
Organic hole-transporting materials for 9.32%-efficiency and stable CsPbBr3 perovskite solar cells†
Yuanyuan
Zhao‡
 a,
Tianshu
Liu‡
a,
Fumeng
Ren‡
b,
Jialong
Duan
a,
Yudi
Wang
*a,
Xiya
Yang
*a,
Qinghua
Li
*b and
Qunwei
Tang
*a
a,
Tianshu
Liu‡
a,
Fumeng
Ren‡
b,
Jialong
Duan
a,
Yudi
Wang
*a,
Xiya
Yang
*a,
Qinghua
Li
*b and
Qunwei
Tang
*a
aInstitute of New Energy Technology, College of Information Science and Technology, Jinan University, Guangzhou 510632, P. R. China. E-mail: wangyd@jnu.edu.cn; xiyayang@jnu.edu.cn; tangqunwei@jnu.edu.cn
bSchool of Physics Science and Technology, Lingnan Normal University, Zhanjiang 524048, P. R. China. E-mail: qhli@hqu.edu.cn
First published on 8th October 2018
Abstract
Cost-effective and stable CsPbBr3-based inorganic perovskite solar cells (PSCs) are regarded as promising candidates for next-generation photovoltaics. However, the large interfacial energy differences at the CsPbBr3/hole-transporting layer lead to serious charge recombination and poor charge extraction kinetics. Herein, we prepare a series of hole-transporting materials (HTMs) to improve hole extraction and to reduce electron–hole recombination at the CsPbBr3/HTM interface. In comparison with the power conversion efficiency (PCE) of 6.10% for an HTM-free device, the CsPbBr3 PSCs with polymeric HTMs such as polythiophene, polypyrrole and polyaniline yield efficiencies of 8.36%, 8.32% and 7.69%, respectively. Similarly, the inorganic PSC with organic small molecule BT-BTH achieves a PCE as high as 9.32% due to the improved hole conductivity. Moreover, the unencapsulated PSC with BT-BTH maintains 94% of its initial efficiency in 70% relative humidity over 80 days.
Introduction
Organic–inorganic hybrid perovskite solar cells (PSCs) have attracted considerable interest due to their low-cost fabrication and high photovoltaic performances.1–3 Grätzel et al. designed the first all-solid state PSCs using CH3NH3PbI3 quantum dots as light harvesting materials, yielding a power conversion efficiency (PCE) of 9.7% under one sun illumination.4 To date, the maximized certified PCE has been 23.3% arising from optimization of the perovskites, processing technology and device structure.5 Although the PCE of hybrid PSCs has exhibited rapid growth over the past few years, the intrinsic instability of organic–inorganic perovskites and iodide diffusion are still challenging for their commercialization.6–10Recently, all-inorganic CsPbBr3 perovskite has been regarded as a promising light-harvester because of its high environmental tolerances and stabilized crystal structure.11,40 However, the first CsPbBr3 based PSC with a classical configuration of FTO/TiO2/CsPbBr3/carbon has a PCE of only 6.7%,12 which is much lower than those of the state-of-the-art hybrid PSCs. There are two main reasons for the low efficiency of CsPbBr3 PSCs. On the one hand, the bandgap as large as 2.3 eV for CsPbBr3 results in a narrow absorption wavelength below 550 nm. On the other hand, the high energy difference at the CsPbBr3/carbon interface (0.6 eV) causes serious electron–hole recombination and poor charge extraction.13 Although the substitution of Br− with I− ions to form mixed CsPbI3−xBrx (x = 0, 1, 2) halides provides a way of reducing perovskite bandgaps and energy differences at interfaces, the environmental tolerances especially in high humidity are also brought down. A solution to this impasse is to set an intermediate energy by introducing a hole-transporting layer (HTL) or coating quantum dots14 between CsPbBr3 and the carbon electrode to reduce interface recombination loss and to promote charge extraction. The low coverage of quantum dots on CsPbBr3 film does not maximize the hole extraction ability, therefore the addition of a HTM is more feasible in charge-transporting theory and solar cell structure.
Hole-transporting materials (HTMs) are good conductors that enable photo-induced holes to be efficiently extracted from the perovskite layer to the back electrodes. This requires HTMs to possess excellent charge-carrier mobility and suitable energy levels to match well with the perovskite halide and back electrode. Small-molecule HTMs made by molecular engineering provide advantages of monodispersity, high purity and good reproducibility in synthesis, and tunable energy levels, charge mobility and hole conductivity in physicochemical performances.
The state-of-the-art HTMs such as 2,2′,7,7′-tetrakis-(N,N-di-p-methoxyphenylamine)-9,9′-spiro biuorene (spiro-MeOTAD) and poly-triarylamine (PTAA) are still economic burdens for their commercial applications in PSCs.15–22 Such HTMs generally need to be doped with lithium ions because of their poor conductivity, however, the strong hygroscopicity of dopant lithium ions simultaneously increases the instability of these HTMs. Alternatively, other cost-effective p-type conducting polymers such as poly 3,4-ethylenedioxythiophene (PEDOT), polypyrrole (PPy) and polyaniline (PANi) can be employed as electron barrier layers without dopant due to their high hole mobility from the conjugated structure. Nevertheless, the long molecular chains and inhomogeneity of polymers are detrimental to film forming properties. Therefore, it is imperative to develop undoped organic molecular materials.
