
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical synthesis of a haemathrin sulfoprotein library reveals enhanced thrombin inhibition following tyrosine sulfation†
Daniel
Clayton
a,
Sameer S.
Kulkarni
a,
Jessica
Sayers
a,
Luke J.
Dowman
a,
Jorge
Ripoll-Rozada
 b,
Pedro José Barbosa
Pereira
b and
Richard J.
Payne
*ac
b,
Pedro José Barbosa
Pereira
b and
Richard J.
Payne
*ac
aSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: richard.payne@sydney.edu.au
bIBMC – Instituto de Biologia Molecular e Celular & Instituto de Investigação e Inovação em Saúde, Universidade do Porto, 4200-135 Porto, Portugal
cAustralian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, The University of Sydney, Sydney, NSW 2006, Australia
First published on 21st September 2020
Abstract
The haemathrins are tick-derived thrombin-inhibiting proteins predicted to be post-translationally sulfated. This study reports the ligation-based assembly of eight homogeneously sulfated variants of haemathrin-1 and haemathrin-2. Functional assays revealed a two orders-of-magnitude enhancement in thrombin-inhibitory potency by tyrosine sulfation, thus reinforcing the crucial role of this post-translational modification for the activity of anticoagulant proteins.
Tyrosine (Tyr, Y) O-sulfation is a post-translational modification (PTM) which is found within many higher organisms such as mammals, plants and insects at an estimated proteome-wide abundance of 1–2%.1–4 Catalyzed by tyrosylprotein sulfotransferase (TPST) enzymes found within the trans-Golgi, this PTM is found exclusively on extracellular or membrane-bound protein targets.5–8 While Tyr sulfation has been discovered as a crucial modification on a number of proteins, including several key players in the inflammatory and immune response, the inherent acid lability of the phenolic sulfate ester means that the modification is difficult to retain on the protein during isolation and/or detect by modern mass spectrometric methods.4,9,10 As such, it is likely that native sulfation of many proteins has been overlooked during isolation or is yet to be identified.10
A key example where this has been the case is the “sialome” of hematophagous organisms. While the archetypal leech-derived hirudin anticoagulants have long been known to be sulfated,11–13 it was not until very recently that Tyr O-sulfation was uncovered as a ubiquitous modification of other salivary anti-inflammatory and anticoagulant proteins from a range of blood feeding organisms.13–17 Specifically, we have recently demonstrated that a number of thrombin inhibiting tick (from Haemaphysalis longicornis,15,17Hyalomma marginatum17 and Dermacentor andersoni17) and mosquito (from Anopheles albimanus and Anopheles gambiae)16 salivary proteins are sulfated. Importantly, it has also been demonstrated that sulfation of these proteins uniformly enhances their thrombin inhibitory potency and anticoagulant activity. As such, it is proposed that post-translational tyrosine sulfation plays a crucial role in prevention of host coagulation and represents a key mechanism for acquisition of a bloodmeal by haematophagus organisms.
Based on this recent work, we sought to identify new anticoagulant sulfoproteins from other species of tick. During a bioinformatics analysis, we identified two 59-residue cysteine-free proteins, called haemathrin-1 (1) and haemathrin-2 (2), derived from the hard-bodied tick Haemaphysalis bispinosa. These proteins have been previously identified via PCR amplification and recombinant variants expressed in bacteria were shown to exhibit thrombin inhibitory activity, albeit in unmodified form.18,19 Based on the sequence similarity to madanin-1, a salivary protein from Haemaphysalis longicornis (known to be sulfated)15 together with the presence of conserved Tyr residues flanked by a number of acidic aspartate (Asp) and glutamate (Glu) amino acids (a known recognition motif for TPSTs),20,21 we postulated that both haemathrin-1 (1) and haemathrin-2 (2) are post-translationally sulfated at Y31 and Y34 (Fig. 1). Herein, we describe the rapid and efficient synthesis of all eight modified variants of the haemathrin-1 (1, 3–5) and haemathrin-2 (2, 6–8) proteins via peptide ligation chemistry. It was envisaged that access to these homogeneously modified proteins would enable the effect of specific tyrosine sulfate modifications on haemathrin thrombin inhibitory activity to be uncovered.
