We next turned our attention to the effects of temperature on the reaction outcome (entries 13, 14, 17). The increase of temperature greatly facilitated product formation by speeding up the reaction rate. Among the examined temperatures, we observed that the starting material was consumed quickly at 140 °C and provided the product with highest yield, 87%. Furthermore, running reaction in different atmospheres also led to various results. In comparison with the reaction under argon, only moderate amount of 3a was observed when the reaction was carried out under pure oxygen or air atmosphere, with 67% and 46% yield, accordingly. Apparently, other gases, especially oxygen, and moisture have momentous effects on the product formation.
The reason of diminished product formation under air or oxygen may be due to the presence of radicals over the course of the reaction. Based on previously known iodine mediated reaction with oxime esters, a plausible mechanism was outlined in Scheme 2. The reaction started with the reduction of acetyl oxime ester 1a by iodine to form the imino-radical A which could be in resonance with α-carbon radical imine B. After incorporated with another iodine radical, B was converted to α-iodoimine intermediate C which could be detected by mass spectroscopy (path a). The nucleophilic attack of the enol-carbon on 4-hydroxycoumarin 2a to the imino carbon of C then generated iodoamine D. Upon electrophilic O-alkylation attack of ketone group on the iodocarbon with the IOAc to I2 promoter recovery, the dihydrofuranocoumarine E was formed and accomplished the formal [3 + 2] annulation step. The intermediate E under acidic catalysis would then undergo olefin formation via elimination of the ammonium group to furnish final furocoumarin product. In addition, the intermediate E could be formed through the intramolecular nucleophilic cyclization of H which was provided by the radical coupling reaction of B and G (path b). The enolic oxygen radical G was generated from 2a with [I+] species as the oxidant via a single-electron-transfer (SET) process.
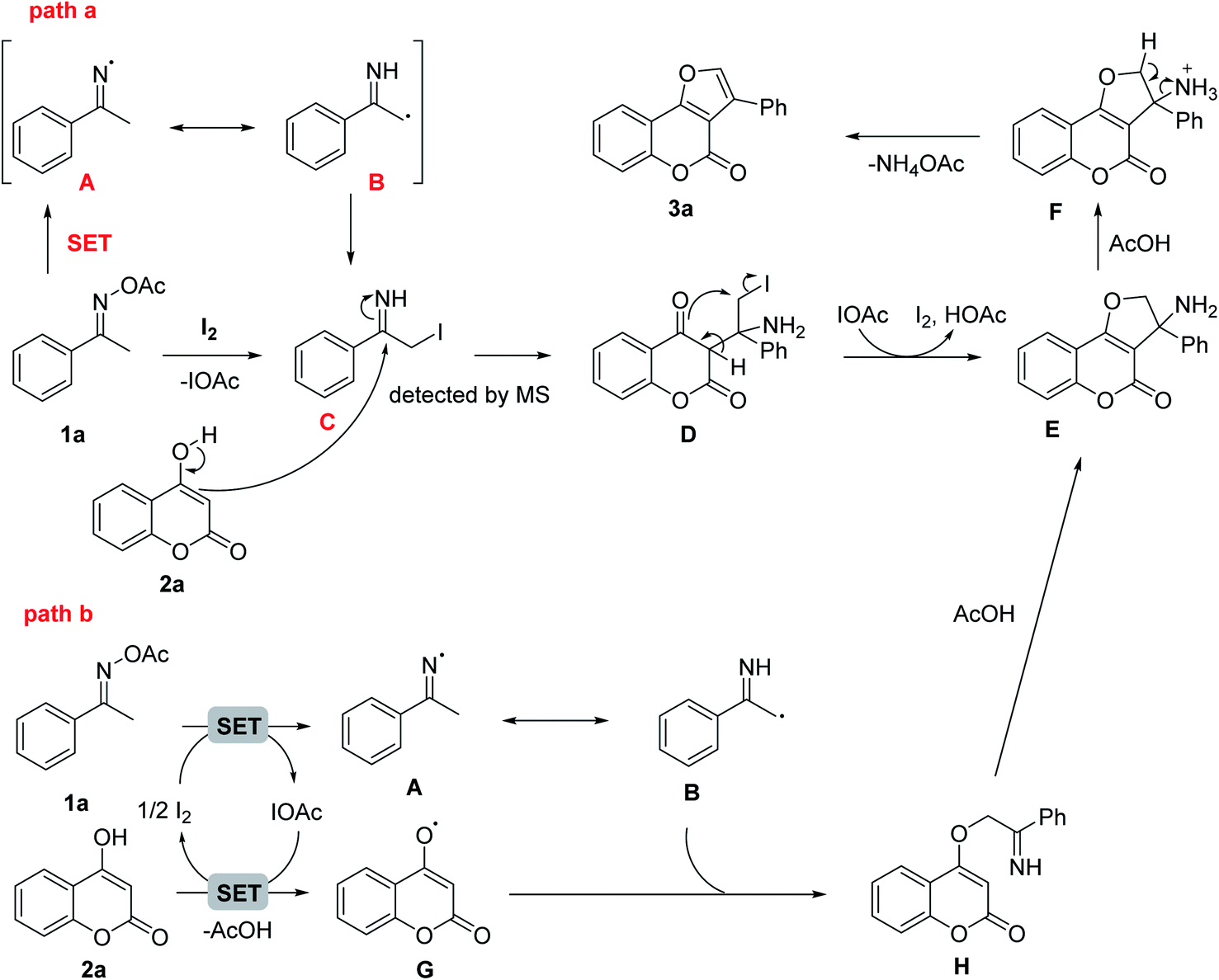 | ||
| Scheme 2 Plausible mechanism of the reaction. | ||
