
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining site-directed spin labeling in vivo and in-cell EPR distance determination†
Pia
Widder
 a,
Julian
Schuck
a,
Daniel
Summerer
*b and
Malte
Drescher
*a
a,
Julian
Schuck
a,
Daniel
Summerer
*b and
Malte
Drescher
*a
aDepartment of Chemistry and Konstanz Research School Chemical Biology (KoRS-CB), University of Konstanz, Konstanz, Germany. E-mail: malte.drescher@uni-konstanz.de
bFaculty of Chemistry and Chemical Biology, TU Dortmund University, Dortmund, Germany. E-mail: daniel.summerer@tu-dortmund.de
First published on 18th February 2020
Abstract
Structural studies on proteins directly in their native environment are required for a comprehensive understanding of their function. Electron paramagnetic resonance (EPR) spectroscopy and in particular double electron–electron resonance (DEER) distance determination are suited to investigate spin-labeled proteins directly in the cell. The combination of intracellular bioorthogonal labeling with in-cell DEER measurements does not require additional purification or delivery steps of spin-labeled protein to the cells. In this study, we express eGFP in E. coli and use copper-catalyzed azide–alkyne cycloaddition (CuAAC) for the site-directed spin labeling of the protein in vivo, followed by in-cell EPR distance determination. Inter-spin distance measurements of spin-labeled eGFP agree with in vitro measurements and calculations based on the rotamer library of the spin label.
The understanding of protein function and structure is crucially linked to the ability to study proteins in their native environment. Effects of not only molecular crowding but also post-translational modifications and the presence of a variety of specific or non-specific interaction partners or chaperones have a great impact on proteins, yet are only incompletely understood through in vitro studies.1
Facing the challenge of highly complex cellular compositions, in-cell electron paramagnetic resonance (EPR) spectroscopy in combination with site-directed spin labeling (SDSL)2 has emerged as a valuable tool to provide structural information3 as many cellular components are diamagnetic and therefore EPR-silent. Pulsed techniques such as double electron–electron resonance (DEER) provide access to long-range distance restraints in the nanometer range by measuring the dipole–dipole interactions between paramagnetic spin labels.4,5
So far, in vivo DEER studies on spin-labeled proteins have either been conducted on outer membrane proteins upon cysteine-based spin labeling on the cellular surface6–8 or relied on the delivery of spin-labeled protein into the cell, e.g. by microinjection into oocytes9–12 or permeabilization of the membrane via hypo-osmotic shock or electroporation.13–18 Non-canonical amino acid (ncAA) incorporation and bioorthogonal in vivo spin labeling offer a more direct and elegant approach for in-cell EPR studies as they may combine expression, labeling and the EPR study of the protein of interest directly inside the same cell without additional delivery steps. While a number of studies on bioorthogonal spin labeling have been published in recent years,19–27 the corresponding DEER measurements were limited to in vitro measurements. Even in cases with confirmed in vivo labeling, purification and concentration of spin-labeled protein were required prior to a DEER measurement. The Steinhoff group has advanced nitroxide spin labeling via copper-catalyzed [3+2] azide–alkyne cycloaddition (CuAAC) and reported labeling of eGFP at one site between the ncAA N-ε-propargyl-L-lysine (PrK) and a nitroxide spin label in E. coli. However, for DEER distance determination, the protein was purified and conventional cysteine labeling with MTSSL was used to introduce the second spin label.24
In our previous work,27 we have identified the ncAA para-ethynyl-phenylalanine (pENF) as a suitable choice for bioorthogonal spin labeling with CuAAC. pENF was incorporated into E. coli thioredoxin with high labeling yields and exhibited favorable linker properties for DEER distance determination in in vitro measurements.
Here, we extend this approach to bioorthogonal double spin labeling and DEER measurements directly in vivo (Fig. 1). We report the incorporation of pENF at two sites of eGFP via amber stop codon suppression in E. coli, develop conditions for CuAAC with an azide-bearing nitroxide spin label and combine it with in-cell DEER distance determination without any additional protein purification step. In-cell inter-spin distances of spin-labeled eGFP are comparable to in vitro measurements and calculated distances based on the rotamer library of the label.