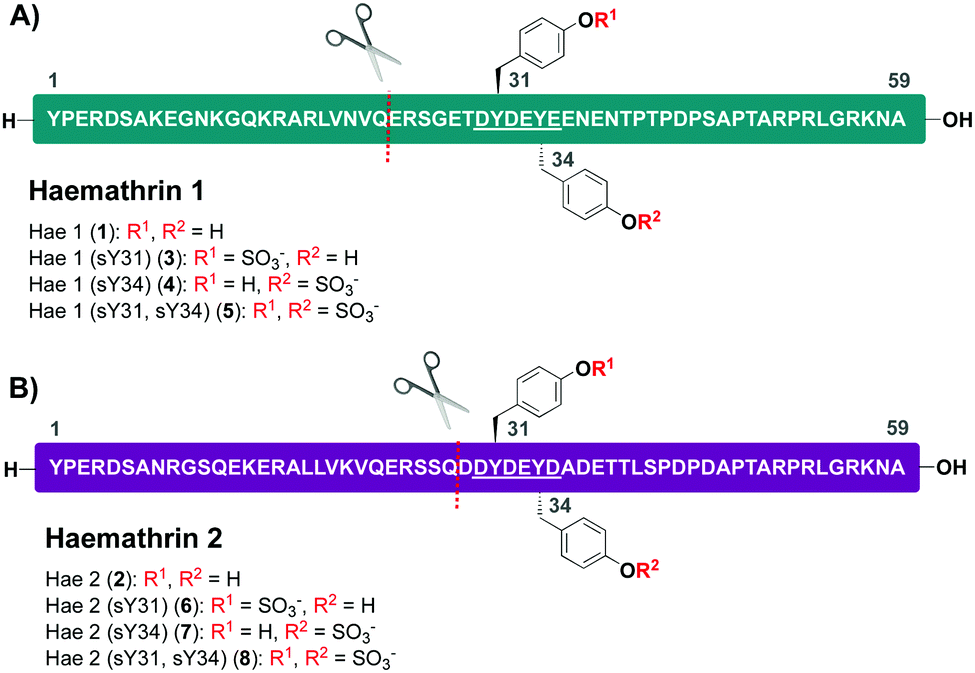 | ||
| Fig. 1 Amino acid sequences and disconnection strategy for (A) haemathrin-1 (1, 3–5) and (B) haemathrin-2 (2, 6–8) sulfoproteins (sY = tyrosine O-sulfate). Ligation junctions are displayed as a red dotted line and acidic recognition sequences for Tyr O-sulfation are underlined. | ||
We proposed that access to this library of sulfated haemathrins could be achieved in a rapid and efficient manner using the additive-free diselenide–selenoester ligation (DSL) methodology recently reported by our group.22 The DSL reaction employs a peptide diselenide dimer fragment bearing an N-terminal selenocystine residue [the oxidized form of selenocysteine (Sec)] and a second peptide fragment bearing a C-terminal selenoester. The reactions proceed cleanly, typically in 1–10 min and can be performed at a range of other ligation junctions through the use of selenylated amino acids.23 Importantly, following the ligation reaction, and without purification, the ligation products can be subjected to in situ deselenization leading to the generation of a native amino acid at the ligation junction [alanine (Ala) in the case of Sec deselenization].24–27 Amongst the selenylated amino acids developed to date, β-selenylated Asp [(β-Se)Asp] and γ-selenylated Glu [(γ-Se)Glu] offer a unique and powerful feature. Specifically, while the deselenization of Sec to Ala typically proceeds to completion over 6–16 hours, in the case of (β-Se)Asp or (γ-Se)Glu, this synthetic transformation is complete within minutes. Importantly, this not only allows proteins to be synthesized on remarkably short time-scales but the rapid nature of the deselenization reactions means that this transformation is chemoselective in the presence of both the thiol and selenol side chains of Cys and Sec residues, respectively.24 We proposed that these features of DSL–deselenization chemistry at (β-Se)Asp or (γ-Se)Glu provided an ideal platform for rapid generation of the target haemathrin sulfoform library (1–8).
While we have previously demonstrated that DSL chemistry is a powerful tool to generate homogeneous sulfoproteins, the use of a hydrophobic neopentyl (nP) sulfate ester-protecting group to prevent acidolysis of the sulfotyrosine (sTyr) residues during synthesis and purification has proven problematic. Specifically, the nP protecting group can lead to reduced solubility of peptide fragments, together with fragment aggregation that can hinder peptide ligation reactions. Moreover, the final nP deprotection step prior to HPLC purification of the sulfoprotein targets proceeds sluggishly, representing the longest reaction step in the synthetic sequence (16 h).17 In order to accelerate access to the haemathrin sulfoform library, we envisaged performing the ligation reactions on peptide fragments with unprotected sTyr residues in this work. Given that DSL reactions are performed at near neutral pH, we proposed that the unprotected sulfate esters would be stable under the reaction conditions and would avoid the aforementioned issues associated with the use of the nP protecting group.
