
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free hypervalent iodine/TEMPO mediated oxidation of amines and mechanistic insight into the reaction pathways†
Ajay H. Bansodeab and
Gurunath Suryavanshi *ab
*ab
aChemical Engineering & Process Development Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune-411008, India. E-mail: gm.suryavanshi@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), New Delhi 110 025, India
First published on 14th September 2018
Abstract
A highly efficient metal free approach for the oxidation of primary and secondary amines to their corresponding aldehydes and ketones using PhI(OAc)2 in combination with a catalytic amount of TEMPO as an oxidizing agent is described. This protocol is rapid and provides diverse products under milder reaction conditions in excellent yields. In addition, the mechanistic study is well demonstrated by spectroscopic methods.
Introduction
Oxidation reactions to access carbonyl functional groups are fundamental transformations and play a most significant role in synthetic organic chemistry. Carbonyl functionalities serve as versatile building blocks in functional group interconversions, and synthesis of complex molecules and are widely present in natural products and biologically active compounds. The conventional way to synthesize aldehydes and ketones involves oxidation of primary and secondary alcohols,1 which has been successfully exploited in academic and industrial research. In an alternative method, amine precursors were also successfully used to access carbonyl compounds due to their ability to undergo oxidation reactions, and their natural and commercial availability. The oxidation of amines is also used as a powerful tool to produce different synthetic intermediates: imines, nitriles, oximes and amides.2 In the last two decades, several protocols reported the synthesis of carbonyl compounds from amines using metal reagents/catalysts such as KMnO4,3 ZnCr2O7,4 nicotinium dichromate,5 palladium,6 copper7 and ruthenium.8 These traditional methods suffer from their own limitations, such as use of stoichiometric amounts of reagents/catalysts, inherent toxicity of metals, high temperature and limited substrate scope.In addition, Voltrova et al. showed that catalytic copper and ascorbic acid in open air condition can be used for oxidation of amines, which is limited to primary amine substrates (eqn (1), Scheme 1).9 De Luca et al. and Davis et al. developed metal free methods using NCS/Et3N and DEAD via an imine intermediate in the classical way. These two methods required harsh reaction conditions and an extra hydrolysis step to access the target carbonyl compound (eqn (2), Scheme 1).10,11
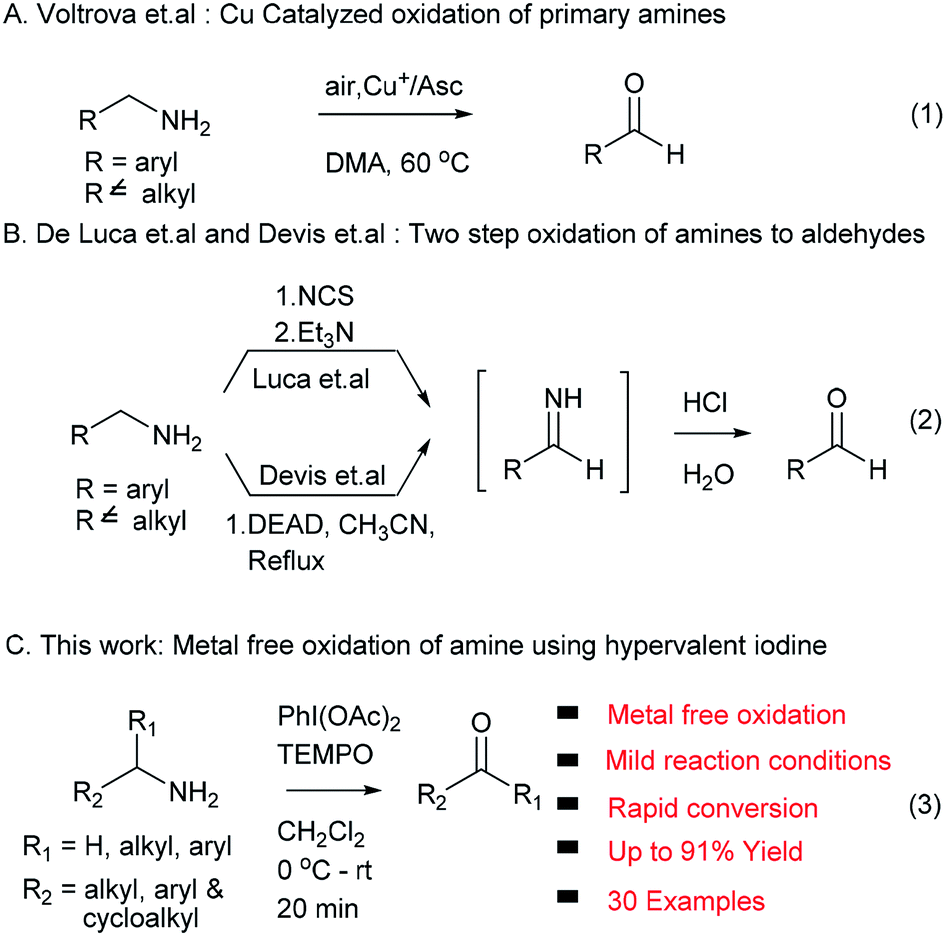 | ||
| Scheme 1 Oxidation of amine to aldehyde. | ||
In continuation of our interest in development of new methodologies using PhI(OAc)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 and to address issues associated with existing protocols for the conversion of amines to carbonyl compounds, herein we report an efficient, rapid, mild, metal free and environment friendly protocol involving non-metallic, less toxic and affordable hypervalent iodine (PhI(OAc)2) in combination with TEMPO as an oxidizing agent for the first time without any external oxygen source (eqn (3), Scheme 1).13,14
12 and to address issues associated with existing protocols for the conversion of amines to carbonyl compounds, herein we report an efficient, rapid, mild, metal free and environment friendly protocol involving non-metallic, less toxic and affordable hypervalent iodine (PhI(OAc)2) in combination with TEMPO as an oxidizing agent for the first time without any external oxygen source (eqn (3), Scheme 1).13,14
Hypervalent iodine compounds such as (diacetoxyiodo)benzene (PhI(OAc)2), [bis(trifluoroacetoxy)] benzene (PhI(OCOCF3)2), Dess–Martin periodinane (DMP), 2-iodoxybenzoic acid (IBX) and their derivatives were extensively used as oxidants and co-oxidants in organic transformations.15,16 Using hypervalent iodine reagent oxidation of amine was well established for conversion to imines,17a–e aldehydes,18a,b ketones and aromatization of pyrrolidine, dihydropyridine rings.19 Very recently, Galletti et al. reported NaIO4/TEMPO mediated conversion of benzylamines to carbonyls via imine formation and in situ hydrolysis using H2O/CH3CN as solvent.20 However, these methods suffer from limitations such as higher temperature, long reaction time and sequence, substrate selectivity and extra step for imine hydrolysis to form an aldehyde.17–20 In this context we envisioned that, PhI(OAc)2 in combination with TEMPO used as an oxidizing agent to convert amine functional groups into corresponding carbonyl compounds at room temperature, with short reaction time and no use of external oxygen source (such as H2O or O2) with excellent yields.