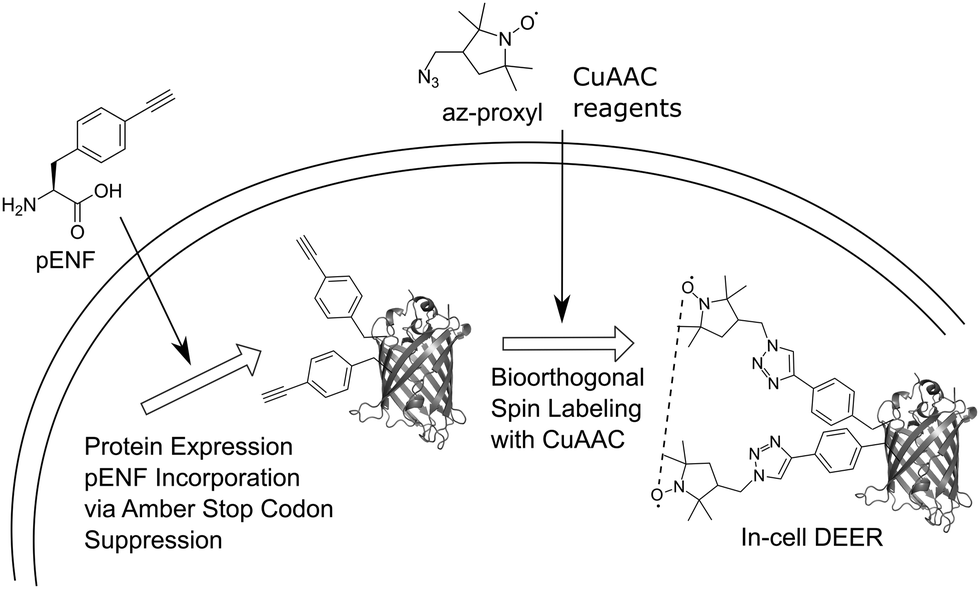 | ||
| Fig. 1 Schematic overview of the in vivo spin labeling approach via copper-catalyzed [3+2] azide–alkyne cycloaddition followed by in-cell EPR distance determination. The ncAA pENF is incorporated site-specifically into eGFP using amber stop codon suppression. The eGFP-expressing E. coli cells are subjected to CuAAC-based spin labeling and subsequent, DEER distance determination is performed directly inside the cell. | ||
Due to the reductive environment of the cell, in-cell EPR studies benefit from the use of reduction-resistant spin labels, such as gadolinium spin labels28 or nitroxides shielded with ethyl groups.29,30 Nevertheless, we decided to use an uncharged, small nitroxide spin labels with a five-membered ring structure and shielding methyl groups in our study because of its known cell permeability.24 EGFP was chosen as a model protein for our study since in vivo CuAAC-based spin labeling of this protein has previously been shown24 and the fluorescence properties of eGFP facilitate in-cell monitoring of the protein. The positions Y39TAG in the β2–β3 loop region and L221TAG in the β11-strand of the barrel were chosen as labeling sites.31 We co-transformed plasmids pBAD_GFP_Y39/L221-TAG and pEVOL_pCNF (encoding a polyspecific Methanocaldococcus jannaschii tRNATyr(CUA)/tyrosyl-tRNA-synthetase (YRS) pair evolved for the genetic encoding of para-cyano-L-phenylalanine, pCNF)32,33 into BL21-Gold (DE3) E. coli, induced the culture and purified Y39/L221pENF eGFP by Ni-NTA chromatography via its C-terminal His6-tag. ESI-MS data confirmed the correct incorporation of pENF at two positions (Fig. S1, ESI†). In vitro labeling reactions were performed as previously described27 with 1 mM CuSO4, 3 mM 2-(4-((bis((1-(tert-butyl)-1H-1,2,3-triazol-4-yl)methyl)amino)methyl)-1H-1,2,3-triazol-1-yl) acetic acid (BTTAA), 1 mM sodium ascorbate (NaAsc) and 1 mM 3-(azidomethyl)-2,2,5,5-tetramethyl-1-pyrrolidinyloxyl (az-proxyl) and yielded Y39/L221pENF-L eGFP with an averaged labeling efficiency of approximately 70% per site based on the ratio of spin concentration to protein concentration. Incorporation of pENF and labeling did not significantly affect the function of eGFP as in vitro excitation and emission fluorescence spectra of Y39/L221pENF before and after labeling did not differ from eGFP wild-type spectra (Fig. S2, ESI†). In vitro DEER measurements of spin-labeled Y39/L221pENF-L eGFP resulted in a narrow distance distribution (HWHM: 0.5 nm) with a mean peak at 2.3 nm (Fig. S3, ESI†) and agree well with the calculated distances based on the crystal structure of eGFP (PDB structure 4EUL) and our previously published rotamer library27 of the pENF-L spin label.