With this in mind, a synthetic strategy was devised by which haemathrin-1 and haemathrin-2 (each 59 amino acid residues long) variants 1–8 could be assembled via ligation of a common N-terminal selenoester fragment with differentially sulfated variants of the C-terminal sequence. Accordingly, suitably positioned amino acid junctions were identified that would be amenable to rapid ligation-deselenization chemistry at (γ-Se)Glu and (β-Se)Asp (Glu-24 in haemathrin-1 and Asp-29 in haemathrin-2, Fig. 1). Syntheses of the C-terminal fragments were achieved using Fmoc-strategy solid-phase peptide synthesis (SPPS) wherein sTyr residues were installed at positions Y31 and Y34 using the commercially available Fmoc-Tyr(SO3nP)-OH building block 9. Selenylated building blocks Boc-(γ-SePMB)Glu-OH (10) and Boc-(β-SePMB)Asp-OH (11) were coupled as the N-terminal residues to afford resin-bound haemathrin-1 (12–15) and haemathrin-2 (16–19) fragments, respectively. Each of the peptides were then deprotected and cleaved from resin using an acidic cocktail followed by PMB deprotection (see ESI,† for full synthetic details).28 The final step to generate the target C-terminal diselenide fragments involved deprotection of the nP sulfate ester(s). While these esters are normally cleaved through the treatment of nucleophilic azide or acetate reagents,17,29 we found that these could be efficiently and cleanly deprotected by simply incubating the fragments in Gnd buffer at 50 °C for 2 hours. Final purification by reverse-phase HPLC then provided the target C-terminal (sulfo)peptide fragments 20–27 as the corresponding diselenide dimers in 3–6% yield based on resin loading (Scheme 1).
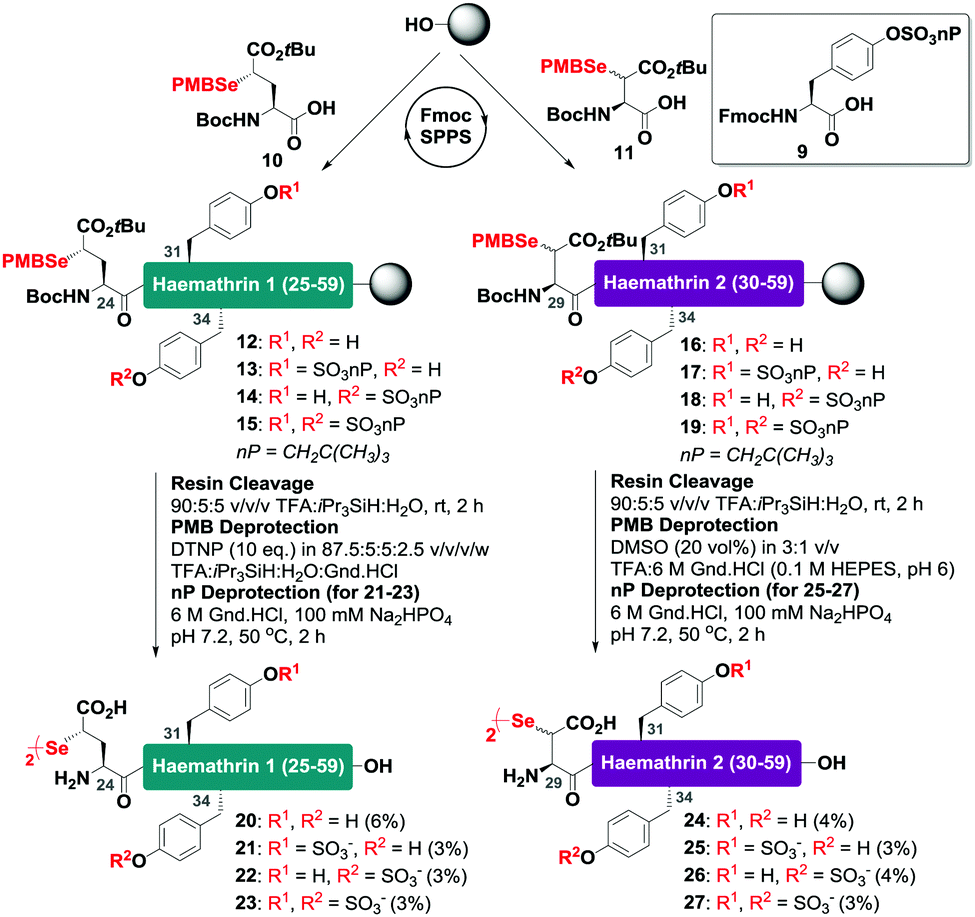 | ||
| Scheme 1 Solid-phase synthesis of the C-terminal diselenide dimer (sulfo)peptides of haemathrin-1 (20–23) and haemathrin-2 (24–27) bearing N-terminal (γ-Se)Glu (β-Se)Asp residues, respectively. | ||
With the C-terminal fragments 20–27 in hand, we next prepared haemathrin-1 (1–23) 28 and haemathrin-2 (1–28) 29 peptide selenoesters. These were both prepared in excellent yield using a side-chain anchoring strategy (see ESI,† for synthetic details and characterization data).30
