
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering a CsPbBr3-based nanocomposite for efficient photocatalytic CO2 reduction: improved charge separation concomitant with increased activity sites†
Xiao-Xuan Guoa,
Shang-Feng Tanga,
Yan-Fei Mua,
Li-Yuan Wua,
Guang-Xing Donga and
Min Zhang *ab
*ab
aInstitute for New Energy Materials and Low-Carbon Technologies, Tianjin University of Technology, Tianjin, 300384, China. E-mail: zm2016@email.tjut.edu.cn
bTianjin Key Laboratory of Organic Solar Cells and Photochemical Conversion, School of Chemistry and Chemical Engineering, Tianjin University of Technology, Tianjin, 300384, China
First published on 25th October 2019
Abstract
Metal-halide perovskite nanocrystals have emerged as one of the promising photocatalysts in the photocatalysis field owing to their low-cost and excellent optoelectronic properties. However, this type of nanocrystals generally displays low activity in photocatalytic CO2 reduction owing to the lack of intrinsic catalytic sites and insufficient charge separation. Herein, we functionalized CsPbBr3 nanocrystals with graphitic carbon nitride, containing titanium-oxide species (TiO-CN) to develop an efficient composite catalyst system for photocatalytic CO2 reduction using water as the electron source. Compared to its congener with pristine CsPbBr3, the introduction of TiO-CN could not only increase the number of active sites, but also led to a swift interfacial charge separation between CsPbBr3 and TiO-CN due to their favorable energy-offsets and strong chemical bonding behaviors, which endowed this composite system with an obviously enhanced photocatalytic activity in the reduction of CO2 to CO with water as the sacrificial reductant. Over 3-fold and 6-fold higher activities than those of pristine CsPbBr3 nanocrystals and TiO-CN nanosheets, respectively, were observed under visible light irradiation. Our study provides an effective strategy for improving the photocatalytic activity of metal-halide perovskite nanocrystals, thus promoting their photocatalytic application in the field of artificial photosynthesis.
Introduction
Sunlight-driven photocatalytic CO2 conversion into high-value added fuels or chemical feedstocks is generally perceived as a promising solution to simultaneously alleviate both energy crisis and environmental problems and has received intense attention in the field of artificial photosynthesis.1–5 In recent years, most of the attention on this technology has been focused on the development of cost-effective photocatalyst systems.6–14 In this regard, semiconductor nanocrystals possess excellent properties, such as finely tunable band gaps, large extinction coefficients and long photogenerated carrier lifetimes, and have been actively pursued as photocatalysts in the field of photocatalysis.15–18 Most recently, metal-halide perovskite nanocrystals, a type of fascinating semiconductor candidates for sunlight harvesting, have also emerged as a class of promising photocatalysts19–31 owing to their high defect tolerance and low-cost as well as the facile processing methods,32–35 which have been dramatically exploited in optoelectronic devices.36–39In the field of photocatalytic CO2 reduction, however, almost all the pristine metal-halide perovskite nanocrystal photocatalysts exhibit poor photocatalytic activity owing to the lack of intrinsic catalytic sites and insufficient charge separation.20,22,24,27,29,31 As a learning from the previous studies on conventional metal chalcogenide semiconductor nanocrystals, loading metal-halide perovskite nanocrystals on some two-dimensional (2D) materials, such as graphene oxide30 or graphitic carbon nitride22 (g-C3N4), is expected to enhance the charge separation efficiency, leading to improved photocatalytic activity in CO2 reduction. However, the lack of intrinsic catalytic sites in these 2D materials also limits the further improvement in their photocatalytic performance in CO2 reduction. Indeed, our preliminary experiment showed that the CsPbBr3@g-C3N4 composite photocatalyst exhibit a little improvement in photocatalytic activity for CO2 reduction compared to pristine CsPbBr3 under the irradiation of visible light. It is known that the polymeric material g-C3N4 is a good support material for single atom40–47 and metal nanoparticles,48–50 and enhanced solar fuel production can be achieved along with the introduction of these metal catalytic sites.
On the basis of the above analysis, to simultaneously increase the number of catalytic sites and improve the charge separation efficiency of the CsPbBr3-based photocatalyst for CO2 reduction, herein, we functionalized the CsPbBr3 nanocrystals with graphitic carbon nitride nanosheets, containing titanium-oxide species (TiO-CN), to develop an efficient composite photocatalytic system (coded as CsPbBr3@TiO-CN) for photocatalytic CO2 reduction. Structural characterization revealed that the CsPbBr3 nanocrystals were closely anchored on 2D TiO-CN via N–Br and O–Br bonding, as discussed below. We further systematically compared its structure, light-harvesting ability, band gap alignment, charge generation and separation, as well as the photocatalytic activity in CO2 reduction, with those of the reference photocatalysts based on pristine CsPbBr3 nanocrystals and TiO-CN nanosheets.
