
A highly stereo-controlled protocol to prepare pipecolic acids based on Heck and cyclohydrocarbonylation reactions†‡
Giada
Arena
a,
Elena
Cini
a,
Elena
Petricci
*a,
Rosario
Randino
b and
Maurizio
Taddei
*a
aDipartimento di Biotecnologie, Chimica e Farmacia, Università di Siena, Via A. Moro 2, 53100 Siena, Italy. E-mail: maurizio.taddei@unisi.it
bDipartimento di Farmacia, Università di Salerno, Via Giovanni Paolo II 132, 84084 Fisciano, Italy
First published on 9th March 2015
Abstract
A novel synthetic protocol based on an indium-mediated glyoxylate allylation, Heck coupling and Rh-catalysed cyclohydrocarbonylation (CHC) was established to access enantiomerically pure polysubstituted pipecolic acids. The key steps are the Heck reaction, which is performed exclusively using a phenone-oxime derived palladacycle and the domino hydroformylation-cyclisation of a styryl derivative obtained from the Heck coupling. The reaction scheme, proceeding with good stereoselectivity, is also suitable for the preparation of substituted piperidine derivatives.
Introduction
L-Pipecolic acid is a non-coded amino acid present in a variety of natural products and as a secondary metabolite derived from bacteria, fungi and plants.1 It is a key element in pharmacologically relevant molecules such as antibiotics,2 anesthetics,3 immunosuppressive and anticancer drugs4 (Fig. 1). Pipecolic acid derivatives also behave as proline analogues, inducing β-turns in peptidomimetics,5 and recently it received considerable attention as a ligand for organocatalytic processes.6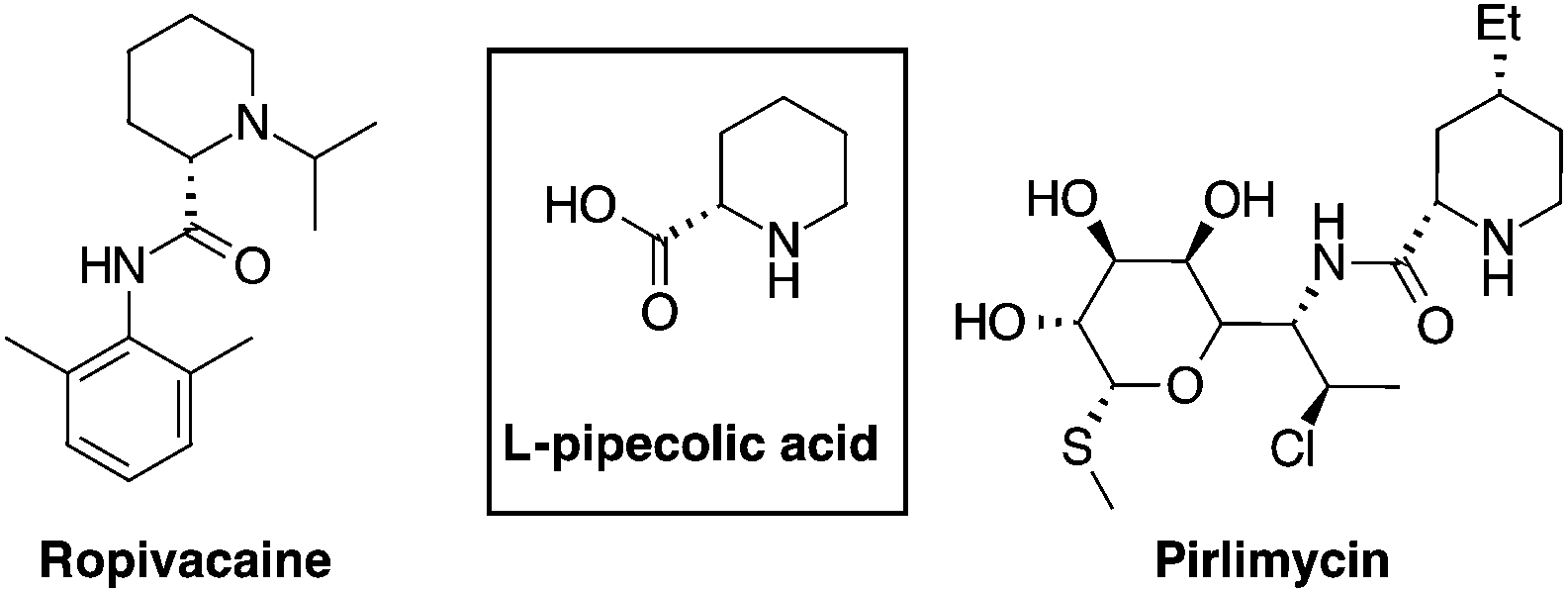 | ||
| Fig. 1 Relevant representative pipecolic acids. | ||
This synthetic and biological versatility stimulated the development of several approaches for the preparation of pipecolic acids including biocatalysis,7 photocatalysis,8 ring closing metathesis,9 chiral aziridine rearrangements10 and many others, which have been recently reviewed.11 Domino hydroformylation-cyclisation of olefins involving a nitrogen nucleophile (cyclohydrocarbonylation, CHC)12 is a powerful method to prepare multifunctional heterocycles such as indolizidine,13 tetrahydroquinolines,14 quinolizidines15 and piperidines.16,17
We sought to expand the scope of our CHC process for the synthesis of piperidine17 with the aim of preparing enantiomerically pure di- and tri-substituted pipecolic acids (Scheme 1).