With the desired N- and C-terminal fragments in hand, attention shifted to the assembly of the haemathrin targets 1–8via DSL-deselenization chemistry (Scheme 2). For the assembly of haemathrin-1 protein 1 and sulfoproteins 3–5, haemathrin-1 (24–59) diselenide dimer fragments 20–23 and peptide selenoester 28 were dissolved in ligation buffer (6 M Gnd·HCl, 0.1 M Na2HPO4, pH 7.2; 2.5 mM final concentration with respect to the diselenide) and the pH of the resulting reaction mixture was adjusted to pH 6 upon mixing (Scheme 2A). Each of the ligation reactions proceeded to completion within 5 min. The reaction mixtures were next extracted with hexane to remove precipitated diphenyl diselenide (DPDS) – an insoluble by-product generated during ligation that quenches the subsequent radical deselenization if not removed. The ligation mixture was next treated with an equal volume of freshly prepared, degassed solution of 250 mM TCEP, 25 mM DTT in ligation buffer to effect deselenization of the γ-Se moiety at the ligation junction, which proceeded to completion within 5 min as assessed by UPLC-MS analysis (Scheme 2B). Importantly, both the DSL and deselenization transformations proceeded cleanly and efficiently when using sulfopeptides with unprotected sTyr residues. Purification of the DSL–deselenization products via reverse-phase HPLC afforded haemathrin-1 (sulfo)proteins 1 and 3–5 in good yields (39–55%) and purities (Scheme 2C). The haemathrin-2 (sulfo)proteins 2 and 6–8 were prepared in a similar manner by reacting diselenide dimer peptides 24–27 bearing N-terminal (β-Se)Asp residues with peptide selenoester 29 under DSL conditions (Scheme 2D). After 5 min the ligation products were treated with TCEP and DTT to facilitate clean deselenization (Scheme 2E). Following purification by reverse-phase HPLC the target haemathrin-2 (sulfo)proteins 2 and 6–8 were isolated in good yield (45–54%) and purities (Scheme 2F).
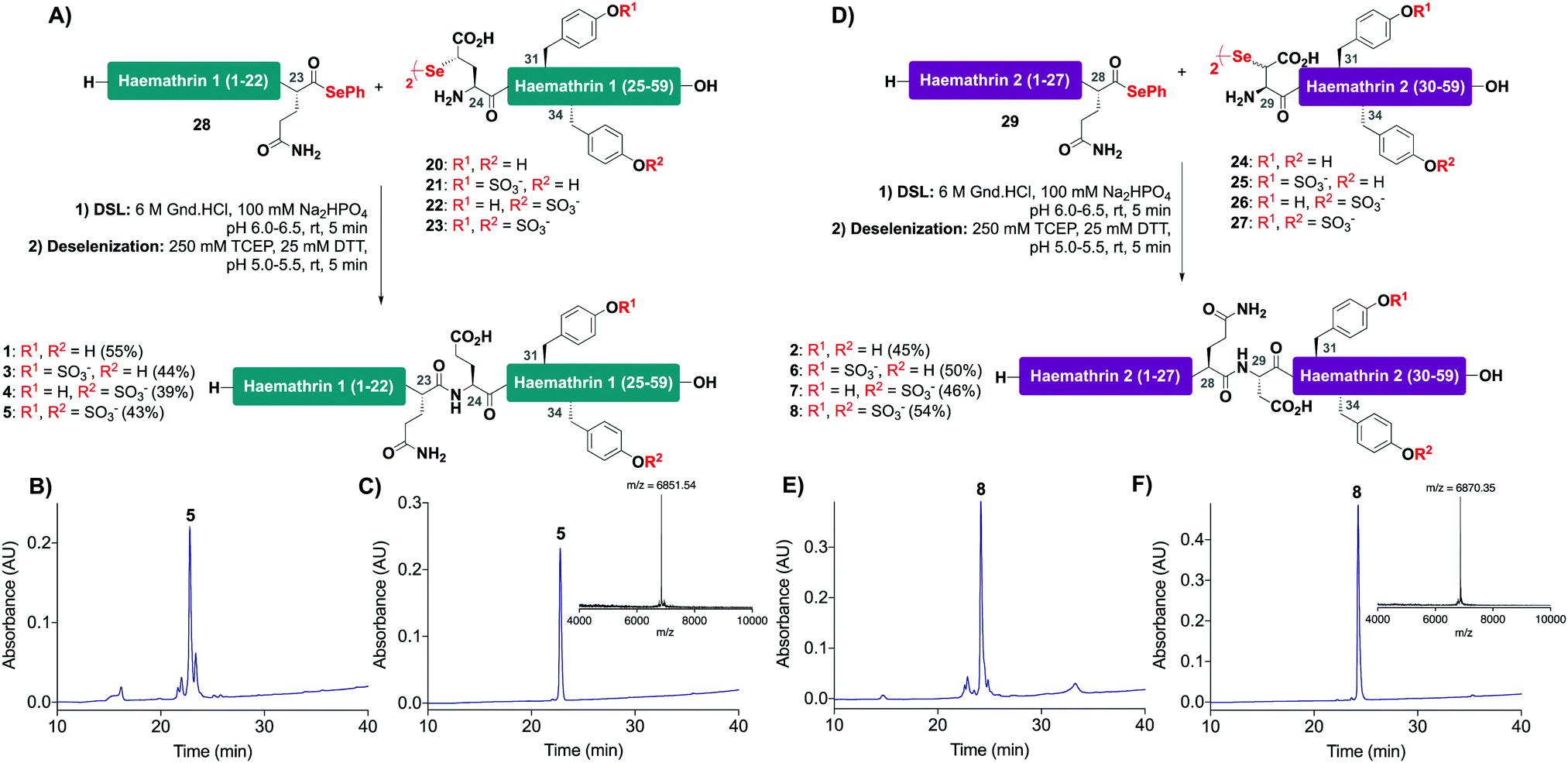 | ||
| Scheme 2 Synthesis of haemathrin-1 and haemathrin-2 (sulfo)proteins 1–8via a one-pot DSL–deselenization approach. (A) One-pot synthesis of haemathrin-1 variants 1 and 3–5; (B) HPLC chromatogram of crude DSL–deselenization reaction to afford doubly sulfated haemathrin-1 5; (C) HPLC chromatogram of purified doubly sulfated haemathrin-1 5 (Rt = 22.8 min, gradient 0–50% B over 30 min, λ = 230 nm) with MALDI-TOF MS inset; (D) one-pot synthesis of haemathrin-2 variants 2 and 6–8via DSL-deselenization; (E) HPLC chromatogram of crude DSL–deselenization reaction to afford doubly sulfated haemathrin-2 8; (F) HPLC chromatogram of purified doubly sulfated haemathrin-2 8 (Rt = 24.2 min, gradient 0–50% B over 30 min, λ = 230 nm) with MALDI-TOF MS inset. | ||