Result and discussion
The 2D nanosheets of TiO-CN were prepared according to the method described in a previous publication,47 which is based on a simple dicyandiamide blowing method.51 The pristine CsPbBr3 nanocrystals were synthesized according to a published procedure using a conventional hot injection approach.52 The CsPbBr3@TiO-CN composite was obtained by simply mixing CsPbBr3 nanocrystals and TiO-CN nanosheets in hexane solvent. The detailed procedure is described in the ESI.† Briefly, CsPbBr3 nanocrystals and TiO-CN nanosheets were first added into hexane solvent. After ultrasonication for 15 minutes and successive stirring for 2 hours, the color of the solution changed from yellow-green to colorless (Fig. S1†), implying that the CsPbBr3 nanocrystals dispersed in hexane were successfully loaded onto the TiO-CN nanosheets. Then, the CsPbBr3@TiO-CN composite was obtained after centrifugation, followed by drying.The morphology and composition of the CsPbBr3 nanocrystals on TiO-CN nanosheets were first evaluated by transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) measurements. As shown in Fig. 1a and b, the CsPbBr3 nanocrystals could be clearly observed on the sheet-like TiO-CN. The average size of the CsPbBr3 nanocrystals on TiO-CN was approximately 15 nm (Fig. 1b), which is similar to that of the as-prepared CsPbBr3 nanocrystals (Fig. S2†). The HRTEM image of the CsPbBr3 nanocrystals on TiO-CN (Fig. 1c) showed a clear lattice spacing of 0.58 nm, corresponding to that of the (100) plane in the monoclinic CsPbBr3 phase.53 As further demonstrated in Fig. 1d, the X-ray diffraction (XRD) pattern of the CsPbBr3 nanocrystals displayed typical diffraction peaks for monoclinic CsPbBr3, which could be well fitted to the standard pattern of JCPDS 00-018-0364. In addition, the XRD pattern of TiO-CN (Fig. 1d) was similar to that of g-C3N4, which is consistent with a previous result.47 Moreover, the diffraction peaks of both CsPbBr3 nanocrystals and TiO-CN nanosheets could be clearly observed in the XRD pattern of the CsPbBr3@TiO-CN composite (Fig. 1d). Furthermore, the TEM image (Fig. S3a†) and element mappings (Fig. S3b–h†) of the CsPbBr3@TiO-CN composite exhibited a uniform dispersion of cesium, lead and bromine, which indicates that the CsPbBr3 nanocrystals were homogeneously loaded on the TiO-CN nanosheets.
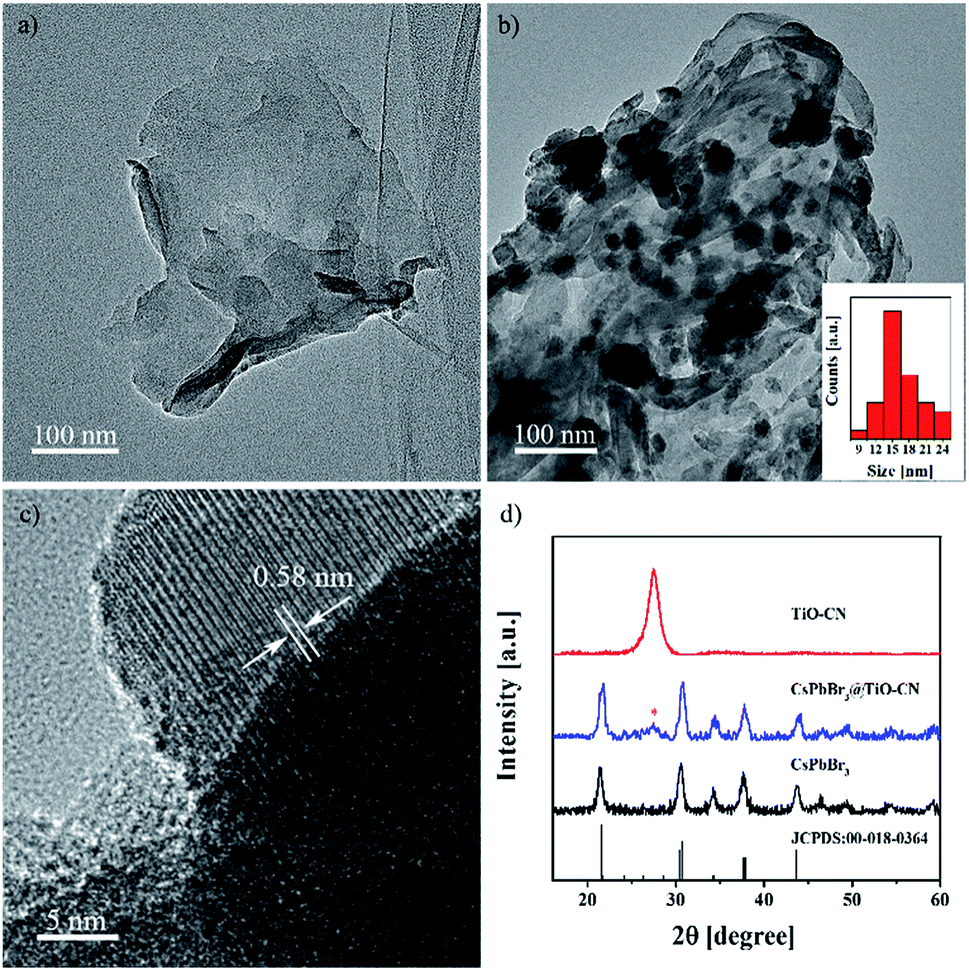 | ||
| Fig. 1 TEM images of TiO-CN (a) without and (b) with CsPbBr3 nanocrystal loading. The black dots in panel b are the CsPbBr3 nanocrystals, and the particle size distribution of CsPbBr3 is given in the inset in panel b. (c) HRTEM image with the lattice spacing of monoclinic CsPbBr3 on TiO-CN. (d) XRD patterns of TiO-CN, CsPbBr3 and CsPbBr3@TiO-CN. | ||
It is widely recognized that the connection pattern between the components in a composite has an important effect on the photogenerated carrier transfer properties of the composite. Therefore, the interaction behavior of the CsPbBr3 nanocrystals and the TiO-CN nanosheets was first investigated with the aid of Fourier transform infrared (FTIR) spectrum measurements. In contrast to pristine TiO-CN with a strong characteristic absorption band at ∼1241 cm−1 (Fig. S4†)54 corresponding to the typical stretching of C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N groups in TiO-CN, the characteristic absorption band in CsPbBr3@TiO-CN displayed an obviously reduced intensity, as presented in Fig. S4.† This phenomenon indicated the existence of a strong bonding interaction between NHx and Br− correspondingly in TiO-CN and CsPbBr3, originating from the easily formed ionic bonding.