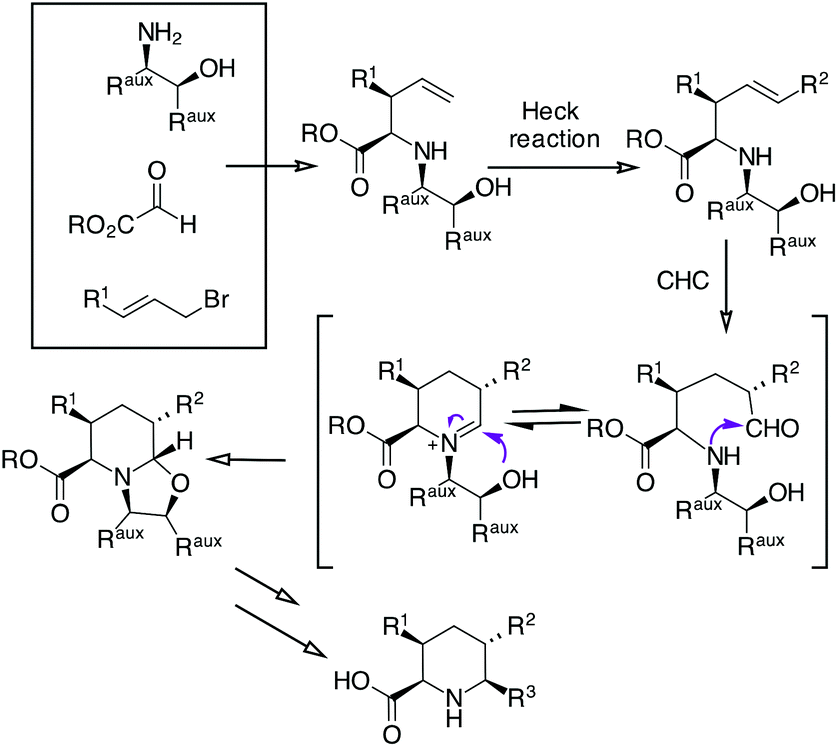 | ||
| Scheme 1 Planned synthesis of polysubstituted pipecolic acids. | ||
Applying an amino alcohol/indium-mediated multicomponent allylation reaction to glyoxylic acid derivatives, we have planned to obtain a 4-pentanoic acid derivative that could be further functionalized by an Heck reaction. Then, a Rh-catalyzed CHC followed by oxazolidine ring opening and chiral auxiliary removal should give 5,6-disubstituted or 3,5,6-trisubstituted pipecolic acids.
Results and discussion
Initially, an allylation reaction with glyoxylic acid derivatives was investigated. Based on our previous result, (1R,2S)-1-amino-2-indanol, denoted as 1, was selected as the chiral auxiliary.17 The imines generated in situ starting from 1 and aldehydes,18 denoted as 2a and 2b, were exposed to indium powder in the presence of allyl or crotyl bromide (namely, 3a or 3b) in alcoholic medium.19 In all the cases, the desired products, namely 4a–c, were obtained in good yields with excellent diastereoselectivities (Scheme 2), confirming that indanol is a good choice as an auxiliary for the enantioselective Barbier-type allylations. The stereochemistry of the product was established by the analysis of the compounds obtained by further cyclisation (refer Table 2).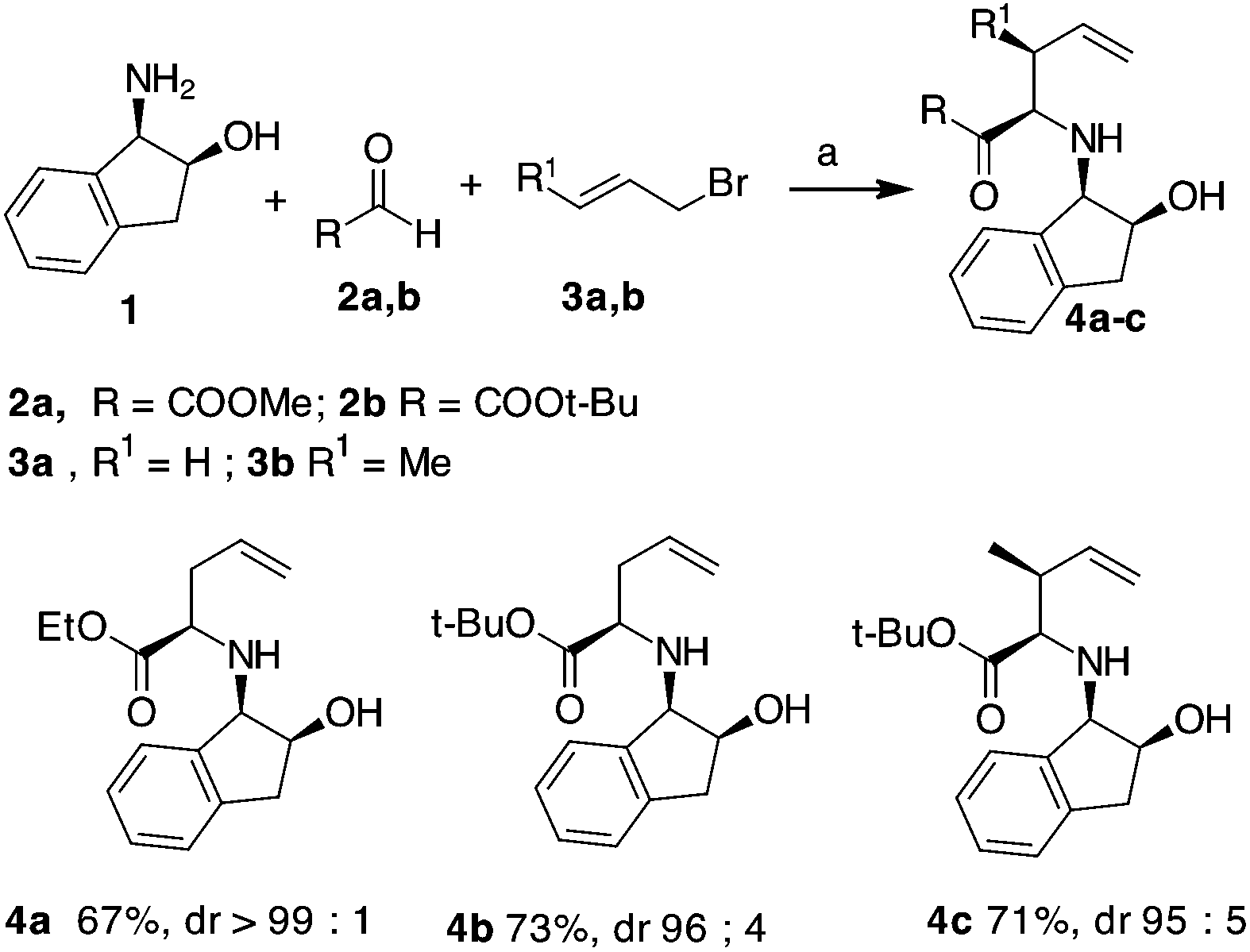 | ||
| Scheme 2 Synthesis of compounds 4a–c. a: In (3 eq.), 3 (2 eq.), EtOH, r.t., 3 h. | ||
A possible functionalization of the allyl derivatives 4a–c was investigated via a Heck Pd-catalysed reaction (Table 1). This protocol is suitable for the synthesis of 5-aryl pipecolic acids and also for 5-aryl piperidine derivatives, which are a series of products that have been scarcely investigated to date.20
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additives/solvent | Conditions | Yield% |
| 1 | Pd(OAc)2 | Et3N (2 eq.), toluene | Reflux, 12 h | — |
| 2 | Pd(OAc)2 | Et3N (2 eq.), PPh3 (4 mol%) toluene | Reflux, 12 h | — |
