DOI:
10.1039/C7QO00042A
(Research Article)
Org. Chem. Front., 2017,
4, 1051-1057
(1S)-(−)-N-Trifluoromethylthio-2,10-camphorsultam and its derivatives: easily available, optically pure reagents for asymmetric trifluoromethylthiolation†
Received
16th January 2017
, Accepted 9th February 2017
First published on 10th February 2017
Abstract
A family of easily scalable, shelf-stable, optically pure reagents (1S)-(−)-N-trifluoromethylthio-2,10-camphorsultam 1a–c was successfully developed. In particular, compound 1c was shown to be an efficient reagent that is capable of transferring chirality to other prochiral nucleophiles such as β-ketoesters, oxindoles and benzofuranones with good to excellent enantioselectivities.
Introduction
Over the past two decades, fluorine and its magic “fluorine effect” in the discovery of new drug molecules have been well recognized in the fields of the pharmaceutical and agrochemical industries.1 Consequently, strategic incorporation of a fluorine atom or a fluoroalkyl group into therapeutic or agrochemical agents has become a routine tool to manipulate the target molecule's physical, chemical and biological properties including conformation, pKa, intrinsic potency, metabolic pathways, and pharmacokinetic properties. In particular, recently, the trifluoromethylthio group (–SCF3) has gained intense interest, mainly due to its high lipophilicity (Hansch's hydrophobicity parameter π = 1.44) that may improve the molecule's trans-membrane permeability.2 Not surprisingly, new reagents and new methods that are able to introduce the trifluoromethylthio group effectively are in high demand.3 As a matter of fact, in the last five years, a number of electrophilic trifluoromethylthiolating reagents,4 as well many transition metal-catalyzed trifluoromethylthiolation reactions5,6 emerged, that now allow for the construction of the C(sp2)–SCF3, C(sp3)–SCF3 or C(sp)–SCF3 bonds efficiently.
Despite these impressive advances in developing the methods for the formation of C–SCF3 bonds, very few methods that could generate optically active compounds with a stereogenic carbon center bearing a trifluoromethylthio group have been reported.7 In 2013, Shen's and Rueping's groups simultaneously reported the first highly enantioselective cinchona alkaloid-catalyzed asymmetric trifluoromethylthiolation of cyclic β-ketoesters with trifluoromethanesulfenate7a or N-trifluoromethylphthalimide7b as the electrophilic SCF3 reagent, respectively. Later on, the asymmetric reactions were successfully extended to trifluoromethylthiolation of oxindoles with excellent enantioselectivity.7e,f Likewise, Liu and Tan discovered that the organocatalytic asymmetric trifluoromethylthiolation of oxindoles can be achieved when an in situ formed electrophilic trifluoromethylthio species generated from trichloroisocyanuric acid (TCCA) and AgSCF3.7d Very recently, Zhao's group reported the first chiral sulphide-catalyzed asymmetric trifluoromethylthiolating lactonization of alkenes with N-trifluoromethylthiodibenzenesulfonimide.7g On the other hand, Gade and co-workers reported the first copper-catalyzed asymmetric trifluoromethylthiolation of cyclic β-ketoesters using the trifluoromethanesulfenate reagent.7c Nevertheless, new and highly enantioselective methods for the construction of a stereogenic carbon center bearing a trifluoromethylthio group are still urgently needed.
Chirality transfer from a chiral reagent to a prochiral substrate to give the product in high enantiomeric excess represents a general and efficient method for the production of optically active compounds.8 In this respect, the design and exploration of optically pure electrophilic trifluoromethylthiolating reagents for asymmetric trifluoromethylthiolation offers a simple and straightforward strategy for the formation of a stereogenic carbon center bearing a trifluoromethylthio group, which would complement the previously reported asymmetric trifluoromethylthiolation methods. Herein, we report the design and synthesis of (1S)-(−)-N-trifluoromethylthio-2,10-camphorsultam9 and its derivatives, a family of optically pure reagents that can efficiently transfer their chirality to prochiral substrates such as β-ketoesters, oxindoles and benzofuran-2(3H)-ones with good to excellent enantioselectivities.