N groups in TiO-CN, the characteristic absorption band in CsPbBr3@TiO-CN displayed an obviously reduced intensity, as presented in Fig. S4.† This phenomenon indicated the existence of a strong bonding interaction between NHx and Br− correspondingly in TiO-CN and CsPbBr3, originating from the easily formed ionic bonding.
The detailed chemical bonding behavior between CsPbBr3 and TiO-CN was further analyzed by performing high-resolution X-ray photoelectron spectroscopy (XPS) measurements. The N 1s spectrum of TiO-CN could be resolved into four peaks, as presented in Fig. 2a. Apart from the three typical peaks at 398.6, 399.5 and 401.2 eV for nitrogen in different chemical environments (C–NC, N–(C)3, and NHx), there was a characteristic peak at 404.5 eV in TiO-CN, which was attributed to the existence of C–N–H in the heterocycles, possessing positive charge localization.55,56 It is worthwhile to note that this characteristic peak at around 404.5 eV disappeared after loading CsPbBr3 on TiO-CN, indicating that the localization of the positive charge can be neutralized by the lone pair of electrons from Br on the surface of CsPbBr3 by forming the chemical bond of C–N–Br. As a result, the N 1s peaks in CsPbB3@TiO-CN showed a perceptible shift to the low-energy region with respect to that of pristine TiO-CN, as observed in Fig. 2a. In addition, the XPS signal for oxygen (Fig. 2b) in the TiO-CN component showed that the binding energy of O 1s in CsPbB3@TiO-CN had clearly negative-shifted in comparison, suggesting the occurrence of a bonding interaction between Br on the CsPbBr3 surface and OH in TiO-CN. Therefore, loading CsPbBr3 on TiO-CN would result in electron deficient Br on the CsPbBr3 surface, which was further demonstrated by the XPS signal of Br 3d. As shown in Fig. 2c, the Br 3d5/2 and Br 3d3/2 peaks appeared at 68.2 and 69.2 eV in pristine CsPbBr3 nanocrystals,57 which positively shifted to 68.4 and 69.4 eV in CsPbBr3@TiO-CN, respectively. Subsequently, the binding energies of Pb 4f7/2 and Pb 4f5/2 in CsPbB3@TiO-CN also correspondingly shifted to high energies from 138.4 and 143.2 eV to 138.6 and 143.5 eV, as illustrated in Fig. 2d.
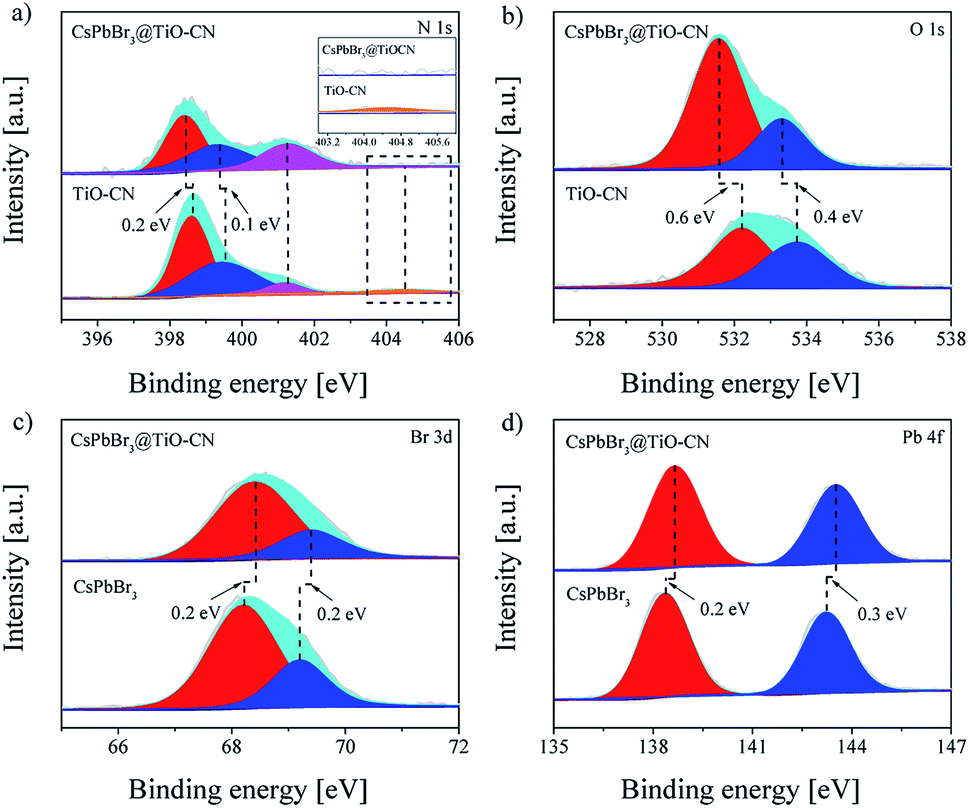 | ||
| Fig. 2 High-resolution XPS spectra of CsPbBr3, TiO-CN and CsPbBr3@TiO-CN: (a) N 1s, (b) O 1s, (c) Br 3d and (d) Pb 4f. | ||
For a typical photocatalyst, a good visible light response is the most basic premise to achieve a visible-light-driven catalytic reaction. Thereby, we first evaluated the light-harvesting capacity of the CsPbBr3@TiO-CN composite by recording the electronic absorption spectra of CsPbBr3@TiO-CN to preliminarily assess its application potential as a photocatalyst for CO2 reduction under the irradiation of visible light. As depicted in Fig. 3a, loading the CsPbBr3 nanocrystals on the TiO-CN nanosheets caused an augmented light response at a desirable longer wavelength compared to pristine TiO-CN owing to the good light-harvesting capacity of CsPbBr3 in the visible region (Fig. 3a). The photoresponse onset wavelength of CsPbBr3@TiO-CN was red-shifted from ∼460 nm to ∼550 nm. Further, the Tauc plots of CsPbBr3 and TiO-CN are given in Fig. 3b and c. The band gaps of CsPbBr3 and TiO-CN were obtained from the intercepts of the extrapolation lines and energy axes as 2.30 and 2.75 eV, respectively.