Result and discussion
To test our hypothesis, initially, p-methoxy benzyl amine (1a) was treated with PhI(OAc)2 (1 equiv.) and TEMPO (1 equiv.) in anhydrous CH2Cl2 for 30 min at 0 °C, which furnished the p-methoxy benzaldehyde (2a) in 40% yield along with 50% of unreacted amine 1a (entry 1), when we raised the reaction temperature to rt, the expected p-methoxy benzaldehyde (2a) was obtained in 65% yield along with decomposition of the starting material 1a in 30 min (entry 2). Hence, we carried out the same reaction for 10 min at 0 °C followed by room temperature for another 10 min, to our delight, this small modification to the reaction conditions afforded the desired oxidation product 2a in the excellent yield of 90% (entry 3). The output of the reaction was not much affected when we reduced the amount of TEMPO (0.5 equiv.), but the yield was dropped to 40% using 0.5 equiv. of PhI(OAc)2 (entries 4 and 5). Further reduction of TEMPO to 0.25 and 0.1 equiv. also furnished the product in good yields (entries 6 and 7). Then we verified the reaction without using TEMPO or PhI(OAc)2, which led to the unchanged starting material (1a) (entries 8 and 9). Ultimately, the reaction conditions of PhI(OAc)2 (1 equiv.) and TEMPO (0.1 equiv.) in anhydrous CH2Cl2 for 10 min at 0 °C followed by room temperature for another 10 min were found to be ideal for this transformation (entry 7) (Table 1).
|
||||
|---|---|---|---|---|
| Entry | PhI(OAc)2 (equiv.) | TEMPO (equiv.) | Temp (time)b | Yieldb |
| a Reaction conditions unless otherwise specified: 1a (1 equiv.), PhI(OAc)2 (1 equiv.), TEMPO (0.1 equiv.), 0 °C to rt, 20 min, anhydrous CH2Cl2.b Isolated yields in percentage.c Starting material (1a) recovered; rt = room temperature; NR: no reaction. | ||||
| 1 | 1 | 1 | 0 °C (30 min) | 40 |
| 2 | 1 | 1 | rt (30 min) | 65 |
| 3 | 1 | 1 | 0 °C to rt (20 min) | 90 |
| 4 | 1 | 0.5 | 0 °C to rt (20 min) | 90 |
| 5 | 0.5 | 1 | 0 °C to rt (20 min) | 40 |
| 6 | 1 | 0.25 | 0 °C to rt (20 min) | 90 |
| 7 | 1 | 0.1 | 0 °C to rt (20 min) | 90 |
| 8 | 1 | — | 0 °C to rt (20 min) | NRc |
| 9 | — | 1 | 0 °C to rt (20 min) | NRc |
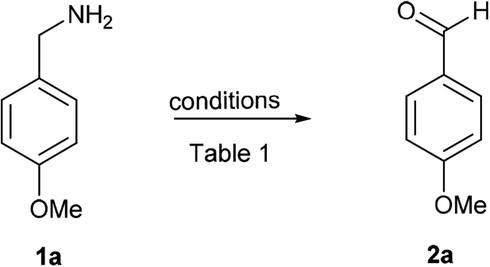
With optimized conditions in hand, initially, the substrate scope for this methodology was examined using a series of diverse aromatic and aliphatic primary amines (Scheme 2). Commercially available aryl and heteroaryl amines possessing electron donating and electron withdrawing substituents were provided corresponding aldehydes in excellent yields without significant discrimination in the output (2b–e). Naphthalen-1-ylmethanamine also well reacted and gave the corresponding aldehyde 2f in good yield of 91%. p-Xylylamine also a good substrate for this reaction, which delivered the terephthalaldehyde (2g) in 85% yield. The benzylamine with dioxolane protection is well tolerated in these reaction conditions and gave 2h in 85% yield. Heteroaryl amines like furan-2-ylmethanamine, (5-bromofuran-2-yl) methanamine and pyridin-3-yl-methanamine were well participated to give 2i, 2j and 2k respectively. To our delight, under identical conditions alkyl and dialkyl substituted amines (3-phenylpropan-1-amine, n-propylamine, n-heptylamines and isobutylamine) well tolerated and furnished corresponding aldehydes in excellent yields (2l–o). Next the reactivity of diverse alpha-disubstituted primary amine derivatives were examined to obtained corresponding ketones. Highly hindered alpha substituted benzyl amine and tetrahydronaphthalene derived amines provided corresponding ketones 2p and 2q in good yields of 83% and 87%. Phenyl and tolyl substituted amines gave 2r and 2s respectively. Cycloalkyl derived amines furnished 2t–v in good yields, even hindered 2-adamantyl amine was converted into 2-adamantanone (2w) in 80% yield.
 | ||
| Scheme 2 Oxidation of primary amines to aldehydes and ketones. Reaction conditions unless otherwise specified: amine 1 (1 equiv.), PhI(OAc)2 (1 equiv.), TEMPO (0.1 equiv.), 0 °C to rt, 20 min, anhydrous CH2Cl2, aPhI(OAc)2 (2 equiv.), TEMPO (0.2 equiv.). | ||