| 3 | Pd(OAc)2 | Et3N (2 eq.), PPh3 (4 mol%), DMF | 90 °C, 12 h | — |
| 4 | Pd(OAc)2 | Et3N (2 eq.), DMF | 20 °C, 20 min | — |
| 5 | Pd(OAc)2 | Et3N (2 eq.), PPh3 (6 mol%), DMF | MW, 120 °C, 60 min | — |
| 6 | Pd(PPh3)4 | Et3N (2 eq.), DMF | 120 °C, 12 h | 15% |
| 7 | Pd(PPh3)4 | DIPEA (2 eq.), DMF | 120 °C, 12 h | 19% |
| 8 | Pd(PPh3)4 | K2CO3 (3 eq.), DMF | 120 °C, 12 h | 12% |
| 9 | Pd(PPh3)4 | K3PO4 (4 eq.), DMF | 120 °C, 12 h | 12% |
| 10 | 5 | Et3N (1.3 eq.), DMF | 120 °C, 12 h | 46% |
| 11 | 5 | Et3N (1.3 eq.), DMF | 120 °C, 20 min | 55% |
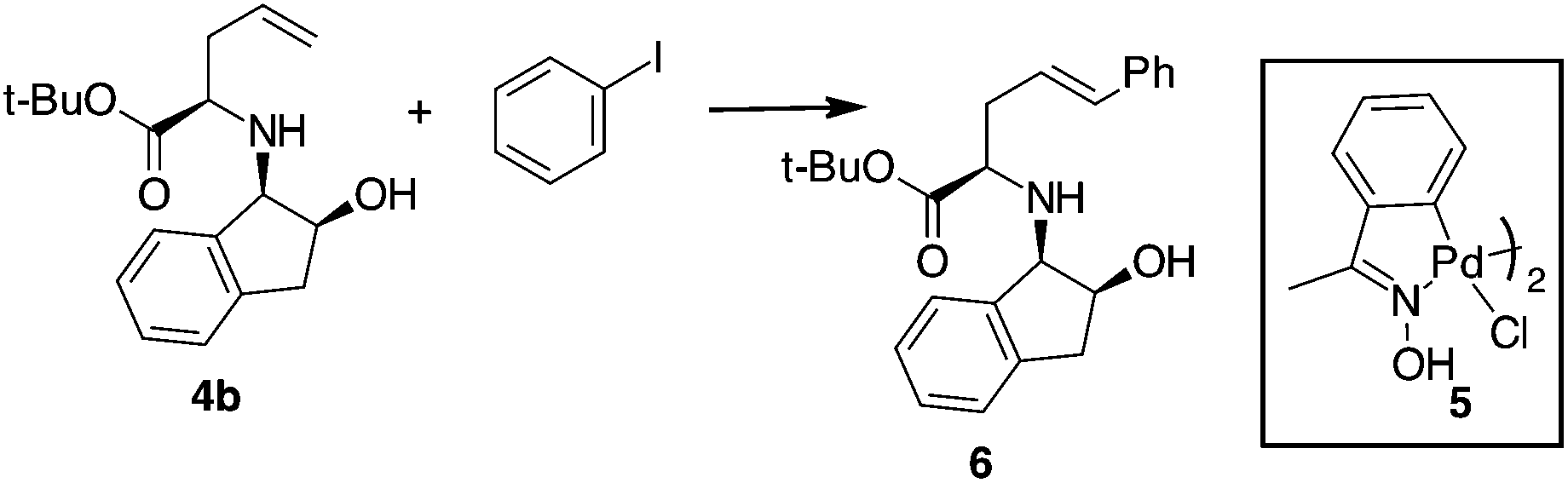
Compound 4b was selected for tuning the conditions of the Heck coupling. When 4b and iodobenzene were subjected to classical Heck reaction conditions using Pd(OAc)2, Et3N, and PPh3 in refluxing toluene for 12 h, no reaction occurred (Table 1, entries 1 and 2). Even using DMF as the solvent at 90 °C or 120 °C, under traditional or microwave (MW) dielectric heating, only the starting materials were recovered (Table 1, entries 3 and 5). A partial conversion into 6 was observed using Pd(PPh3)4, Et3N in DMF at 120 °C for 12 h (Table 1, entry 6). However, no improvements were obtained when the base was changed from Et3N to DIPEA, K2CO3 or K3PO4 (Table 1, entries 7–9). After exploring different Pd sources and ligands, the best reaction conditions were finally identified using the Pd catalyst 5, without phosphorous ligands, in the presence of Et3N in DMF as the solvent. This palladacycle can be easily prepared from the accessible p-methoxy acetophenone oxime and Li2PdCl4.21 It gave excellent results, especially when used immediately after isolation. The reaction of 4b with iodobenzene and Pd catalyst 5 gave the desired internal alkene 6 in good yield with a stereoselectivity toward the E-isomer.22 Moreover, while Pd(PPh3)4 gave effective conversion in 4% mol amounts, catalyst 5 gave product 6 when used in 1% mol amount with respect to the starting compound 4b.