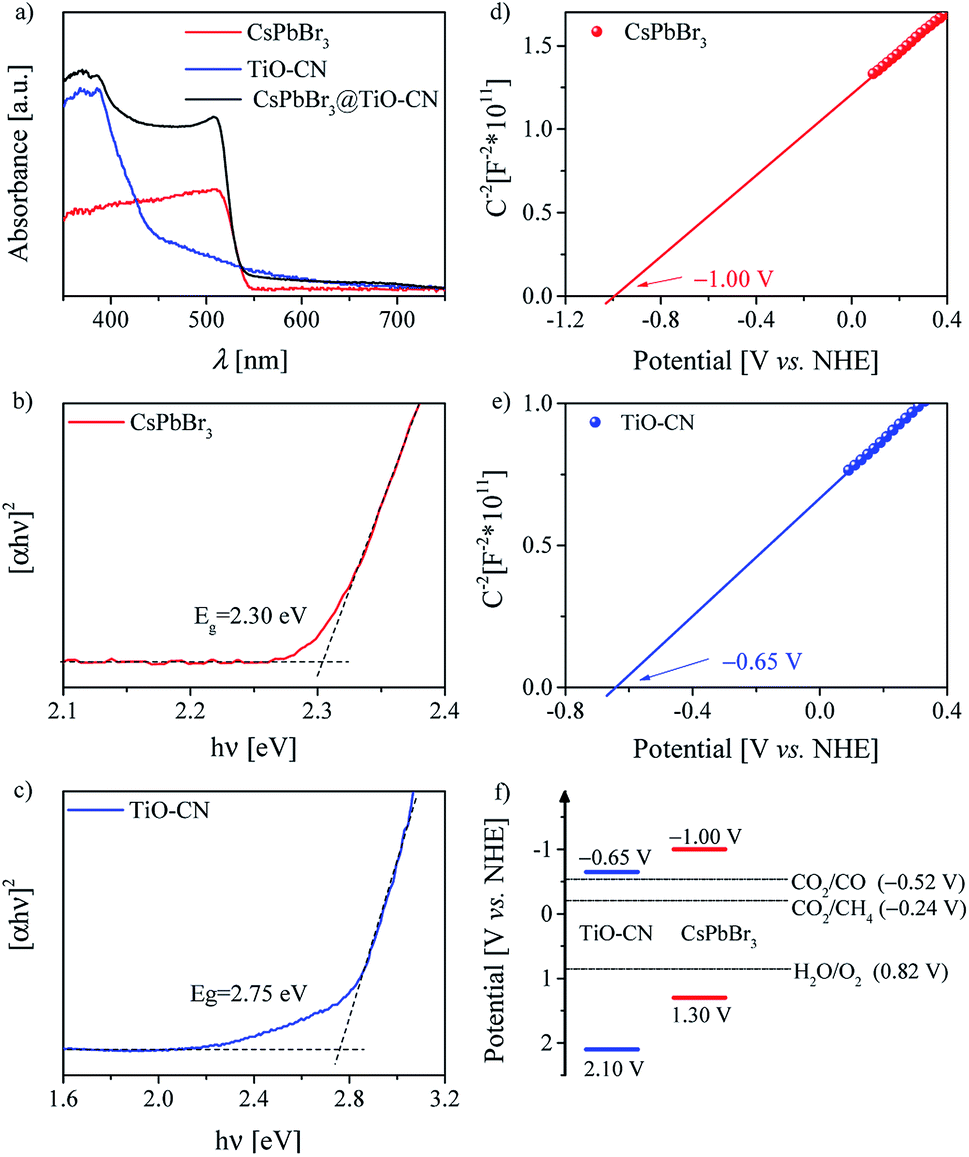 | ||
| Fig. 3 (a) UV/Vis diffuse reflectance spectra of CsPbBr3, TiO-CN and CsPbBr3@TiO-CN. Tauc plots of (b) CsPbBr3 and (c) TiO-CN. Mott–Schottky plots of (d) CsPbBr3 and (e) TiO-CN. (f) Schematic illustration of the band structures of CsPbBr3 and TiO-CN derived from the results of flat-band potential and UV/Vis diffuse reflectance spectroscopy measurements. | ||
The energy-offset between the conduction band edges of CsPbBr3 and TiO-CN plays a pivotal role in determining the dynamic process of photogenerated electron transfer. Since the conduction band edge potential is practically associated with the flat band potential in n-type semiconductors,58 herein, we employed electrochemical measurements to evaluate the conduction band edge of CsPbBr3 and TiO-CN by constructing the Mott–Schottky plots, as depicted in Fig. 3d and e. The Mott–Schottky plots of both CsPbBr3 and TiO-CN clearly exhibited positive slopes, indicating that CsPbBr3 and TiO-CN were n-type semiconductors. The flat band potentials of CsPbBr3 and TiO-CN were determined from the corresponding intercepts of the linear plot region with the potential axis to be −1.00 and −0.65 V vs. the normal hydrogen electrode (NHE), respectively, which indicate that the photogenerated electrons in the CsPbBr3 nanocrystals were transferred to the TiO-CN nanosheets, and the reduction reaction of CO2 to CO (−0.52 V vs. NHE) by the photogenerated electrons in TiO-CN is thermodynamically permitted. In addition, the corresponding valence band edge potentials of CsPbBr3 and TiO-CN could be derived from their band gaps and conduction-band edge potentials, as 1.30 and 2.10 V vs. NHE, as illustrated in Fig. 3f, indicating that the photogenerated holes in the TiO-CN nanosheets could be transferred to the CsPbBr3 nanocrystals, and the photogenerated holes in the CsPbBr3 nanocrystals had enough driving force to trigger the water oxidation reaction to generate O2 (0.82 V vs. NHE).