A major concern of in vivo applications of CuAAC is copper-mediated cytotoxicity. Copper ions are linked to the formation of reactive oxygen species (ROS)34 and are known to impair cytoplasmic proteins with Fe–S clusters due to their thiophilic tendency.35 However, it has been shown that this toxicity is greatly reduced by adding chelators to the copper species with BTTAA being a ligand that shows especially promising properties for in vivo CuAAC applications.36 To investigate the effects of in vivo CuAAC on E. coli, we monitored the bacterial growth immediately after CuAAC treatment as well as the long-term growth behavior with a plate sensitivity assay (Fig. 2). Cells were treated with the labeling reagents copper(II)sulfate, BTTAA and sodium ascorbate (in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1 ratio), diluted, and the optical density of the cells at 600 nm (O.D. 600) was measured over time and compared to an untreated control sample (Fig. 2A and Fig. S4, ESI†). The growth rate of E. coli cells treated with CuAAC labeling reagents under labeling conditions did not differ from the untreated controls (only under far higher copper concentrations of 10 mM a reduction of growth rate was observed). In addition, we investigated the long-term growth behavior of treated and untreated cells by plating out diluted E. coli suspensions onto LB agar plates and incubating overnight at 37 °C. After counting the number of colony-forming units, we concluded that CuAAC treatment did not impact the number of viable cells (Fig. 2B and Fig. S5, ESI†). Finally, to evaluate the integrity of the bacterial membrane after CuAAC labeling, we performed dead cell/alive cell discrimination via FACS analysis (Fig. 2C and Fig. S6, ESI†). Events were pre-gated based on their forward (FSC-A) and sideward scattering (SSC-A) to exclude cell debris and buffer crystals. E. coli cells were then gated based on SSC-A and eGFP fluorescence, and the fraction of dead cells was detected via staining with propidium iodide (PI). Untreated cells and cells treated with 75% ethanol were used as controls to determine the level of PI intensity characteristic for live and dead cells, respectively. We did not observe an increase in the dead cell fraction in the CuAAC-treated compared to the untreated cells. For higher copper(II)sulfate concentrations of 30 mM, FACS data suggested the onset of copper-mediated cell death. The FACS analysis was conducted approximately 60–65 minutes after the start of the labeling reaction, the time point at which samples intended for in-cell EPR distance determination were frozen in liquid nitrogen as part of the DEER sample preparation. We therefore expect a similar level of integrity as seen in FACS measurements for the respective DEER samples.
:3:1 ratio), diluted, and the optical density of the cells at 600 nm (O.D. 600) was measured over time and compared to an untreated control sample (Fig. 2A and Fig. S4, ESI†). The growth rate of E. coli cells treated with CuAAC labeling reagents under labeling conditions did not differ from the untreated controls (only under far higher copper concentrations of 10 mM a reduction of growth rate was observed). In addition, we investigated the long-term growth behavior of treated and untreated cells by plating out diluted E. coli suspensions onto LB agar plates and incubating overnight at 37 °C. After counting the number of colony-forming units, we concluded that CuAAC treatment did not impact the number of viable cells (Fig. 2B and Fig. S5, ESI†). Finally, to evaluate the integrity of the bacterial membrane after CuAAC labeling, we performed dead cell/alive cell discrimination via FACS analysis (Fig. 2C and Fig. S6, ESI†). Events were pre-gated based on their forward (FSC-A) and sideward scattering (SSC-A) to exclude cell debris and buffer crystals. E. coli cells were then gated based on SSC-A and eGFP fluorescence, and the fraction of dead cells was detected via staining with propidium iodide (PI). Untreated cells and cells treated with 75% ethanol were used as controls to determine the level of PI intensity characteristic for live and dead cells, respectively. We did not observe an increase in the dead cell fraction in the CuAAC-treated compared to the untreated cells. For higher copper(II)sulfate concentrations of 30 mM, FACS data suggested the onset of copper-mediated cell death. The FACS analysis was conducted approximately 60–65 minutes after the start of the labeling reaction, the time point at which samples intended for in-cell EPR distance determination were frozen in liquid nitrogen as part of the DEER sample preparation. We therefore expect a similar level of integrity as seen in FACS measurements for the respective DEER samples.