Our discovery was highly interesting because the nitrogen of oxime normally participates in the ring formation to generate N-containing heteocycles and the α-keto carbon of the oxime esters often plays as a nucleophile in nucleophilic attacks; however, this has not been observed in our case. To evaluate the feasibility of our proposed mechanism, we conducted mechanistic studies in which several control experiments were performed. Firstly, TEMPO was introduced to the reaction mixtures in addition to all other reagents to confirm a radical involment. The reaction was indeed completely suppressed without any trace of the furocoumarin 3a (Scheme 3a). This result supported the proposed radical process via SET process. Secondly, no formation of 3a was also observed when acetophenone was used instead of O-acetyl ketoxime starting material (Scheme 3b). The oxime undoubtedly played a key role as a radical component and a strong electrophilic partner in the reaction condition. The ketone, on the other hand, was unlikely to be electrophilic enough for the nucleophilic attack from 4-hydroxycoumarin. This turned out to be an appropriate rational when we used two equivalents of ammonium acetate together with acetophenone which could generate the imine in situ, 3a was isolated in 23% yield (Scheme 3c). The last puzzle in our proposed mechanism was C detected by mass spectroscopy. The iodoimine C may be the key intermediate for this methodology since the iodo-substitution has made the α-carbon of the amino group become more electrophilic and facilitated the ring closure approach from keto-oxygen of the coumarin ring.23 This was consistent with the results of reactions in the absence of 4-hydroxycoumarin (Scheme 3d). Interestingly, reaction path b is likely to be not favorable as shown in Scheme 3e. Particularly, reactions of 4-hydroxycoumarin in deuterated 1,4-dioxane resulted in less than 5% of H/D exchange. It was reported that hydrogen abstraction of ethers by free radicals in liquid phase was significant.24 However, more further mechanistic experiments are still needed to confirm the reaction path.
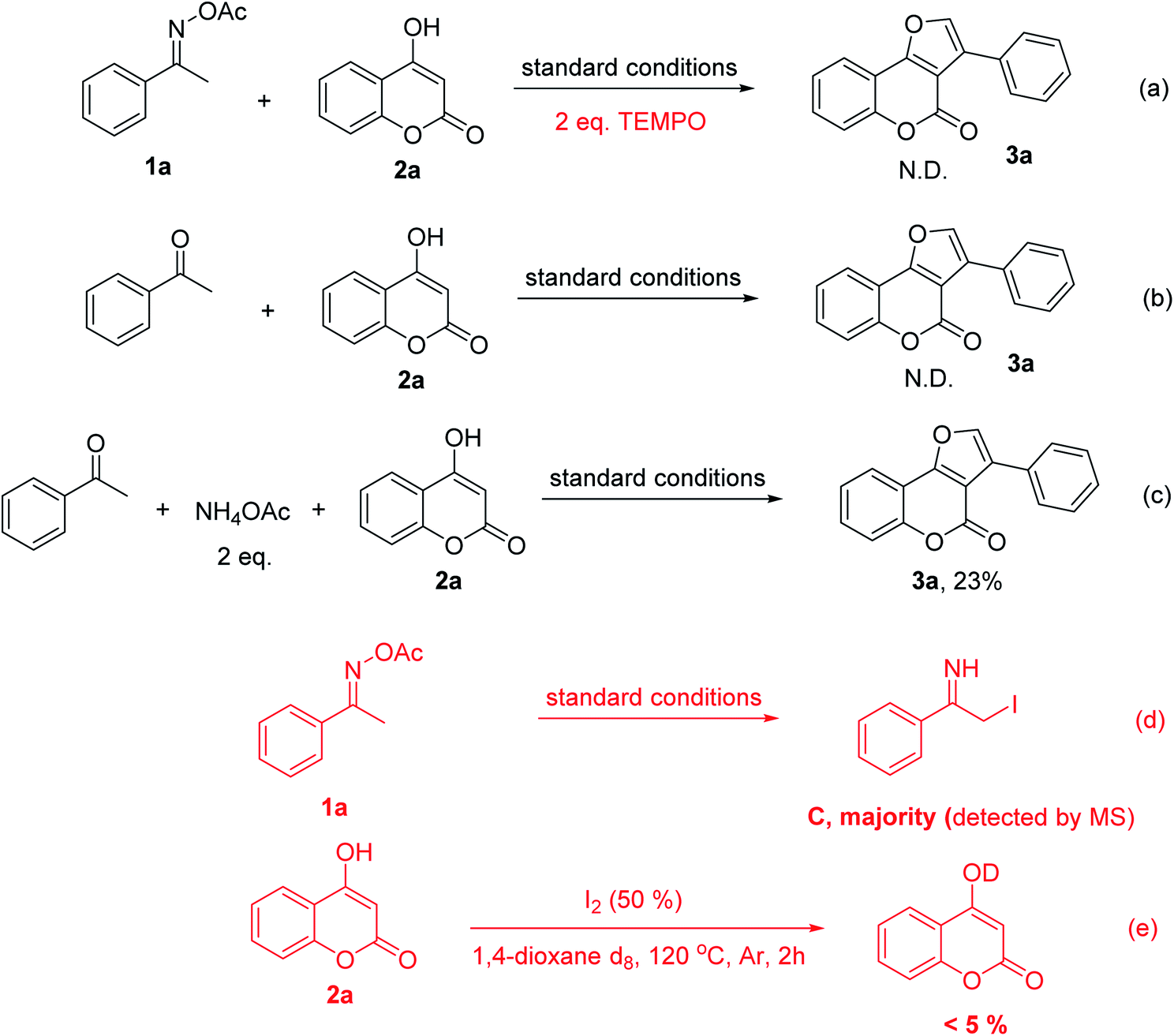 | ||
| Scheme 3 Control experiments. | ||