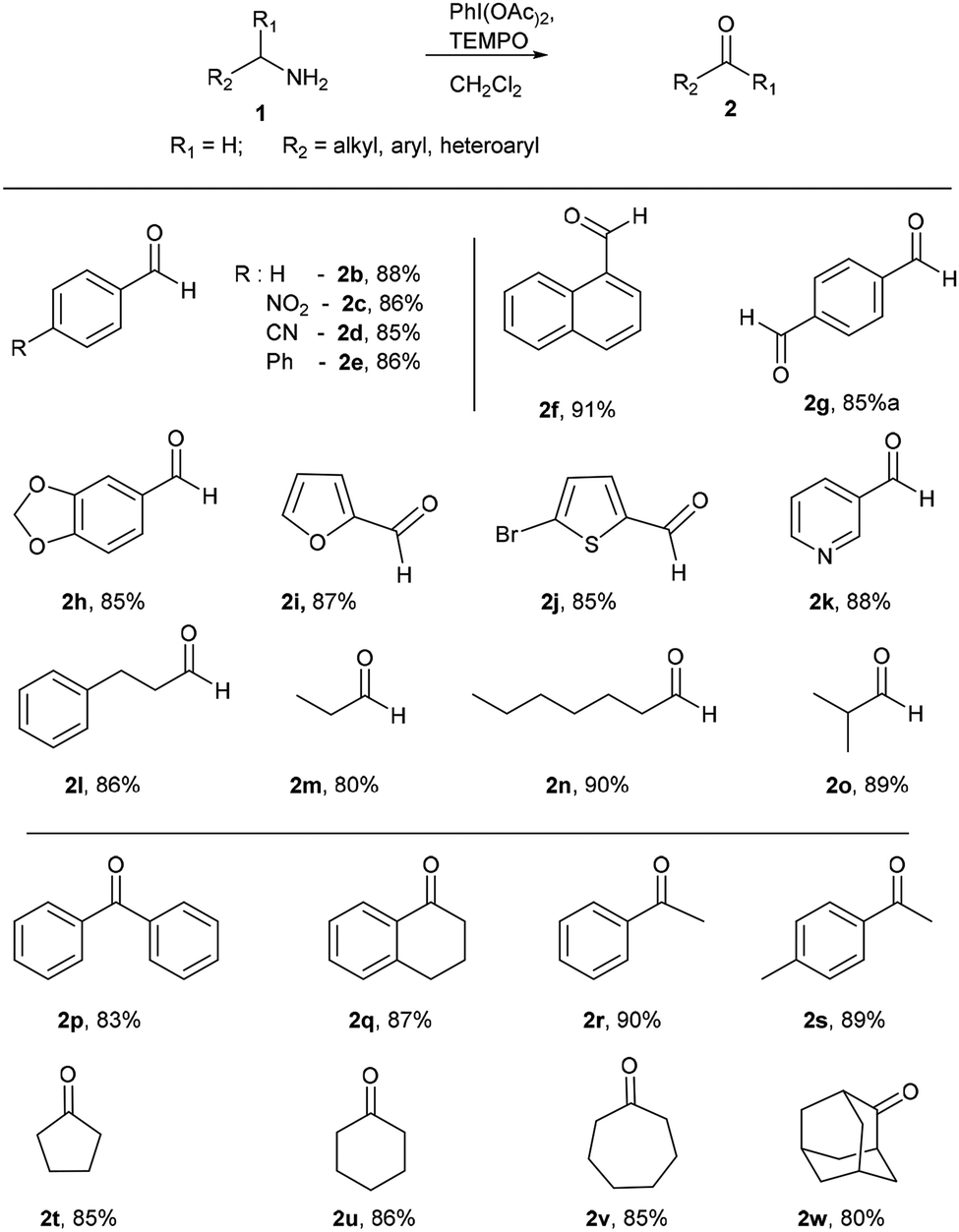
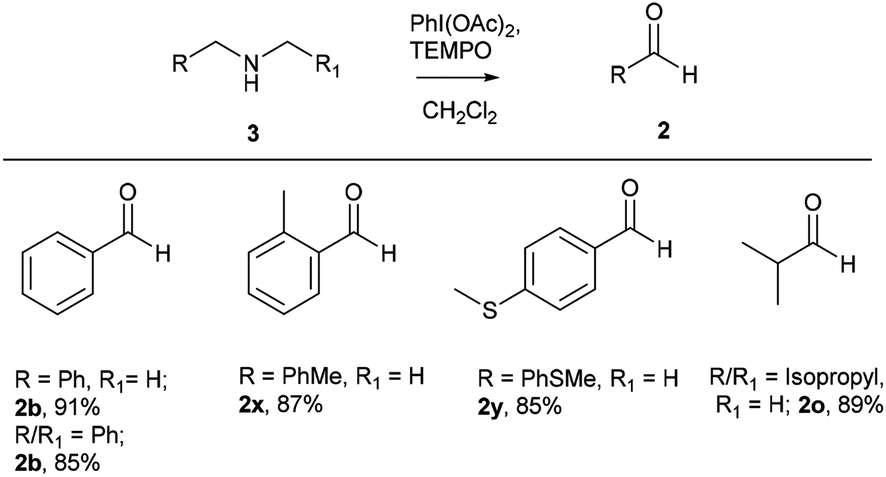
After successful studies involving primary amines, we were curious to know the reactivity of secondary amines in this oxidation protocol. Several alkyl–aryl and dialkyl amines were subjected to slightly modified reaction conditions of 2 equiv. of PhI(OAc)2 and 0.2 equiv. of TEMPO in dry CH2Cl2. The oxidations of N-methyl-aryl amine substrates were quite interesting, which would generate possible two products of aryl-aldehydes and formaldehyde based on the choice of oxidation sites. To our surprise, aryl-aldehydes were obtained exclusively in good yield (2b, 2x and 2y), which could be due to the more reactivity of the benzylic position of substrates. This selectivity is in contrast with other oxidizing agents used in earlier reports.3–11 Isobutyl-methylamine also well reacted under identical conditions to provide corresponding isobutyraldehyde 2o in excellent yield of 89% (Scheme 3). Oxidation of symmetrical amines such as dibenzylamine using PhI(OAc)2 (1 equiv) and TEMPO (0.1 equiv) in dry CH2Cl2 provided the mixture of benzaldehyde (2b) and benzyl amine (1b) in 1:1 ratio in the good yield of 90% yield. Equivalent results were obtained in the oxidation of diisobutylamine (7) and gave isobutyraldehyde (2o) and isobutylamine (1o) in 1:1 ratio in excellent yield of 85% (Scheme 4).
 | ||
| Scheme 3 Oxidation of secondary amines to aldehydes. Reaction conditions unless otherwise specified: amine 1 (1 equiv.), PhI(OAc)2 (2 equiv), TEMPO (0.2 equiv), anhydrous CH2Cl2, 0 °C to rt, 20 min. | ||
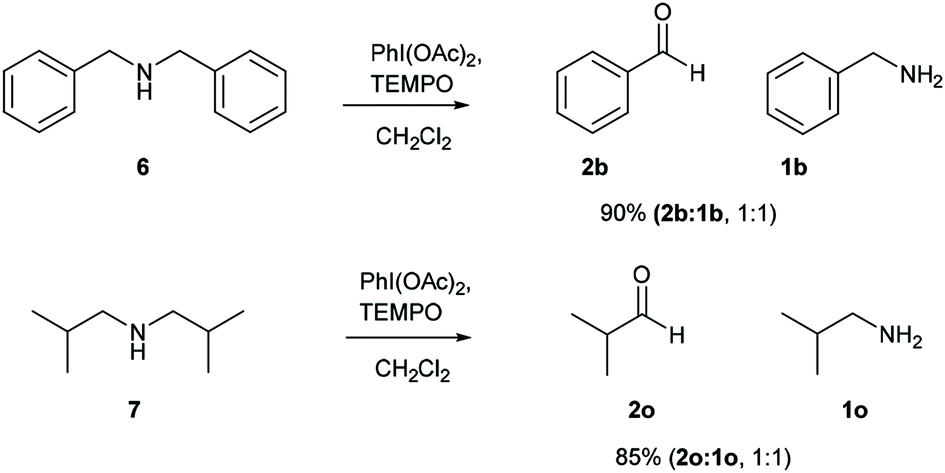 | ||
| Scheme 4 Comparative study on symmetrical secondary amines. Reaction conditions unless otherwise specified: amine 1 (1 equiv.), PhI(OAc)2 (1 equiv.), TEMPO (0.1 equiv.), anhydrous CH2Cl2, 0 °C to rt, 20 min. | ||
Based on earlier reports21 and results obtained in this work, a plausible reaction mechanism was proposed. Initially, TEMPO was converted into oxoammonium species A using PhI(OAc)2 as oxidant. The reaction of benzylamine (1) with oxoammonium species A could provide the intermediate B via attack of lone pair of nitrogen from benzyl amine on electron deficient nitrogen of oxoammonium A. Then, intermediate B could undergo reductive elimination which provide the reactive imine C through the abstraction of proton from benzylic position of intermediate B. Further PhI(OAc)2 was used for oxidation of hydroxyamine species I to regenerate oxoammonium species A and re-enters in catalytic cycle, expels iodobenzene and acetic acid which was used in further reaction sequence. Imine C then converted into corresponding aldehyde via two probable pathways. In the path-I, imine intermediate C will react with acetic acid and forms the amino acetate intermediate D. Intermediate D would react with another 1 mole of acetic acid to give the corresponding acetate intermediate E, which further reacts to give desired aldehyde 2 via releasing the acetic anhydride and ammonia. In an alternate path II, imine C will react with benzylamine (1b) with exertion of ammonia to provide secondary imine F,22 which will react with acetic acid to provide the amino-acetate intermediate G. Under acidic condition as in the path I, intermediate G delivers the aldehyde 2 and benzylamine which re-enters the catalytic cycle.
To further understand the proposed mechanistic sequence, a few supporting experiments were carried out and described in Scheme 5. To verify, the probable formation of amino acetate intermediate D, we have prepared the same using known literature procedure23 and subjected to standard reaction conditions of PhI(OAc)2 (1 equiv.) and TEMPO (0.1 equiv.). To our delight, the expected aldehyde 2 was isolated in 90% yield. The possible hydrolysis of imine intermediate C, which could directly deliver the aldehyde without the intervention of path I and/or path II was established by performing the reaction of amino acetate or amine under optimized conditions using 4 Å-MS.