To investigate the effect of sulfation on the biological activity of the proteins, we next assessed synthetic (sulfo)proteins 1–8 for their inhibitory activity against human α-thrombin via a kinetic assay employing the chromogenic substrate, Tos-Gly-Pro-Arg-p-nitroanilide (Fig. 2). The non-sulfated haemathrin-1 (1) and haemathrin-2 (2) exhibited similar potency against the activity of α-thrombin, Ki = 210 and 290 nM (cf. IC50 values of 40–46 μM reported for the recombinant material).19 Interestingly, sulfation at Y31 was found to impart a dramatic improvement in activity (ca. 1.5 orders of magnitude) for both singly sulfated variants of haemathrin-1 3 (Ki = 13 nM) and haemathrin-2 6 (Ki = 18 nM). Sulfation at Y34 in 4 (Ki = 58 nM) and 7 (Ki = 24 nM) still led to a marked (but less pronounced) improvement in thrombin inhibitory activity. However, sulfation at both Y31 and Y34 in haemathrin-1 5 and haemathrin-2 8 led to a further improvement in activity (two orders-of-magnitude over the unsulfated proteins 1 and 2) with inhibition constants of 2.4 and 1.9 nM, respectively. In addition to screening against α-thrombin, we also assessed the inhibitory activity of the unsulfated (1 and 2) and doubly sulfated (5 and 8) haemathrins against human γ-thrombin, a thrombin variant with a disrupted exosite I, an important binding surface for many polypeptide thrombin inhibitors, e.g., the leech-derived hirudin (see ESI,† for details). These experiments revealed that each of these proteins exhibited similar inhibition constants against both α- and γ-thrombin, suggesting a thrombin inhibitory mode via exosite II binding, in addition to the catalytic site. This proposal is further supported by the sequence homology between the haemathrins and madanin-1, a salivary protein from the hard tick Haemaphysalis longicornis which has been validated as an exosite II binding inhibitor of α-thrombin using X-ray crystallography.15 Overall, these results highlight a vital role of sulfation in the enhancement of thrombin inhibitory activity of the haemathrins and, akin to other tick-derived salivary proteins, likely plays a role in the effective acquisition of a bloodmeal from the host.