In this work, we present the synthesis of an organic small molecule BT-BTH [2-(3,5-bis(5-(5-hexylthiophen-2-yl)thiophen-2-yl)thiophen-2-yl)-3,5-bis(5-(5-hexylthiophen-2-yl)thiophen-2-yl)thiophene] as a HTM for an inorganic CsPbBr3 PSC. A maximum PCE of 9.32% is obtained for the BT-BTH based device; alternatively, PEDOT, PPy and PANi are also synthesized as HTMs, yielding PCEs of 8.36%, 8.32% and 7.69% in the corresponding solar cells, respectively. Besides, the long-term stability of the BT-BTH based device free of any encapsulation maintains 94% of the initial efficiency in 70% RH humidity over 80 days. The advantages of high efficiency, good stability and low cost of CsPbBr3 PSCs demonstrate that small molecule BT-BTH and large conjugated polymers are promising HTMs for inorganic PSCs.
Experimental
Synthesis of PEDOT, PPy and PANi
PEDOT, PPy and PANi were synthesized by an in situ chemical oxidative polymerization method. The synthesis of PEDOT was carried out in a three-necked flask filled with high-purity nitrogen. Anhydrous FeCl3 (8.94 g, 0.055 mol) was dissolved in 180 mL of chloroform, thiophene monomer was added slowly to the flask and the mixture was agitated for 24 h at room temperature. The molar ratio of FeCl3 to thiophene monomer was controlled at 2.3. After polymerization, the PEDOT was rinsed with methanol, 1 M hydrochloric acid and deionized water three times. Finally, the solid powders were filtrated and dried at 60 °C for 24 h.23,247.788 g of FeCl3·6H2O was dissolved in 58 mL of deionized water, then 1 mL of pyrrole monomer was added slowly to the above solution, and the mixture was stirred for 24 h at 5 °C. After polymerization, the PPy powders were collected, rinsed by deionized water and vacuum dried at 60 °C for 24 h.25 10 mg of PPy powders was dispersed in 1 mL of chloroform to make the precursor for depositing the HTL.
(NH4)2S2O8 aqueous solution was obtained by dissolving 2.912 g of (NH4)2SO4 in 100 mL of 1 M hydrochloric acid solution. PANi was prepared by dropwise adding 20 mL of (NH4)2S2O8 solution into 20 mL of a hydrochloric acid solution of aniline at 0 °C for 3 h.26 Then the PANi powders were rinsed with deionized water, ethanol, and 2 M HCl solution, and dried at 60 °C for 24 h. The FTIR spectra of BT-BTH, PEDOT, PPy and PANi are shown in Fig. S3 (ESI†).
Synthesis of BT-BTH
The synthesis routes of BT-BTH and the analysis of 1H NMR and 13C NMR are described in Fig. S1 (ESI†). In detail, intermediate compound 2 was obtained according to the following method. 16.6 g of 2,2′-bithiophene was dissolved in 150 mL of anhydrous tetrahydrofuran (THF), and 44 mL of n-butyl lithium (n-BuLi) and 18.15 g of bromohexane were slowly added into the mixture under an argon atmosphere. After agitating for 10 h, the reactant was cooled down and then extracted by ether. Finally, the mixture was dried and distilled under reduced pressure to obtain a colorless liquid after rinsing three times with saturated NaHCO3 and NaCl solution. Under the same conditions, compound 2 was treated with n-BuLi, followed by the addition of tri-n-butyltin chloride, which afforded the compound 3 in 80% yield. Formation of the compound 5 was achieved by the bromination reaction with 2,2′-bithiophene and chloroform solution of HOAC. The coupling reaction of compound 3 and compound 5 yielded the target molecule BT-BTH in 70% yield.27,28Preparation of a TiO2 anode