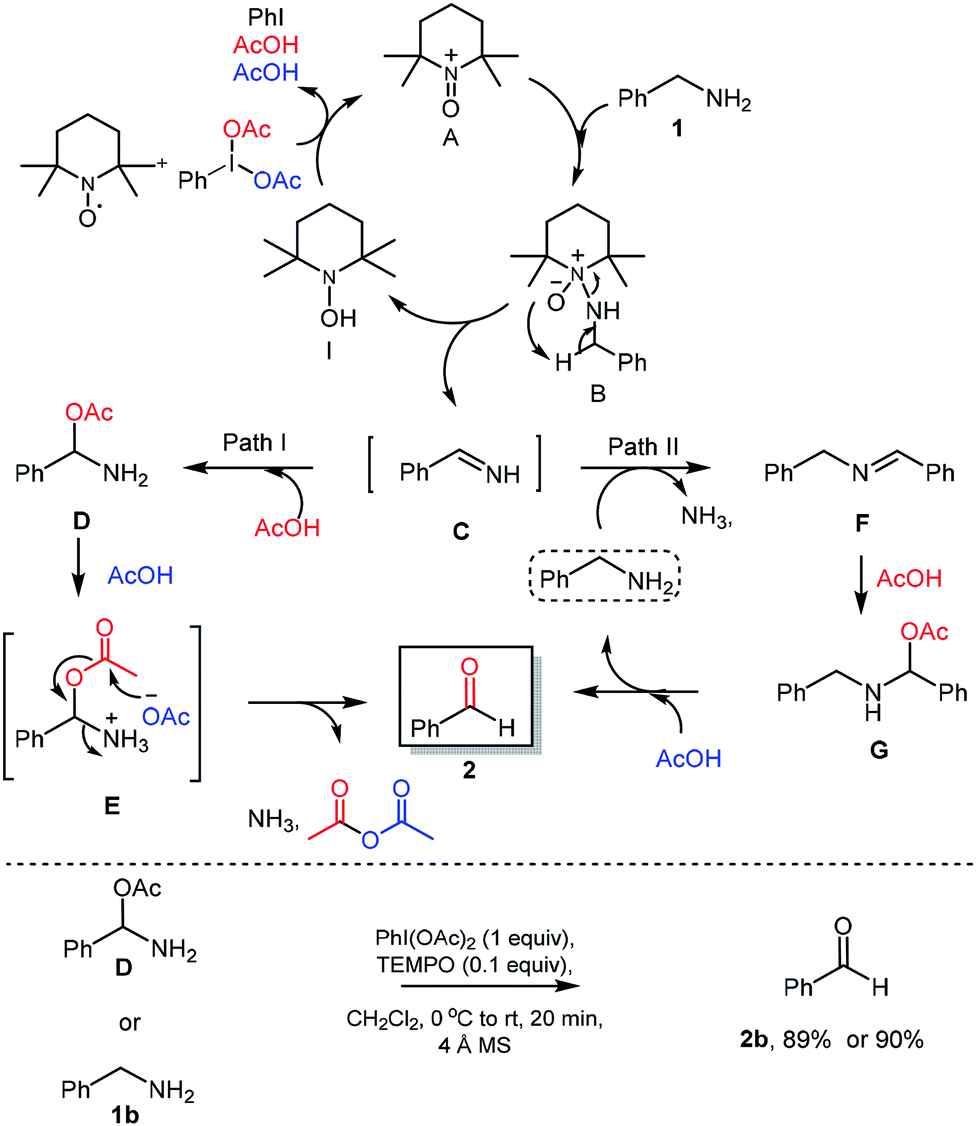 | ||
| Scheme 5 Plausible reaction mechanism & control experiments. | ||
The formation of imine C and amino acetate D intermediates and release of acetic anhydride and ammonia as by-products in this reaction was established by time dependent 1H NMR analysis. A standard reaction using benzyl amine in a sealed NMR tube in CDCl3 under inert atmosphere was studied.
Analysis of 1H NMR at 0 h without adding reagents were analysed, showed peaks at 1.5 (s, –NH2), 3.97(s, benzylic –CH2), 7.2–7.6 (m, aromatic 5H), belongs to benzyl amine. To the same NMR tube, PhI(OAc)2 (1 equiv.) and TEMPO (0.1 equiv.) were added at 0 °C, as soon as the addition was completed, 1H NMR was recorded, in which we found that, peak at 3.97 ppm belongs to benzylic proton of benzyl amine was disappeared and new peaks for intermediates were clearly indicative. We found out a singlet at 10.1 ppm, which clearly show that the peak of benzaldehyde proton and doublet at 7.98 ppm is for ortho proton of benzaldehyde. In addition, we found signals related to imine intermediates, amino acetate intermediate as well while performing the analysis (as shown in Fig. 1).
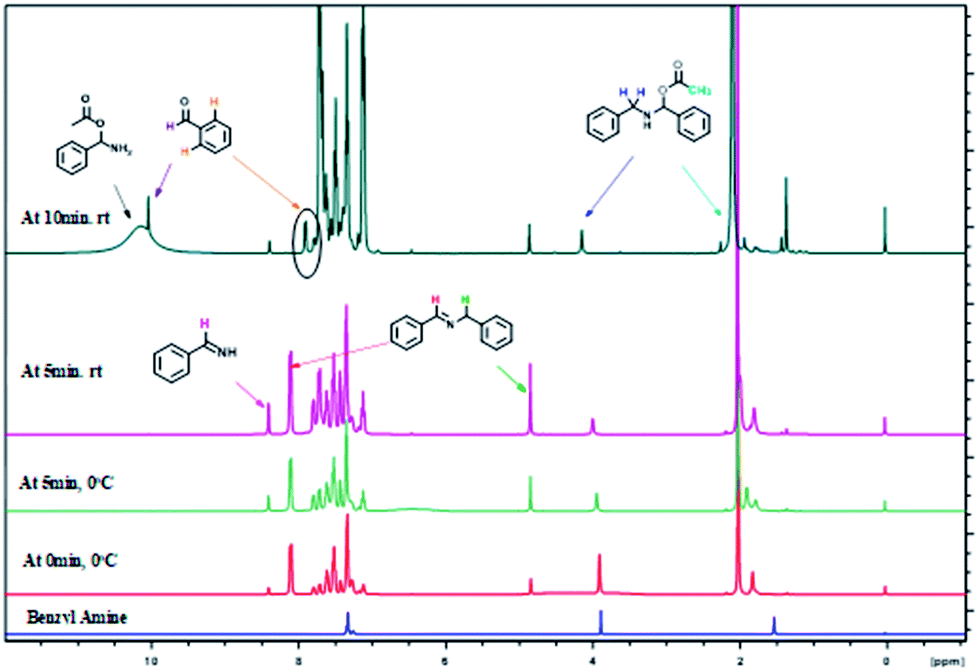 | ||
| Fig. 1 Time dependent 1H NMR experiment for mechanistic determination. | ||
In addition, we carried out GC-MS study to prove our hypothesis that oxygen is not coming from out sources and it is from PhI(OAc)2. We performed reaction in air tight GC-MS vial to avoid the contamination with atmospheric oxygen. To our delight, we found out peak corresponding to benzaldehyde and intermediate F. Generation of benzaldehyde in reaction mixture itself, without additional hydrolysis clearly indicates that no external oxygen source is required and PhI(OAc)2 is acting as an oxygen source in this reaction. In addition, release of ammonia was detected using well-known ammonia test.
Initially, we took 1H NMR of our intermediate amino acetate D in CDCl3, which showed peaks at 2.4 ppm (s, –CH3), 7.68 ppm (s, benzylic –CH), 10.22 ppm (bs, NH2). In same NMR tube we added PhI(OAc)2 (1 equiv.) and TEMPO (0.1 equiv.) at 0 °C, as soon as addition completed we took 1H NMR. We found out that peak at 2.4 (s, –CH3), 7.68 (s, benzylic –CH), 10.22 (bs, NH2) was disappeared and new peaks at 2.10 ppm for acetic acid, 2.2 ppm for acetic anhydride are clearly indicative (Fig. 2).