After investigating the reaction mechanism, we directed our effort to explore the generality and scope of this versatile I2-triggered formal [3 + 2] annulation using optimized conditions. To our expectation, the reaction had a very broad substrate scope in regard to both substituted 4-hydroxycoumarin and oxime esters. Specifically, the reaction of 4-hydroxycoumarin with a wide range of acetophenone oximes afforded aromatic-substituted furocoumarins (3a–3h) in moderate to excellent yields. Both electron-donating and electron-withdrawing groups were well tolerated. Changing the positions of substituents on the benzene ring is possible, the products were still generated effectively, from 66% to 87% yield. The p-fluorophenyl substrate, however, worked sluggishly with only 26% yield of the product formation (3i). Interestingly, cyclohexanone also tolerable in reaction conditions, afforded furocoumarin 3j in 73% yield. This showed that the method is not limited to only aromatic ketoximes. Moreover, the reaction surprisingly worked well with more steric oxime esters. When different propiophenone acetyloximes reacted with 4-hydroxycoumarin 1, disubstituted furocoumarins (3k–3m) were produced from moderate to high yields. In fact, steric hindrance did not expose any barrier for the product formation, with diphenylfurocoumarin 3n could be obtained in very good yield (82%). With regard to 4-hydroxycoumarin scope, it was realized that the electronic nature of substituents has a certain effect on the reaction outcome. The halogenated electron-deficient substrates tend to give products with lower yield than the methoxy electron-rich substrates (compared to 3o–3p and 3r–3s). Especially, when both 4-hydroxycoumarin and acetophenone oxime bearing the electron-withdrawing groups, the furocoumarin was obtained with only 40% yield (product 3q). Similarly, product 3t with a free-hydroxyl group was also isolated in 39% yield. Nevertheless, although obtained in low yield, this product shows that a protected functional group was not necessary for a successful product generation. This also indicates that the radical scavenging property of free hydroxy groups did not totally suppress product formation in the current reaction conditions (Scheme 4).