Fluorine-doped tin oxide (FTO) glass was etched by Zn powders and HCl for a desirable strip pattern and further rinsed with acetone, ethanol, and deionized water. The compact TiO2 (c-TiO2) layer was deposited on FTO glass by spin-coating an ethanol solution of titanium isopropoxide (0.5 M) and diethanol amine (0.5 M) at 7000 rpm for 30 s and annealing in air at 500 °C for 2 h. The mesoscopic TiO2 (m-TiO2) layer was then deposited by spin-coating a TiO2 paste at 2000 rpm for 30 s and annealed in air at 450 °C for 30 min. TiO2 paste was prepared according to our previous work.29 Then the m-TiO2 film was immersed in an aqueous solution of 0.04 M TiCl4 at 70 °C for 30 min, cleaned with deionized water and ethanol, and finally annealed at 450 °C for another 30 min.Assembly of solar cells
An N,N-dimethylformamide (DMF) solution of 1.0 M PbBr2 was spin-coated onto the m-TiO2 layer at 2000 rmp for 30 s, followed by heating at 80 °C for 30 min. Then 90 μL of 0.07 M CsBr methanol solution was coated onto FTO/c-TiO2/m-TiO2/PbBr2 at 2000 rmp for 30 s and heated at 250 °C for 5 min. The CsBr was repeatedly spin-coated four times to form a CsPbBr3 layer.30,31 The HTM solution in chlorobenzene was spin-coated at 2000 rmp for 30 s and then heated at 80 °C for 10 min. Finally, the carbon back electrode was deposited on the CsPbBr3 layer by doctor-blade coating carbon ink and heating at 90 °C for 10 min.Photovoltaic measurements and characterization
The current density–voltage (J–V) curves were measured using a solar simulator (Newport, Oriel Class A, 91195A) under AM 1.5G simulated solar illumination (100 mW cm−2, calibrated by a standard silicon solar cell). The photoelectric performances of each device were measured at least ten times to control the experimental error within ±5%. The crystal structure of a perovskite material was tested by X-ray diffraction (PHILIPSPW1800 diffractometer with Cu-Kα radiation), and the morphologies of the perovskite film and solar cell were characterized by field-emission scanning electron microscopy (FESEM, Japan Hitachi field emission S4800). The steady-state photoluminescence was recorded by an FLS920 all functional fluorescence spectrometer. The incident-photo-to-current conversion efficiency (IPCE) was characterized by a power source (Newport 300 W xenon lamp, 66920) with a monochromator (Newport Cornerstone 260) in the wavelength range of 300–1000 nm. The absorption spectra were recorded on an ultraviolet-visible spectrophotometer (UV-3600, Shimadzu) at room temperature. The time-resolved PL (TRPL) spectra were obtained on a time-resolved fluorescence spectrometer (Horiba Jobin Yvon, FL).Results and discussion
Fig. 1a represents the preparation routes for an all-inorganic PSC device. The CsPbBr3 perovskite layer is prepared by using a multi-step solution-processable method developed by our group: one layer of PbBr2 and sequentially four successive layers of CsBr are spin-coated. Due to the large concentration difference between saturated PbBr2 solution in DMF and CsBr methanol solution, the state-of-the-art one-step or two-step methods are inapplicable to make a high-purity CsPbBr3 layer. The excessive PbBr2 can be balanced by multiple CsBr2 layers, resulting in a complete transformation of PbBr2 and CsBr into perovskite-structured CsPbBr3 phase (Fig. S2, ESI†). The stability of the perovskite is obviously promoted thanks to removal of all organic components. Fig. 1b shows a scanning electron microscopy (SEM) image of an inorganic CsPbBr3 layer, which depicts a dense and uniform surface with a grain size of around 700 nm. HTMs are spin-coated onto the CsPbBr3 perovskite layer to modify the CsPbBr3/carbon interface and the corresponding SEM images of CsPbBr3 film covered with BT-BTH, PEDOT, PPy, and PANi HTMs are shown in Fig. 1c–f. BT-BTH, PEDOT, and PPy materials have a larger grain size and higher surface coverage compared with PANi, contributing to facile charge transport and reduced energy loss at the interface.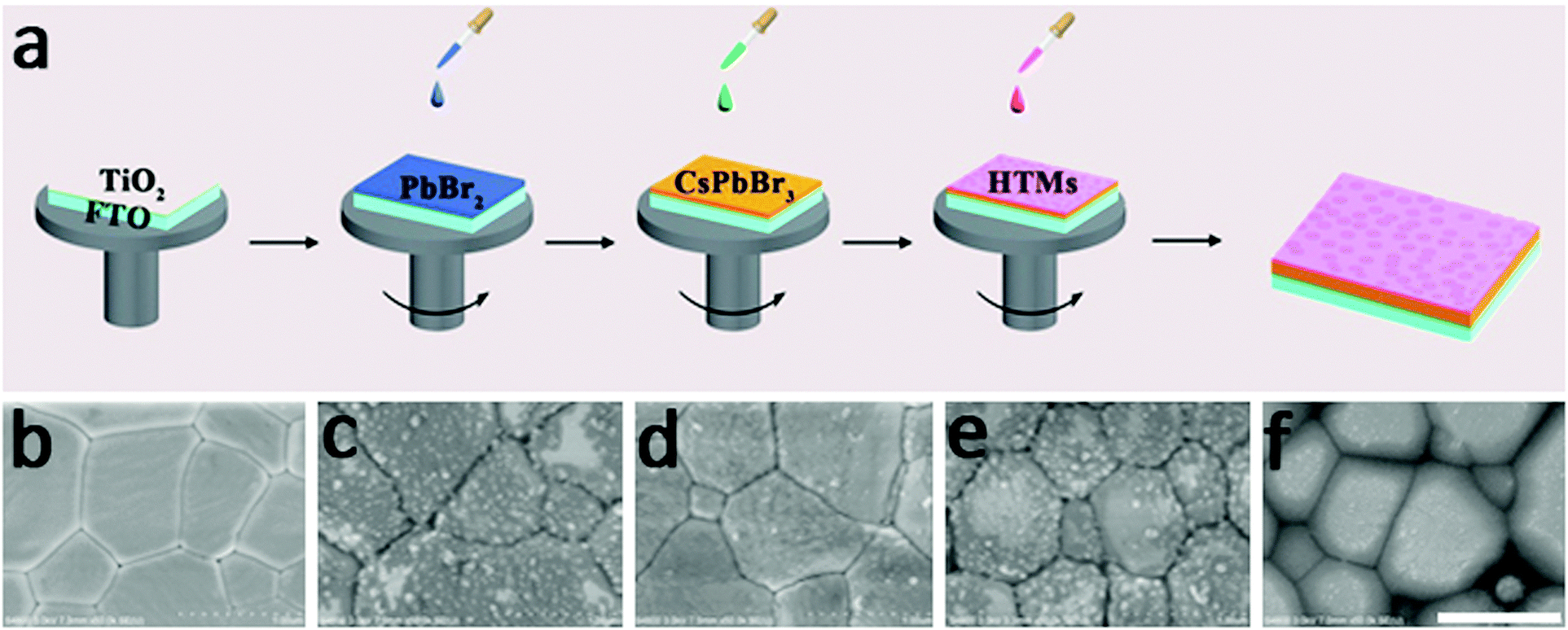 | ||