Results and discussion
Preparation of (1S)-(−)-N-trifluoromethylthio-2,10-camphorsultam and N-trifluoromethylthio-oxazolidin-2-one and their derivatives
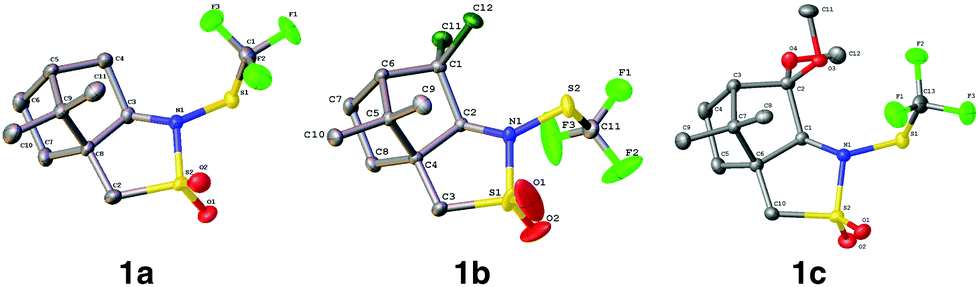
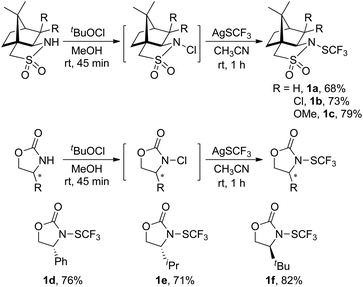
(1S)-(−)-N-Trifluoromethylthio-2,10-camphorsultam 1a could be easily synthesized via a two-step procedure, as shown in Fig. 1. Reaction of commercially available (1S)-(−)-2,10-camphorsultam with tert-butyl hypochlorite in methanol at room temperature for 45 min yielded (1S)-(−)-N-chloro-2,10-camphorsultam in quantitative yield. Without further purification, the in situ generated (1S)-(−)-N-chloro-2,10-camphorsultam was then further treated with AgSCF3 in CH3CN at room temperature for 1 h to give (1S)-(−)-N-trifluoromethylthio-2,10-camphorsultam 1a in 68% yield. The reaction can be easily scaled up to a quantity of 4.7 g without erosion of the yield. Likewise, two analogous compounds 1b and 1c were prepared in 73% and 79% yield, respectively. In addition, compounds 1d–f derived from oxazolidin-2-one were synthesized by the same method in 71–82% yields, as shown in Fig. 1. Compounds 1a–f were fully characterized by 1H, 13C, and 19F NMR spectroscopy. The structures of compounds 1a–c were further unambiguously confirmed by X-ray analysis of their single crystals (Fig. 2).
 |
| | Fig. 1 Preparation of (1S)-(−)-N-trifluoromethylthio-2,10-camphorsultam and N-trifluoromethylthio-oxazolidin-2-one and their derivatives. | |
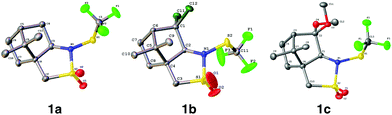 |
| | Fig. 2 ORTEP diagrams of compounds 1a–c. | |
Asymmetric trifluoromethylthiolation of β-ketoesters with reagent 1c
To probe whether compounds 1a–f can efficiently transfer their chirality to other substrates, we first studied the reactions of β-ketoesters with compounds 1a–f. The reaction of β-ketoester 2a derived indanone with compound 1a was chosen initially as a model reaction to optimize the reaction parameters. A quick screening of the base disclosed that the desired trifluoromethylthio-β-ketoester 3a was generated in higher yield when the reaction was conducted in THF using K2CO3 as the base than those using Cs2CO3, Li2CO3, K3PO4 or DMAP as the base (Scheme 1, entries 1–5). Next, the effect of the solvents was studied. Reactions in THF occurred much faster than those in CH2Cl2, Et2O or toluene (Scheme 1, entries 6–9). For example, reaction of β-ketoester 2a with compound 1a occurred in 78% yield with 77% ee after 12 h at −20 °C while reactions in CH2Cl2, Et2O or toluene occurred in less than 10% conversion under otherwise identical conditions. Under the current reaction conditions (K2CO3 as the base, THF as the solvent, −20 °C, 12 h), we compared the reactivity of compounds 1a–f. Interestingly, reactions of compounds 1a–c derived from (1S)-(−)-camphorsultam were much faster and with higher enantioselectivity than reactions of compounds 1d–f derived from oxazolidin-2-one (Scheme 1, entries 7, 10–14). In addition, compound 1c with two methoxy groups reacted faster and with higher enantioselectivity than its analogs 1a–b (Scheme 1, entries 7, 10 and 11). With these promising results in mind, we further optimized the reaction conditions using compound 1c as the electrophilic trifluoromethylthiolating reagent. It was found that reactions using 0.1 equivalent of K2CO3 as the base worked equally efficient, while reactions conducted at −40 °C occurred slowly but with higher enantioselectivity (Scheme 1, entry 16). Finally, full conversion of the substrate was achieved by elongating the reaction time to 72 h (Scheme 1, entry 18).