We speculated that the formation of chemical bonds and favorable energy-offsets between CsPbBr3 and TiO-CN would facilitate the photogenerated electron transfer from CsPbBr3 to TiO-CN in the CsPbBr3@TiO-CN composite. To check out this, steady state photoluminescence (PL) measurements were firstly recorded to investigate the photoexcited electron transfer processes in the CsPbBr3@TiO-CN composite. Herein, pristine CsPbBr3 was selected as the reference. As depicted in Fig. 4a, the pristine CsPbBr3 nanocrystals exhibited a strong PL peak at 519 nm, and loading CsPbBr3 on the TiO-CN surface significantly reduced the PL intensity, indicating the occurrence of a swift photogenerated electron transfer from CsPbBr3 to TiO-CN. Furthermore, we recorded the time-resolved photoluminescence (TRPL) traces of CsPbBr3 and CsPbBr3@TiO-CN samples to scrutinize the photogenerated electron transfer dynamics using the time-correlated single photon counting technique (TCSPC), as shown in Fig. 4b. An excitation wavelength of 450 nm was picked to selectively excite CsPbBr3. The dynamic PL trace of pristine CsPbBr3 (red symbols) in Fig. 4b can be ascribed to the deactivation of the optically generated excitons in CsPbBr3 through radiative and nonradiative processes. There was an obviously accelerated PL decay (blue symbols) after loading CsPbBr3 on the surface of TiO-CN, indicating the opening of a rapid electron transfer channel at the interface between CsPbBr3 and TiO-CN due to their favourable energy-offset. These traces could be fitted to a multi-exponential function, and the fitting parameters are listed in Table S1.† The value of the amplitude-averaged lifetime of the pristine CsPbBr3 nanocrystals was ∼14.8 ns, which is obviously larger than ∼8.4 ns for CsPbBr3@TiO-CN composites.
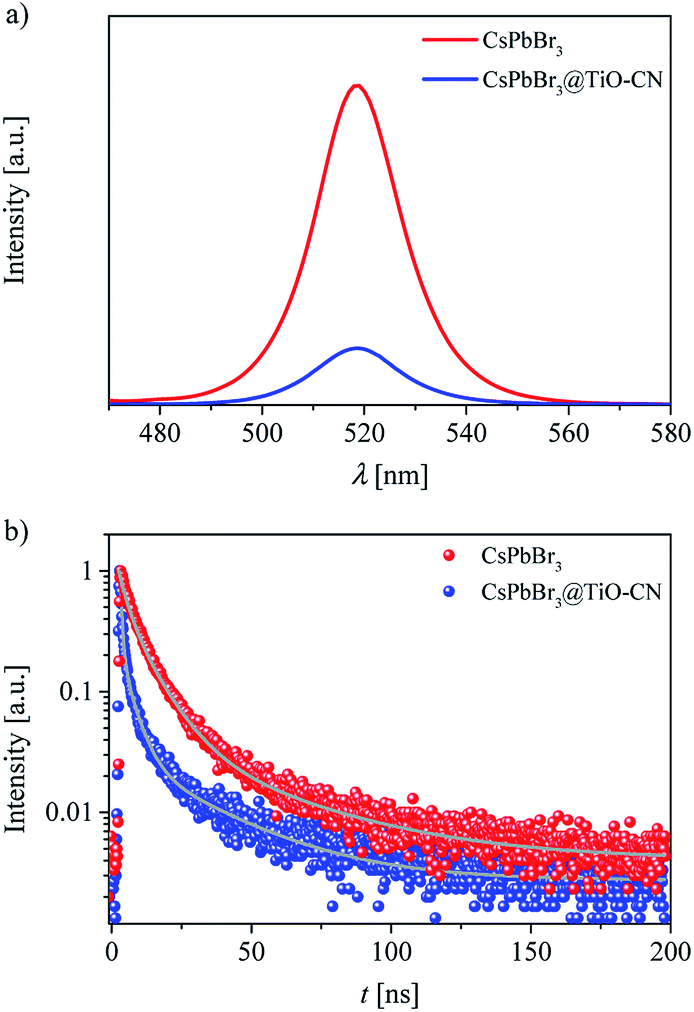 | ||
| Fig. 4 (a) Photoluminescence spectra of CsPbBr3 and CsPbBr3@TiO-CN. (b) Time-resolved photoluminescence decays of CsPbBr3 and CsPbBr3@TiO-CN. The gray curves are the fitting lines based on a multi-exponential function. | ||
The efficient charge separation between CsPbBr3 and TiO-CN was further elucidated by electrochemical impedance spectroscopy (EIS) and photocurrent response measurements. As depicted in Fig. S5a,† the semicircular arc of the CsPbBr3@TiO-CN composite exhibited an obviously smaller diameter in the Nyquist plot compared to those of pristine CsPbBr3 and TiO-CN, indicating the improvement in the separation efficiency of photogenerated carriers in CsPbBr3@TiO-CN. In addition, a remarkably increased photocurrent density could also be detected, as presented in Fig. S5b,† which was over 2 and 3 times higher than those of the pristine CsPbBr3 and TiO-CN counterparts, respectively, confirming that combining CsPbBr3 and TiO-CN substantially improved the photogenerated carrier separation efficiency. These phenomena are in line with the results of the abovementioned steady state and transient PL measurements.