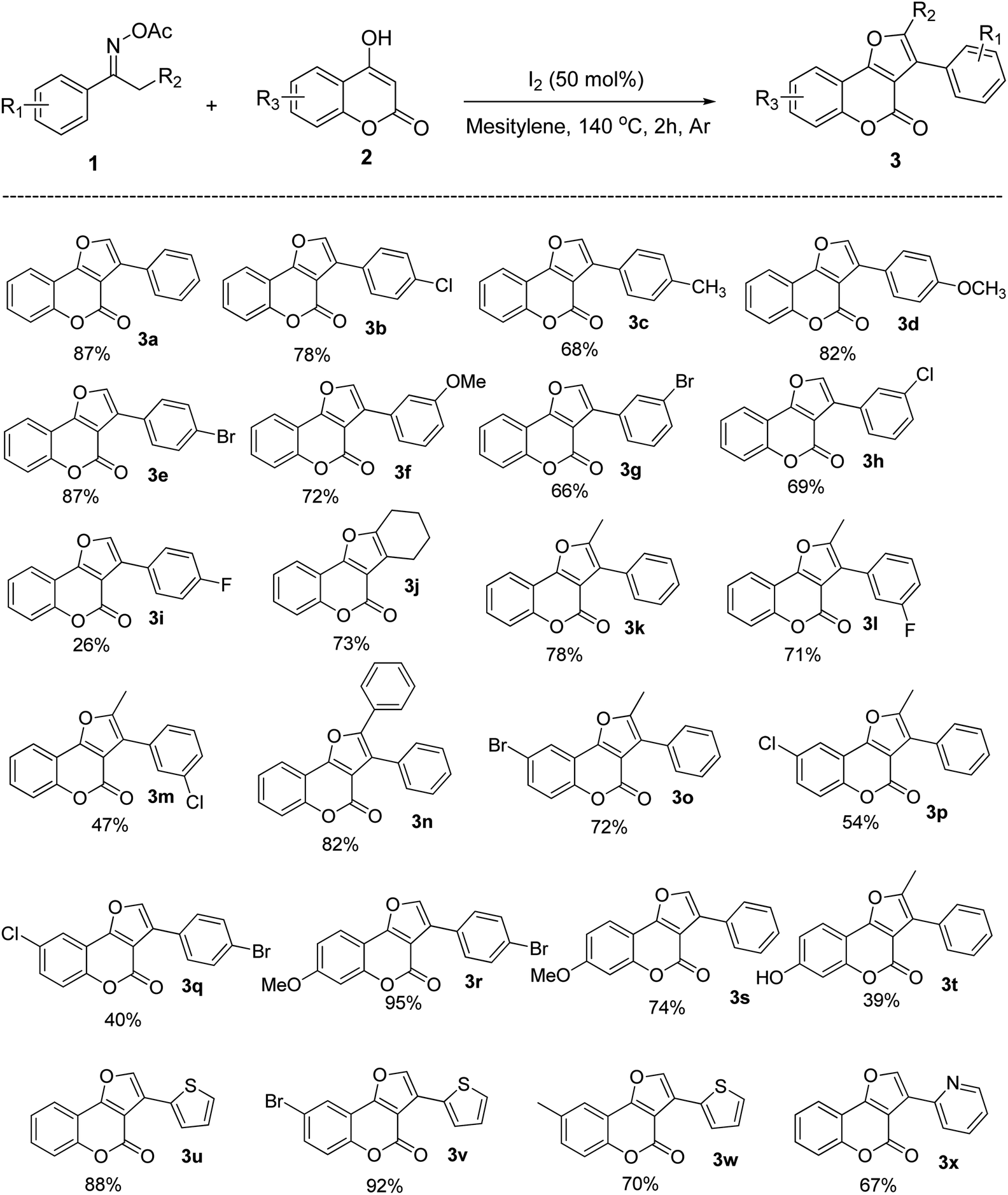 | ||
| Scheme 4 Substrates scope of the reaction.a aReaction condition: 1 (0.15 mmol), 2 (0.3 mmol), I2 (50 mol%) in mesitylene (1.5 mL) at 140 °C under Ar for 2 h. | ||
Finally, the heteroaromatic substrates were well-tolerated in this I2-promoted formal [3 + 2] annulation. The furocoumarins carrying thiophenyl groups (3u–3w) and pyridinyl groups (3x) were obtained in high to excellent yields. Since heteroatoms usually coordinate with metals and make the metal-catalyzed reaction proceed sluggishly,25 these results demonstrate the superior of our method compared to those with metal catalysts and heterocyclic substrates.