| Fig. 1 (a) The diagrammatic sketch for making TiO2, PbBr2, CsPbBr3 and HTM layers. (b) Top-view SEM image of the CsPbBr3 layer. Top-view SEM pictures of CsPbBr3 film covered with (c) BT-BTH, (d) PEDOT, (e) PPy and (f) PANi. Scale bar: 1 μm. | ||
The inorganic PSC has a device configuration of FTO/c-TiO2/m-TiO2/CsPbBr3/HTL/carbon, as shown in Fig. 2a. The cross-sectional SEM images in Fig. 2b and Fig. S4 (ESI†) indicate a typical multi-layered structure with average thicknesses of 200 nm, 400 nm and 15 μm for c-TiO2/m-TiO2, CsPbBr3 and the carbon electrode (Fig. S5, ESI†), respectively. Furthermore, the rough surface for either FTO, c-TiO2/m-TiO2 or CsPbBr3 is expected to increase the adhesion of the next layer and to reduce the charge loss at cell interfaces. The fluctuant CsPbBr3 grains lead to a rough HTM surface, but the thickness of the HTL in the valley or peak is almost the same. The homogeneous distribution of HTMs may maximize the hole extraction from CsPbBr3 film. The J–V characteristics of the PSCs with different HTMs are measured under simulated AM 1.5G solar illumination. The optimal concentration is 3 mg mL−1 by optimization of the solution concentration of HTMs (Fig. S6 and S7, ESI†). Fig. 2c displays J–V curves of all-inorganic CsPbBr3 PSCs under a light intensity of 100 mW cm−2 and the photovoltaic data including short-circuit density (Jsc), open-circuit voltage (Voc), fill factor (FF), and PCE are summarized in Table 1. The HTM-free device achieves a PCE of 6.10% (Jsc = 5.93 mA cm−2, Voc = 1.33 V, FF = 77.5%), which is comparable to that reported by Jin's group.12 After adding a HTM between the CsPbBr3 layer and carbon electrode, the photovoltaic data are significantly improved. A maximum PCE of 9.32% is obtained for the BT-BTH based CsPbBr3 PSC and the related parameters of Jsc, Voc, and FF are 7.56 mA cm−2, 1.50 V, and 81.9%, respectively. Moreover, the CsPbBr3 PSCs tailored with PEDOT, PPy and PANi HTMs present PCEs of 8.36%, 8.33% and 7.69%, respectively. The improved PCE by setting the HTL at the perovskite/carbon interface may be attributed to the reduced electron–hole recombination and therefore improved charge extraction, which will be discussed in the next section.
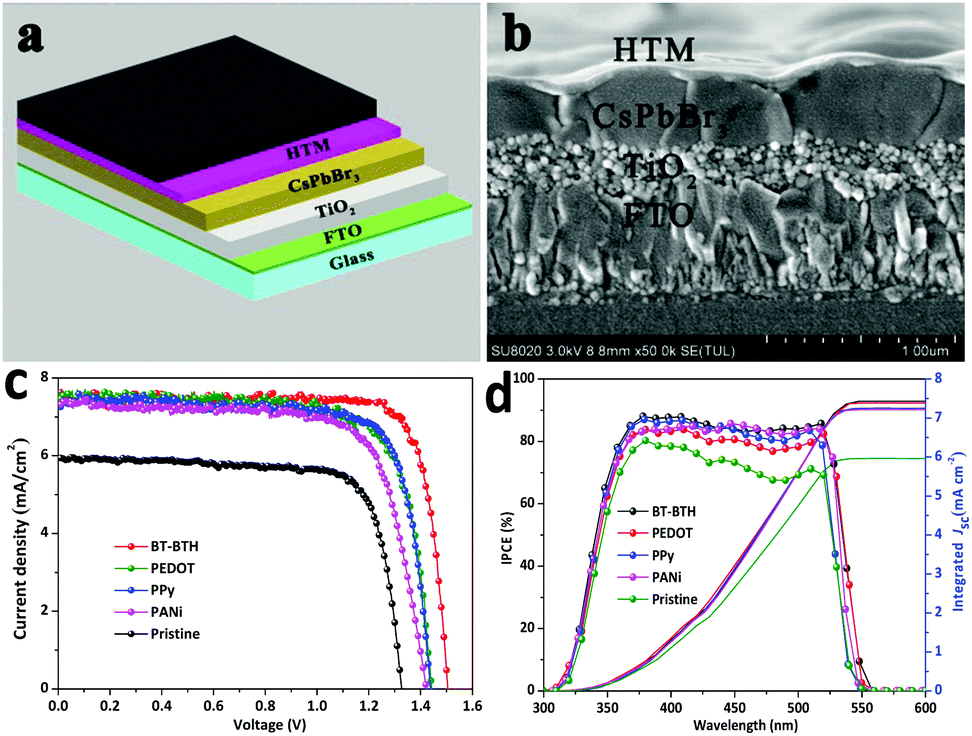 | ||
| Fig. 2 (a) Illustration of an inorganic PSC device. (b) Cross-sectional SEM image of a multilayer structure with FTO/TiO2/CsPbBr3/HTM configuration. (c) J–V plots and (d) IPCE spectra of these inorganic PSCs. | ||
| Devices with HTMs | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| BT-BTH | 1.50 (1.51) | 7.56 (7.60) | 81.9 (80.0) | 9.32 (9.25) |
| PEDOT | 1.44 (1.45) | 7.56 (7.55) | 76.7 (76.1) | 8.36 (8.48) |
| PPy | 1.44 (1.42) | 7.37 (7.30) | 78.6 (77.5) | 8.33 (8.30) |
| PANi | 1.42 (1.40) | 7.33 (7.30) | 73.8 (73.2) | 7.69 (7.89) |
| HTM-free | 1.33 (1.35) | 5.93 (5.98) | 77.5 (75.4) | 6.10 (6.37) |
IPCE spectra in Fig. 2d are performed to cross-check the photoelectric behaviours of these inorganic CsPbBr3 PSCs. It is noticeable that these inorganic CsPbBr3 PSCs exhibit a narrower light response in a wavelength range of 325–550 nm compared with the state-of-the-art organic–inorganic PSCs.15,32–35 In the current work, the lower PCE of CsPbBr3 solar cells is due to a large bandgap (2.3 eV) of CsPbBr3. The maximal IPCE value of a pristine device is around 78.3%, and it upgrades to 88.3%, 86.6%, 85.6% and 84.2% for BT-BTH, PEDOT, PPy and PANi based PSCs, respectively. The integrated Jsc values are 7.48, 7.38, 7.25, 7.21 and 5.96 mA cm−2 for BT-BTH, PEDOT, PPy, and PANi based PSCs and the pristine device, which is consistent with the result of the J–V characteristics tests (Fig. 2c).
