
The facile and stereoselective synthesis of pyrrolidine β-amino acids via copper(I)-catalyzed asymmetric 1,3-dipolar cycloaddition†
Fu-Sheng
He
,
Cong-Shan
Li
,
Hua
Deng
,
Xing
Zheng
,
Zhong-Tao
Yang
and
Wei-Ping
Deng
*
School of Pharmacy and Shanghai Key Laboratory of New Drug Design, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: weiping_deng@ecust.edu.cn
First published on 8th October 2016
Abstract
A highly efficient copper(I)-catalyzed asymmetric 1,3-dipolar cycloaddition of azomethine ylides with β-phthaliminoacrylate esters was developed under mild conditions, providing the desired pyrrolidine β-amino acid derivatives in high yields (up to 98%) with excellent diastereo- and enantioselectivities (dr >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, ee up to >99%). Subsequent deprotection and hydrogenolysis of the corresponding cycloadducts could deliver highly functionalized and biologically important free pyrrolidine β-amino acids in a straightforward and efficient pathway.
:1, ee up to >99%). Subsequent deprotection and hydrogenolysis of the corresponding cycloadducts could deliver highly functionalized and biologically important free pyrrolidine β-amino acids in a straightforward and efficient pathway.
Conformationally constrained heterocyclic β-amino acids and their derivatives are key elements of peptides and many bioactive products.1 As an interesting class of five-membered N-heterocyclic compounds, pyrrolidine β-amino acids have received considerable attention among synthetic and medicinal chemists in the past few decades because of their important biological activities.2–4 For instance, β-aminopyrrolidinecarboxylic acid (APC) together with β-aminocyclopentanecarboxylic acid (ACPC) have been utilized as building blocks for the construction of β-peptides that possess antimicrobial activities,2 while A-87380 and A-192558 are influenza neuraminidase inhibitors.3 In addition, a series of compounds containing a pyrrolidine β-amino acid framework have been evaluated as DPP-IV inhibitors,4a TACE inhibitors4b and factor Xa inhibitors (Fig. 1).4c Due to their potential application in chemical biology and drug discovery, numerous synthetic methods have been developed for the synthesis of functionalized pyrrolidine β-amino acids, especially in an enantiomerically pure form.1,5 However, the majority of synthetic routes suffered from tedious synthetic steps through chiral auxiliary-based asymmetric synthesis, and a narrow substrate scope, thus greatly limiting their further application in a drug discovery scenario. Therefore, the development of efficient catalytically asymmetric access to functionalized pyrrolidine β-amino acids is still highly desirable.
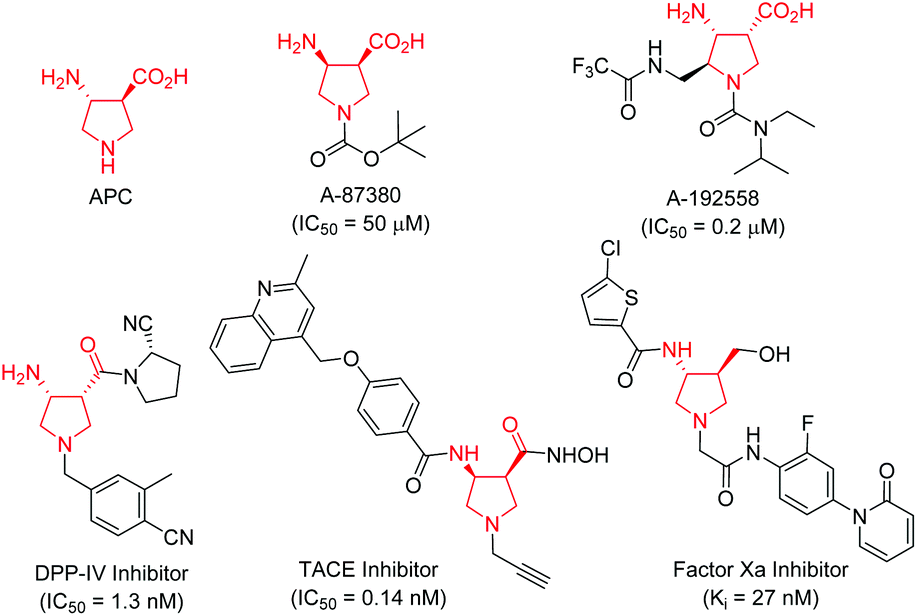 | ||
| Fig. 1 Selected examples of biologically active pyrrolidine β-amino acids. | ||