Efficient charge transfer and separation between CsPbBr3 and TiO-CN in the CsPbBr3@TiO-CN composite improved the photocatalytic performance, which was demonstrated by the following photocatalytic measurements. The photocatalytic CO2 reduction reactions were carried out in CO2-saturated ethyl acetate/water solution without additional sacrificial reductants under Xe-lamp irradiation with a 400 nm filter at the light intensity of 100 mW cm−2. Herein, the reaction systems contained a very small amount of water since the metal-halide perovskite is unstable when exposed to a large amount of water.31 For all the evaluated pristine and composite photocatalyst samples, results from the chromatographic analysis showed that the main product from CO2 reduction was CO accompanied by a very small amount of CH4, and no other reduction products such as H2 were detected. The product yield of CO was plotted as a function of the photocatalysts, as presented in Fig. 5a. With pristine CsPbBr3 as the photocatalyst, the yield of CO was only 38 μmol g−1 after 10 h of irradiation, which is similar to the poor photocatalytic activity commonly observed in the recent reports on pristine CsPbBr3-based photocatalysts.20,22,24,27,29,31 On the other hand, pristine TiO-CN also showed very inferior photocatalytic activity with a low CO yield of 19 μmol g−1 from CO2 reduction under the same irradiation time, as presented in Fig. 5a, which may be ascribed to weak light absorption in the visible region and inferior charge separation.
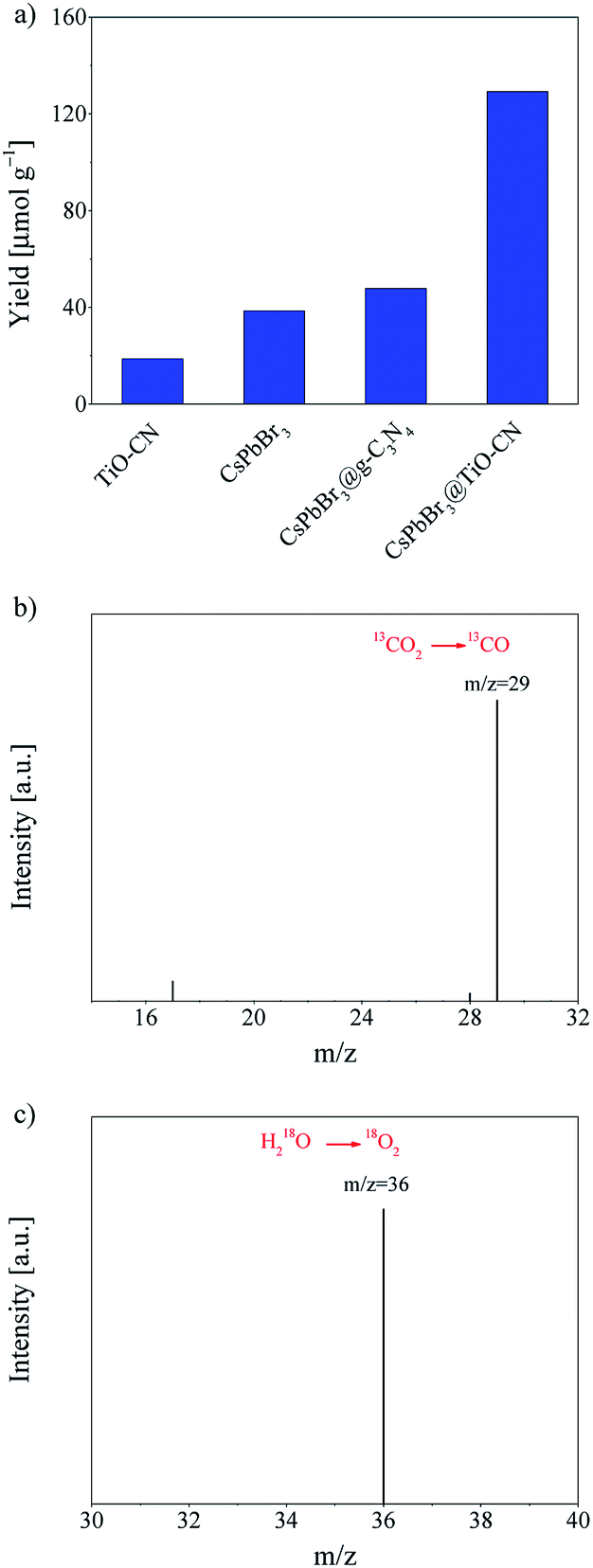 | ||
| Fig. 5 (a) The yields of CO generated from the photocatalytic CO2 reduction reactions with TiO-CN, CsPbBr3, CsPbBr3@g-C3N4 and CsPbBr3@TiO-CN photocatalysts after 10 h of irradiation under a 300 W Xe-lamp with the light intensity of 100 mW cm−2. Gas chromatograms and mass spectra (GC-MS) of the solar-driven (b) reduction of 13CO2 to 13CO (m/z = 29) and (c) oxidation of H218O to 18O2 (m/z = 36) using CsPbBr3@TiO-CN as the photocatalyst. | ||
It should be noted that combining CsPbBr3 with TiO-CN indeed boosts the photocatalytic activity in CO2 reduction to CO owing to improved charge separation and an increased number of catalytic sites. As shown in Fig. 5a, the CsPbBr3@TiO-CN composite exhibited a significantly improved yield of 129 μmol g−1 for CO formation after 10 h of irradiation, which is over 3 and 6 times higher than those of pristine CsPbBr3 and TiO-CN, respectively. We also found that the CsPbBr3 content on the surface TiO-CN plays a pivotal role in the photocatalytic performance of CsPbBr3@TiO-CN composite, exhibiting a volcano-type trend, as depicted in Fig. S6.† A lower loading amount of CsPbBr3 on TiO-CN led to weak light absorption, resulting in a lower yield of CO product for CsPbBr3@TiO-CN. The lower yield of CO product for CsPbBr3@TiO-CN with a higher loading amount of CsPbBr3 may be ascribed to the increased amount of CsPbBr3 without chemical bonding with TiO-CN, resulting in accelerated charge recombination through the surface defect states in CsPbBr3. Moreover, to assess the superiority of titanium implantation in g-C3N4, we prepared a control sample with CsPbB3 nanocrystals anchored on 2D g-C3N4 (coded as CsPbBr3@g-C3N4) without titanium implantation. Fig. 5a shows that the yield of CO from CO2 reduction in the case of CsPbBr3@g-C3N4 was only little higher compared to that of pristine CsPbBr3 under the same light irradiation condition, which is about 3 times lower than that of the CsPbBr3@TiO-CN composite owing to the lack of effective catalytic sites on g-C3N4, unlike TiO-CN.
The origin of the reduction product CO was further confirmed by a control experiment. In an isotope labelling experiment with 13CO2 replacing 12CO2 as the feedstock, gas chromatography/mass spectrometry analysis could clearly identify the major reaction product 13CO (m/z = 29), as shown in Fig. 5b, confirming that the CO was mainly generated from visible-light-driven CO2 reduction and not the photo-oxidation of ethyl acetate. On the other hand, the oxidation product O2 could also be detected along with CO in the chromatographic analysis results, which is consistent with the common observation in CsPbBr3-based photocatalyst systems.20,27,31 To further verify the origin of the oxidation product O2, an isotope trace experiment with H218O was performed in a CO2-saturated ethyl acetate/H218O solution with CsPbBr3@TiO-CN as the photocatalyst. A major mass signal of 18O2 with an m/z value of 36 could be clearly detected, as presented in Fig. 5c, indicating that O2 originates from the water oxidation reaction by consuming the photogenerated holes in the CsPbBr3@TiO-CN composite, which demonstrates that water is the main electron source for the photocatalytic reduction of CO2 to CO.
Conclusions
In summary, we successfully fabricated a CsPbBr3@TiO-CN composite based on a facile process, which can be employed as an efficient photocatalyst for visible-light-driven CO2 reduction using water as the electron source. This composite photocatalyst could achieve a significant improvement in CO conversion yield from CO2, which is over 3 and 6 times higher than those of pristine CsPbBr3 and TiO-CN, respectively. Microstructure characterization revealed that the CsPbBr3 nanocrystals were closely anchored on 2D TiO-CN via N–Br and O–Br bonding. Combining CsPbBr3 with TiO-CN led to an obviously enhanced of photogenerated carrier separation efficiency, as disclosed by the steady-state and transient photoluminescence data, as well as electrochemical measurements. We also observed the pivotal role of the metal anion in enhancing the photocatalytic activity of the catalyst in CO2 reduction, providing a clear clue for further design of metal-halide perovskite-based photocatalysts through material engineering.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Natural Science Foundation of Tianjin City (17JCJQJC43800), National Key R&D Program of China (2017YFA0700104), NSFC (21790052) and the 111 Project (D17003).Notes and references
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef.
- K. K. Sakimoto, A. B. Wong and P. Yang, Science, 2016, 351, 74–77 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- K. C. Christoforidis and P. Fornasiero, ChemCatChem, 2019, 11, 368–382 CrossRef CAS.
- X. Chen and F. Jin, Front. Energy, 2019, 13, 207–220 CrossRef.
- D. C. Grills, M. Z. Ertem, M. McKinnon, K. T. Ngo and J. Rochford, Coord. Chem. Rev., 2018, 374, 173–217 CrossRef CAS.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS.
- C. Li, Y. Xu, W. Tu, G. Chen and R. Xu, Green Chem., 2017, 19, 882–899 RSC.
- H. Abdullah, M. M. R. Khan, H. R. Ong and Z. Yaakob, J. CO2 Util., 2017, 22, 15–32 CrossRef CAS.
- Z.-F. Huang, J. Song, L. Pan, X. Zhang, L. Wang and J.-J. Zou, Adv. Mater., 2015, 27, 5309–5327 CrossRef CAS.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS.
- J. Wang, T. Xia, L. Wang, X. Zheng, Z. Qi, C. Gao, J. Zhu, Z. Li, H. Xu and Y. Xiong, Angew. Chem., Int. Ed., 2018, 57, 16447–16451 CrossRef CAS PubMed.
- M. F. Kuehnel, C. D. Sahm, G. Neri, J. R. Lee, K. L. Orchard, A. J. Cowan and E. Reisner, Chem. Sci., 2018, 9, 2501–2509 RSC.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS.
- R. D. Harris, S. B. Homan, M. Kodaimati, C. He, A. B. Nepomnyashchii, N. K. Swenson, S. C. Lian, R. Calzada and E. A. Weiss, Chem. Rev., 2016, 116, 12865–12919 CrossRef CAS.
- X. Zhu, Y. Lin, J. S. Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843 CrossRef.
- L.-Y. Wu, Y.-F. Mu, X.-X. Guo, W. Zhang, Z.-M. Zhang, M. Zhang and T.-B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS.
- X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2019, 141, 733–738 CrossRef CAS.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 CrossRef CAS.
- Y.-F. Xu, X.-D. Wang, J.-F. Liao, B.-X. Chen, H.-Y. Chen and D.-B. Kuang, Adv. Mater. Interfaces, 2018, 5, 1801015 CrossRef.
- Y.-F. Xu, M.-Z. Yang, H.-Y. Chen, J.-F. Liao, X.-D. Wang and D.-B. Kuang, ACS Appl. Energy Mater., 2018, 1, 5083–5089 CrossRef CAS.
- Y. Wu, P. Wang, X. Zhu, Q. Zhang, Z. Wang, Y. Liu, G. Zou, Y. Dai, M.-H. Whangbo and B. Huang, Adv. Mater., 2018, 30, 1704342 CrossRef.
- H. Huang, H. Yuan, K. P. F. Janssen, G. Solís-Fernández, Y. Wang, C. Y. X. Tan, D. Jonckheere, E. Debroye, J. Long, J. Hendrix, J. Hofkens, J. A. Steele and M. B. J. Roeffaers, ACS Energy Lett., 2018, 3, 755–759 CrossRef CAS.
- Z.-C. Kong, J.-F. Liao, Y.-J. Dong, Y.-F. Xu, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- S. Park, W. J. Chang, C. W. Lee, S. Park, H.-Y. Ahn and K. T. Nam, Nat. Energy, 2016, 2, 16185 CrossRef.
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS.
- K. Chen, X. Deng, G. Dodekatos and H. Tüysüz, J. Am. Chem. Soc., 2017, 139, 12267–12273 CrossRef CAS.
- J. Hou, S. Cao, Y. Wu, Z. Gao, F. Liang, Y. Sun, Z. Lin and L. Sun, Chem.–Eur. J., 2017, 23, 9481–9485 CrossRef CAS.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS.
- L. Polavarapu, B. Nickel, J. Feldmann and A. S. Urban, Adv. Energy Mater., 2017, 7, 1700267 CrossRef.
- X. Li, F. Cao, D. Yu, J. Chen, Z. Sun, Y. Shen, Y. Zhu, L. Wang, Y. Wei, Y. Wu and H. Zeng, Small, 2017, 13, 1603996 CrossRef.
- Q. Zhang and Y. Yin, ACS Cent. Sci., 2018, 4, 668–679 CrossRef CAS.
- L. N. Quan, F. P. G. de Arquer, R. P. Sabatini and E. H. Sargent, Adv. Mater., 2018, 30, 1801996 CrossRef.
- M. Ahmadi, T. Wu and B. Hu, Adv. Mater., 2017, 29, 1605242 CrossRef PubMed.
- S. A. Veldhuis, P. P. Boix, N. Yantara, M. Li, T. C. Sum, N. Mathews and S. G. Mhaisalkar, Adv. Mater., 2016, 28, 6804–6834 CrossRef CAS PubMed.
- S. Tian, Z. Wang, W. Gong, W. Chen, Q. Feng, Q. Xu, C. Chen, C. Chen, Q. Peng, L. Gu, H. Zhao, P. Hu, D. Wang and Y. Li, J. Am. Chem. Soc., 2018, 140, 11161–11164 CrossRef CAS.
- P. Huang, J. Huang, S. A. Pantovich, A. D. Carl, T. G. Fenton, C. A. Caputo, R. L. Grimm, A. I. Frenkel and G. Li, J. Am. Chem. Soc., 2018, 140, 16042–16047 CrossRef CAS.
- X. Wang, W. Wang, M. Qiao, G. Wu, W. Chen, T. Yuan, Q. Xu, M. Chen, Y. Zhang, X. Wang, J. Wang, J. Ge, X. Hong, Y. Li, Y. Wu and Y. Li, Sci. Bull., 2018, 63, 1246–1253 CrossRef CAS.
- M. Ou, S. Wan, Q. Zhong, S. Zhang and Y. Wang, Int. J. Hydrocarbon Eng., 2017, 42, 27043–27054 CAS.
- W. Liu, L. Cao, W. Cheng, Y. Cao, X. Liu, W. Zhang, X. Mou, L. Jin, X. Zheng, W. Che, Q. Liu, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 9312–9317 CrossRef CAS.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS.
- S. Tang, X. Yin, G. Wang, X. Lu and T. Lu, Nano Res., 2019, 12, 457–462 CrossRef CAS.
- Y. Zhu, T. Wang, T. Xu, Y. Li and C. Wang, Appl. Surf. Sci., 2019, 464, 36–42 CrossRef CAS.
- G. Shi, L. Yang, Z. Liu, X. Chen, J. Zhou and Y. Yu, Appl. Surf. Sci., 2018, 427, 1165–1173 CrossRef CAS.
- H. Li, Y. Gao, Z. Xiong, C. Liao and K. Shih, Appl. Surf. Sci., 2018, 439, 552–559 CrossRef CAS.
- X. Lu, K. Xu, S. Tao, Z. Shao, X. Peng, W. Bi, P. Chen, H. Ding, W. Chu, C. Wu and Y. Xie, Chem. Sci., 2016, 7, 1462–1467 RSC.
- J. Li, L. Xu, T. Wang, J. Song, J. Chen, J. Xue, Y. Dong, B. Cai, Q. Shan, B. Han and H. Zeng, Adv. Mater., 2017, 29, 1603885 CrossRef.
- J. Song, J. Li, L. Xu, J. Li, F. Zhang, B. Han, Q. Shan and H. Zeng, Adv. Mater., 2018, 30, 1800764 CrossRef.
- M. Ou, S. Wan, Q. Zhong, S. Zhang, Y. Song, L. Guo, W. Cai and Y. Xu, Appl. Catal., B, 2018, 221, 97–107 CrossRef CAS.
- J. Fu, B. Zhu, C. Jiang, B. Cheng, W. You and J. Yu, Small, 2017, 13, 1603938 CrossRef.
- P. Yang, H. Ou, Y. Fang and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 3992–3996 CrossRef CAS.
- J. Liang, C. Wang, Y. Wang, Z. Xu, Z. Lu, Y. Ma, H. Zhu, Y. Hu, C. Xiao, X. Yi, G. Zhu, H. Lv, L. Ma, T. Chen, Z. Tie, Z. Jin and J. Liu, J. Am. Chem. Soc., 2016, 138, 15829–15832 CrossRef CAS.
- C. Baumanis and D. W. Bahnemann, J. Phys. Chem. C, 2008, 112, 19097–19101 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data and experimental details. See DOI: 10.1039/c9ra07236e |
| This journal is © The Royal Society of Chemistry 2019 |