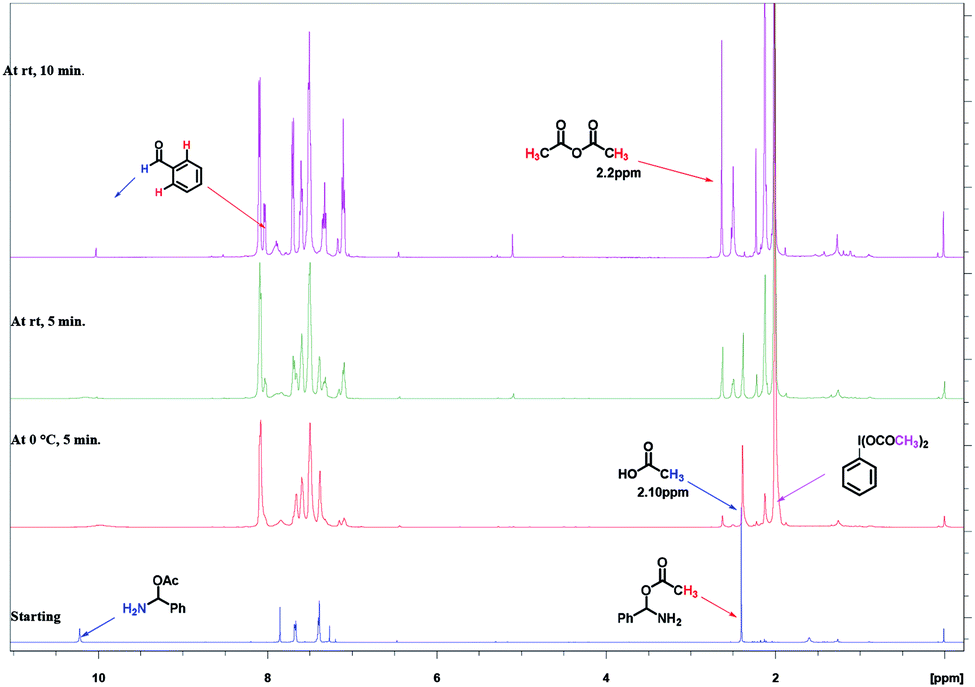 | ||
| Fig. 2 Time dependent 1H NMR experiment for mechanistic determination on intermediate. | ||
Our main goal is to find out in which form –NH2 is going out either ammonia or amide. To prove our side product, we carried out ammonia trapping experiment. In this we took two neck RB (A) equipped with magnetic needle contenting Benzyl amine (500 mg, 4.67 mmol) and dry CH2Cl2 (5 mL). Other side single neck RB (B) having water and small pieces of pH paper. To connect this two RB's, we used silicon pipe having one side connecter connected to two neck RB (A) and other side dropper which dipped in single neck RB (B) (as shown in picture below). Reaction start with addition of PhI(OAc)2 (1.5 g, 4.67 mmol) and TEMPO (72 mg, 0.467 mmol) at 0 °C for 10 minutes followed by 10 minutes at room temperature. When we did reaction at 0 °C for 10 minutes neither bubbling nor color change observed in RB-B. When we increase temperature form 0 °C to room temperature of RB-A and within 5 minutes in RB-B bubbling as well as color change of pH paper from yellow to purple was observed. With time span of 5 to 10 minutes at room temperature vigorous bubbling and color changed to purple. In addition to this intense smell of ammonia was observed. Bubbling and color change of pH paper yellow to purple clearly indicate formation of ammonia (pH: 11.6 and purple on pH paper) (Fig. 3).
 | ||
| Fig. 3 Mechanistic study (ammonia trapping experiments). | ||
Experimental section
General procedure
All reaction carried out under nitrogen atmosphere with dry solvents kept anhydrous condition. Anhydrous CH2Cl2 was distilled from P2O5. Yields are calculated by chromatographical isolation. Thin layer chromatography was performed using commercially prepared silica gel plates, and visualization was effect with short wavelength UV light (254 nm). Column chromatography performed on 100–200 mesh size silica gel.The 1H and 13C NMR spectra were recorded on Bruker advance 500 (1H 500 MHz, 13C 125 MHz) or Bruker advance 400 (1H 400 MHz, 13C 100 MHz) or Bruker advance 200(1H 200 MHz, 13C 50 MHz), otherwise mentioned. Deuterated solvent CDCl3 + CCl4 (70:30) were used as internal standard and singlet at 96.1 ppm in 13C NMR corresponds to carbon of CCl4. Solvent signal was used as reference for 13C NMR (CDCl3, 77.0 ppm) or 13C NMR (DMSO-d6, 40.0 ppm). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiple, br = broad. Chemical shifts are reported in ppm and referenced to residual solvent peak or TMS. Coupling constants are reported in hertz. High resolution mass spectra were recorded on a double focusing magnetic sector mass spectrometer using EI at 70 eV. The GC analysis were done using Agilent 7890A GC coupled with mass detector. All reagents were used as obtained commercially. The PhI(OAc)2 and TEMPO were purchased by Sigma Aldrich.
p-Anisaldehyde (2a)24. 54 mg (90%); colourless oil; 1H NMR (200 MHz, CDCl3, ppm): δ 3.89 (s, 3H), 6.99 (m, J = 8.84 Hz, 2H), 7.83 (m, J = 8.84 Hz, 2H), 9.88 (s, 1H); 13C NMR (50 MHz, CDCl3, ppm): δ 55.47, 114.28, 130.06, 131.93, 164.54, 190.39; HRMS (ESI) calcd for C8H8O2 [M + H]+ 137.0597; found 137.0599.
Benzaldehyde (2b)24. 194 mg, 88%; colourless oil; 1H NMR (200 MHz, CDCl3, ppm): δ 7.46–7.68 (m, 4H), 7.78–7.98 (m, 2H), 10.02 (s, 1H); 13C NMR (50 MHz, CDCl3, ppm): δ 128.9, 129.7, 134.3, 136.5, 191.9; HRMS (ESI) calcd for C7H6O [M + H]+ 107.0491; found 107.0495.
4-Nitrobenzaldehyde (2c)28. 20 mg, 86%; white solid; 1H NMR (200 MHz, CDCl3, ppm): δ 8.09 (m, J = 8.84 Hz, 2H), 8.41 (m, J = 8.72 Hz, 2H), 10.17 (s, 1H); 13C NMR (50 MHz, CDCl3, ppm): δ 124.3, 130.5, 140.1, 151.2, 190.3; HRMS (ESI) calcd for C7H5NO3 [M + H]+ 152.0342; found 152.0703.
4-Formylbenzonitrile (2d)26. 50 mg, 85%; white solid; 1H NMR (200 MHz, CDCl3, ppm): δ 7.83–7.92 (m, 2H), 7.97–8.05 (m, 2H), 10.11 (s, 1H); 13C NMR (50 MHz, CDCl3, ppm) δ 117.6, 117.7, 129.9, 132.9, 138.8, 190.7; HRMS (ESI) calcd for C8H5NO [M + H]+ 132.0444; found 132.0443.
4-Phenylbenzaldehyde (2e)27. 85 mg, 86%; white solid; 1H NMR (200 MHz, CDCl3, ppm): δ 7.38–7.52 (m, 3H), 7.58–7.67 (m, 2H), 7.69–7.78 (m, 2H), 7.89–7.98 (m, 2H), 10.04 (s, 1H); 13C NMR (50 MHz, CDCl3): δ 127.4, 127.7, 128.5, 129.1, 130.3, 135.3, 139.8, 147.2, 191.93; HRMS (ESI) calcd for C13H10O [M + H]+ 183.0804; found 183.0804.
1-Naphthaldehyde (2f)24. 195 mg, 91%; colourless oil; 1H NMR (200 MHz, DMSO-d6, ppm): δ 7.54–7.84 (m, 3H), 7.96–8.39 (m, 3H), 9.18 (d, J = 8.34 Hz, 1H), 10.42 (s, 1H); 13C NMR (101 MHz, DMSO-d6, ppm): δ 124.6, 125.9, 127.4, 129.2, 129.5, 130.2, 131.3, 133.8, 135.7, 137.2, 194.9; HRMS (ESI) calcd for C11H8O [M + H]+ 157.0648; found 157.0647.
Terephthalaldehyde (2g)32. 100 mg, 85%; white solid; 1H NMR (200 MHz, CDCl3, ppm): δ 8.06 (s, 4H), 10.14 (s, 2H); 13C NMR (50 MHz, CDCl3, ppm): δ 130.1, 140.0, 191.1; HRMS (ESI) calcd for C8H6O2 [M + H]+ 135.0441; found 135.0439.
Piperonal (2h)29. 45 mg, 85%; white solid; 1H NMR (500 MHz, CDCl3, ppm): δ 6.03–6.11 (m, 2H), 6.88–6.96 (m, 1H), 7.29–7.35 (m, 1H) 7.36–7.44 (m, 1H), 9.77–9.83 (m, 1H); 13C NMR (126 MHz, CDCl3, ppm): δ 102.0, 106.9, 108.3, 128.49, 131.9, 148.7, 153.0, 189.9; HRMS (ESI) calcd for C8H6O3 [M + H]+ 151.0390; found 151.0390.
Furfural (2i)24. 43 mg, 87%; colourless oil; 1H NMR (200 MHz, CDCl3, ppm): δ 6.62 (dd, J = 3.54, 1.64 Hz, 1H) 7.26 (d, J = 3.54 Hz, 1H), 7.61–7.79 (m, 1H), 9.67 (s, 1H); 13C NMR (50 MHz, CDCl3, ppm): δ 112.5, 120.7, 147.9, 153, 177.6; GC MS (ESI) calcd for C5H4O2 [M]+ 97; found 97.