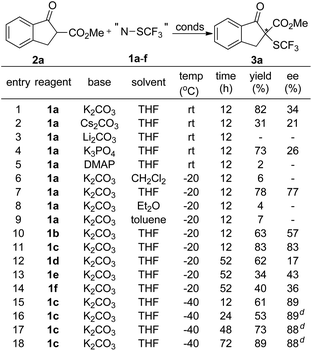 |
| | Scheme 1 Optimization of the conditions for the reactions of β-ketoester 2a with compounds 1a–f.a,b,c aReaction conditions: β-ketoester 2a (0.050 mmol), reagents 1a–f (0.055 mmol), base (1.1 mmol) in 0.5 mL of solvent; byields were determined by 19F NMR spectroscopy in the presence of trifluoromethylbenzene as the internal standard; cthe ee values were determined by HPLC analysis on a chiral stationary phase. d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 0.1 equivalent of K2CO3 was used. 0.1 equivalent of K2CO3 was used. | |
Scope of the asymmetric trifluoromethylthiolation of β-ketoesters with reagent 1c
With the optimum reaction conditions in hand, the stage was set to explore the scope and the limitation of the asymmetric trifluoromethylthiolation reactions. A variety of β-ketoesters 2a–o derived from indanone or tetralone were subjected to the optimized conditions and the results are summarized in Scheme 2. In general, reactions of six-membered ring β-ketoesters occurred with higher enantioselectivities than those of five-membered ring β-ketoesters. Reactions of six-membered ring β-ketoesters typically generated the desired trifluoromethylthiolated compounds in 94–98% ee while reactions of five-membered ring β-ketoesters gave the desired products in 79–92% ee. In addition, reactions of less nucleophilic β-ketoesters with an electron-withdrawing group such as fluoride, chloride, or bromide occurred much slower than those with electron-donating groups. For examples, reactions of β-ketoesters 2g–h and 2o required 32 h at −25 °C to convert completely to the desired products (Scheme 2, 3g–h and 3o). The effects of the steric hindrance of the ester group of β-ketoesters on the enantioselectivity of the reaction were not significant since the enantioselectivities increased from 88% ee to 92% ee when the ester group of β-ketoester was changed from the methyl group to the adamantyl group (Scheme 2, 3a–d). Thus, methyl esters of the six-membered ring β-ketoesters reacted with reagent 1c to form the corresponding trifluoromethylthiolated products in 94–98% ee (Scheme 2, 3i–o). As a comparison, organo-catalytic or transition-metal catalyzed asymmetric trifluoromethylthiolation of the six-membered ring β-ketoesters typically required the use of steric-bulky tert-butyl or adamantyl esters to achieve excellent enantioselectivity.7 The absolute configuration of compound 3n was established to be R as determined by analysis of the X-ray diffraction data of its single crystal (Fig. 3). The configurations of the rest of the β-ketoesters were assigned based on the same mechanism assumption.
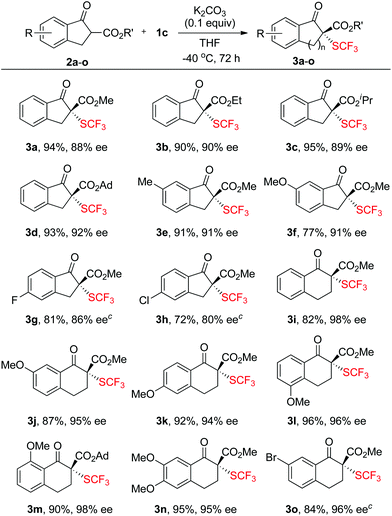 |
| | Scheme 2 Scope of the reactions of β-ketoesters 2a–n with compound 1c. a,b aReaction conditions: β-ketoester 2a (0.50 mmol), reagent 1c (0.55 mmol), K2CO3 (0.050 mmol) in 2.5 mL of THF at −40 °C for 72 h; bisolated yields; the ee values were determined by HPLC analysis on a chiral stationary phase; creactions were conducted at −25 °C for 32 h. | |
 |
| | Fig. 3 ORTEP diagram of compound 3n. | |
Scope of the asymmetric trifluoromethylthiolation of oxindoles with reagent 1c or 1b
Having established that compound 1c is an effective reagent for chirality transfer, we then tried to expand the scope of reagent 1c for asymmetric trifluoromethylthiolation reaction to other carbon-based nucleophiles. Oxindole represents one of the privileged structural units in many biologically active natural products and the development of highly enantioselective methods for trifluoromethylthiolation of oxindoles would be of great interest to medicinal chemists. As a matter of fact, three different organo-catalytic methods for asymmetric trifluoromethylthiolation of oxindoles have been reported recently.7d–f Interestingly, when the reaction of compound 1c and N-methyl oxindole was conducted in THF at −40 °C using 0.1 equivalent of K2CO3 as the base, moderate enantioselectivity of the desired product was observed. Instead, switching the base from K2CO3 to Cs2CO3 and the solvent from THF to Et2O led to a significant improvement of the enantioselectivity. As summarized in Scheme 3, a variety of 3-aryloxindoles reacted with compound 1c to give 3-trifluoromethylthio-3-aryloxindoles in excellent yields and with excellent enantioselectivities except for one case in which a cyanomethyl substituent was in the para-position of the 3-aryl group (Scheme 3, 4j). For this particular substrate, using compound 1b as the chiral electrophilic trifluoromethylthiolating reagent greatly improved the enantioselectivity from 57% ee to 94% ee (Scheme 3, 4j). A few other examples also support that reactions of oxindoles with compound 1b occurred with higher enantioselectivities than those with compound 1c (Scheme 3, 4a, 4c and 4j). Because of the mild reaction conditions, various common functional groups such as the fluoride, chloride, bromide, cyano or ester group were tolerant. Nevertheless, reaction of 3-alkylated oxindoles with compound 1b occurred with moderate enantioselectivity (Scheme 3, 4p).