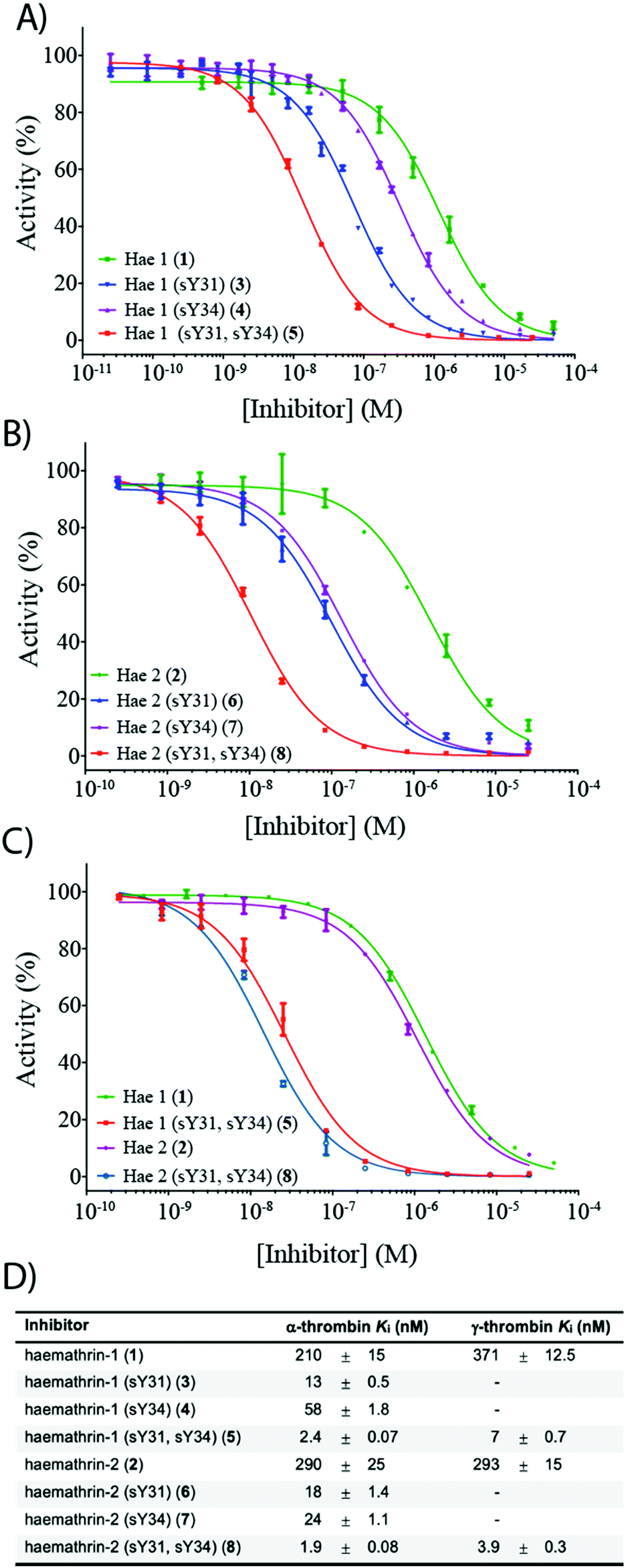 | ||
| Fig. 2 Dose–response curves for the inhibition of (A) α-thrombin by haemathrin-1 (sulfo)proteins 1, 3–5; (B) α-thrombin by haemathrin-2 (sulfo)proteins 2, 6–8 and (C) γ-thrombin by unsulfated and doubly sulfated haemathrin-1 and -2 proteins 1, 2, 5 and 8 using an amidolytic assay. Error bars represent S.E.M. values; (D) inhibition constants (Ki, ±S.E.M.) for haemathrin-1 and -2 (sulfo)proteins. Data were determined by fitting the inhibited steady-state velocities to the Morrison model.31 | ||
Conclusions
In conclusion, we have verified the critical role of Tyr sulfation on the thrombin inhibitory activity of haemathrin-1 and haemathrin-2 – two anticoagulant proteins from the tick species Haemaphysalis bispinosa – through the ligation-based synthesis of a sulfoprotein library. In order to accelerate access to these homogeneously sulfated proteins, we employed DSL chemistry at (β-Se)Asp or (γ-Se)Glu with fully unprotected sulfopeptide fragments followed by in situ deselenization. Both of these reactions proceeded to completion within 5 min making this synthetic approach an extremely fast and efficient means for accessing homogeneously modified proteins by chemical synthesis. Access to this sulfoprotein library enabled the determination of key structure–activity relationships. Specifically, sulfation at Y31 was shown to provide a significant enhancement in thrombin inhibitory activity, while sulfation at both Y31 and Y34 led to the most potent activity, with inhibition constants two orders-of-magnitude lower than those determined for the unmodified proteins. Taken together, the results of this study emphasize the critical role of Tyr sulfation on protein function, as well as its involvement in haematophagous activity within a multitude of organisms, many of which are significant vectors for human disease. In addition, the synthetic approach employed here should find use in the generation of a wide range of other site-specifically modified proteins, including but not limited to sulfation. As such, these synthetic tools will expedite our ability to probe the critical role of PTMs on the structure and biological function of proteins.Conflicts of interest
The authors declare no conflicts of competing financial interest(s).Acknowledgements
The authors would like to acknowledge funding from the National Health and Medical Research Council Investigator Grant Scheme APP1174941 (to R. J. P.) and the John A. Lamberton Research Scholarship (to L. J. D. and J. S.). This work was funded in part by national funds through Fundação para a Ciência e a Tecnologia (Portugal) in the form of Contract DL 57/2016/CP1355/CT0011 (to J. R.-R.). The authors also thank Dr Nicholas Proschogo and Dr Cody Szczepina for their assistance with analytical mass spectrometry and liquid chromatography.Notes and references
- W. B. Huttner, Sulphation of Tyrosine Residues – A Widespread Modification of Proteins, Nature, 1982, 299, 273–276 Search PubMed.
- J. W. Kehoe and C. R. Bertozzi, Tyrosine sulfation: a modulator of extracellular protein–protein interactions, Chem. Biol., 2000, 7, 57–61 Search PubMed.
- M. J. Stone, S. Chuang, X. Hou, M. Shoham and J. Z. Zhu, Tyrosine sulfation: an increasingly recognised post-translational modification of secreted proteins, New Biotechnol., 2009, 25, 299–317 Search PubMed.
- M. J. Stone and R. J. Payne, Homogeneous sulfopeptides and sulfoproteins: synthetic approaches and applications to characterize the effects of tyrosine sulfation on biochemical function, Acc. Chem. Res., 2015, 48, 2251–2261 Search PubMed.
- P. A. Baeuerle and W. B. Huttner, Tyrosine sulfation is a trans-Golgi-specific protein modification, J. Cell Biol., 1987, 105, 2655–2664 Search PubMed.
- R. Beisswanger, D. Corbeil, C. Vannier, C. Thiele, U. Dohrmann, R. Kellner, K. Ashman, C. Niehrs and W. B. Huttner, Existence of distinct tyrosylprotein sulfotransferase genes: molecular characterization of tyrosylprotein sulfotransferase-2, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11134–11139 Search PubMed.
- C. Niehrs and W. B. Huttner, Purification and characterization of tyrosylprotein sulfotransferase, EMBO J., 1990, 9, 35–42 Search PubMed.
- Y. Ouyang, W. S. Lane and K. L. Moore, Tyrosylprotein sulfotransferase: purification and molecular cloning of an enzyme that catalyzes tyrosine O-sulfation, a common posttranslational modification of eukaryotic
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) proteins, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2896–2901 Search PubMed.
proteins, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2896–2901 Search PubMed. - R. P. Bhusal, S. R. Foster and M. J. Stone, Structural basis of chemokine and receptor interactions: key regulators of leukocyte recruitment in inflammatory responses, Protein Sci., 2020, 29, 420–432 Search PubMed.
- J. W. C. Maxwell and R. J. Payne, Revealing the Functional Roles of Tyrosine Sulfation using Synthetic Sulfopeptides and Sulfoproteins, Curr. Opin. Chem. Biol., 2020, 58, 72–85 Search PubMed.
- S. R. Stone and J. Hofsteenge, Kinetics of the inhibition of thrombin by hirudin, Biochemistry, 1986, 25, 4622–4628 Search PubMed.
- C. C. Liu and P. G. Schultz, Recombinant expression of selectively sulfated proteins in Escherichia coli, Nat. Biotechnol., 2006, 24, 1436–1440 Search PubMed.
- Y. S. Hsieh, L. C. Wijeyewickrema, B. L. Wilkinson, R. N. Pike and R. J. Payne, Total synthesis of homogeneous variants of hirudin P6: a post-translationally modified anti-thrombotic leech-derived protein, Angew. Chem., Int. Ed., 2014, 53, 3947–3951 Search PubMed.
- C. Franck, S. R. Foster, J. Johansen-Leete, S. Chowdhury, M. Cielesh, R. P. Bhusal, J. P. Mackay, M. Larance, M. J. Stone and R. J. Payne, Semisynthesis of an evasin from tick saliva reveals a critical role of tyrosine sulfation for chemokine binding and inhibition, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12657–12664 Search PubMed.
- R. E. Thompson, X. Liu, J. Ripoll-Rozada, N. Alonso-Garcia, B. L. Parker, P. J. Pereira and R. J. Payne, Tyrosine Sulfation Modulates Activity of Tick-Derived Thrombin Inhibitors, Nat. Chem., 2017, 9, 909–917 Search PubMed.
- E. E. Watson, X. Liu, R. E. Thompson, J. Ripoll-Rozada, M. Wu, I. Alwis, A. Gori, C.-T. Loh, B. L. Parker, G. Otting, S. Jackson, P. J. B. Pereira and R. J. Payne, Mosquito-Derived Anophelin Sulfoproteins Are Potent Antithrombotics, ACS Cent. Sci., 2018, 4, 468–476 Search PubMed.