5-Bromo-2-thiophenecarboxaldehyde (2j). 15 mg, 85%; colourless oil; 1H NMR (200 MHz, CDCl3, ppm) δ 7.18–7.23 (m, 1H), 7.53–7.57 (m, 2H), 9.78–9.80 (m, 1H); 13C NMR (50 MHz, CDCl3, ppm): δ 125.0, 131.5, 136.7, 145.2, 181.81; HRMS (ESI) calcd for C5H3BrOS [M + H]+ 190.9161; found 190.9162.
3-Pyridinecarboxaldehyde (2k)25. 200 mg, 88%; colourless oil; 1H NMR (200 MHz, CDCl3, ppm): δ 7.51 (dd, J = 7.71, 4.93 Hz, 1H), 8.20 (d, J = 7.83 Hz, 1H), 8.87 (dd, J = 4.61, 1.07 Hz, 1H), 9.03–9.15 (m, 1H), 10.14 (s, 1H); 13C NMR (50 MHz, CDCl, ppm) δ 124.1, 131.4, 135.8, 152.1, 154.7, 190.8; HRMS (ESI) calcd for C6H5NO [M + H]+ 108.0444; found 108.0446.
3-Phenyl propan-1-al (2l)26. 40 mg, 86%; colourless oil; 1H NMR (200 MHz, CDCl3, ppm): δ 2.70–2.81 (m, 2H), 2.89–2.99 (m, 2H), 7.14–7.25 (m, 5H), 9.79 (t, J = 1.26 Hz, 1H); 13C NMR (50 MHz, CDCl3, ppm): δ 28.2, 45.3, 126.3, 128.3, 128.4, 128.5, 128.6, 140.3, 201.1; HRMS (ESI) calcd for C9H10O [M + Na]+ 157.0624; found 157.0648.
Propionaldehyde (2m). 100 mg, 80% {isolated by distillation}; colourless oil; 1H NMR (200 MHz, CDCl3, ppm) δ 1.11 (t, J = 7.39 Hz, 3H), 2.39–2.56 (m, 2H), 9.80 (s, 1H); 13C NMR (50 MHz, CDCl3, ppm): δ 6, 37.2, 203; GC-MS (ESI) calcd for C3H6O [M] 58.04; found 58.0.
Heptaldehyde (2n)23. 143 mg, 90%; colourless oil; 1H NMR (500 MHz, CDCl3, ppm): δ 0.90 (t, J = 6.48 Hz, 3H), 1.29–1.37 (m, 6H), 1.57–1.69 (m, 2H), 2.42 (td, J = 7.25, 1.53 Hz, 2H), 9.77 (s, 1H); 13C NMR (126 MHz, CDCl3, ppm): δ 14, 22.1, 22.4, 28.8, 31.5, 43.9, 202.3; HRMS (ESI) calcd for C7H14O [M + H]+ 115.1117; found 115.0868.
Isobutyraldehyde (2o)27. 150 mg, 89%; colourless oil; 1H NMR (500 MHz, CDCl3, ppm): δ 1.09–1.11 (m, 6H), 2.40 (dtd, J = 14.11, 7.06, 7.06, 1.14 Hz, 1H), 9.57–9.80 (m, 1H); 13C NMR (126 MHz, CDCl3, ppm): δ 15.4, 40.9, 204.5; GC-MS (ESI) calcd for C4H8O [M] 73.0; found 73.0.
Benzophenone (2p)23. 150 mg, 83%; white solid; 1H NMR (200 MHz, CDCl3, ppm): δ ppm 7.41–7.60 (m, 6H), 7.75–7.84 (m, 4H); 13C NMR (50 MHz, CDCl3, ppm): δ 128.2, 130.1, 132.3, 137.7, 196.4; HRMS (ESI) calcd for C13H11O [M + H]+ 183.0804; found 183.0803.
Tetralone (2q)30. 75 mg, 87%; colourless oil; 1H NMR (200 MHz, CDCl3,ppm): δ 2.04–2.25 (m, 2H), 2.56–2.72 (m, 2H), 2.97 (t, J = 6.06 Hz, 2H), 7.16–7.35 (m, 2H), 7.38–7.51 (m, 1H), 8.02 (dd, J = 7.77, 1.20 Hz, 1H); 13C NMR (50 MHz, CDCl3, ppm): δ 23.32, 29.77, 39.13, 126.65, 127.28, 128.68, 132.68, 133.30, 144.30, 197.88; HRMS (ESI) calcd for C10H10O [M + H]+ 147.0804; found 147.0806.
Acetophenone (2r)24. 112 mg, 90%; colourless oil; 1H NMR (200 MHz, CDCl3, ppm): δ 2.60 (s, 3H) 7.37–7.58 (m, 3H), 7.89–8.00 (m, 2H); 13C NMR (50 MHz, CDCl3, ppm): δ 26.50, 128.30, 128.53, 133.00, 137.19, 197.60; HRMS (ESI) calcd for C8H8O [M + H]+ 121.0648; found 113.0651.
4-Methylacetophenone (2s)28. 59 mg, 89%; colourless oil; 1H NMR (400 MHz, CDCl3, ppm): δ 2.40 (s, 3H), 2.55 (s, 3H), 7.23 (m, J = 7.93 Hz, 2H), 7.83 (m, J = 7.93 Hz, 2H); 13C NMR (101 MHz, DMSO-d6, ppm): δ 21.6, 26.4, 128.4, 129.2, 134.8, 143.6, 197.3; HRMS (ESI) calcd for C9H10O [M + H]+ 135.0804; found 135.0807.
Cyclopentanone (2t)30. 100 mg, 85%; colourless oil; 1H NMR (400 MHz, CDCl3, ppm) δ 1.84–2.02 (m, 4H), 2.12 (t, J = 7.32 Hz, 4H); 13C NMR (101 MHz, CDCl3, ppm) δ 23.2, 38.3, 220.7; GC-MS (ESI) calcd for C5H8O [M] 85.0; found 85.0.
Cyclohexanone (2u)26. 200 mg, 86%; colourless oil; 1H NMR (200 MHz, CDCl3, ppm) δ 1.67–1.79 (m, 2H), 1.80–1.97 (m, 4H), 2.27–2.39 (m, 4H); 13C NMR (50 MHz, CDCl3, ppm) δ 25.1, 27.0, 41.91, 211.39; GC-MS (ESI) calcd for C6H10O [M] 98.0; found 98.0.
Cycloheptanone (2v)30. 45 mg, 85%; colourless oil; 1H NMR (200 MHz, CDCl3, ppm): δ 1.56–1.87 (m, 8H), 2.38–2.63 (m, 4H); 13C NMR (50 MHz, CDCl3, ppm): δ 24.36, 30.46, 43.80, 214.64; HRMS (ESI) calcd for C7H12O [M + H]+ 113.0961; found 113.0965.
2-Adamantanone (2w)31. 90 mg, 80%; white solid; 1H NMR (400 MHz, CDCl3, ppm): δ 1.9 (br. s, 2H), 2.0 (m, 5H), 2.1 (m, 5H), 2.6 (br. s, 2H); 13C NMR (50 MHz, CDCl3, ppm): δ 27.5, 36.3, 39.3, 47.0, 218.5; HRMS (ESI) calcd for C10H14O [M + H]+ 173.0937; found173.0123.
o-Tolualdehyde (2x)28. 50 mg, 87%; colourless oil; 1H NMR (200 MHz, CDCl3, ppm): δ 2.70 (s, 3H), 7.23–7.56 (m, 3H), 7.83 (d, J = 7.33 Hz, 1H), 10.30 (s, 1H); 13C NMR (50 MHz, CDCl3, ppm): δ 19.6, 126.3, 131.8, 132.1, 133.7, 134.2, 140.6, 192.9; HRMS (ESI) calcd for C8H8O [M + H]+ 121.0648; found 121.648.