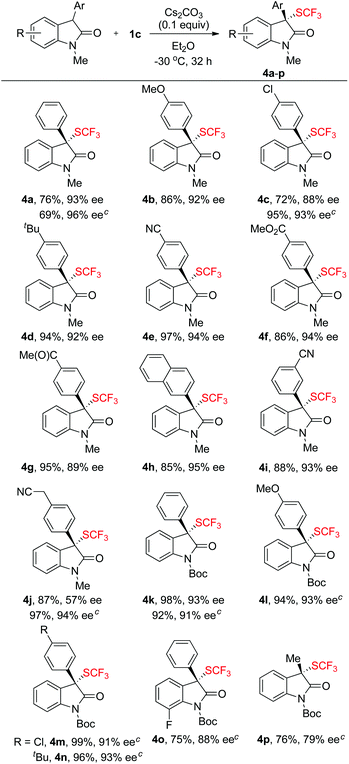 |
| | Scheme 3 Scope of the reactions of oxindoles with compound 1c or 1b. a,b aReaction conditions: oxindole (0.50 mmol), reagent 1c (0.55 mmol), Cs2CO3 (0.050 mmol) in 2.5 mL of Et2O at −30 °C for 32 h; bisolated yields; the ee values were determined by HPLC analysis on a chiral stationary phase; creactions were conducted with compound 1b. | |
The absolute configuration of the optically active compound 4i was established to be S as determined by analysis of the X-ray diffraction data of its single crystal (Fig. 4). The configurations of other oxindole derivatives were assigned based on the assumption that the reaction proceeded via a similar mechanistic pathway.
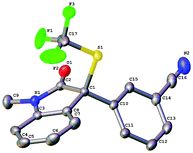 |
| | Fig. 4 ORTEP diagram of compound 4i. | |
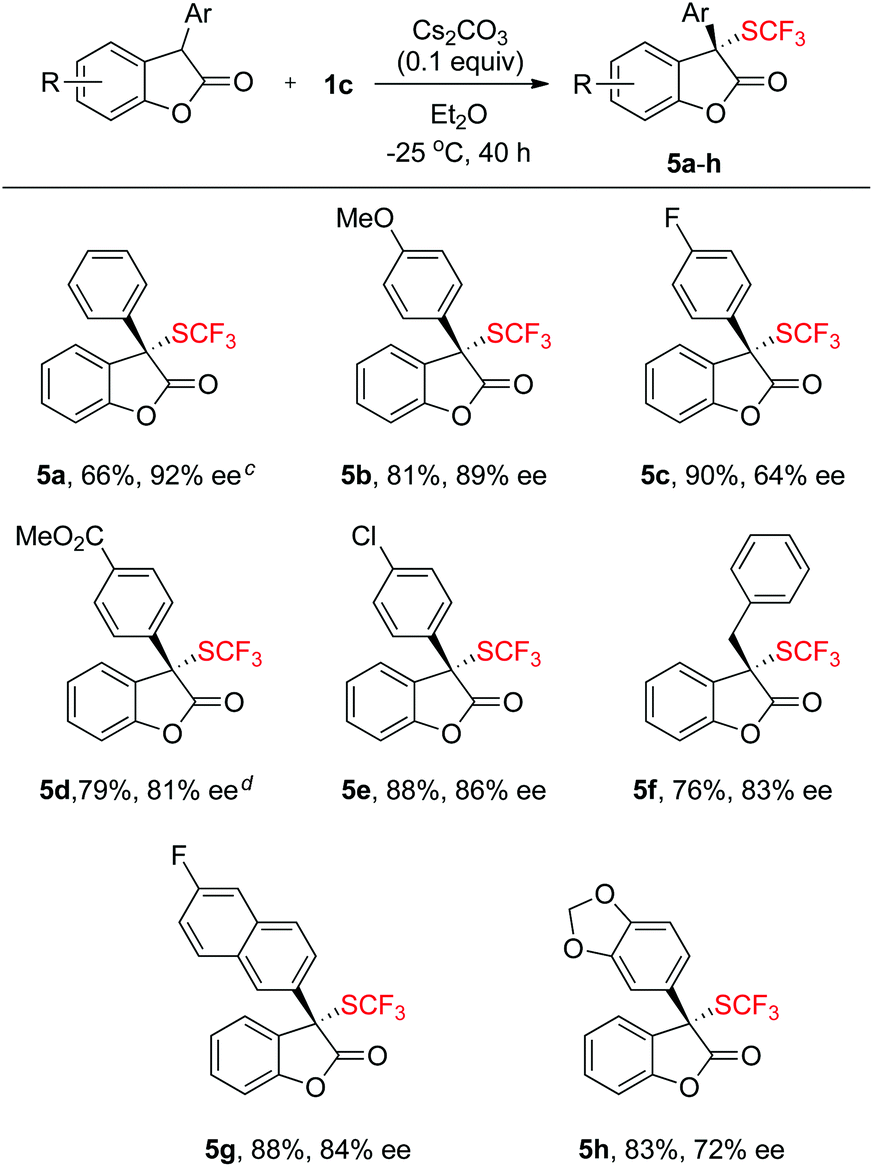
Scope of the asymmetric trifluoromethylthiolation of benzofuran-2(3H)-ones with reagent 1c
To further expand the scope of the asymmetric trifluoromethylthiolation of compound 1c, we studied the reactions of 2-aryl or 2-alkyl benzofuran-2(3H)-ones with compound 1c, as summarized in Scheme 4. In general, good to excellent enantioselectivities (64–92% ee) were achieved for the reactions of 2-aryl or 2-alkyl benzofuran-2(3H)-ones with compound 1c when the reactions were conducted at −25 °C for 40 h using Cs2CO3 as the base. Reaction of methyl 4-(2-oxo-2,3-dihydrobenzofuran-3-yl)benzoate with compound 1c was slow at −25 °C but occurred to full conversion after 32 h at 10 °C to give the corresponding product in 81% ee (Scheme 4, 5d). Reaction of alkyl substituted oxindoles occurred also smoothly to give the desired product in 76% yield and 83% ee (Scheme 4, 5f). Likewise, the absolute configuration of compound 5g was established to be S as determined by analysis of the X-ray diffraction data of its single crystal (Fig. 5). The configurations of the rest of the lactones were assigned based on the same mechanism assumption.