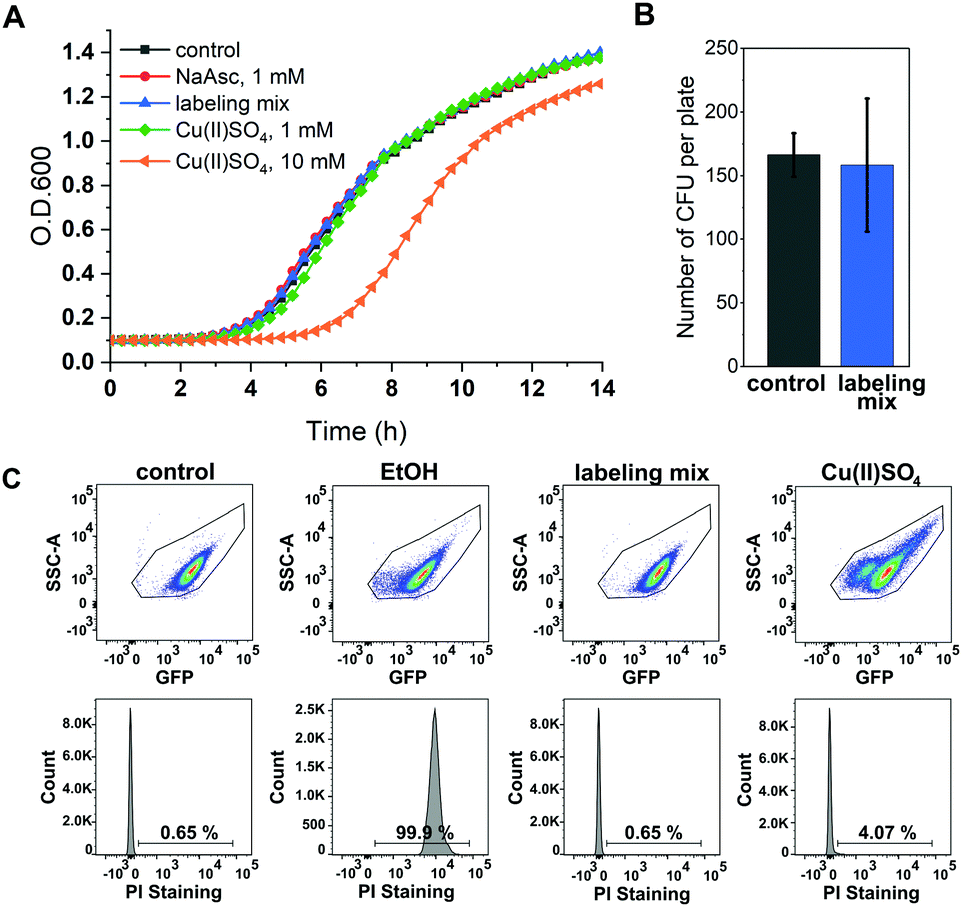 | ||
| Fig. 2 Toxicity of CuAAC labeling reagents for E. coli. (A) E. coli growth after treatment with CuAAC reagents: 1 mM sodium ascorbate (red), labeling mix Cu(II)SO4, BTTAA, NaAsc (1/3/1 mM) (blue), 1 mM Cu(II)SO4 (green), 10 mM Cu(II)SO4 (orange), untreated control (gray) (n = 2) (B) plate sensitivity assay. Diluted E. coli suspensions of cells treated with Cu(II)SO4, BTTAA, NaAsc (1/3/1 mM) (blue), and untreated control sample (gray) were plated, incubated overnight and the number of colony-forming units (CFU) was counted (n = 3). (C) Dead cell/alive cell identification with flow cytometry. E. coli cells expressing Y39/L221pENF eGFP were either treated with ethanol (EtOH), subjected to CuSO4, BTTAA, NaAsc (1/3/1 mM, labeling mix) or treated with 30 mM Cu(II)SO4. Untreated cells (control) served as alive control sample. Dead cell detection was facilitated by PI staining and the indicated numbers represent the percentage of dead cells. | ||
Next, we were interested in studying possible nitroxide reduction inside E. coli (Fig. 3). Proxyl-based spin labels as employed in this study are prone to fast biological reduction and it has been shown that they can be rendered EPR-inactive on a minute timescale.29 However, it is possible to prolong the time scale on which nitroxide signal is still detectable by adding a large excess of the spin label to the cells and thereby practically overloading cellular reduction mechanisms.9,30
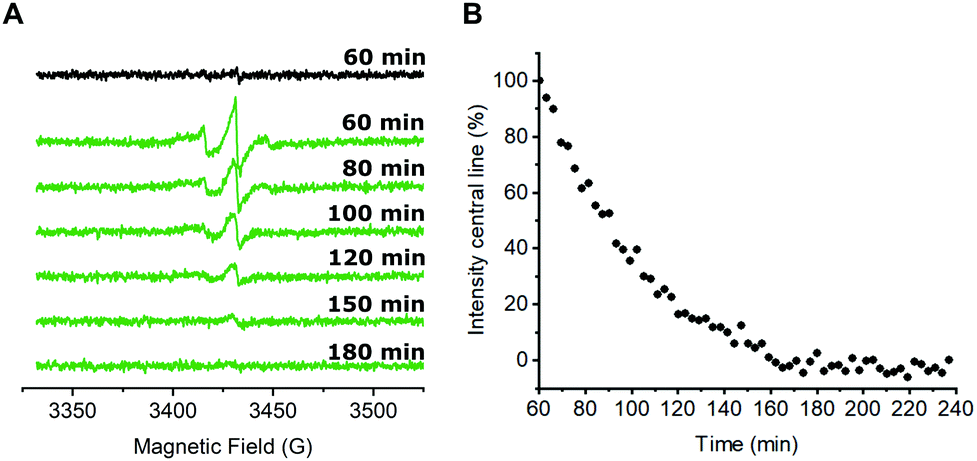 | ||
| Fig. 3 In-cell nitroxide stability after CuAAC spin labeling. (A) CW EPR spectra of E. coli cells expressing the tRNATyr(CUA)/YRS pair for the incorporation of pENF as well as eGFP wild-type (black) or Y39/L221pENF eGFP (green) after in vivo CuAAC spin labeling. A detailed explanation is given in the text. Spectra were averaged over 5 scans. Indicated time points refer to the passed time after the addition of the nitroxide labeling reagent to the cells. t = 60 min marks the end of the labeling procedure and the start of the EPR measurement. (B) For a qualitative description of the nitroxide reduction, the amplitude of the centerline from averaged CW spectra was plotted against the time. The signal intensity at t = 60 min was set to 100%. | ||