4-(Methylthio)benzaldehyde (2y)29. 30 mg, 85%; colourless oil; 1H NMR (200 MHz, CDCl3, ppm): δ 2.50 (s, 3H), 7.26–7.32 (m, 2H), 7.71–7.78 (m, 2H), 9.89 (s, 1H); 13C NMR (50 MHz, CDCl3, ppm): δ 14.7, 125.2, 130, 133, 147.9, 191.2; HRMS (ESI) calcd for C8H8OS [M + H]+ 153.0369; found 153.367.
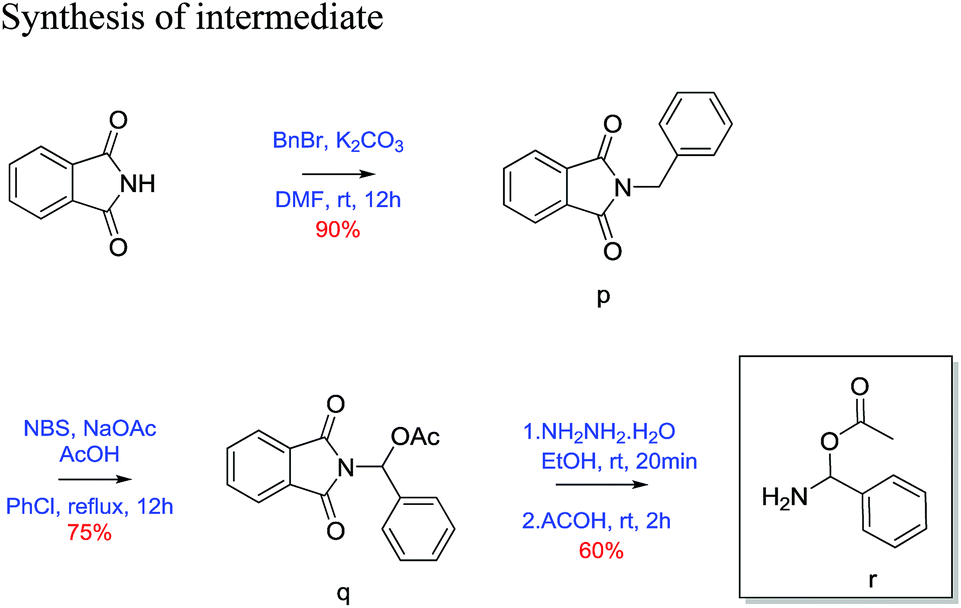
2-Benzylisoindoline-1,3-dione (p)23. Phthalimide (1 g, 6.79 mmol) was taken in round bottom flask equipped with magnetic stirrer and dissolved in dry DMF (25 mL). K2CO3(1.87 g, 13.59 mmol) and benzyl bromide (1.162 g, 6.79 mmol) was added to above reaction mixture. Then the reaction mixture was stirred for 12 h at room temperature. Reaction was quenched with H2O (12 mL) and extracted with ethyl acetate (3 × 15 mL). The combined organic layer wash with brine and dried over Na2SO4. The crude product was then purified by column chromatography using silica (100–200 mesh) and 20% ethyl acetate/pet. ether as eluent gave 90% yield of benzyl protected phthalimide as white solid. 30 mg; 1H NMR (200 MHz, CDCl3, ppm): δ 4.82 (s, 2H), 7.35 (m, 5H), 7.67 (m, 2H), 7.82 (m, 2H); 13C NMR (50 MHz, CDCl3, ppm): δ 41.63, 123.3, 127.8, 128.6, 128.7, 132.2, 134.0, 136.4, 168.0; HRMS (ESI) calcd for C8H8OS [M + H]+ 238.0863; found 238.0862.
(1,3-Dioxoisoindolin2-yl) (phenyl)methyl acetate (q)23. 2-Benzylisoindoline-1,3-dione (1.5 g, 6.3 mol) was dissolved in 25 mL of chlorobenzene. N-Bromosuccinamide (1.68 g, 9.4 mol), sodium acetate (0.77 g, 9.4 mol) and acetic acid (0.54 mL, 9.4 mol) was added to above reaction mixture. Then reaction mixture was refluxed with constant stirring for 12 h. After completion of reaction, reaction mixture evaporated on reduced pressure then extracted with ethyl acetate (3 × 15 mL). Combined organic layer wash with brine and dried over Na2SO4. Crude product was purified by column chromatography using silica gel (100–200 mesh) and 10% ethyl acetate/pet. ether as eluent. Desired product as a white solid in 75% yield. (30 mg); 1H NMR (200 MHz, CDCl3, ppm): δ 2.2 (s, 3H), 7.4 (m, 3H), 7.6 (d, J = 7.25 Hz, 2H), 7.7 (s, 1H), 7.8 (m, 2H), 7.9 (m, 2H); 13C NMR (50 MHz, CDCl3, ppm): δ 20.8, 74.2, 123.8, 126.4, 128.5, 129.0, 131.6, 134.5, 135.1, 166.3, 169.3; HRMS (ESI) calcd for C8H8OS [M + Na]+ 318.0737; found 318.0733.
Amino(phenyl)methyl acetate (r)23. (1,3-dioxoisoindolin2-yl)-(phenyl)methyl acetate (1 g, 3.38 mol) was taken in two neck round bottom flasks and 20 mL of ethanol was added. To above solution hydrazine hydrate (0.17 g, 3.38 mol) was added and refluxed for 20 minutes. After 20 minutes acetic acid (0.58 mL, 5.1 mol) was added and further refluxed for 2 h. Then EtOH was evaporated on reduced pressure and extracted with ethyl acetate (3 × 15 mL). Combined organic layer wash with brine and dried over Na2SO4. Crude product was purified using silica gel (100–200 mesh) and 10% ethyl acetate/pet. ether as eluent. Desired product was obtained as white solid in 60% yield (30 mg). 1H NMR (200 MHz, CDCl3, ppm): δ 2.4 (s, 3H), 7.4 (m, 3H), 7.7 (dd, J = 7.06, 2.48 Hz, 2H), 7.9 (s, 1H), 10.2 (s, 1H); 13C NMR (50 MHz, CDCl3, ppm): δ 20.3, 127.1, 128.7, 130.0, 133.9, 143.7, 174.0; HRMS (ESI) calcd for C8H8OS [M + Na]+ 188.0682; found 188.2123.
Conclusions
In conclusion, we have developed a rapid and metal-free oxidation protocol to access carbonyl compounds from primary and secondary amines using PhI(OAc)2 in combination with catalytic amount of TEMPO as an eco-friendly oxidation without the need of external oxygen source under mild conditions. In addition, we established the mechanistic pathway, with aid of control experiments, time-dependent 1H NMR and GC-MS analyses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. H. B. thanks, UGC, New Delhi, India for the award of research fellowship. The authors are thankful to Mr Pramod Mali (Centre for NMR Facility, CSIR-NCL) for assistance with NMR Study.Notes and references
- (a) G. Tojo and M. Fernandez, Oxidation of Alcohols to Aldehydes and Ketones, Springer, New York, 1st edn, 2006 Search PubMed; (b) R. A. Sheldon and I. W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef; (c) J. M. Vatèle, Synlett, 2006, 2055–2058 CrossRef; (d) T. Dohi, K. Fukushima, T. Kamitanaka, K. Morimoto, N. Takenagab and Y. Kita, Green Chem., 2012, 14, 1493–1501 RSC.
- (a) K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, J. Am. Chem. Soc., 2004, 126, 5192–5201 CrossRef PubMed; (b) G. Chu and C. Li, Org. Biomol. Chem., 2010, 8, 4716–4719 RSC; (c) P. Galletti, F. Funiciello, R. Soldati and D. Giacomini, Adv. Synth. Catal., 2015, 357, 1840–1848 CrossRef; (d) V. V. Patil, E. M. Gayakwad and G. S. Shankarling, J. Org. Chem., 2016, 81, 781–786 CrossRef PubMed; (e) V. V. Patil and G. S. Shankarling, J. Org. Chem., 2015, 80, 7876–7883 CrossRef PubMed; (f) X. Wu, C. B. Bheeter, H. Neumann, P. H. Dixneuf and M. Beller, Chem. Commun., 2012, 48, 12237–12239 RSC; (g) J. W. Kim, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2008, 47, 9249–9251 (Angew. Chem., Int. Ed., 2008, 120, 9389–9391) CrossRef PubMed; (h) Y. Wang, H. Kaboyashi, K. Yamaguchi and N. Mizuno, Chem. Commun., 2012, 48, 2642–2644 RSC.