On the other hand, the catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides with activated dipolarophiles has become one of the most powerful tools for preparing enantioenriched polysubstituted pyrrolidines.6,7 Over the past decade, much effort has been devoted to the development of novel catalytic systems, and the exploration of a variety of new dipolarophiles. However, unlike the commonly utilized activated alkenes7,8 such as acrylates,7b,8a–c enones,7c–f,8d,e maleates,8f–j fumarates,8k maleimides,8l,m nitroalkenes,8n–t vinyl sulfones8u,v and α-heteroatom substituted alkenes,8w–z β-heteroatom anchored electron-deficient alkenes have rarely been explored.9 In connection with our continuing efforts in developing catalytic asymmetric transformations of azomethine ylides,8x–z,10 we have recently reported a rapid access to 3,4-diaminopyrrolidines via a newly designed 4-iodo-DHIPOH ligand/Cu(I) catalyzed 1,3-dipolar cycloaddition of azomethine ylides with β-phthalimidonitroethene (Scheme 1a).10d Inspired by this work, we further envisioned whether pyrrolidine β-amino acids could be produced in a concise manner via asymmetric 1,3-dipolar cycloaddition of azomethine ylides with β-amino substituted acrylates serving as new dipolarophiles. Nevertheless, β-amino substituted acrylates as dipolarophiles could suffer from low reactivity due to the electron-donating nitrogen substituent at the β-position, thus making the catalytic asymmetric cycloaddition process more challenging. To address this issue, a phthalimide group for amino protection could be employed to reduce its electron-donating properties and thus enhance the reaction activity. Herein, we report the first example of Cu(I)-catalyzed 1,3-dipolar cycloaddition of azomethine ylides with β-phthaliminoacrylate esters11 to generate pyrrolidine β-amino esters in high yields with excellent diastereo- and enantioselectivities, which can be easily converted into highly functionalized free pyrrolidine β-amino acids (Scheme 1b).
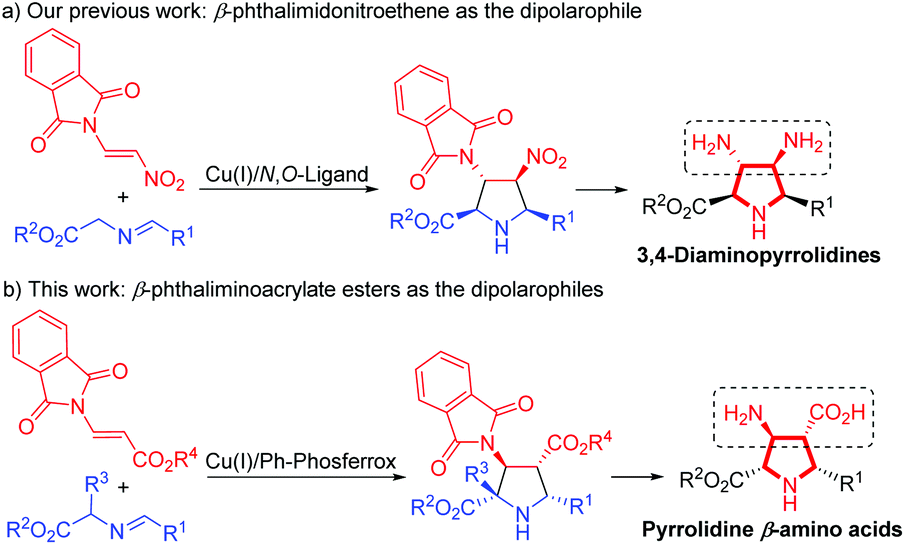 | ||
| Scheme 1 Catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides with β-heteroatom alkenes. | ||
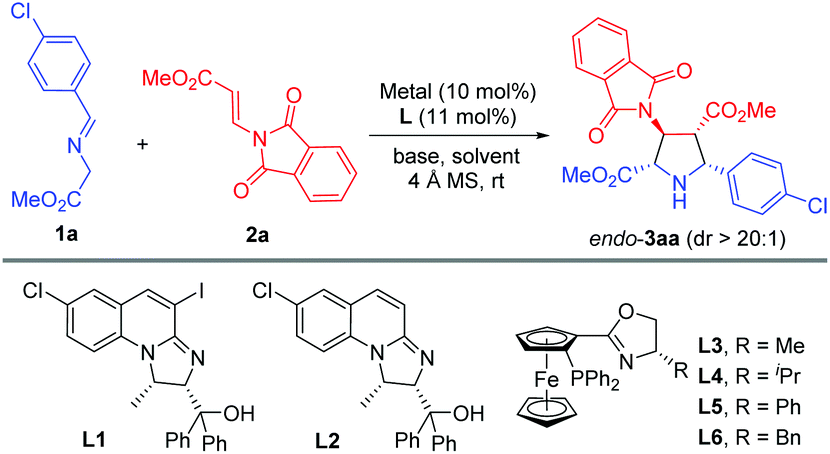
Initially, imino ester 1a and β-phthaliminoacrylate methyl ester 2a were chosen as model substrates for probing this reaction (Table 1). We commenced our investigation using CuBF4 (10 mol%)/N,O-ligands L1 or L2 (11 mol%) as the catalyst system, in the presence of 20 mol% of Et3N as the base in CH2Cl2 at rt, given that these conditions had proven to be very effective in our previous 1,3-dipolar cycloaddition.10d However, the expected cycloaddition did not occur under these reaction conditions (Table 1, entries 1 and 2). Pleasingly, when AgOAc was used instead of CuBF4, changing 20 mol% of Et3N into two equivalents of K2CO3 in THF, the reaction took place after 24 h and gave the desired cycloadduct 3aa in low yield (30%) with excellent diastereoselectivity (dr >20:1) and moderate enantioselectivity (51% ee) (Table 1, entry 3). Encouraged by this result, we decided to investigate the effect of chiral ligands including N,O-ligand L2 and planar-chiral ferrocene P,N-ligands (Phosferrox) L3–L6 (Table 1, entries 4–8). To our delight, all the tested ligands promoted this reaction smoothly within 2 h and Phosferrox ligand L5 exhibited the optimal reaction outcome, delivering the corresponding cycloadduct 3aa in 93% yield and 86% ee (Table 1, entry 7). Further switching the metal source from Ag(I) back to Cu(I) had a remarkable improvement in enantioselectivity (Table 1, entries 9 and 10), with CuBF4 giving the optimal results (96% yield, 98% ee) (Table 1, entry 9). The effect of solvent was then examined, and THF showed the best result (Table 1, entries 11 and 12). Furthermore, lowering the temperature to 0 °C afforded 3aa in 96% yield with extremely high enantioselectivity (>99% ee) (Table 1, entry 13). Finally, when a lower catalyst loading (5 mol%) was employed at this temperature (0 °C), 3aa was obtained with almost the same excellent outcome (95% yield, >99% ee), which was identified as the optimal reaction conditions (Table 1, entry 14).
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal/L | Base | Solvent | Yieldb (%) | eec (%) |
|
a Unless otherwise stated, reactions were performed with 1a (0.15 mmol), 2a (0.10 mmol) in 1 mL of solvent (C = 0.1 M). CuBF4 = Cu(CH3CN)4BF4, CuPF6 = Cu(CH3CN)4PF6.
b Isolated yield.
c >20:1 dr was determined by crude 1H NMR spectroscopy, and ee was determined by chiral HPLC analysis.
d In 24 h.
e In 2 h.
f In 3 h.
g
T = 0 °C.
h 5 mol% cat, 0.2 mmol scale.
|
|||||
| 1d | CuBF4/L1 | Et3N | CH2Cl2 | Trace | |
| 2d | CuBF4/L2 | Et3N | CH2Cl2 | Trace | |
| 3d | AgOAc/L1 | K2CO3 | THF | 30 | −51 |
| 4e | AgOAc/L2 | K2CO3 | THF | 89 | −67 |
| 5e | AgOAc/L3 | K2CO3 | THF | 91 | 75 |
| 6e | AgOAc/L4 | K2CO3 | THF | 88 | 64 |
| 7e | AgOAc/L5 | K2CO3 | THF | 93 | 86 |
| 8e | AgOAc/L6 | K2CO3 | THF | 90 | 83 |
| 9e | CuBF4/L5 | K2CO3 | THF | 96 | 98 |
| 10e | CuPF6/L5 | K2CO3 | THF | 91 | 93 |
| 11d | CuBF4/L5 | K2CO3 | CH2Cl2 | 70 | 98 |
| 12d | CuBF4/L5 | K2CO3 | Et2O | 81 | 97 |
| 13f,g | CuBF4/L5 | K2CO3 | THF | 96 | >99 |
| 14f,g,h | CuBF4/L5 | K2CO3 | THF | 95 | >99 |
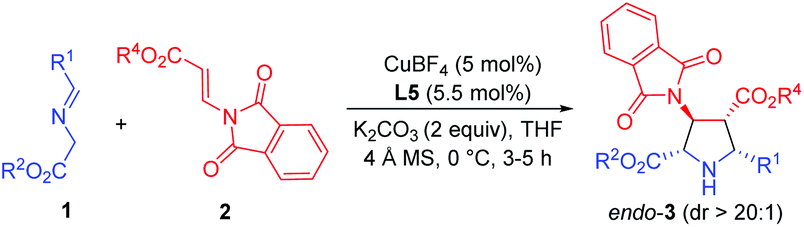
With the optimized reaction conditions in hand, the generality and substrate scope of this process were examined, and the results are shown in Table 2. To begin with, the ester groups of β-phthaliminoacrylate esters (2a–2d) were varied (Me, Et, tBu, Bn), giving the corresponding products (3aa–3ad) in high yields (91–96%) with high diastereoselectivities (dr >20:1) and excellent enantioselectivities (99 to >99% ee) (Table 2, entries 1–4). By changing the ester groups of imino esters from methyl to ethyl and tert-butyl groups (1b–1c) led to a decrease in enantioselectivity of the desired cycloadducts (3ba–3ca) (96% ee and 92% ee, respectively) (Table 2, entries 5 and 6 versus entry 1). Then, a variety of imino esters (1d–1m) derived from aryl aldehydes bearing electron-deficient (Table 2, entries 7–10), electron-rich (Table 2, entries 12–15), and electron-neutral substituents (Table 2, entries 10 and 16) on the aryl rings reacted smoothly with β-phthaliminoacrylate methyl ester (2a), affording the corresponding products (3da–3ma) in high yields (92–98%) with high diastereoselectivities (dr >20:1) and excellent enantioselectivities (94 to >99% ee). Additionally, the heteroaryl imino esters (1n–1o) derived from 2-thenaldehyde and 2-furylaldehyde worked efficiently in this transformation, resulting in the desired cycloadducts (3na–3oa) in >99% ee and 98% ee, respectively (Table 2, entries 17 and 18). Notably, less reactive aliphatic-substituted imino ester 1p was also tolerated in this reaction and the desired adduct 3pa was obtained in good yield (87%) with excellent diastereoselectivity (dr >20:1) and enantioselectivity (98% ee) in the presence of a stronger base Cs2CO3 (Table 2, entry 19).
a
|
|
||||
|---|---|---|---|---|
| Entry | R1/R2 (1) | R4 (2) | Yieldb (%) | eec (%) |
|
a All reactions were performed with 1 (0.30 mmol), and 2 (0.20 mmol) in 2 mL of THF.
b Isolated yield.
c >20:1 dr was determined by crude 1H NMR spectroscopy, and ee was determined by chiral HPLC analysis.
d Cs2CO3 was used instead of K2CO3 and the reaction completed in 3 h, but no product was obtained under standard reaction conditions after 12 h.
|
||||
| 1 | p-ClC6H4/Me (1a) | Me (2a) | 95 (3aa) | >99 |
| 2 | p-ClC6H4/Me (1a) | Et (2b) | 91 (3ab) | 99 |
| 3 | p-ClC6H4/Me (1a) | t Bu (2c) | 94 (3ac) | >99 |
| 4 | p-ClC6H4/Me (1a) | Bn (2d) | 96 (3ad) | >99 |
| 5 | p-ClC6H4/Et (1b) | Me (2a) | 91 (3ba) | 96 |
| 6 | p-ClC6H4/tBu (1c) | Me (2a) | 92 (3ca) | 92 |
| 7 | o-ClC6H4/Me (1d) | Me (2a) | 96 (3da) | >99 |
| 8 | m-ClC6H4/Me (1e) | Me (2a) | 92 (3ea) | 95 |
| 9 | p-BrC6H4/Me (1f) | Me (2a) | 98 (3fa) | 99 |
| 10 | p-CNC6H4/Me (1g) | Me (2a) | 97 (3ga) | >99 |
| 11 | Ph/Me (1h) | Me (2a) | 96 (3ha) | >99 |
| 12 | o-MeC6H4/Me (1i) | Me (2a) | 95 (3ia) | >99 |
| 13 | m-MeC6H4/Me (1j) | Me (2a) | 93 (3ja) | 94 |
| 14 | p-MeC6H4/Me (1k) | Me (2a) | 95 (3ka) | 99 |
| 15 | p-MeOC6H4/Me (1l) | Me (2a) | 97 (3la) | >99 |
| 16 | 2-Naphthyl/Me (1m) | Me (2a) | 94 (3ma) | 98 |
| 17 | 2-Thienyl/Me (1n) | Me (2a) | 93 (3na) | >99 |
| 18 | 2-Furyl/Me (1o) | Me (2a) | 81 (3oa) | 98 |
| 19d | Cy/Me (1p) | Me (2a) | 87 (3pa) | 98 |
Remarkably, α-methyl imino ester 4 derived from alanine was also examined for the construction of pyrrolidine β-amino ester bearing one quaternary and three contiguous tertiary stereogenic centers. Due to the lower reactivity, we made some additional optimization for this cyclization process, including the use of Cs2CO3 instead of K2CO3 as the base and the rise of the temperature from 0 °C to rt. The reaction proceeded smoothly, furnishing the corresponding pyrrolidine 5a in 83% yield, >20:1 dr, and >99% ee (Scheme 2).
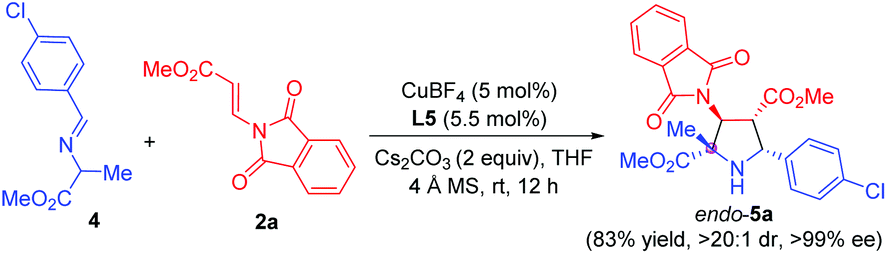 | ||
| Scheme 2 Asymmetric 1,3-dipolar cycloaddition of imino ester 4 derived from alanine with β-phthaliminoacrylate methyl ester 2a. | ||
To further demonstrate the synthetic utility of this process, the asymmetric 1,3-dipolar cycloaddition between 1a and 2a was carried out on a gram scale with lower catalyst loading (1 mol%), giving 3aa in 99% yield with >20:1 dr and >99% ee. The absolute configuration of 3aa was unequivocally determined to be (2S,3S,4R,5R) by single crystal X-ray diffraction of the corresponding N-tosylated amide 6 (Scheme 3).12 The further transformation of the corresponding cycloadducts by employing 3ad as an example into free pyrrolidine β-amino acids was demonstrated as follows. The phthalimide group of 3ad was removed in the presence of ethylenediamine to afford the free amine 7 in good yield (83%) without loss of the ee value, which was then subjected to a catalytic hydrogenation condition to deliver optically pure free pyrrolidine β-amino acid 8 in 96% yield (Scheme 4).
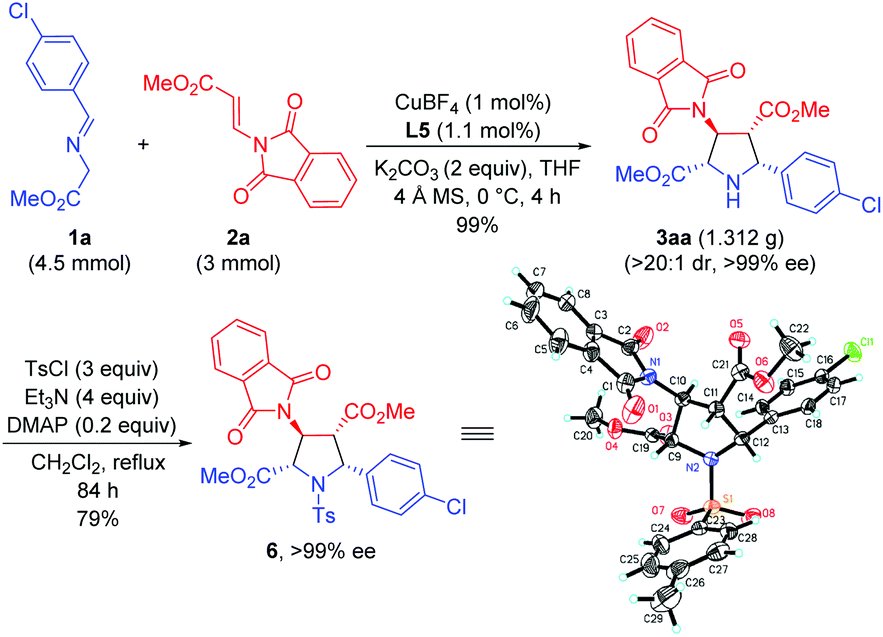 | ||
| Scheme 3 Gram scale synthesis and the absolute configuration determination of the cycloadduct derivative 6. | ||
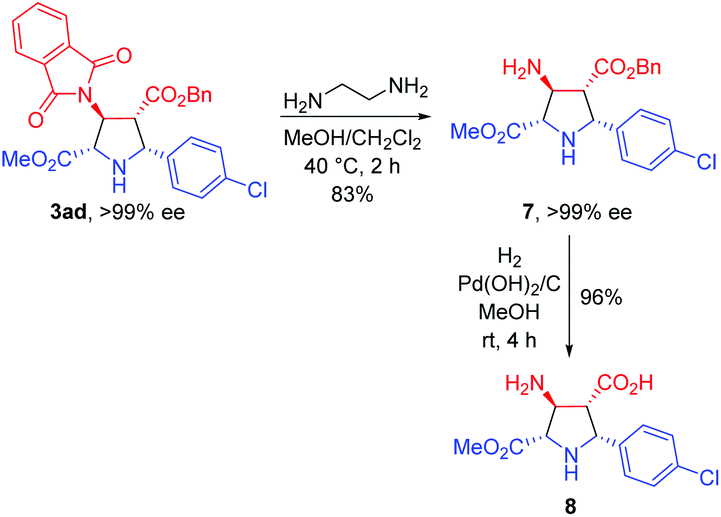 | ||
| Scheme 4 Deprotection and hydrogenolysis of cycloadduct 3ad to pyrrolidine β-amino acid 8. | ||
Conclusions
In summary, we have developed a facile and stereoselective synthesis of a series of structurally novel and biologically important pyrrolidine β-amino acid derivatives via catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides with β-phthaliminoacrylate esters. The Cu(I)/Ph-Phosferrox catalytic system exhibited excellent efficiency even in 1 mol% catalyst loading, providing the corresponding cycloadducts in high yields (up to 98%) with excellent stereoselectivity control (dr >20:1, ee up to >99%). Importantly, highly functionalized free pyrrolidine β-amino acids could be easily obtained from the cycloadducts by simple deprotection and hydrogenolysis, showing great potential in organic synthesis and drug discovery. Further application of this protocol in drug discovery is still underway in our laboratory.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (no. 21372074 and 21572053).Notes and references
- (a) L. Kiss and F. Fülöp, Chem. Rev., 2014, 114, 1116–1169 CrossRef CAS PubMed and references cited therein; (b) L. Kiss, E. Forró and F. Fülöp, Amino Acids, Peptides and Proteins in Organic Chemistry: Origins and Synthesis of Amino Acid, Wiley-VCH, Weinheim, 2009, pp. 367–409 Search PubMed.
- (a) E. A. Porter, X. Wang, H.-S. Lee, B. Weisblum and S. H. Gellman, Nature, 2000, 404, 565–565 CrossRef CAS PubMed; (b) J. D. Sadowsky, W. D. Fairlie, E. B. Hadley, H.-S. Lee, N. Umezawa, Z. Nikolovska-Coleska, S. Wang, D. C. Huang, Y. Tomita and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 139–154 CrossRef CAS PubMed.
- (a) G. T. Wang, Y. Chen, S. Wang, R. Gentles, T. Sowin, W. Kati, S. Muchmore, V. Giranda, K. Stewart and H. Sham, J. Med. Chem., 2001, 44, 1192–1201 CrossRef CAS PubMed; (b) M. E. Bunnage, S. G. Davies, P. M. Roberts, A. D. Smith and J. M. Withey, Org. Biomol. Chem., 2004, 2, 2763–2776 RSC.
- (a) J. Corbett, K. Dirico, W. Song, B. Boscoe, S. Doran, D. Boyer, X. Qiu, M. Ammirati, M. VanVolkenburg and R. McPherson, Bioorg. Med. Chem. Lett., 2007, 17, 6707–6713 CrossRef CAS PubMed; (b) G. R. Ott, N. Asakawa, Z. Lu, R.-Q. Liu, M. B. Covington, K. Vaddi, M. Qian, R. C. Newton, D. D. Christ, J. M. Traskos, C. P. Decicco and J. J. W. Duan, Bioorg. Med. Chem. Lett., 2008, 18, 694–699 CrossRef CAS PubMed; (c) K. G. Zbinden, L. Anselm, D. W. Banner, J. Benz, F. Blasco, G. Décoret, J. Himber, B. Kuhn, N. Panday, F. Ricklin, P. Risch, D. Schlatter, M. Stahl, S. Thomi, R. Unger and W. Haap, Eur. J. Med. Chem., 2009, 44, 2787–2795 CrossRef CAS PubMed.
- For selected examples, see: (a) X. Wang, J. F. Espinosa and S. H. Gellman, J. Am. Chem. Soc., 2000, 122, 4821–4822 CrossRef CAS; (b) H.-S. Lee, P. R. LePlae, E. A. Porter and S. H. Gellman, J. Org. Chem., 2001, 66, 3597–3599 CrossRef CAS PubMed; (c) V. K. Aggarwal, S. Roseblade and R. Alexander, Org. Biomol. Chem., 2003, 1, 684–691 RSC; (d) R. Hanselmann, J. Zhou, P. Ma and P. N. Confalone, J. Org. Chem., 2003, 68, 8739–8741 CrossRef CAS PubMed.
- For recent reviews about asymmetric 1,3-dipolar cycloaddition reactions of azomethine ylides, see: (a) L. M. Stanley and M. P. Sibi, Chem. Rev., 2008, 108, 2887–2902 CrossRef CAS PubMed; (b) Q.-A. Chen, D.-S. Wang and Y.-G. Zhou, Chem. Commun., 2010, 46, 4043–4051 RSC; (c) J. Adrio and J. C. Carretero, Chem. Commun., 2011, 47, 6784–6794 RSC; (d) R. Narayan, M. Potowski, Z.-J. Jia, A. P. Antonchick and H. Waldmann, Acc. Chem. Res., 2014, 47, 1296–1310 CrossRef CAS PubMed; (e) E. E. Maroto, M. Izquierdo, S. Reboredo, J. Marco-Martínez, S. Filippone and N. Martín, Acc. Chem. Res., 2014, 47, 2660–2670 CrossRef CAS PubMed; (f) J. Adrio and J. C. Carretero, Chem. Commun., 2014, 50, 12434–12446 RSC; (g) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed.
- For selected very recent examples of catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides, see: (a) K. Liu, Y. Xiong, Z.-F. Wang, H.-Y. Tao and C.-J. Wang, Chem. Commun., 2016, 52, 9458–9461 RSC; (b) E. M. Arpa, M. Gonzalez-Esguevillas, A. Pascual-Escudero, J. Adrio and J. C. Carretero, J. Org. Chem., 2016, 81, 6128–6135 CrossRef CAS PubMed; (c) Z. M. Zhang, B. Xu, S. Xu, H. H. Wu and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 6324–6328 CrossRef CAS PubMed; (d) H. Xu, C. Golz, C. Strohmann, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2016, 55, 7761–7765 CrossRef CAS PubMed; (e) V. H. Lauridsen, L. Ibsen, J. Blom and K. A. Jørgensen, Chem. – Eur. J., 2016, 22, 3259–3263 CrossRef CAS PubMed; (f) J. Y. Li, H. Y. Kim and K. Oh, Adv. Synth. Catal., 2016, 358, 984–993 CrossRef CAS; (g) M.-C. Tong, X. Chen, H.-Y. Tao and C.-J. Wang, Angew. Chem., Int. Ed., 2013, 52, 12377–12380 CrossRef CAS PubMed and references cited therein.
- For selected examples of catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides with various activated alkenes, see: (a) S. Saito, T. Tsubogo and S. Kobayashi, J. Am. Chem. Soc., 2007, 129, 5364–5365 CrossRef CAS PubMed; (b) H. Y. Kim, H. J. Shih, W. E. Knabe and K. Oh, Angew. Chem., Int. Ed., 2009, 48, 7420–7423 CrossRef CAS PubMed; (c) Y. Yamashita, T. Imaizumi and S. Kobayashi, Angew. Chem., Int. Ed., 2011, 50, 4893–4896 CrossRef CAS PubMed; (d) J. Hernández-Toribio, R. G. Arrayás, B. Martín-Matute and J. C. Carretero, Org. Lett., 2009, 11, 393–396 CrossRef PubMed; (e) M. Potowski, C. Merten, A. P. Antonchick and H. Waldmann, Chem. – Eur. J., 2015, 21, 4913–4917 CrossRef CAS PubMed; (f) J. M. Longmire, B. Wang and X. Zhang, J. Am. Chem. Soc., 2002, 124, 13400–13401 CrossRef CAS PubMed; (g) W. Zeng and Y.-G. Zhou, Org. Lett., 2005, 7, 5055–5058 CrossRef CAS PubMed; (h) W. Zeng, G.-Y. Chen, Y.-G. Zhou and Y.-X. Li, J. Am. Chem. Soc., 2007, 129, 750–751 CrossRef CAS PubMed; (i) X.-H. Chen, W.-Q. Zhang and L.-Z. Gong, J. Am. Chem. Soc., 2008, 130, 5652–5653 CrossRef CAS PubMed; (j) C.-J. Wang, G. Liang, Z.-Y. Xue and F. Gao, J. Am. Chem. Soc., 2008, 130, 17250–17251 CrossRef CAS PubMed; (k) A. S. Gothelf, K. V. Gothelf, R. G. Hazell and K. A. Jørgensen, Angew. Chem., Int. Ed., 2002, 41, 4236–4238 CrossRef CAS; (l) C. Nájera, M. D. G. Retamosa and J. M. Sansano, Org. Lett., 2007, 9, 4025–4028 CrossRef PubMed; (m) M. Martín-Rodríguez, C. Najera, J. M. Sansano, A. de Cózar and F. P. Cossío, Chem. – Eur. J., 2011, 17, 14224–14233 CrossRef PubMed; (n) X. X. Yan, Q. Peng, Y. Zhang, K. Zhang, W. Hong, X. L. Hou and Y. D. Wu, Angew. Chem., Int. Ed., 2006, 45, 1979–1983 CrossRef CAS PubMed; (o) T. Arai, A. Mishiro, N. Yokoyama, K. Suzuki and H. Sato, J. Am. Chem. Soc., 2010, 132, 5338–5339 CrossRef CAS PubMed; (p) T. Arai, N. Yokoyama, A. Mishiro and H. Sato, Angew. Chem., Int. Ed., 2010, 49, 7895–7898 CrossRef CAS PubMed; (q) H. Y. Kim, J.-Y. Li, S. Kim and K. Oh, J. Am. Chem. Soc., 2011, 133, 20750–20753 CrossRef CAS PubMed; (r) R. Narayan, J. O. Bauer, C. Strohmann, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 12892–12896 CrossRef CAS PubMed; (s) X.-F. Bai, T. Song, Z. Xu, C.-G. Xia, W.-S. Huang and L.-W. Xu, Angew. Chem., Int. Ed., 2015, 54, 5255–5259 CrossRef CAS PubMed; (t) J.-Y. Li, H. Y. Kim and K. Oh, Org. Lett., 2015, 17, 1288–1291 CrossRef CAS PubMed; (u) T. Llamas, R. G. Arrayás and J. C. Carretero, Org. Lett., 2006, 8, 1795–1798 CrossRef CAS PubMed; (v) G. Liang, M.-C. Tong and C.-J. Wang, Adv. Synth. Catal., 2009, 351, 3101–3106 CrossRef CAS; (w) M. González-Esguevillas, J. Adrio and J. C. Carretero, Chem. Commun., 2013, 49, 4649–4651 RSC; (x) Z. Wang, S. Luo, S. Zhang, W.-L. Yang, Y.-Z. Liu, H. Li, X. Luo and W.-P. Deng, Chem. – Eur. J., 2013, 19, 6739–6745 CrossRef CAS PubMed; (y) Z. Wang, X. Yu, B. X. Tian, D. T. Payne, W.-L. Yang, Y.-Z. Liu, J. S. Fossey and W.-P. Deng, Chem. – Eur. J., 2015, 21, 10457–10465 CrossRef CAS PubMed; (z) W.-L. Yang, F.-F. Tang, F.-S. He, C.-Y. Li, X. Yu and W.-P. Deng, Org. Lett., 2015, 17, 4822–4825 CrossRef CAS PubMed.
- (a) A. Awata and T. Arai, Angew. Chem., Int. Ed., 2014, 53, 10462–10465 CrossRef CAS PubMed; (b) Q.-L. Yang, M.-S. Xie, C. Xia, H.-L. Sun, D.-J. Zhang, K.-X. Huang, Z. Guo, G.-R. Qu and H.-M. Guo, Chem. Commun., 2014, 50, 14809–14812 RSC; (c) A. L. Gerten and L. M. Stanley, Org. Chem. Front., 2016, 3, 339–343 RSC; (d) D.-J. Zhang, M.-S. Xie, G.-R. Qu, Y.-W. Gao and H.-M. Guo, Org. Lett., 2016, 18, 820–823 CrossRef CAS PubMed.
- (a) Y.-H. Shi, Z. Wang, B. Hu, M. Wang, J. S. Fossey and W.-P. Deng, Org. Lett., 2011, 13, 6010–6013 CrossRef CAS PubMed; (b) M. Wang, Y.-H. Shi, J.-F. Luo, W. Du, X.-X. Shi, J. S. Fossey and W.-P. Deng, Catal. Sci. Technol., 2011, 1, 100–103 RSC; (c) M. Wang, Z. Wang, Y.-H. Shi, X.-X. Shi, J. S. Fossey and W.-P. Deng, Angew. Chem., Int. Ed., 2011, 50, 4897–4900 CrossRef CAS PubMed; (d) F.-S. He, H. Zhu, Z. Wang, M. Gao, X. Yu and W.-P. Deng, Org. Lett., 2015, 17, 4988–4991 CrossRef CAS PubMed; (e) C.-Y. Li, W.-L. Yang, X. Luo and W.-P. Deng, Chem. – Eur. J., 2015, 21, 19048–19057 CrossRef CAS PubMed; (f) F.-F. Tang, W.-L. Yang, X. Yu and W.-P. Deng, Catal. Sci. Technol., 2015, 5, 3568–3575 RSC; (g) W.-L. Yang, Y.-Z. Liu, S. Luo, X. Yu, J. S. Fossey and W.-P. Deng, Chem. Commun., 2015, 51, 9212–9215 RSC; (h) F.-S. He, J.-H. Jin, Z.-T. Yang, X. Yu, J. S. Fossey and W.-P. Deng, ACS Catal., 2016, 6, 652–656 CrossRef CAS; (i) W.-L. Yang, C.-Y. Li, W.-J. Qin, F.-F. Tang, X. Yu and W.-P. Deng, ACS Catal., 2016, 6, 5685–5690 CrossRef CAS.
- To date, β-phthaliminoacrylate esters have only been utilized as an active Michael acceptor in Rh(I)-catalyzed asymmetric arylation, see: T. Nishimura, J. Wang, M. Nagaosa, K. Okamoto, R. Shintani, F.-Y. Kwong, W.-Y. Yu, A. S. Chan and T. Hayashi, J. Am. Chem. Soc., 2009, 132, 464–465 CrossRef PubMed.
- CCDC 1495843 (6) contains the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1495843. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00512h |
| This journal is © the Partner Organisations 2017 |