In case of CuAAC-based spin labeling, Kucher et al. have already reported on the successful in vivo nitroxide labeling at a single labeling site,24 while sufficient double-labeling in vivo has yet to be proven. We limited the labeling times for the in vivo labeling approach to 40 minutes at room temperature, followed by additional washing steps to remove unbound nitroxide spin labels (see ESI† for details). In total, approximately 60 minutes passed between the addition of the nitroxide spin labeling reagent to E. coli cells and the start of the EPR measurement. Time-dependent EPR spectra are shown in Fig. 3A. For a qualitative description of the signal reduction, we plotted the amplitude of the nitroxide center field peak against the time (Fig. 3B). Upon performing the CuAAC-based spin labeling procedure, E. coli cells expressing Y39/L221pENF eGFP show a typical nitroxide EPR spectrum that prevails up to 120 minutes after the addition of azido-proxyl spin labeling reagent. In principle, EPR signals in E. coli can stem from several possible species. Besides spin-labeled Y39/L221pENF-L eGFP, the free spin label from the labeling reaction, as well as labeled pENF, both in free form and esterified to tRNATyr(CUA) might contribute to the signal. To limit off-target labeling, protein-expressing cells were transferred to fresh LB medium without pENF and protein expression was prolonged for 90 minutes prior to CuAAC labeling. In addition, approximately 9% of E. coli proteins are terminated with an amber stop codon (TAG)37 and pENF might also eventually be incorporated into these proteins. To investigate potential sources of signal, we co-transformed E. coli cells with plasmids for eGFP wild-type and the tRNATyr(CUA)/YRS pair and expressed eGFP wild-type in the presence of pENF in the medium. In this scenario, the EPR signal after CuAAC labeling procedure can only arise from free or tRNA-esterified spin-labeled pENF or from labeled pENF incorporated at amber sites of off-target proteins. However, we did only observe neglectable signal intensities in this sample (Fig. 3A, black). In addition, we tested our washing protocol after CuAAC-based spin labeling with E. coli cells treated with CuAAC labeling reagents and azido-proxyl in the absence of pENF and found that free spin-label is effectively removed from the bacterial solution (Fig. S7, ESI†). We therefore assumed that the main contribution to the signal stems from spin-labeled Y39/L221pENF-L eGFP. The CW EPR spectra of Y39/L221pENF-L after in vitro or in vivo labeling were not identical which might be attributed to different ratios of spin labels attached to the individual labeling sites or differences in the mobility of the spin label within the cytoplasm (Fig. S8, ESI†).
For pulsed EPR experiments, E. coli cells expressing Y39/L221pENF eGFP were labeled as described above and shock-frozen in liquid 60 minutes after the start of the labeling reaction. The overall protein content was estimated by Western Blot analysis to be in the range of 200–800 μM with a spin concentration of approximately 20 μM, resulting in less than 5% EPR-active spin label per site. Echo-detected field sweeps of E. coli samples after CuAAC spin labeling contained spectral contributions from Cu(II) species as a result of the incomplete removal of the catalyst after labeling (Fig. S9, ESI†). The phase memory time of the spins was reduced to 0.66 μs for protonated cellular samples compared to 2.44 μs from the in vitro experiment in partially deuterated aqueous solution (Fig. S10, ESI†). The dipolar evolution time of in-cell DEER measurements was limited to 1.3 μs due to the reduced phase memory time in the cellular environment, but still long enough to provide reliable information about distance distributions in the range of the in vitro experiment.4 For background correction, we recorded in-cell DEER traces with the two singly labeled eGFP variants Y39pENF-L and L221pENF-L eGFP which could be completely described by a homogeneous background model (Fig. S11, ESI†). A similar background model was then used for the background correction for doubly-labeled eGFP. The form factor and the derived distance distribution were compared to the in vitro measurement of Y39/L221pENF-L eGFP and the expected distances based on the rotamer library (Fig. 4 and Fig. S12, ESI†).38 The shape of the in-cell form factor was strikingly similar to the in vitro measurement and both experimental distance distributions contained the main distance constraint at 2.3 nm, while a second maximum at 2.6 nm could not be evaluated in the in-cell experiment. The in-cell measurement yielded a low modulation depth of roughly 2%, compared to 38% for the in vitro sample. We suspect that the large background contribution is a result of incomplete labeling of eGFP in the cellular environment as well as fast nitroxide reduction during the labeling procedure, while the presence of unbound spin label only plays a minor role.
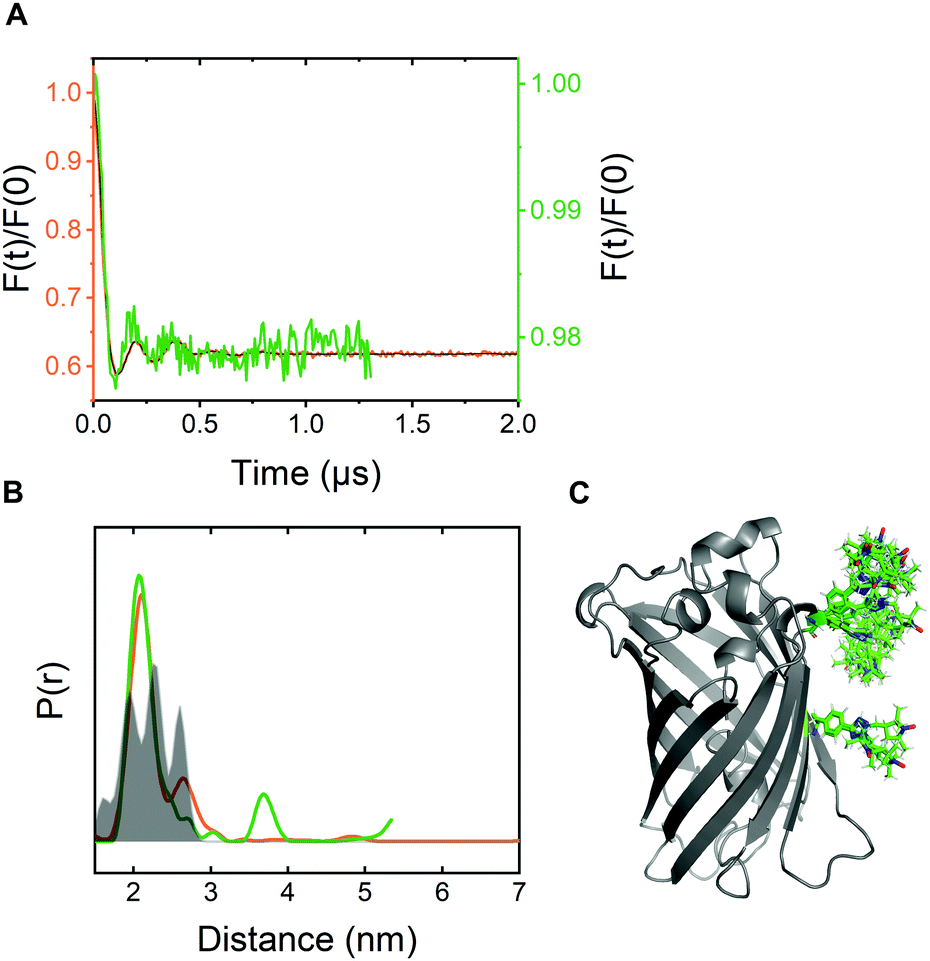 | ||
| Fig. 4 In-cell DEER distance determination after bioorthogonal CuAAC spin labeling (A) Comparison of the form factors of Y39/L221pENF-L eGFP from DEER measurements conducted in vitro (orange) or in vivo (green). The modulation depth decreased from 38% for in vitro DEER to approximately 2% for in vivo DEER. (B) Derived distance distribution from form factors for DEER measurements in vitro (orange) and in vivo (green). The gray area indicates the expected distance distribution based on MMM38 calculations with the rotamer library of pENF-L and a crystal structure of eGFP (C) The crystal structure of eGFP with indicated labeling sites and expected label conformers based on the rotamer library of pENF-L. | ||