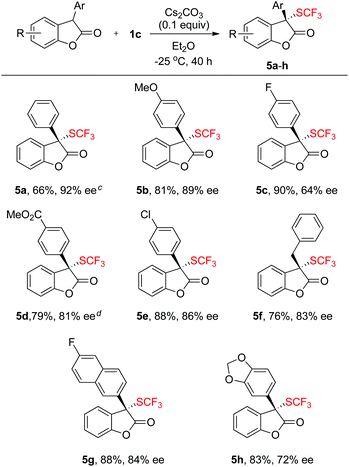 |
| | Scheme 4 Scope of the reactions of benzofuran-2(3H)-ones with compound 1c. a,b aReaction conditions: benzofuran-2(3H)-ones (0.5 mmol), reagent 1c (0.55 mmol), Cs2CO3 (0.05 mmol) in Et2O (2.5 mL) at −25 °C for 40 h; bisolated yields; the ee values were determined by HPLC analysis on a chiral stationary phase; creactions were conducted at −35 °C for 40 h; dreactions were conducted at 10 °C for 32 h. | |
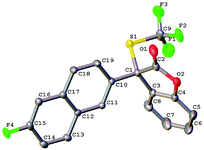 |
| | Fig. 5 ORTEP diagram of compound 5g. | |
Experimental
General procedure for the preparation of optically active electrophilic trifluoromethylthiolating reagent 1a
To a solution of (1S)-(−)-2,10-camphorsultam (2.15 g, 10.0 mmol) in methanol (15.0 mL) and in a 50 mL round-bottom flask covered with aluminum foil was added tert-butylhypochlorite (2.71 g, 25.0 mmol) under an argon atmosphere. The solution was then stirred at room temperature for 45 min. The solution was evaporated under reduced pressure to give (1S)-(−)-N-chloro-2,10-camphorsultam quantitatively, which was used directly without further purification.10
To an oven dried 500 mL Schlenk flask equipped with a stir bar was added dry (1S)-(−)-N-chloro-2,10-camphorsultam (2.50 g, 10.0 mmol). The flask was fitted with a glass stopper and evacuated and refilled with argon three times. Dry MeCN (30.0 mL) was injected into the flask, then AgSCF3 (2.49 g, 12.0 mmol) was added. The mixture was stirred vigorously at room temperature for 1 h. The CH3CN was then evaporated under reduced pressure. And the residue was extracted with CH2Cl2 (3 × 30.0 mL). The solution was combined and dried over anhydrous Na2SO4. The solvent was evaporated under vacuum and the residue was purified by recrystallization from a solution of THF and petroleum ether to give (1S)-(−)-N-trifluoromethylthio-2,10-camphorsultam 1a as a white solid (2.14 g, 68% yield).