4. Conclusions
In conclusion, we have successfully developed an I2-mediated formal [3 + 2] annulation between two highly versatile starting materials, 4-hydroxycoumarin and acetyl oxime esters. The obtained furocoumarin products were highly applicable due to their potential biological activities. Our reaction was proposed through a radical mechanism which was supported by control experiments and observed intermediates. The ability of iodine to activate the N–O bond of the oximes and form a α-iodoimine intermediate was considered decisive for the ring-closing step which established the furano moiety of furocoumarins. This study also assisted our future protocols to employment of more steric and functional groups tolerance. We are in the process of exploring this efficient iodine catalyst system in the formation of other bioactive heterocycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Vietnam National Foundation for Science and Technology Development (NAFOSTED) is acknowledged for financial support via grant no. 104.01-2018.326.References
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 Search PubMed
.
-
(a) M.-S. Chang, Y.-C. Yang, Y.-C. Kuo, Y.-H. Kuo, C. Chang, C.-M. Chen and T.-H. Lee, J. Nat. Prod., 2005, 68, 11 Search PubMed
-
(a) G. J. Keating and R. O'Kennedy, The Chemistry and Occurrence of Coumarins. Coumarins: Biology, Applications and Mode of Action; R. O'Kennedy and R. D. Thornes, John Wiley&Sons, Chichester, New-York, 1997, p. 23 Search PubMed
- R. Sancho, N. Marquez, G. Gomez-Gonzalo, M. Calzado, G. Bettoni, M. T. Coiras, J. Alcami, M. Lopez-Cabrera, G. Appendino and E. Munoz, J. Biol. Chem., 2004, 279, 37349 Search PubMed
- J. J. Serrano-Pérez, M. Merchán and L. Serrano-Andrés, J. Phys. Chem. B, 2008, 112, 14002 Search PubMed
- For the review of 4-hydroxycoumarin chemistry, see: M. M. Abdou, Recent advances in 4-hydroxycoumarin chemistry. Part 2: Scaffolds for heterocycle moleculardiversity, Arabian J. Chem., 2019, 112, 974 Search PubMed
-
(a) J. A. Froggett, M. H. Hockley and R. B. J. Titman, Chem. Res., 1997, 30–31 Search PubMed
- R. S. Lamani, N. S. Shetty, R. R. Kamble and I. A. M Khazi, Eur. J. Med. Chem., 2008, 44, 2828 Search PubMed
- D. Garella, A. Barge, D. Upadhyaya, Z. Rodriguez, G. Cravotto and G. Palmisano, Synth. Commun., 2010, 40, 120 Search PubMed
-
(a) Y. R. Lee, M. W. Byun and B. S. Kim, Bull. Korean Chem. Soc., 1998, 10, 1080 Search PubMed
- V. Cadierno, J. Diez, J. Gimeno and N. Nebra, J. Org. Chem., 2008, 73, 5852 Search PubMed
-
(a) T. A. To, Y. H. Vo, A. T. Nguyen, A. N. Q. Phan, T. Truong and N. T. S. Phan, Org. Biomol. Chem., 2018, 16, 5086 Search PubMed
-
(a) H. Leutbecher, G. Greiner, R. Amann, U. Beifuss, J. Conrad and A. Stolz, Org. Biomol. Chem., 2011, 9, 2667 Search PubMed
-
(a) S. K. Panja, J. Ghosh, S. Maiti and C. Bandyopadhyay, J. Chem. Res., 2012, 36, 222 Search PubMed
- A. A. Tabolin and S. L. Ioffe, Chem. Rev., 2014, 114, 5426 Search PubMed
- H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155 Search PubMed
- H. Huang, J. Cai, L. Tang, Z. Wang, F. Li and G.-J. Deng, J. Org. Chem., 2016, 81, 1499 Search PubMed
- H. Huang, J. Cai, H. Xie, J. Tan, F. Li and G. J. Deng, Org. Lett., 2017, 19, 3743 Search PubMed
- Y. Xia, J. Cai, H. Huang and G. J. Deng, Org. Biomol. Chem., 2018, 16, 124 Search PubMed
-
(a) Q. Gao, H. Yan, M. Wu, J. Sun, X. Yan and A. Wu, Org. Biomol. Chem., 2018, 16, 2342 Search PubMed
-
(a) A. Monga, S. Bagchia and A. Sharma, New J. Chem., 2018, 42, 1551 Search PubMed
- T. Ogura, A. Miyoshi and M. Koshi, Phys. Chem. Chem. Phys., 2007, 9, 5133 Search PubMed
- A. W. Erian, S. M. Sherif and H. M. Gaber, Molecules, 2003, 8, 793 Search PubMed
- R. S. Davidson, Q. Rev. Chem. Soc., 1967, 21, 249 Search PubMed
-
(a) Y. Makida, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2012, 51, 4122 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2026801. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra07566c |
| This journal is © The Royal Society of Chemistry 2020 |