- E. E. Watson, J. Ripoll-Rozada, A. C. Lee, M. C. L. Wu, C. Franck, T. Pasch, B. Premdjee, J. Sayers, M. F. Pinto, P. M. Martins, S. P. Jackson, P. J. B. Pereira and R. J. Payne, Rapid assembly and profiling of an anticoagulant sulfoprotein library, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13873–13878 Search PubMed.
- W. Jablonka, M. Kotsyfakis, D. M. Mizurini, R. Q. Monteiro, J. Lukszo, S. K. Drake, J. M. C. Ribeiro and J. F. Andersen, Identification and mechanistic analysis of a novel tick-derived inhibitor of thrombin, PLoS One, 2015, 10, e0133991 Search PubMed.
- R. K. Brahma, G. Blanchet, S. Kaur, R. M. Kini and R. Doley, Expression and characterization of haemathrins, madanin-like thrombin inhibitors, isolated from the salivary gland of tick Haemaphysalis bispinosa (Acari: Ixodidae), Thromb. Res., 2017, 152, 20–29 Search PubMed.
- F. Monigatti, E. Gasteiger, A. Bairoch and E. Jung, The sulfinator: predicting tyrosine sulfation sites in protein sequences, Bioinformatics, 2002, 18, 769–770 Search PubMed.
- T. Teramoto, Y. Fujikawa, Y. Kawaguchi, K. Kurogi, M. Soejima, R. Adachi, Y. Nakanishi, E. Mishiro-Sato, M.-C. Liu, Y. Sakakibara, M. Suiko, M. Kimura and Y. Kakuta, Crystal structure of human tyrosylprotein sulfotransferase-2 reveals the mechanism of protein tyrosine sulfation reaction, Nat. Commun., 2013, 4, 1572 Search PubMed.
- N. J. Mitchell, L. R. Malins, X. Liu, R. E. Thompson, B. Chan, L. Radom and R. J. Payne, Rapid Additive-Free Selenocystine–Selenoester Peptide Ligation, J. Am. Chem. Soc., 2015, 137, 14011–14014 Search PubMed.
- S. S. Kulkarni, J. Sayers, B. Premdjee and R. J. Payne, Rapid and efficient protein synthesis through expansion of the native chemical ligation concept, Nat. Rev. Chem., 2018, 2, 0122 Search PubMed.
- N. J. Mitchell, J. Sayers, S. S. Kulkarni, D. Clayton, A. M. Goldys, J. Ripoll-Rozada, P. J. B. Pereira, B. Chan, L. Radom and R. J. Payne, Accelerated Protein Synthesis via One-Pot Ligation-Deselenization Chemistry, Chem, 2017, 2, 703–715 Search PubMed.
- X. Wang, J. Sanchez, M. Stone and R. J. Payne, Sulfation of Human Cytomegalovirus Protein UL22A Enhances Binding to the Chemokine RANTES, Angew. Chem., Int. Ed., 2017, 56, 8490–8494 Search PubMed.
- J. Sayers, P. M. T. Karpati, N. J. Mitchell, A. M. Goldys, S. M. Kwong, N. Firth, B. Chan and R. J. Payne, Construction of Challenging Proline–Proline Junctions via Diselenide–Selenoester Ligation Chemistry, J. Am. Chem. Soc., 2018, 140, 13327–13334 Search PubMed.
- X. Wang, L. Corcilius, B. Premdjee and R. J. Payne, Synthesis and Utility of β-Selenophenylalanine and β-Selenoleucine in Diselenide–Selenoester Ligation, J. Org. Chem., 2020, 85, 1567–1578 Search PubMed.
- S. S. Kulkarni, E. E. Watson, B. Premdjee, K. W. Conde-Frieboes and R. J. Payne, Diselenide–Selenoester Ligation for Chemical Protein Synthesis, Nat. Protoc., 2019, 14, 2229–2257 Search PubMed.
- L. S. Simpson and T. S. Widlanski, A Comprehensive Approach to the Synthesis of Sulfate Esters, J. Am. Chem. Soc., 2006, 128, 1605–1610 Search PubMed.
- C. C. Hanna, S. S. Kulkarni, E. E. Watson, B. Premdjee and R. J. Payne, Solid-phase synthesis of peptide selenoesters via a side-chain anchoring strategy, Chem. Commun., 2017, 53, 5424–5427 Search PubMed.
- J. W. Williams and J. F. Morrison, The kinetics of reversible tight-binding inhibition, Methods Enzymol., 1979, 63, 437–467 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00146e |
| This journal is © The Royal Society of Chemistry 2020 |