General procedure for asymmetric trifluoromethylthiolation of β-ketoesters
β-Ketoester 2a (95.1 mg, 0.500 mmol, 1.0 equiv.), potassium carbonate (7.90 mg, 0.0500 mmol, 0.1 equiv.) and reagent 1c (206 mg, 0.550 mmol, 1.1 equiv.) were added into a flame-dried Schlenk tube. The tube was put into liquid nitrogen, and dry tetrahydrofuran (2.5 mL) was added under an argon atmosphere. The resulting solution was stirred at −40 °C for 72 h (or −25 °C for 32 h). After the reaction was complete as monitored by 19F NMR spectroscopy, the reaction was quenched by addition of HCl (2.0 M). The mixture was extracted with Et2O (3 × 10.0 mL). The organic phase was separated and dried over anhydrous Na2SO4. After filtration, the solvent was removed under vacuum. The residue was purified by flash chromatography to give the trifluoromethylthiolated product 3a (R)-methyl-1-oxo-2-((trifluoromethyl)thio)-2,3-dihydro-1H-indene-2-carboxylate 3a as a white solid (136 mg, 94% yield). HPLC: (IE-3 (0.46 × 25 cm, 3 μm), hexane/iPrOH = 97/3, 0.7 mL min−1, 214 nm), tR (major) = 12.13 min, tS (minor) = 12.93 min (88% ee); [α]25D = −99.2 (c = 0.110, CHCl3, 88% ee).
General procedure for asymmetric trifluoromethylthiolation of oxindoles with reagent 1c
Oxindole (112 mg, 0.500 mmol, 1.0 equiv.), cesium carbonate (16.3 mg, 0.0500 mmol, 0.1 equiv.) and compound 1c (206 mg, 0.550 mmol, 1.1 equiv.) were added into a flame-dried Schlenk tube. The tube was put into liquid nitrogen. Dry ethyl ether (2.5 mL) was added under an argon atmosphere. The resulting solution was stirred at −30 °C for 32 h. After the reaction was complete as monitored by 19F NMR spectroscopy, the reaction was quenched with HCl (2.0 M). The mixture was extracted with Et2O (3 × 10.0 mL). The combined organic phase was separated and dried over anhydrous Na2SO4. After filtration, the solvent was removed under vacuum. The residue was purified by flash chromatography to give (S)-1-methyl-3-phenyl-3-((trifluoromethyl)thio)indolin-2-one 4a as a white solid (122 mg, 76% yield). HPLC: (AD-H (0.46 × 25 cm, 5 μm), hexane/iPrOH = 95/5, 0.7 mL min−1, 214 nm), tS (major) = 9.83 min, tR (minor) = 9.04 min (93% ee); [α]25D = +102.4 (c = 0.140, CHCl3, 93% ee).
General procedure for asymmetric trifluoromethylthiolation of benzofuranones with compound 1c
Benzofuranone (105 mg, 0.500 mmol, 1.0 equiv.), cesium carbonate (16.3 mg, 0.0500 mmol, 0.1 equiv.) and compound 1c (206 mg, 0.550 mmol, 1.1 equiv.) were added into a flame-dried Schlenk tube. The tube was put into liquid nitrogen. Dry ethyl ether (2.5 mL) was added under an argon atmosphere. The resulting solution was stirred at −25 °C for 40 h. After the reaction was complete as monitored by 19F NMR spectroscopy, the reaction was quenched with HCl (2.0 M). The mixture was extracted using Et2O (3 × 10.0 mL). The organic phase was separated and dried over anhydrous Na2SO4. After filtration, the solvent was removed under vacuum. The residue was purified by flash chromatography to give (S)-3-phenyl-3-((trifluoromethyl)thio)benzofuran-2(3H)-one 5a as a colorless oil (102 mg, 66% yield). HPLC: (IE-3 (0.46 × 25 cm, 3 μm), hexane/iPrOH = 98/2, 0.7 mL min−1, 214 nm), tS (major) = 8.25 min, tR (minor) = 7.93 min (92% ee); [α]25D = +92.9 (c = 0.050, CHCl3, 92% ee).