In conclusion, we have demonstrated CuAAC-based spin labeling of an alkyne-bearing protein in vivo without apparent toxicity in E. coli. Moreover, we observe minimal background signal from nitroxides not attached to our target protein positions. Stability of the nitroxide spin label is a limiting factor of in vivo labeling that requires a large excess of spin label reagent and high cellular protein concentrations. Despite fast nitroxide reduction in the cytoplasm, double labeling of Y39/L221pENF eGFP and subsequent in-cell DEER measurements were performed that argue for a similar conformation that eGFP adopts in the E. coli cytoplasm and in vitro. Taken together, our approach combines natural translation, folding, and processing of a target protein with bioorthogonal double labeling and DEER distance measurements directly in the natural environment of a bacterial cell. It overcomes the necessity for introducing spin-labeled proteins into cells, e.g. via electroporation or hypo-osmotic shock and thus represents a new access point to in-cell EPR studies of protein structure and function.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank P. G. Schultz for plasmid pEVOL_pCNF. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement number: 772027—SPICE—ERC-2017-COG) and from the Deutsche Forschungsgemeinschaft (SFB 969, Project C3).References
- C. Kyne and P. B. Crowley, FEBS J., 2016, 283, 3016–3028 CrossRef CAS PubMed.
- W. L. Hubbell, C. J. López, C. Altenbach and Z. Yang, Curr. Opin. Struct. Biol., 2013, 23, 725–733 CrossRef CAS PubMed.
- G. Jeschke, Emerging Top. Life Sci., 2018, 2, 9–18 CrossRef CAS.
- G. Jeschke, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS PubMed.
- G. Jeschke, ChemPhysChem, 2002, 3, 927–932 CrossRef CAS PubMed.
- B. Joseph, A. Sikora, E. Bordignon, G. Jeschke, D. S. Cafiso and T. F. Prisner, Angew. Chem., Int. Ed., 2015, 54, 6196–6199 CrossRef CAS PubMed.
- B. Joseph, A. Sikora and D. S. Cafiso, J. Am. Chem. Soc., 2016, 138, 1844–1847 CrossRef CAS PubMed.
- S. Dunkel, L. P. Pulagam, H. J. Steinhoff and J. P. Klare, Phys. Chem. Chem. Phys., 2015, 17, 4875–4878 RSC.
- K. Singewald, M. J. Lawless and S. Saxena, J. Magn. Reson., 2019, 299, 21–27 CrossRef CAS PubMed.
- M. J. Lawless, A. Shimshi, T. F. Cunningham, M. N. Kinde, P. Tang and S. Saxena, ChemPhysChem, 2017, 18, 1653–1660 CrossRef CAS PubMed.
- M. Qi, A. Groß, G. Jeschke, A. Godt and M. Drescher, J. Am. Chem. Soc., 2014, 136, 15366–15378 CrossRef CAS PubMed.
- R. Igarashi, T. Sakai, H. Hara, T. Tenno, T. Tanaka, H. Tochio and M. Shirakawa, J. Am. Chem. Soc., 2010, 132, 8228–8229 CrossRef CAS PubMed.
- Y. Yang, F. Yang, Y.-J. Gong, J.-L. Chen, D. Goldfarb and X.-C. Su, Angew. Chem., Int. Ed., 2017, 56, 2914–2918 CrossRef CAS PubMed.
- Y. Yang, F. Yang, X.-Y. Li, X.-C. Su and D. Goldfarb, J. Phys. Chem. B, 2019, 123(5), 1050–1059 CrossRef CAS PubMed.
- F. C. Mascali, H. Y. Ching, R. M. Rasia, S. Un and L. C. Tabares, Angew. Chem., Int. Ed., 2016, 55, 11041–11043 CrossRef CAS PubMed.
- A. Martorana, G. Bellapadrona, A. Feintuch, E. Di Gregorio, S. Aime and D. Goldfarb, J. Am. Chem. Soc., 2014, 136, 13458–13465 CrossRef CAS PubMed.
- A. Dalaloyan, A. Martorana, Y. Barak, D. Gataulin, E. Reuveny, A. Howe, M. Elbaum, S. Albeck, T. Unger, V. Frydman, E. H. Abdelkader, G. Otting and D. Goldfarb, ChemPhysChem, 2019, 20, 1860–1868 CrossRef CAS PubMed.
- F. X. Theillet, A. Binolfi, B. Bekei, A. Martorana, H. M. Rose, M. Stuiver, S. Verzini, D. Lorenz, M. van Rossum, D. Goldfarb and P. Selenko, Nature, 2016, 530, 45–50 CrossRef CAS PubMed.
- M. R. Fleissner, E. M. Brustad, T. Kálai, C. Altenbach, D. Cascio, F. B. Peters, K. Hideg, S. Peuker, P. G. Schultz and W. L. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21637–21642 CrossRef CAS PubMed.
- C. T. Loh, K. Ozawa, K. L. Tuck, N. Barlow, T. Huber, G. Otting and B. Graham, Bioconjugate Chem., 2013, 24, 260–268 CrossRef CAS PubMed.
- L. Garbuio, E. Bordignon, E. K. Brooks, W. L. Hubbell, G. Jeschke and M. Yulikov, J. Phys. Chem. B, 2013, 117, 3145–3153 CrossRef CAS PubMed.
- E. H. Abdelkader, A. Feintuch, X. Yao, L. A. Adams, L. Aurelio, B. Graham, D. Goldfarb and G. Otting, Chem. Commun., 2015, 51, 15898–15901 RSC.
- E. G. Evans and G. L. Millhauser, Methods Enzymol., 2015, 563, 503–527 CAS.
- S. Kucher, S. Korneev, S. Tyagi, R. Apfelbaum, D. Grohmann, E. A. Lemke, J. P. Klare, H. J. Steinhoff and D. Klose, J. Magn. Reson., 2017, 275, 38–45 CrossRef CAS.
- A. Kugele, T. S. Braun, P. Widder, L. Williams, M. J. Schmidt, D. Summerer and M. Drescher, Chem. Commun., 2019, 55, 1923–1926 RSC.
- A. Kugele, B. Silkenath, J. Langer, V. Wittmann and M. Drescher, ChemBioChem, 2019, 20, 2479–2484 CrossRef CAS PubMed.
- P. Widder, F. Berner, D. Summerer and M. Drescher, ACS Chem. Biol., 2019, 14, 839–844 CrossRef CAS PubMed.
- A. Feintuch, G. Otting and D. Goldfarb, Methods Enzymol., 2015, 563, 415–457 CAS.
- G. Karthikeyan, A. Bonucci, G. Casano, G. Gerbaud, S. Abel, V. Thome, L. Kodjabachian, A. Magalon, B. Guigliarelli, V. Belle, O. Ouari and E. Mileo, Angew. Chem., Int. Ed., 2018, 57, 1366–1370 CrossRef CAS PubMed.
- S. Bleicken, T. E. Assafa, H. Zhang, C. Elsner, I. Ritsch, M. Pink, S. Rajca, G. Jeschke, A. Rajca and E. Bordignon, ChemistryOpen, 2019, 8, 1057 CrossRef CAS PubMed.
- M. Ormo, A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien and S. J. Remington, Science, 1996, 273, 1392–1395 CrossRef CAS PubMed.
- K. C. Schultz, L. Supekova, Y. Ryu, J. Xie, R. Perera and P. G. Schultz, J. Am. Chem. Soc., 2006, 128, 13984–13985 CrossRef CAS PubMed.
- D. D. Young, T. S. Young, M. Jahnz, I. Ahmad, G. Spraggon and P. G. Schultz, Biochemistry, 2011, 50, 1894–1900 CrossRef CAS PubMed.
- D. C. Kennedy, C. S. McKay, M. C. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993–18001 CrossRef CAS PubMed.
- S. Chillappagari, A. Seubert, H. Trip, O. P. Kuipers, M. A. Marahiel and M. Miethke, J. Bacteriol., 2010, 192, 2512–2524 CrossRef CAS PubMed.
- M. Yang, A. S. Jalloh, W. Wei, J. Zhao, P. Wu and P. R. Chen, Nat. Commun., 2014, 5, 4981 CrossRef CAS PubMed.
- J. Xie and P. G. Schultz, Methods, 2005, 36, 227–238 CrossRef CAS PubMed.
- J. Gunnar, Protein Sci., 2018, 27, 76–85 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp05584c |
| This journal is © the Owner Societies 2020 |