Comparable results were obtained under traditional heating (120 °C, 12 h, Table 1, entry 10) or under MW dielectric heating (120 °C, 20 min Table 1, entry 11). The best conditions found were successfully applied to 4a and 4c and also to other homoallylic derivatives 4d–g prepared, as previously reported (Scheme 3). In all the examples investigated, the use of palladacyle 5 allowed the isolation of the coupling products in good yield, even when substituted and hetero-aryl iodides were involved (Scheme 3). All the products were formed with excellent diastereoselectivities, except in the cases of compounds 16 and 17, where a long aliphatic chain was present in the homo-allyl position. The substituent present on the allyl position seemed to have a minor impact in the E/Z ratio of the Heck product compared to the substituent that is farther from the reaction centre (compare yields and dr of products 15 and 17 in Scheme 3). Compounds 8, 15 and 17 were isolated as single diastereoisomers, suggesting that the Pd-mediated isomerisation of the double bond did not occur.
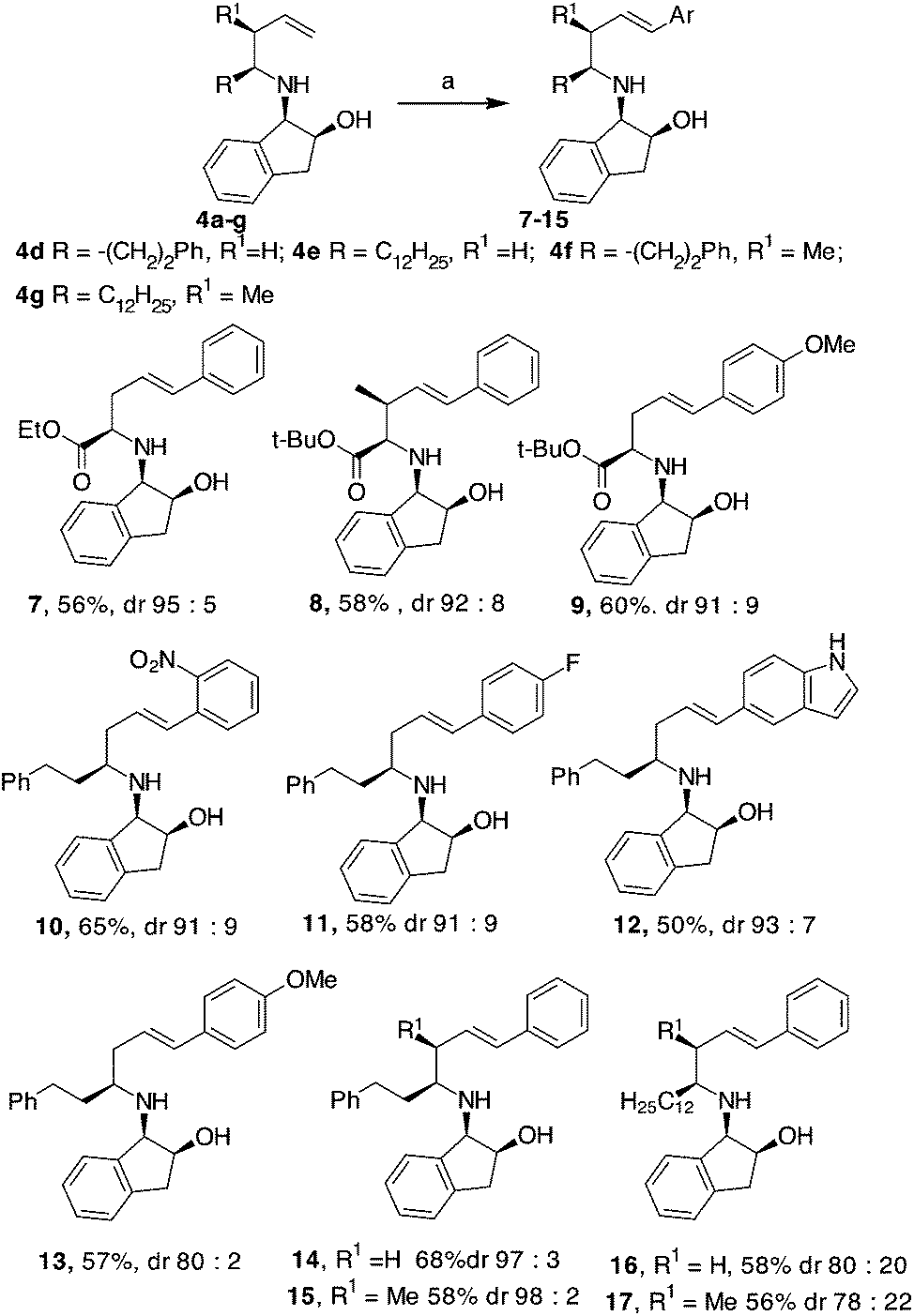 | ||
| Scheme 3 Heck reaction. a: 5 (0.01 mol%), Et3N (1.3 eq.), ArI (1 eq.), DMF, MW, 120 °C, 20 min. | ||
Some selected terminal and internal alkenes synthesized (compounds 4a–c, 14 and 16) were then subjected to CHC conditions: 7 bar of CO/H2 in the presence of [Rh(acac)(CO)2] and BIPHEPHOS (6,6′-[(3,3′-di-tert-butyl-5,5′-dimethoxy-1,1′-biphenyl-2,2′-diyl)bis(oxy)]bis(dibenzo[d,f][1,3,2]dioxa-phosphepin) in THF for 12 h at 70 °C (Table 2).
|
|
|||
|---|---|---|---|
| Entry | sm | Conditionsa | R, R1, R2, product, yield%, dr |
a Reaction conditions: H2–CO 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, [Rh(acac)(CO)2] (2 mol%), ligand (4 mol%), THF, 70 °C, 12 h.
b Reaction performed under MW dielectric heating (70 °C for 2 h.) :1, [Rh(acac)(CO)2] (2 mol%), ligand (4 mol%), THF, 70 °C, 12 h.
b Reaction performed under MW dielectric heating (70 °C for 2 h.)
|
|||
| 1 | 4a | H2–CO (7 bar), BIPHEPHOS | -CO2Et, H, H, 18, 67%, >99:1 |
| 2 | 4b | H2–CO (7 bar), BIPHEPHOS | -CO2t-Bu, H, H, 19 56%, 95:5 |
| 3 | 4c | H2–CO (7 bar), BIPHEPHOS | -CO2t-Bu, Me, H, 20, 58%, 50:50 |
| 4 | 4d | H2–CO (7 bar), BIPHEPHOS | -(CH2)2Ph, Me, H, 21, 56%, dr 96; 5 |
| 5 | 6 | H2–CO (7 bar), BIPHEPHOS | -CO2t-Bu, H, Ph, 22, 15%, 96:4 |
| 6b | 6 | H2–CO (7 bar), BIPHEPHOS |
22, 13%, 92:8 |
| 7 | 6 | H2–CO (7 bar), (EtO)3P |
22, 12%, 95:5 |
| 8 | 6 | H2–CO (30 bar), BIPHEPHOS |
22, 62%, 95:5 |
| 9 | 14 | H2–CO (30 bar), (EtO)3P | -(CH2)2Ph, H, Ph, 23, 58%, 96; 5 |
| 10 | 15 | H2–CO (30 bar), (EtO)3P | -(CH2)2Ph, Me, Ph, 24, 58%, 85:15 |
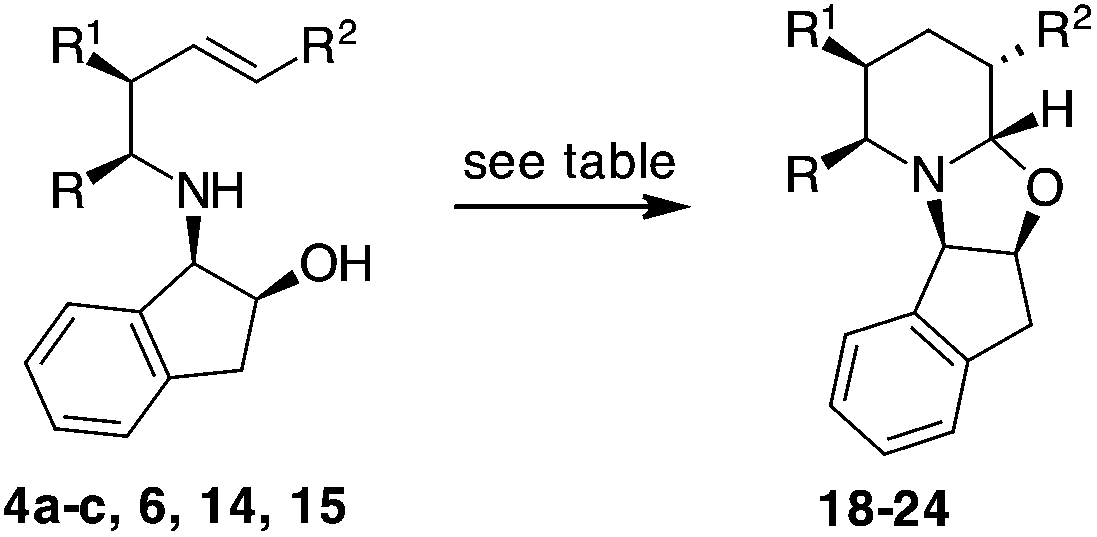
To our delight, 4a and 4b provided bicyclic products 18 and 19 in satisfactory yields and with high diastereoselectivities (Table 2, entries 1 and 2), respectively.17 The presence of the methyl group did not affect the yield and diastereoselectivity in the CHC reaction of 4d, in which a Ph-CH2CH2-chain replaces a t-butoxycarbonyl moiety. On the contrary, when compound 4c, carrying a methyl group in the allyl position, was subjected to the same conditions, product 20 (R1 = Me) was obtained with an acceptable yield of 58% but as a 1:1 mixture of diastereomers (Table 2, entry 3). Probably the contemporary presence of an ester in the C-2 position and an alkyl group in the C-3 position influenced the stability of the chair-like intermediates in the CHC reaction. A lower energy difference between the two possible conformations (both having an alternate equatorial/axial disposition of the methyl and t-butyl ester group) generates no preference for the attack of the OH group.
With internal alkenes 6, 14 and 15, the first hydroformylation is expected to occur with the insertion of the aldehyde onto the benzylic position and to exclusively afford a six-membered ring through OH cyclisation.23 Effectively, when compound 6 was exposed to the same CHC conditions developed so far, a bicyclic product 22 was obtained as a single product, albeit in low yield (Table 2, entry 5). With an aim of improving the reaction, MW dielectric heating was applied and triethylphosphite was employed as the ligand; however, the reaction did not show any improvement (Table 2, entries 6 and 7). Because the main constituent of the crude reaction mixture was the unreacted alkene 6, we decided to increase the syngas pressure to as high as 30 bars. Under these conditions, product 22 was finally isolated in an acceptable yield with an excellent diastereomeric ratio of 95:5 (Table 2, entry 8). Analogously, 14 was readily transformed into 23 with acceptable yield and high selectivity (Table 2, entry 9). However, when the more hindered compound 14 was subjected to the same conditions that were applied to 6, the yield of the CHC product decreased. In this case, the use of triethylphosphite (a less hindered ligand as compared to BIPHEPHOS) was necessary to obtain product 24 in 85:15 dr and with comparable yield in comparison to 23 (Table 2, entry 10). This result could be related to the contemporary presence of the methyl group in the C-3 position as well as the phenyl moiety in the C-5 position. The absolute configuration of the product was established on the basis of X-ray analysis of a crystal derived from compound 22 (Fig. 2).
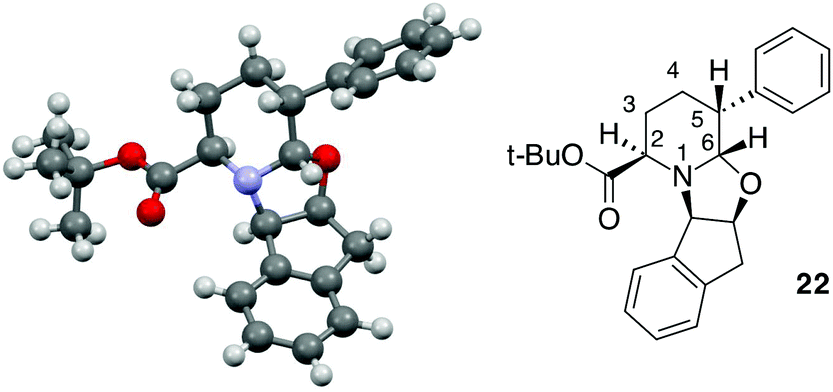 | ||
| Fig. 2 X-ray structure of compound 22. | ||
A trans relationship between the hydrogen atoms at the C-2 position and C-5 position was revealed, whereas at the ring junction, the hydrogen at the C-6 position was in a cis-conformation with respect to the hydrogen at the C-5 position and a trans-conformation with respect to the hydrogen at the C-2 position (Fig. 2). The relative configuration at the C-3 position (for compounds 20, 21 and 24) was established by NOE experiments. Finally, to test the applicability of the proposed protocols for the synthesis of substituted pipecolic acid derivatives, the introduction of a substituent in the position 6 and the removal of the chiral auxiliary were attempted using compounds 18, 19 and 22 (Table 3).
|
|
|||
|---|---|---|---|
| Entry | sm | Conditions | Product, Yield%, dr |
| a Stereochemistry not investigated. b A large excess of MeMgBr (10 eq.) was employed. Probably the intermediate OH was transformed into the halogen derivative, which further reduced under Grignard reaction conditions. | |||
| 1 | 18 | MeMgBr, Et2O, r.t., 30 min or 2 h |
|
| 2 | 18 | MeMgBr, Et2O, −78 °C, 30 minb |
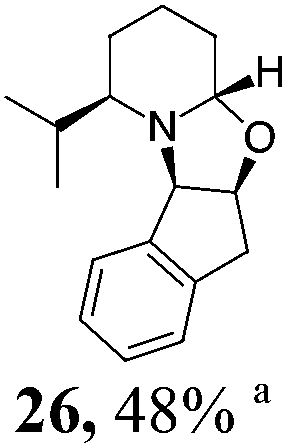
|
| 3 | 18 | Zn, allyl bromide, TMSCl, EtOH, 1 h | 18 |
| 4 | 18 | Zn, ethyl bromoacetate, Et2O–toluene 1 h | 18 |
| 5 | 19 | MeMgBr, Et2O, 0 °C, 30 min |
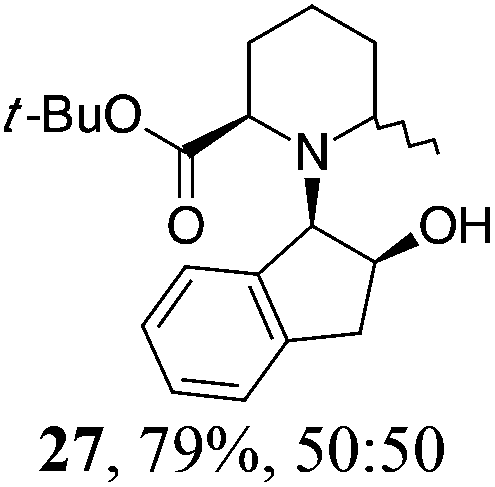
|
| 7 | 19 | i-BuMgBr, Et2O, 0 °C, 30 min |
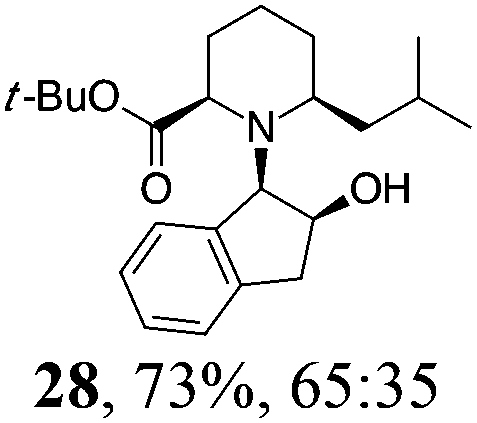
|
| 8 | 22 | BuMgBr, Et2O, r.t., 2 h |
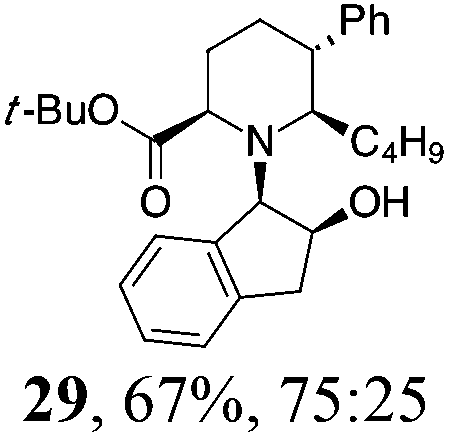
|
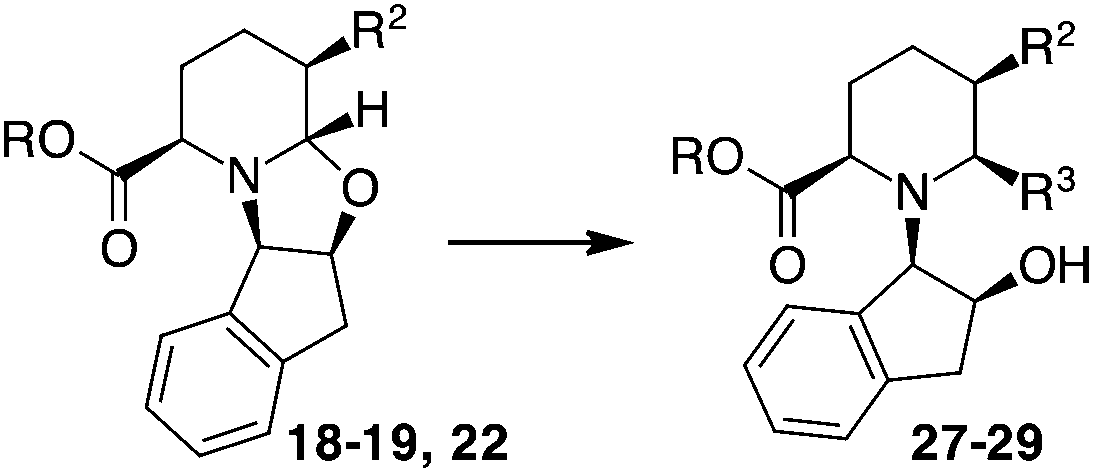
Initial efforts for promoting the oxazolidine ring-opening in methyl ester 18 were fruitless because the reaction of methyl magnesium bromide gave compounds 25 and 26 derived from the attack of the Grignard either on the C-6 position or the ethyl ester moiety (Table 3, entry 1) and a further OH reduction (Table 3, entry 2). Speculating that the hemiaminal carbon (at the C-6 position) would have a reactivity similar to a carbonyl group, 18 was subjected to classic Barbier conditions with allyl bromide (3 eq.) and Zn(0) (2 eq.) in EtOH for 4 h (Table 3, entry 3). However, this attempt was ineffective because both starting materials were recovered. Moreover, when the protocol was repeated on ethyl bromoacetate in the presence of TMSCl as the activating agent for Zn(0) in a mixture of Et2O–toluene, the reaction did not occur (Table 3, entry 4). However, the more bulky t-butyl ester group present in 19 and 22 prevented the nucleophilic attack on the carbonyl present on the position 2. Treating these compounds with different Grignard reagents, the desired products 28 and 29 were isolated in good yields (Table 3, entries 5–9).24 Probably the presence of the t-butoxycarbonyl group, which can principally chelate the Mg, may explain the lower selectivity observed on these substrates when compared with oxazolidine ring opening in substrates carrying an alkyl group at position 2.17
Compounds 28 and 29, after separation from other diastereoisomers by column chromatography, were subjected to the removal of the chiral auxiliary using hydrogen and Pd, followed by t-butyl ester hydrolysis in acidic conditions (TFA, Scheme 4).
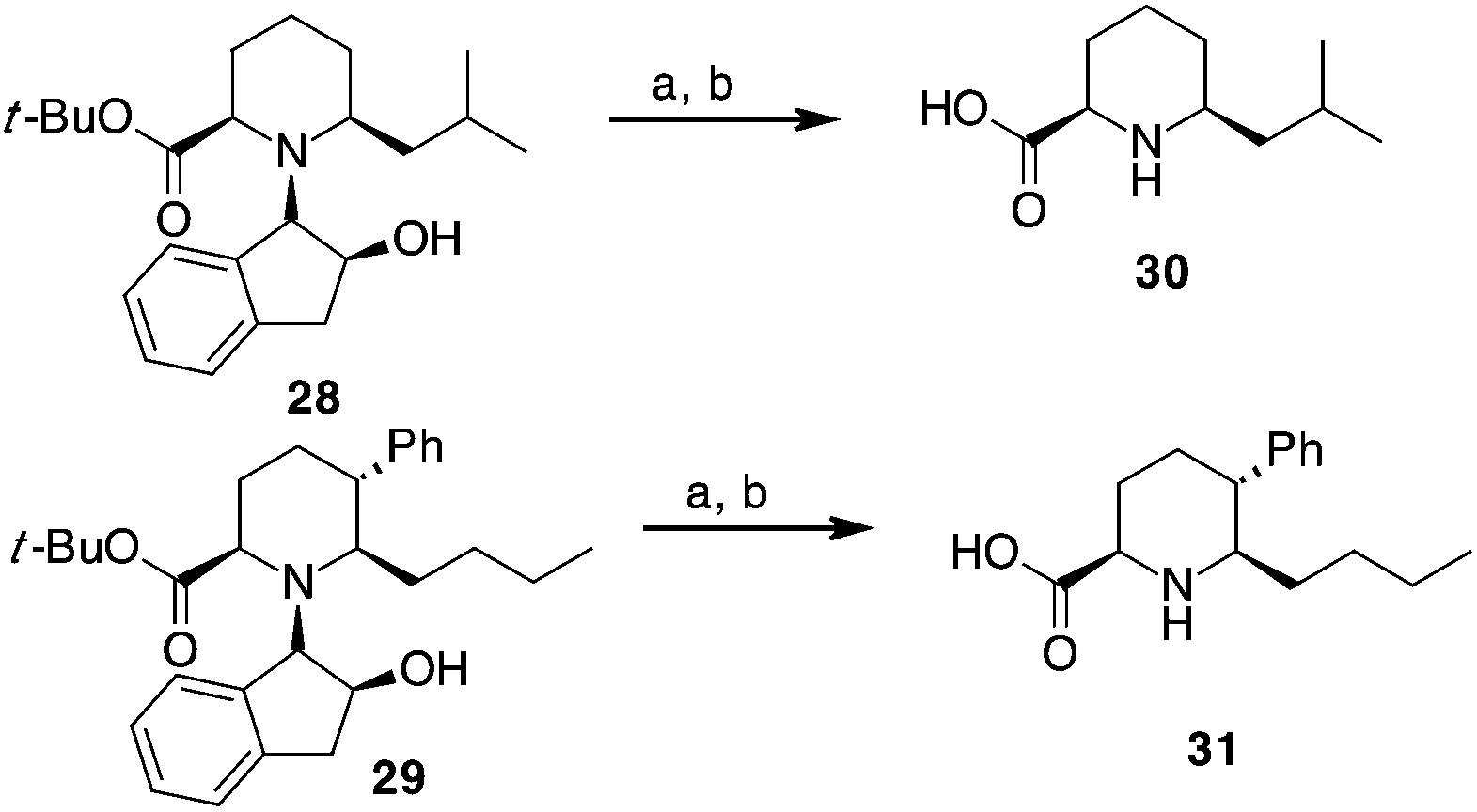 | ||
| Scheme 4 Synthesis of pipecolic acid derivatives 30 and 31. a: H2 (10 bar), Pd(OH)2/C, MeOH, r.t. 10 h. b: TFA, CH2Cl2, r.t., 4 h. | ||
Compound 30 was obtained in 70% yield over the last two steps (dr 76:24), while a better stereoselectivity (>99:1) with 61% yield was observed for trisubstituted compound 31.
Conclusions
Once again CHC reaction proved to be a useful transformation in the stereoselective synthesis of heterocyclic compounds. A good protocol for the functionalization of 4-pentenoic acid derivatives via Heck reaction followed by CHC was developed to obtain bicyclic products with good yields and excellent selectivities. The final transformation of the CHC products into the corresponding pipecolic acids demonstrates the potential of the protocol proposed. The CHC of internal alkenes still needs further studies, especially in terms of the diastereoselectivity observed in some substrates substituted at the C-3 position. Moreover, the proposed synthetic scheme permits to access new interesting building blocks that have not been available to date, and it is potentially interesting for the synthesis of new biologically active molecules or organocatalysts.Acknowledgements
The authors thanks Prof. G. Giorgi (University of Siena) for help in X-ray analysis. The partial financial support from Chemo-Chemessentia (Novara, Italy) is gratefully acknowledged.Notes and references
- V. Vranova, L. Lojkova, K. Rejsek and P. Formanek, Chirality, 2013, 25, 823–831 CrossRef CAS PubMed.
- M. Tandon, D. L. Coffen, P. Gallant, D. Keithb and M. A. Ashwella, Bioorg. Med. Chem. Lett., 2004, 14, 1909–1911 CrossRef CAS PubMed.
- H. Tsuchiya, T. Ueno and M. Mizogami, Bioorg. Med. Chem., 2011, 19, 3410–3415 CrossRef CAS PubMed.
- S. Nobili, I. Landini, B. Giglioni and E. Mini, Curr. Drug Targets, 2006, 7, 861 CrossRef CAS.
- (a) H. Pellissier, Adv. Synth. Catal., 2012, 354, 237 CrossRef CAS; (b) S. Hanessian and l. Auzzas, Acc. Chem. Res., 2008, 41, 1241–1251 CrossRef CAS PubMed; (c) P. H.-Y. Cheoug, H. Zhang, R. Thayumanavan, F. Tanaka, K. N. Houk and C. F. Barbas III, Org. Lett., 2006, 8, 811 CrossRef PubMed; (d) D. I. Peter and M. Lionel, Angew. Chem., Int. Ed., 2004, 39, 5138 Search PubMed.
- S. Mohapatra, S. Bhakta, N. Baral and S. Nayak, Res. Chem. Intermed., 2014 DOI:10.1007/s11164-014-1550-8 , press on-line.
- (a) A. Soler, X. Garrabou, K. Hernández, M. L. Gutiérrez, E. Busto, J. Bujons, T. Parella, J. Joglar and P. Clapés, Adv. Synth. Catal., 2014, 356, 3007–3024 CrossRef CAS; (b) S. Begliomini, L. Sernissi, D. Scarpi and E. G. Occhiato, Eur. J. Org. Chem., 2014, 5448–5455 CrossRef CAS.
- B. Pal, S. Ikeda, H. Kominami, Y. Kera and B. Ohtani, J. Catal., 2003, 217, 152–159 CAS.
- X. Ginesta, M. A. Pericás and A. Rivera, Tetrahedron Lett., 2002, 43, 779–782 CrossRef CAS.
- S. P. Chavan, L. B. Khairnar, P. N. Chavan and D. B. Kalbhor, Tetrahedron: Asymmetry, 2014, 25, 1246–1251 CrossRef CAS PubMed.
- A. A. Cant and A. Sutherland, Synthesis, 2012, 1935–1950 CAS.
- (a) E. Petricci and E. Cini, Top. Curr. Chem., 2013, 342, 117–150 CrossRef; (b) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed; (c) G. Varchi and I. Ojima, Curr. Org. Chem., 2006, 10, 1341–1362 CrossRef CAS; (d) W.-H. Chiou, S.-Y. Lee and I. Ojima, Can. J. Chem., 2005, 83, 681–692 CrossRef CAS.
- E. Airiau, C. Chemin, N. Girard, G. Lonzi, A. Mann, E. Petricci, J. Salvadori and M. Taddei, Synthesis, 2010, 2901 CAS.
- T. O. Vieira and H. Alper, Chem. Commun., 2007, 2710 RSC.
- E. Airiau, T. Spangenberg, N. Girard, B. Breit and A. Mann, Org. Lett., 2010, 12, 528–531 CrossRef CAS PubMed.
- E. Airiau, N. Girard, M. Pizzeti, J. Salvadori, M. Taddei and A. Mann, J. Org. Chem., 2010, 75, 8670–8673 CrossRef CAS PubMed.
- G. Arena, N. Zill, J. Salvadori, N. Girard, A. Mann and M. Taddei, Org. Lett., 2011, 13, 2294 CrossRef CAS PubMed.
- Z. Yao, J. Bhaumik, S. Dhanalekshmi, M. Ptaszek, P. A. Rodriguez and J. S. Lindsey, Tetrahedron, 2007, 63, 10657–10670 CrossRef CAS PubMed.
- T. Vilaivan, C. Winotapan, C. Vorawit, T. Shinada and Y. Ohfune, J. Org. Chem., 2005, 70, 3464 CrossRef CAS PubMed.
- T. Kimura, K. Kawano, E. Doi, N. Kitazawa, M. Takaishi, K. Ito, T. Kaneko, T. Sasaki, N. Sato, T. Miyagawa and H. Hagiwara, U.S. Pat. Appl. Publ, US 20070117798 A1 20070524, 2007 Search PubMed.
- E. Alacid and C. Najera, Chem. Record., 2006, 6, 117–132 CrossRef CAS PubMed.
- E. Alacid and C. Najera, Adv. Synth. Catal., 2008, 350, 1316–1322 CrossRef CAS . The coupling constants of the olefin protons (J = 16.0 Hz) confirmed the formation of E-isomer.
- I. del Rio, O. Pàmies, P. W. N. M. van Leeuwen and C. Clavier, J. Organomet. Chem., 2000, 608, 115–120 CrossRef CAS.
- (a) E. Poupon, D. Francois, N. Kunesch and H.-P. Husson, J. Org. Chem., 2004, 69, 3836–3841 CrossRef CAS PubMed; (b) A. R. Katritzky, G. Qiu, B. Yang and P. J. Steel, J. Org. Chem., 1998, 63, 6699–6703 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Professor Ei-ichi Negishi on his 80th Birthday. |
| ‡ Electronic supplementary information (ESI) available: General experimental procedures and product characterization. CCDC 1045261. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00025d |
| This journal is © the Partner Organisations 2015 |