Conclusions
In summary, a family of easily scalable, shelf-stable, optically pure reagents (1S)-(−)-N-trifluoromethylthio-2,10-camphorsultam 1a–c was successfully developed. Among them, compound 1c was shown to be an efficient reagent that is capable of transferring its chirality to other prochiral nucleophiles, as demonstrated by its reactions with β-ketoesters, oxindoles and benzofuranones with good to excellent enantioselectivities. The studies for further applications of reagent 1c toward the construction of a stereogenic tertiary carbon center bearing a trifluoromethylthio group are undergoing currently in our laboratory.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (21625206, 21632009, 21372247, 21572258, and 21421002) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000).
Notes and references
-
(a)
R. Filler, Biomedical Aspests of Fluorine Chemistry, Ko-dansha, Tokyo, 1982 Search PubMed
 ;
(b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC ;
(c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed ;
(d) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed ;
(e) J. Wang, M. Sánchez-Roselló, J. Aceña, C. Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed .
;
(b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC ;
(c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed ;
(d) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed ;
(e) J. Wang, M. Sánchez-Roselló, J. Aceña, C. Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed .
-
(a) A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525 CrossRef CAS ;
(b) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS ;
(c) G. Landelle, A. Panossian and F. R. Leroux, Curr. Top. Med. Chem., 2014, 14, 941 CrossRef CAS PubMed .
-
(a) F. Leroux, P. Jeschke and M. Schlosser, Chem. Rev., 2005, 105, 827 CrossRef CAS PubMed ;
(b) B. Manteau, S. Pazenok, J.-P. Vors and F. R. Leroux, J. Fluorine Chem., 2010, 131, 140 CrossRef CAS ;
(c) V. N. Boiko, Beilstein J. Org. Chem., 2010, 6, 880 CrossRef PubMed ;
(d) G. Landelle, A. Panossian, S. Pazenok, J.-P. Vors and F. R. Leroux, Beilstein J. Org. Chem., 2013, 9, 2476 CrossRef PubMed ;
(e) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed ;
(f) X.-X. Shao, C.-F. Xu, L. Lu and Q. Shen, Acc. Chem. Res., 2015, 48, 1227 CrossRef CAS PubMed ;
(g) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed ;
(h) C.-F. Ni, M.-Y. Hu and J.-B. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed .
- Selected examples for transition metal-catalyzed or -mediated trifluoromethylthiolation methods:
(a) G. Teverovskiy, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 7312 CrossRef CAS PubMed ;
(b) C. P. Zhang and D. A. Vicic, J. Am. Chem. Soc., 2012, 134, 183 CrossRef CAS PubMed ;
(c) C. Chen, Y. Xie, L.-L. Chu, R.-W. Wang, X.-G. Zhang and F.-L. Qing, Angew. Chem., Int. Ed., 2012, 51, 2492 CrossRef CAS PubMed ;
(d) Z. Weng, W. He, C. Chen, R. Lee, D. Dan, Z. Lai, D. Kong, Y. Yuan and K.-W. Huang, Angew. Chem., Int. Ed., 2013, 52, 1548 CrossRef CAS PubMed ;
(e) G. Danoun, B. Bayarmagnai, M. F. Grünberg and L. J. Gooßen, Chem. Sci., 2014, 5, 1312 RSC ;
(f) R. Pluta, P. Nikolaienko and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 150 CrossRef PubMed ;
(g) K. Kang, C.-F. Xu and Q. Shen, Org. Chem. Front., 2014, 1, 294 RSC ;
(h) W.-Y. Yin, Z.-F. Wang and Y. Huang, Adv. Synth. Catal., 2014, 356, 2998 CrossRef CAS ;
(i) J. Sheng, S.-Y. Li and J. Wu, Chem. Commun., 2014, 50, 578 RSC ;
(j) H.-Y. Xiong, T. Besset, D. Cahard and X. Pannecoucke, J. Org. Chem., 2015, 80, 4204 CrossRef CAS PubMed ;
(k) G. Yin, I. Kalvet, U. Englert and F. Schoenebeck, J. Am. Chem. Soc., 2015, 137, 4164 CrossRef CAS PubMed ;
(l) G. Yin, I. Kalvet and F. Schoenebeck, Angew. Chem., Int. Ed., 2015, 54, 6809 CrossRef CAS PubMed .