- S. S. Rawalay and H. Shechter, J. Org. Chem., 1967, 32, 3129–3131 CrossRef.
- S. Sabhani and M. F. Maleki, Synlett, 2010, 383–386 CrossRef.
- S. Sobhani, S. Aryanejad and M. F. Maleki, Helv. Chim. Acta, 2012, 95, 613–617 CrossRef.
- S. Chandrasekhar, G. P. K. Reddy, C. Nagesh and C. R. Reddy, Tetrahedron Lett., 2007, 48, 1269–1271 CrossRef.
- S. Naya, T. Niwa, R. Negishi, H. Kobayashi and H. Ada, Angew. Chem., Int. Ed., 2014, 53, 13894–13897 (Angew. Chem., Int. Ed., 2014, 126, 14114–14117) CrossRef PubMed.
- N. Iqbal and E. J. Cho, Adv. Synth. Catal., 2015, 357, 2187–2192 CrossRef.
- J. Srogl and S. Voltrova, Org. Lett., 2009, 11, 843–845 CrossRef PubMed.
- S. Gaspa, A. Porcheddu, A. Valentoni, S. Garroni, S. Enzo and L. De Luca, Eur. J. Org. Chem., 2017, 5519–5526 CrossRef.
- G. Wang, G. P. de Silva, N. E. Wiebe, G. M. Fehra and R. L. Davis, RSC Adv., 2017, 7, 48848–48852 RSC.
- S. S. Chavan, B. D. Rupanawar, R. B. Kamble, A. M. Shelke and G. M. Suryavanshi, Org. Chem. Front., 2018, 5, 544–548 RSC.
- Recent reviews: (a) H. Togo and M. Katohgi, Synlett, 2001, 565–581 CrossRef; (b) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523–2584 CrossRef PubMed; (c) P. J. Stang, J. Org. Chem., 2003, 68, 2997–3008 CrossRef PubMed; (d) H. Tohma and Y. Kita, Adv. Synth. Catal., 2004, 346, 111–124 CrossRef; (e) R. M. Moriarty, J. Org. Chem., 2005, 70, 2895–2903 Search PubMed; (f) R. D. Richardson and T. Wirth, Angew. Chem., Int. Ed., 2006, 45, 4402–4404 CrossRef PubMed.
- (a) V. V. Zhdankin and P. Stang, Chem. Rev., 2008, 108, 5299–5358 CrossRef PubMed; (b) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef PubMed; (c) M. Ochiai and K. Miyamoto, Eur. J. Org. Chem., 2008, 4229–4239 CrossRef; (d) T. Dohi and Y. Kita, Chem. Commun., 2009, 2073–2085 RSC; (e) M. Uyanik and K. Ishihara, Chem. Commun., 2009, 2086–2099 RSC; (f) L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235–2262 CrossRef; (g) V. V. Zhdankin, J. Org. Chem., 2011, 76, 1185–1197 CrossRef PubMed; (h) E. A. Merritt and B. Olofsson, Synthesis, 2011, 4, 517–538 Search PubMed.
- (a) Hypervalent Iodine Chemistry, in Top. Curr. Chem., ed. T. Wirth, Springer, Berlin, 2003, p. 224 Search PubMed; (b) T. Wirth, Organic Synthesis Highlights V, Wiley-VCH, Weinheim, 2003, p. 144 Search PubMed; (c) T. Wirth, Angew. Chem., Int. Ed., 2001, 40, 2812–2814 CrossRef; (d) V. V. Zhdankin, Curr. Org. Synth., 2005, 2, 121 CrossRef.
- (a) T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656–3665 CrossRef PubMed; (b) U. Ladziata and V. V. Zhdankin, ARKIVOC, 2006, 4, 26 Search PubMed; (c) M. Ochiai, Chem. Rec., 2007, 7, 12–23 CrossRef PubMed; (d) M. A. Ciufolini, N. A. Braun, S. Canesi, M. Ousmer, J. Chang and D. Chai, Synthesis, 2007, 3759–3772 CrossRef; (e) U. Ladziata and V. V. Zhdankin, Synlett, 2007, 527–537 Search PubMed; (f) S. Quideau, L. Pouysegu and D. Deffieux, Synlett, 2008, 467–495 CrossRef.
- (a) K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, J. Am. Chem. Soc., 2004, 126, 5192–5201 CrossRef PubMed; (b) K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, Angew. Chem., Int. Ed., 2003, 42, 4077–4082 CrossRef PubMed; (c) J. N. Moorthy and I. Neogi, Tetrahedron Lett., 2011, 52, 3868–3871 CrossRef; (d) R. M. Moriarty, T. E. Hopkins, I. Prakash and R. K. Vaid, Synth. Commun., 1990, 20, 2353–2357 CrossRef; (e) P. Moller and D. M. Gilbert, Tetrahedron, 1988, 44, 7171–7175 CrossRef.
- (a) R. M. Moriarty, R. K. Vaid and M. P. Duncan, Tetrahedron Lett., 1988, 29, 6913–6916 CrossRef; (b) S. Desjardkins, G. Jacquemot and S. Canesi, Synlett, 2012, 23, 1497–1500 CrossRef.
- (a) S. Narayana Murthy and Y. V. D. Nageswar, Tetrahedron Lett., 2011, 52, 4481–4484 CrossRef; (b) J. S. Yadav, B. V. S. Reddy, A. K. Basak, G. Baishya and A. Venkat Narsaiah, Synthesis, 2006, 3, 0451–0454 CrossRef.
- P. Galletti, G. Martelli, G. Prandini, C. Colucci and D. Giacomini, RSC Adv., 2018, 8, 9723–9730 RSC.
- (a) M. F. Semmenlhack and C. R. Schmid, J. Am. Chem. Soc., 1983, 105, 6732–6734 CrossRef; (b) M. Angelin, M. Hermansson, H. Dong and O. Ramström, Eur. J. Org. Chem., 2006, 4323–4326 CrossRef; (c) A. D. Mico, R. Margarita, L. Parlanti, A. Vescoci and G. Piancatelli, J. Org. Chem., 1997, 62, 6974–6977 CrossRef; (d) A. E. J. de Nooy, A. C. Besemer and H. van Bekkum, Synthesis, 1996, 1153–1176 CrossRef.
- B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef.
- S. Cho, H. Kim, C. Ahn, J. R. Falck and D. Shin, Tetrahedron Lett., 1999, 40, 8215–8217 CrossRef.
- A. Wang and H. Jiang, J. Org. Chem., 2010, 75, 2321–2326 CrossRef PubMed.
- Q. Feng and Q. Song, J. Org. Chem., 2014, 79, 1867–1871 CrossRef PubMed.
- E. Barragan and A. Bugarin, J. Org. Chem., 2017, 82, 1499–1506 CrossRef PubMed.
- S. H. Christensen, E. P. K. Olsen, J. Rosenbaum and R. Madsen, Org. Biomol. Chem., 2015, 13, 938–945 RSC.
- W. Han, B. Liu, J. Chen and Q. Zhou, Synlett, 2017, 28, 835–840 CrossRef.
- M. Iinuma, K. Moriyama and H. Togo, Eur. J. Org. Chem., 2014, 772–780 CrossRef.
- J. Ma, C. Hong, Y. Wan, M. Li, X. Hu, W. Mo, B. Hu, N. Sun, L. Jin and Z. Shen, Tetrahedron Lett., 2017, 58, 652–657 CrossRef.
- Y. Matsukawa, T. Hirashita and S. Araki, Tetrahedron, 2017, 73, 6052–6056 CrossRef.
- S. Wu, L. Chen, B. Yin and Y. Li, Chem. Commun., 2015, 51, 9884–9887 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07451h |
| This journal is © The Royal Society of Chemistry 2018 |