Fig. S8a (ESI†) shows the PCE distribution of 10 random devices tailored with different HTMs. It can be concluded that all these inorganic PSCs have high reproducibility within ±5% and the BT-BTH based PSC exhibits reasonable performance distribution and high reproducibility. Steady-state photocurrent outputs are monitored at the point of maximum power for an illumination period of 100 s, as shown in Fig. S8b (ESI†). The BT-BTH, PEDOT and pristine PSCs achieve stabilized PCEs of 9.1%, 8.2% and 6.0%, respectively. The differences between PCEs obtained from steady-state and dynamic-state characterization are mainly attributed to the hysteresis effect in the devices.
In order to investigate the recombination reaction of photo-generated charges at the CsPbBr3/HTL interface, we study the variation of Jsc and Voc for inorganic PSCs with and without HTMs as a function of the graded-regulated incident light intensity, as shown in Fig. 3a and b. The interior charge recombination mechanism can be understood according to the following equation: Jsc ∝ Iα (α ≤ 1),36 where I represents light intensity and α is an exponential factor to evaluate charge recombination. An α value closer to 1 means lower levels of charge recombination or limited space charge effects. According to the fitting result (Fig. 3a), the α value of the pristine PSC is 0.902, while it increases to 0.994, 0.948, 0.927, and 0.920 for the BT-BTH, PEDOT, PPy, and PANi based PSCs, respectively. This suggests a reduced radiative recombination owing to a higher hole extraction ability of small molecular BT-BTH. Additionally, Fig. 3b displays the dependence of Voc upon light intensity of the PSC devices. The relationship between Voc and light intensity follows Voc = εkT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(I)/q + constant,37 where ε is the ideality factor, k is the Boltzmann constant, T is the absolute temperature, and q is elementary charge. According to the Shockley–Read–Hall recombination mechanism, the slopes of the Voc–I curves represent the degree of monomolecular recombination. The slope value is 1.848 kT/e for the pristine PSC, and it reduced to 1.479 kT/e, 1.576 kT/e, 1.658 kT/e, and 1.678 kT/e for the BT-BTH, PEDOT, PPy, and PANi based PSCs, respectively. Both investigations on Jsc and Voc demonstrate a suppressed charge recombination by employing conductive polymers as HTMs for inorganic CsPbBr3 PSC applications.
ln(I)/q + constant,37 where ε is the ideality factor, k is the Boltzmann constant, T is the absolute temperature, and q is elementary charge. According to the Shockley–Read–Hall recombination mechanism, the slopes of the Voc–I curves represent the degree of monomolecular recombination. The slope value is 1.848 kT/e for the pristine PSC, and it reduced to 1.479 kT/e, 1.576 kT/e, 1.658 kT/e, and 1.678 kT/e for the BT-BTH, PEDOT, PPy, and PANi based PSCs, respectively. Both investigations on Jsc and Voc demonstrate a suppressed charge recombination by employing conductive polymers as HTMs for inorganic CsPbBr3 PSC applications.
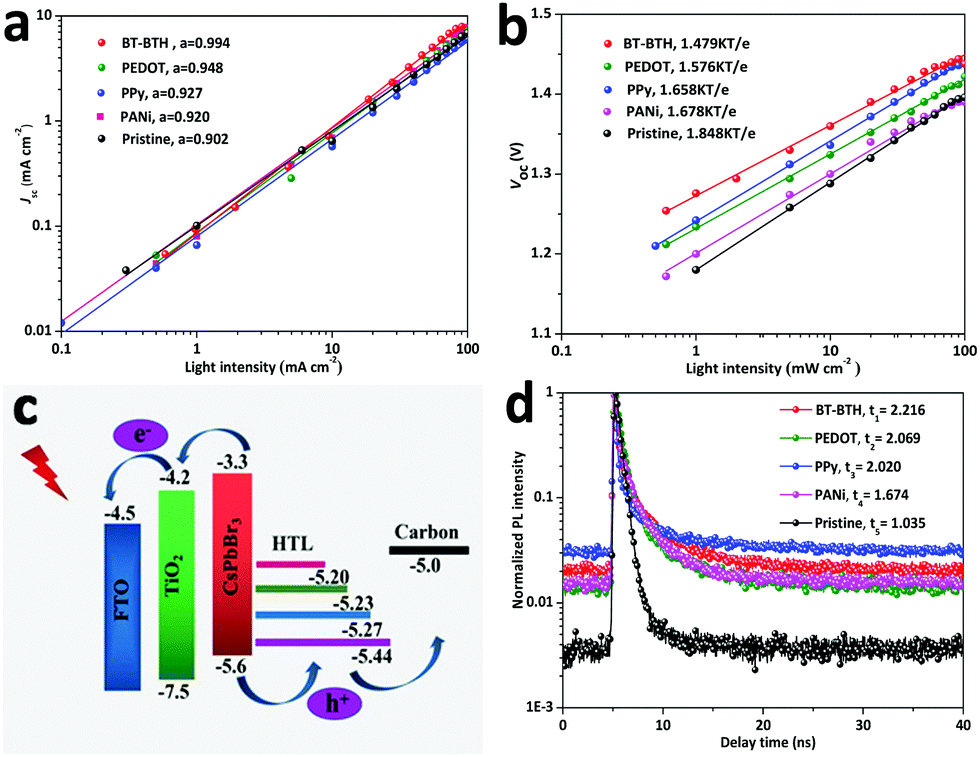 | ||
| Fig. 3 The plots of (a) Jsc and (b) Vocversus light intensity. (c) Diagram of energy level alignments for inorganic CsPbBr3 PSCs. (d) TRPL decay curves for HTM-free and HTM tailored PSC devices. | ||
The diagram of energy level alignments for the inorganic CsPbBr3 PSC is shown in Fig. 3c. The corresponding HOMO energy level values are calculated according to cyclic voltammetry curves (Fig. S9, ESI†). And specific values are determined to be −5.44, −5.27, −5.23 and −5.20 eV for BT-BTH, PEDOT, PPy and PANi, respectively. Under sunlight illumination, the perovskite layer absorbs photons to produce electrons and holes. Photo-induced electrons transfer from their conduction band (CB) to the CB of TiO2, while holes are collected by the carbon electrode. However, the high energy level difference (ΔE = 0.6 eV) at the CsPbBr3/carbon interface results in serious electron–hole recombination.38 HTMs can effectively reduce the energy level difference by setting an intermediate energy band between CsPbBr3 and carbon. BT-BTH has the lowest energy level difference from CsPbBr3, therefore it can best inhibit charge recombination and accelerate holes extraction.
Steady-state photoluminescence (PL) and the time-resolved PL (TRPL) spectra are characterized to investigate the charge-transporting performance. PL spectra of the inorganic PSC with and without polymeric HTMs are shown in Fig. S10 (ESI†). All films exhibit similar characteristic peaks at 535 nm, which is consistent with previous reports.39–41 It should be noted that polymeric HTMs show more efficient PL quenching than the pristine PSC, especially in the case of the BT-BTH HTM tailored solar cell. The intensity of PL decreases in the following order: pristine > PANi > PPy > PEDOT > BT-BTH. The smallest PL intensity for BT-BTH means an optimal hole extraction ability. Besides, the values of electron lifetime are extracted from TRPL, as shown in Fig. 3d, and the FTO/TiO2/CsPbBr3/BT-BTH film presents the longest electron lifetime of 2.22 ns, which indicates the most effective hole-injection rate. Furthermore, the space-charge limited current (SCLC) tests (Fig. S11, ESI†) also indicate an increased hole mobility. All of these results manifest the promoting effect of introducing BT-BTH, PEDOT, PPy, and PANi in all-inorganic CsPbBr3 PSCs in accelerating charge extraction and therefore enhancing photovoltaic performances.
High efficiency,42,43 improved stability44,45 and low cost are three criteria to evaluate the practical applications of a new-type solar cell. The cell efficiency of 9.32% is in a very high level in comparison with the highest record for a CsPbBr3 PSC reported in our previous literature.46 Using the solution-processable method, the cost of PSCs is regarded as half that of commercial silicon solar cells, therefore the stability of these CsPbBr3 PSCs seems to be crucial to determine their potential commercialization. Therefore, the humidity tolerances of the BT-BTH and PEDOT based inorganic PSCs were investigated to assess their applicability. The devices without any encapsulation are measured by storing in an atmosphere of 70% RH in air at 20 °C. Fig. 4 presents the normalized PCE, Jsc, Voc, and FF as a function of exposure time. Both devices are relatively stable and the PCE of the BT-BTH based device still maintains 94% of the initial efficiency over 80 days compared with 92% for the pristine device. The improved stability along with high solar cell efficiency demonstrates polymeric HTMs to be promising for high-performance CsPbBr3 PSC platforms.
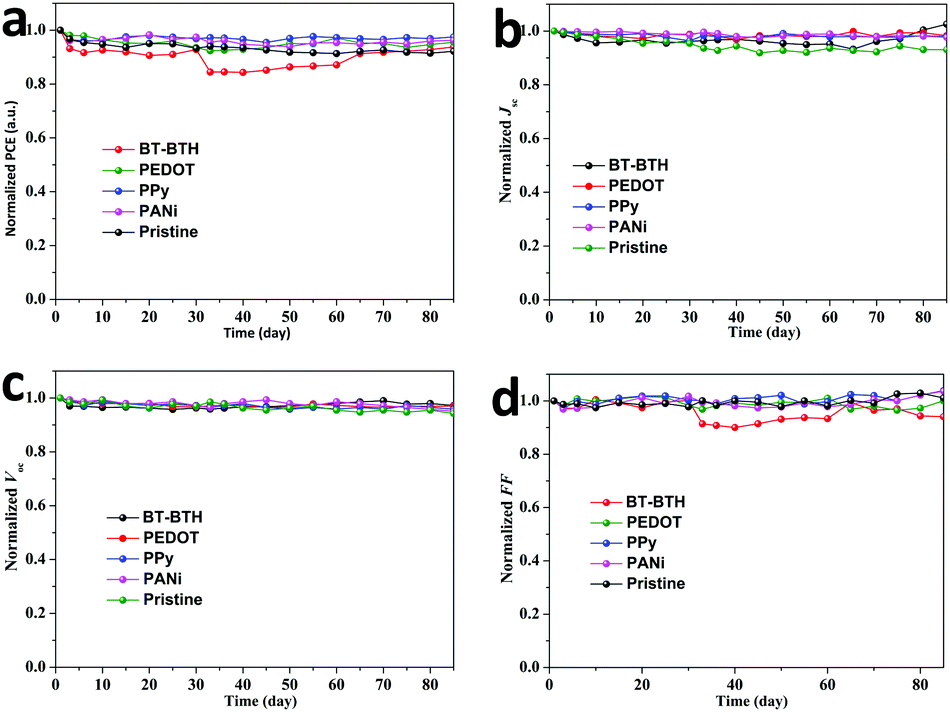 | ||
| Fig. 4 The normalized stability for (a) PCE, (b) Jsc, (c) Voc and (d) FF for the HTM-free solar cell and the PSCs with BT-BTH, PEDOT, PPy and PANi as HTMs. The stability of these CsPbBr3 PSCs free of encapsulation is measured at a temperature of 20 °C and 70% relative humidity. | ||
Conclusions
In summary, we present the feasibility of using either small molecular BT-BTH or conjugated PEDOT, PPy, and PANi as an HTM material for inorganic CsPbBr3 PSCs incorporating HTLs to reduce charge-hole recombination at the CsPbBr3/carbon interface for high-performance. The BT-BTH based device achieves a maximum PCE as high as 9.32% under one sun illumination, which is higher than 8.36%, 8.32%, 7.69%, and 6.10% for PEDOT, PPy, PANi and HTM-free inorganic PSCs, respectively. Detailed study demonstrates a reduced electron–hole recombination and improved charge extraction at cell interfaces. Moreover, the unencapsulated PSCs tailored with polymeric HTMs present improved humidity tolerance in high humidity over 80 days. These cost-effective and facile HTMs provide new opportunities of increasing the PCE outputs of inorganic CsPbBr3 PSCs with sacrificing long-term stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (21503202 and 61774139) and the Fundamental Research Funds for the Central Universities (11618409).Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- H. Zhang, J. Mao, H. X. He, D. Zhang, H. L. Zhu, F. X. Xie, K. S. Wong, M. Grätzel and W. C. H. Choy, Adv. Energy Mater., 2015, 5, 1501354 CrossRef.
- H. S. Li, Y. S. Li, Y. M. Li, J. J. Shi, H. Y. Zhang, X. Xu, J. H. Wu, H. J. Wu, Y. H. Luo, D. M. Li and Q. B. Meng, Nano Energy, 2017, 42, 222–231 CrossRef CAS.
- H. S. Kim, C. R. Lee, J. H. Im, K. B. Lee, T. Moehl, A. Marchioro, S. J. Moon, R. Humphry-Baker, J. H. Yum, J. E. Moser, M. Grätzel and N. G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- NREL Chart http://www.nrel.gov/pv/assets/images/efficiency-chart.png.
- Q. W. Han, Y. S. Bai, J. Liu, K. Z. Du, T. Y. Li, D. Ji, Y. H. Zhou, C. Y. Cao, D. Shin, J. Ding, A. D. Franklin, J. T. Glass, J. S. Hu, M. J. Therien, J. Liu and D. Mitzi, Energy Environ. Sci., 2017, 10, 2365–2371 RSC.
- E. J. Juarezperez, Z. Hawash, S. R. Raga, L. K. Ono and Y. B. Qi, Energy Environ. Sci., 2016, 9, 3406–3410 RSC.
- Z. Wang, Z. J. Shi, T. T. Li, Y. H. Chen and W. Huang, Angew. Chem., Int. Ed., 2017, 56, 1190–1212 CrossRef CAS PubMed.
- Y. K. Ren, X. H. Ding, Y. H. Wu, J. Zhu, T. Hayat, A. Alsaedi, Y. F. Xu, Z. Q. Li, S. F. Yang and S. Y. Dai, J. Mater. Chem. A, 2017, 5, 20327–20333 RSC.
- Y. K. Ren, X. Q. Shi, X. H. Ding, J. Zhu, T. Hayat, A. Alsaedi, Z. Q. Li, X. X. Xu, S. F. Yang and S. Y. Dai, Inorg. Chem. Front., 2018, 5, 348–353 RSC.
- J. L. Duan, D. W. Dou, Y. Y. Zhao, Y. D. Wang, X. Y. Yang, H. W. Yang, B. L. He and Q. W. Tang, Mater. Today Energy, 2018, 10, 146–152 CrossRef.
- J. Liang, C. X. Wang, Y. R. Wang, Z. R. Xu, Z. P. Lu, Y. Ma, H. F. Zhu, Y. Hu, C. C. Xiao, X. Yi, G. Y. Zhu, H. L. Lv, L. B. Ma, T. Chen, Z. X. Tie, Z. Jin and J. Liu, J. Am. Chem. Soc., 2016, 138, 15829–15832 CrossRef CAS PubMed.
- J. Ding, Y. Y. Zhao, J. L. Duan, B. L. He and Q. W. Tang, ChemSusChem, 2018, 11, 1–7 CrossRef.
- H. Z. Xu, J. L. Duan, Y. Y. Zhao, Z. B. Jiao, B. L. He and Q. W. Tang, J. Power Sources, 2018, 399, 76–82 CrossRef CAS.
- S. Mabrouk, M. M. Zhang, Z. H. Wang, M. Liang, B. Bahrami, Y. G. Wu, J. H. Wu, Q. Q. Qiao and S. F. Yang, J. Mater. Chem. A, 2018, 6, 7950–7958 RSC.
- S. H. Peng, T. W. Huang, G. Gollavelli and C. S. Hsu, J. Mater. Chem. C, 2017, 5, 5193–5198 RSC.
- J. Kim, N. Park, J. S. Yun, S. Huang, M. A. Green and A. W. Y. HoBaillie, Sol. Energy Mater. Sol. Cells, 2017, 162, 41–46 CrossRef CAS.
- M. C. Jung and Y. B. Qi, Org. Electron., 2016, 31, 71–76 CrossRef CAS.
- Z. Hawash, L. K. Ono and Y. B. Qi, Adv. Mater. Interfaces, 2016, 3, 1600117 CrossRef.
- S. Carli, J. P. C. Baena, G. Marianetti, N. Marchetti, M. Lessi, A. Abate, S. Caramori, M. Gratzel, F. Bellina, C. A. Bignozzi and A. Hagfeldt, ChemSusChem, 2016, 9, 657–661 CrossRef CAS PubMed.
- D. Q. Bi, B. Xu, P. Gao, L. C. Sun, M. Graetzel and A. Hagfeldt, Nano Energy, 2016, 23, 138–144 CrossRef CAS.
- P. Ananthajothi and P. Venkatachalam, J. Solid State Electrochem., 2016, 20, 2633–2642 CrossRef CAS.
- F. Wu, J. Z. Cheng, R. J. Chen, S. X. Wu, L. Li, S. Chen and T. Zhao, J. Phys. Chem. C, 2011, 115, 6057–6063 CrossRef CAS.
- E. Eren, E. Aslan and A. U. Oksuz, Polym. Eng. Sci., 2015, 54, 2632–2640 CrossRef.
- M. A. Garakani, S. Abouali, J. Cui and J. K. Kim, Mater. Chem. Front., 2018, 2, 1481–1488 RSC.
- J. Yang, Y. Liu, S. Liu, L. Li, C. Zhang and T. X. Liu, Mater. Chem. Front., 2017, 1, 251–268 RSC.
- S. X. Sun, Y. Huo, M. M. Li, X. W. Hu, H. J. Zhang, Y. W. Zhang, Y. D. Zhang, X. L. Cheng, Z. F. Shi, X. Gong, Y. S. Chen and H. L. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 19914–19922 CrossRef CAS PubMed.
- G. Gong, N. Zhao, D. B. Ni, J. Y. Chen, Y. Shen, M. K. Wang and G. L. Tu, J. Mater. Chem. A, 2016, 4, 3661–3666 RSC.
- Q. W. Tang, J. Wang, B. L. He and P. Z. Yang, Nano Energy, 2017, 33, 266–271 CrossRef CAS.
- J. L. Duan, T. Y. Hu, Y. Y. Zhao, B. L. He and Q. W. Tang, Angew. Chem., Int. Ed., 2018, 57, 5746–5749 CrossRef CAS PubMed.
- J. L. Duan, Y. Y. Zhao, B. L. He and Q. W. Tang, Angew. Chem., Int. Ed., 2018, 57, 3787–3791 CrossRef CAS PubMed.
- B. B. Zong, W. Y. Fu, H. J. Liu, L. W. Huang, H. Bala, X. D. Wang, G. Sun, J. L. Cao and Z. Y. Zhang, J. Alloys Compd., 2018, 748, 1006–1012 CrossRef CAS.
- Z. Y. Liu, B. Sun, X. Y. Liu, J. H. Han, H. B. Ye, Y. X. Tu, C. Chen, T. L. Shi, Z. R. Tang and G. L. Liao, J. Mater. Chem. A, 2018, 6, 7409–7419 RSC.
- Y. Liu, H. Zhang, Y. P. Zhang, B. Xu, L. J. Liu, G. Chen, C. Im and W. J. Tian, J. Mater. Chem. A, 2018, 6, 7922–7932 RSC.
- L. Li, Y. Q. Wang, X. L. Deng, L. H. Nie, Z. D. Cui and C. W. Shi, J. Nanosci. Nanotechnol., 2018, 18, 5095–5100 CrossRef CAS PubMed.
- K. Chen, Q. Hu, T. H. Liu, L. C. Zhao, D. Y. Luo, J. Wu, Y. F. Zhang, W. Zhang, F. Liu, T. P. Russell, R. Zhu and Q. H. Gong, Adv. Mater., 2016, 28, 10718 CrossRef CAS PubMed.
- A. Pockett, G. E. Eperon, T. Peltola, H. J. Snaith, A. Walker, L. M. Peter and P. J. Cameron, J. Phys. Chem. C, 2015, 119, 3456–3465 CrossRef CAS.
- A. Mei, X. Li, L. Liu, Y. Rong, M. Xu, M. Hu, J. Chen, Y. Yang, M. Grätzel and H. Han, Science, 2014, 345, 295–298 CrossRef CAS PubMed.
- X. S. Zhan, Z. W. Jin, J. R. Zhan, D. L. Bai, H. Bian, K. Wang, J. Sun, Q. Wang and S. F. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 7145–7154 CrossRef PubMed.
- Z. Y. Liu, B. Sun, X. Y. Liu, J. H. Han, H. B. Ye, T. L. Shi, Z. R. Tang and G. L. Liao, Nano-Micro Lett., 2018, 10, 34–46 CrossRef.
- X. W. Chang, W. P. Li, L. Q. Zhu, H. C. Liu, H. F. Geng, S. S. Xiang, J. M. Liu and H. N. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 33649–33655 CrossRef CAS PubMed.
- M. L. Cai, N. Ishida, X. Li, X. D. Yang, T. Noda, Y. Z. Wu, F. X. Xie, H. Naito, D. Fujita and L. Y. Han, Joule, 2018, 2, 296–306 CrossRef CAS.
- J. Zhang, D. L. Bai, Z. W. Jin, H. Bian, K. Wang, J. Sun, Q. Wang and S. Z. Liu, Adv. Energy Mater., 2018, 8, 1703246 CrossRef.
- T. Niu, R. Munir, J. B. Li, D. Barrit, X. Zhang, H. L. Hu, Z. Yang, A. Amassian, K. Zhao and S. Z. Liu, Adv. Mater., 2018, 30, 1706576 CrossRef PubMed.
- Y. Wang, T. Y. Zhang, F. Xu, Y. H. Li and Y. X. Zhao, Sol. RRL, 2018, 2, 1700180 CrossRef.
- J. L. Duan, Y. Y. Zhao, X. Y. Yang, Y. D. Wang, B. L. He and Q. W. Tang, Adv. Energy Mater., 2018 DOI:10.1002/aenm.201802346.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00337h |
| ‡ These authors made equal contributions to this work. |
| This journal is © the Partner Organisations 2018 |