- Reviews for electrophilic trifluoromethylthiolating reagents:
(a) F. Toulgoat, S. Alazet and T. Billard, Eur. J. Org. Chem., 2014, 2415 CrossRef CAS ;
(b) H. Chachignon and D. Cahard, Chin. J. Org. Chem., 2016, 34, 445 CrossRef CAS ;
(c) S. Barata-Vallejo, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 7150 RSC .
- Selected examples for trifluoromethylthiolation with electrophilic trifluoromethylthiolating reagents:
(a) A. Ferry, T. Billard, B. R. Langlois and E. Bacqué, J. Org. Chem., 2008, 73, 9362 CrossRef CAS PubMed ;
(b) S. Alazet, L. Zimmer and T. Billard, J. Fluorine Chem., 2015, 171, 78 CrossRef CAS ;
(c) Y.-D. Yang, A. Azuma, E. Tokunaga, M. Yamasaki, M. Shiro and N. Shibata, J. Am. Chem. Soc., 2013, 135, 8782 CrossRef CAS PubMed ;
(d) X.-X. Shao, X.-Q. Wang, T. Yang, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 3457 CrossRef CAS PubMed ;
(e) E. V. Vinogradova, P. Müller and S. L. Buchwald, Angew. Chem., Int. Ed., 2014, 53, 3125 CrossRef CAS PubMed ;
(f) S. Munavalli, D. K. Rohrbaugh, D. I. Rossman, F. J. Berg, G. W. Wagner and H. D. Durst, Synth. Commun., 2000, 30, 2847 CrossRef CAS ;
(g) C. Xu, B. Ma and Q. Shen, Angew. Chem., Int. Ed., 2014, 53, 9316 CrossRef CAS PubMed ;
(h) P.-P. Zhang, M. Li, X.-S. Xue, C.-F. Xu, Q.-C. Zhao, Y.-F. Liu, H.-Y. Wang, Y.-L. Guo, L. Lu and Q. Shen, J. Org. Chem., 2016, 81, 7486 CrossRef CAS PubMed .
-
(a) T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2013, 52, 12856 CrossRef CAS PubMed ;
(b) X.-Q. Wang, T. Yang, X.-L. Cheng and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 12860 CrossRef CAS PubMed ;
(c) Q.-H. Deng, C. Rettenmeier, H. Wadepohl and L. H. Gade, Chem. – Eur. J., 2014, 20, 93 CrossRef CAS PubMed ;
(d) X.-L. Zhu, J.-H. Xu, D.-J. Cheng, L.-J. Zhao, X.-Y. Liu and B. Tan, Org. Lett., 2014, 16, 2192 CrossRef CAS PubMed ;
(e) M. Rueping, X. Liu, T. Bootwicha, R. Pluta and C. Merkens, Chem. Commun., 2014, 50, 2508 RSC ;
(f) T. Yang, Q. Shen and L. Lu, Chin. J. Chem., 2014, 32, 678 CrossRef CAS ;
(g) X. Liu, R. An, X.-L. Zhang, J. Luo and X. D. Zhao, Angew. Chem., Int. Ed., 2016, 55, 5846 CrossRef CAS PubMed .
-
(a) I. Farnadez and N. Khiar, Chem. Rev., 2003, 103, 3651 CrossRef PubMed ;
(b) J. M. Brunel, Chem. Rev., 2007, 107, PR1 CrossRef CAS .
- Review about camphorsultam-based optically pure reagents:
(a) M. M. Heravi and V. Zadsirjan, Tetrahedron: Asymmetry, 2014, 25, 1061 CrossRef CAS . Reports on camphorsultam-based optically pure fluorinating reagents:
(b) E. Differding, G. W. Ruegg and R. W. Lang, Tetrahedron Lett., 1991, 32, 1779 CrossRef CAS ;
(c) F. A. Davis, P. Zhou, C. K. Murphy, G. Sundarababu, H. Qi, W. Han, R. M. Przeslawski, B.-C. Chen and P. J. Carroll, J. Org. Chem., 1998, 63, 2273 CrossRef CAS ;
(d) Y. Takeuchi, Z. Liu, A. Satoh, T. Shiragami and N. Shibata, Chem. Pharm. Bull., 1999, 47, 1730 CrossRef CAS ;
(e) Z.-P. Liu, N. Shibata and Y. Takeuchi, J. Org. Chem., 2000, 65, 7583 CrossRef CAS PubMed .
- L. Duhamel, G. Ple and P. Angibaud, Synth. Commun., 1993, 23, 2423 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, analytical data, computational details, NMR data of compounds 1a–f, 3a–n, 4a–p, 5a–h and X-ray crystallography data of 1a–c, 4i and 5c. CCDC 1527557–1527562. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qo00042a |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence