
Mechanistic studies and optimisation of a Pd-catalysed direct arylation reaction using phosphine-free systems†
Junpei
Kuwabara
*,
Masaru
Sakai
,
Qiao
Zhang
and
Takaki
Kanbara
*
Tsukuba Research Center for Interdisciplinary Materials Science (TIMS), Graduate School of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba 305-8573, Japan. E-mail: kuwabara@ims.tsukuba.ac.jp; kanbara@ims.tsukuba.ac.jp
First published on 18th March 2015
Abstract
The initial step of a Pd-catalysed phosphine-free direct arylation reaction was investigated to provide mechanistic insight, leading to optimisation of the catalytic reaction. On the basis of our observations, the amount of the Pd(OAc)2 catalyst was reduced to 1 mol%, affording a good yield of the desired product in combination with a reduction in the amount of by-products.
Introduction
Dehydrohalogenative cross-coupling reactions, so-called direct arylation reactions, have recently been recognised as efficient synthetic protocols. This is because such synthetic routes to the direct functionalisation of C–H bonds can result in a decrease in both the number of reaction steps and the amount of reaction by-products.1–4 A representative Pd-catalysed direct arylation proceeds via (i) oxidative addition of the aryl halide to Pd(0), (ii) C–H bond cleavage of the (hetero)aryl compounds and (iii) reductive elimination, resulting in C–C bond formation and regeneration of the Pd(0) species.5 This process is similar to conventional cross-coupling reactions, such as the Kumada–Tamao, Suzuki–Miyaura and Negishi reactions, however in the case of direct arylation, transmetallation of the organometallic reagent is replaced by C–H bond cleavage.6,7 As both of these reactions begin from a Pd(0) species, Pd(0) precatalysts such as Pd2(dba)3 (dba = dibenzylideneacetone) can be utilized in the reaction. In addition, Pd(II) precatalysts such as Pd(OAc)2 and PdCl2 are also common choices, owing to their high stability to air and moisture.8–10 These Pd(II) precatalysts are reduced to a catalytically active Pd(0) species by either organometallic reagents8 or phosphine ligands11–14 in the initial step of the catalytic reaction. Recently, it was reported that Pd(II) precatalysts promoted direct arylation reactions in the absence of a phosphine ligand.15–19 However, in these reactions, the pathway to the Pd(0) species from a Pd(II) precatalyst is unclear due to the absence of potential reductants, organometallic reagents and phosphine ligands. Mechanistic studies to elucidate this initial step are important for optimisation of the catalytic reaction, as mechanistic insights can potentially lead to reduced catalyst loadings and minimisation of potential side reactions. We therefore focused on mechanistic studies of the initial step of the direct arylation reaction using a phosphine-free Pd catalyst. We also report herein optimisation of the reaction conditions based on insight gained from these mechanistic studies.Results and discussion
We initially investigated the reaction of Pd(OAc)2 as a Pd(II) precatalyst in the absence of any phosphine ligands. We expected that a thiophene substrate bearing a reactive C–H bond would react with Pd(OAc)2, and thus we attempted the reaction of Pd(OAc)2 with 2,2′-bithiophene under similar conditions to the catalytic reaction.17 The reaction of Pd(OAc)2 with 5 equivalents of 2,2′-bithiophene in DMAc (dimethylacetamide) in the presence of K2CO3 at 100 °C afforded quaterthiophene in 24% isolated yield, with the formation of Pd black (Scheme 1). We expected that the formation of quaterthiophene resulted from reductive elimination from a diaryl Pd complex, which was generated by the reaction of Pd(OAc)2 with 2 equivalents of 2,2′-bithiophene (Scheme 2 and S1†).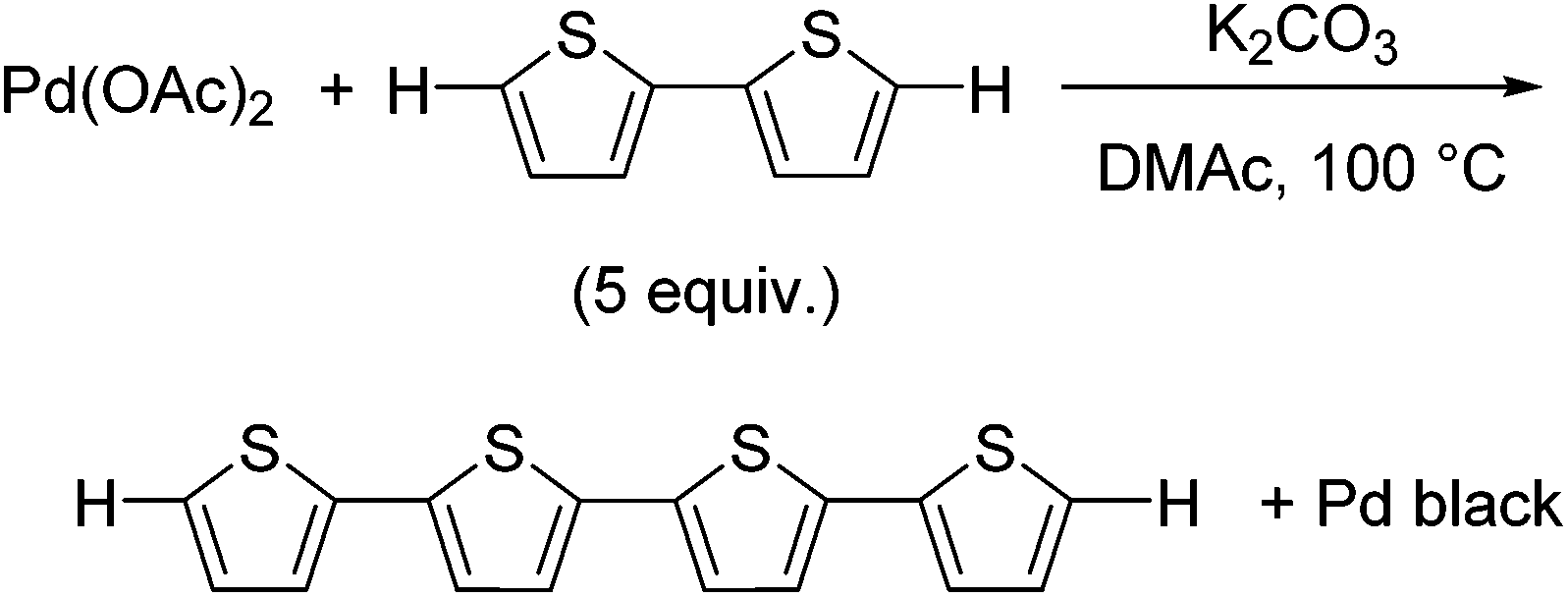 | ||
| Scheme 1 Formation of quaterthiophene. | ||
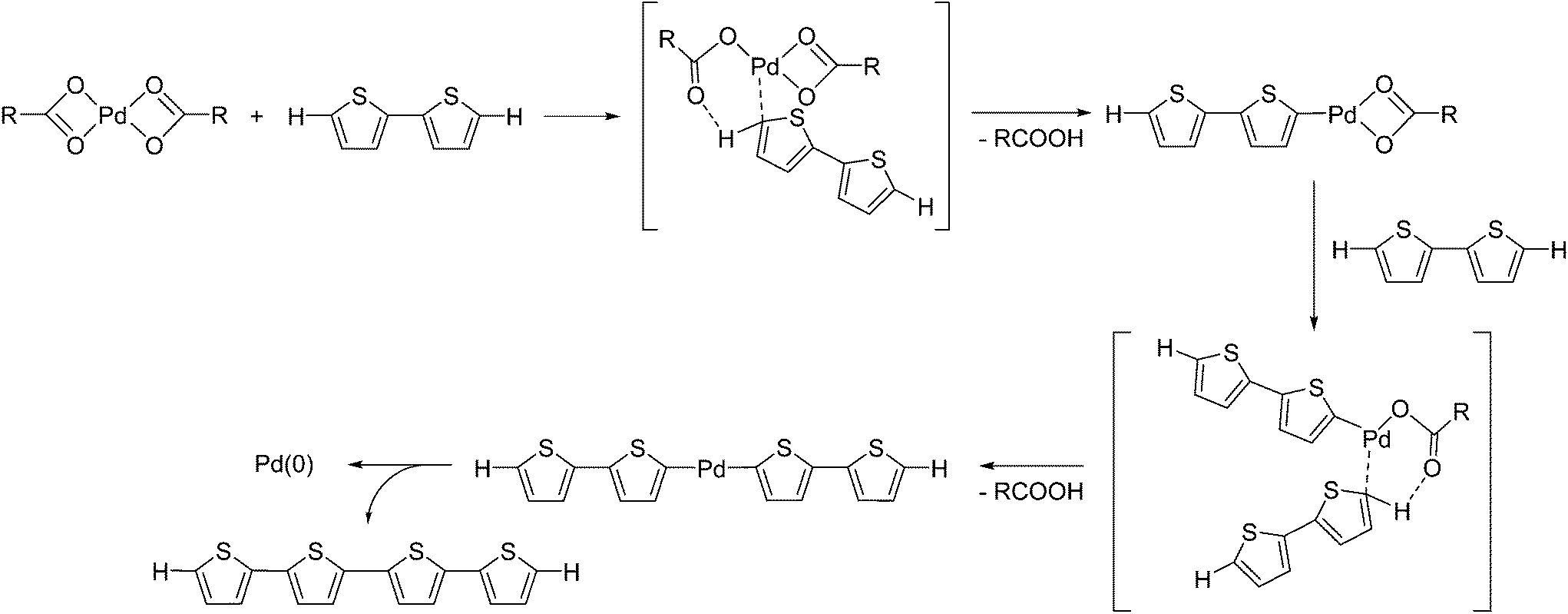 | ||
| Scheme 2 A proposed reaction mechanism for the formation of Pd(0) and quaterthiophene. | ||
Itahara reported a similar oxidative homocoupling of thiophenes with Pd(OAc)2 in stoichiometric quantities to give the corresponding dimers.20 It was proposed that the reaction of Pd(OAc)2 with 2,2′-bithiophene was expected to proceed via a concerted metallation deprotonation (CMD) pathway.21,22 Indeed, DFT calculations by Sakaki et al., revealed that the CMD pathway is indeed a reasonable mechanism for the preparation of aryl–Pd–aryl complexes from Pd(OAc)2 and a thiophene.23
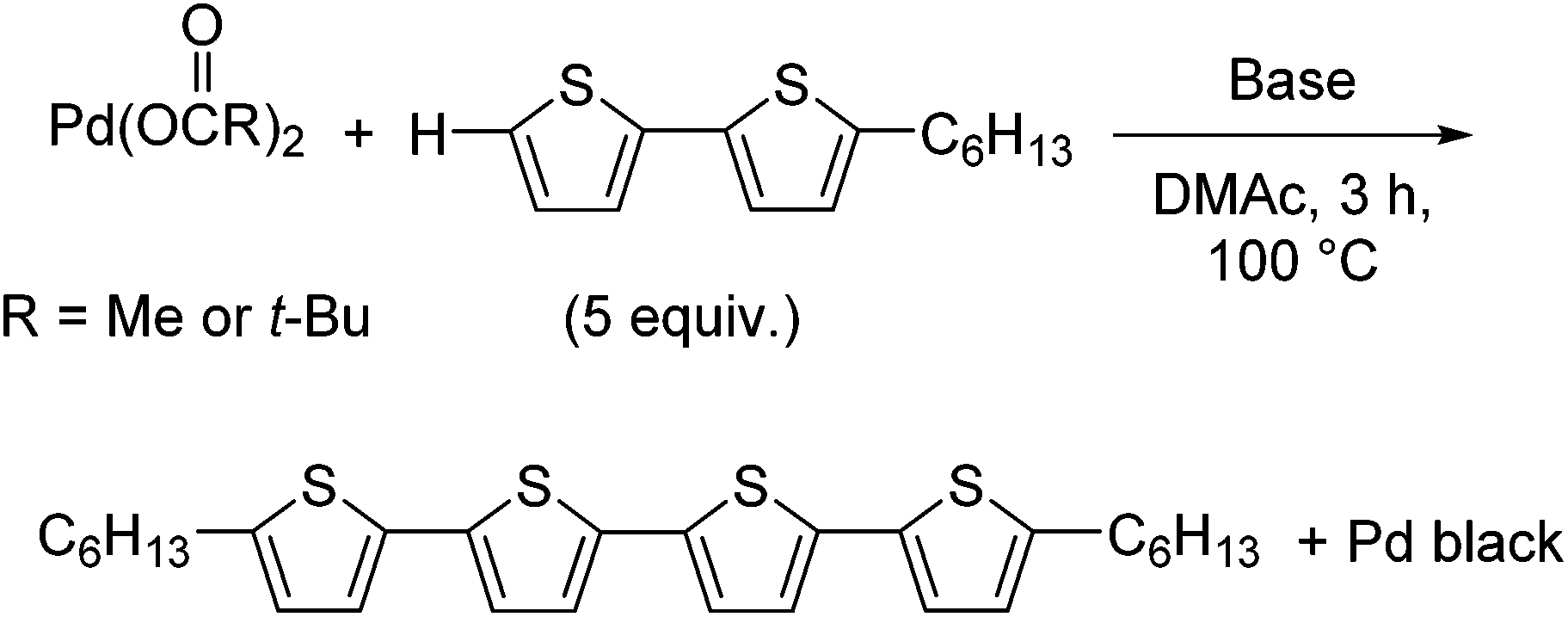
Generally, it is expected that a Pd(0) species is more likely than Pd black to be generated from Pd(II) via the oxidative homocoupling of thiophene substrates. However, as shown in Scheme 1, we observed that Pd black precipitated out of the reaction mixture instead of the expected formation of a Pd(0) complex. This was likely due to the presence of a high concentration of Pd(0) without a stabilising ligand present for complex formation. We expected that the efficiency of quaterthiophene formation would relate to the efficiency of Pd(0) formation, and thus in order to explore suitable conditions for the formation of Pd(0) complexes, the amount of quaterthiophene formed in the reaction was used as an indicator for Pd(0) formation. For the purpose of our investigation, 5-hexyl-2,2′-bithiophene was chosen as the substrate instead of 2,2′-bithiophene (Table 1), as the reaction of 2,2′-bithiophene resulted in the formation of a mixture of oligomers, formed by further coupling reactions of the product. Under the same conditions as reported in Scheme 1, 5,5‴-dihexylquaterthiophene was thus obtained in 43% yield (Table 1, entry 1). The use of KOPiv (potassium pivalate) instead of K2CO3 increased the yield of 5,5‴-dihexylquaterthiophene to 73% (entry 2). We also found that the reaction of Pd(OPiv)2 gave a better yield (51%) than that of Pd(OAc)2 (entry 3), with the difference in reactivity being particularly obvious in the early stages of the reaction (Fig. S1†). These results suggest that the acetato ligand exchanges with the pivalate to form Pd(OPiv)2, which forms Pd(0) more efficiently than Pd(OAc)2. It has been proposed that the higher basicity of the pivalato ligand than the acetato ligand may contribute to a smooth CMD pathway.21,22 It is also well known that the presence of a pivalate source accelerates the catalytic reaction in direct arylations.24 However, it is difficult to rule out the possibility that the decomposition of DMAc may contribute to the formation of Pd(0).25 The high conversion of the bithiophene derivative in dimerization supports the theory of a strong contribution of the oxidative homocoupling of thiophene substrates. In the reaction of a monothiophene derivative, namely 2-butylthiophene, bearing a less-reactive C–H bond compared to a bithiophene derivative,26 the corresponding dimer was formed in only 24% yield (Scheme S2†). This result suggests that the direct arylation of a substrate with poor reactivity tends to lower the efficiency of the formation of catalytically active Pd(0).
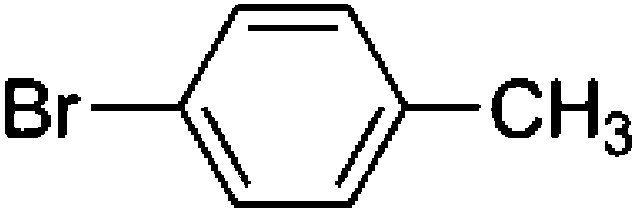
To confirm that the generated Pd(0) species serves as the catalytically active species, the reaction in the presence of a coupling partner, 1-bromo-4-octylbenzene, was conducted (Scheme 3). In the presence of K2CO3, the sub-stoichiometric reaction of Pd(OPiv)2, 2-butylthiophene and 1-bromo-4-octylbenzene yielded 5,5′-dibutyl-2,2′-bithiophene as a homocoupling product (15% based on Pd) and 2-butyl-5-(4-octylphenyl)thiophene as a cross-coupling product (14% based on Pd) (Scheme 3). This result supports the hypothesis that the oxidative homocoupling reaction of the thiophene substrate affords a Pd(0) species, which serves as a catalyst in the cross-coupling reaction. In addition, the formation of 4,4′-dioctyl-1,1′-biphenyl (12% based on Pd) was also observed as the other homocoupling product (Fig. S2†). This product is expected to be formed via the disproportionation of the mono-aryl-Pd species formed after the oxidative addition (Scheme S3†).28,29 Ozawa and Hartwig reported the formation of biphenyl derivatives from isolated mono-aryl-Pd complexes, via disproportionation.16,27 Although these biphenyl derivatives are generally formed only as minor products in coupling reactions, the reaction outlined in Scheme 3 afforded 4,4′-dioctyl-1,1′-biphenyl in notable quantities (12%). As the ratio of the Pd species to the thiophene substrate in the stoichiometric reaction is much higher than that in the catalytic reaction, disproportionation between the Pd species competes with the reaction between the Pd species and the thiophene substrate. In order to suppress the undesired reaction, the Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :thiophene ratio should be kept as low as possible.
:thiophene ratio should be kept as low as possible.
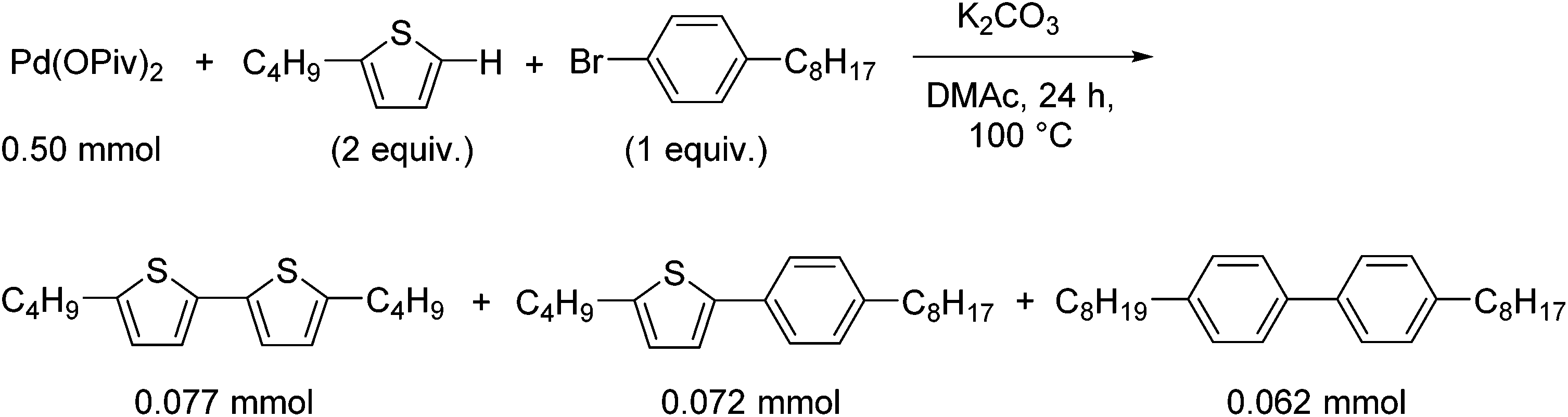 | ||
| Scheme 3 Sub-stoichiometric reaction. | ||
The mechanistic studies thus provided three main insights: (1) the pivalato ligand is effective in the formation of Pd(0); (2) the thiophene substrate is consumed in the initial step of the catalytic reaction; and (3) the potential side reaction via disproportionation of a mono-aryl-Pd species must be suppressed by keeping the Pd:thiophene ratio as low as possible. Based on these observations, optimisation of the catalytic reaction was conducted. In the model reaction, Pd(OAc)2 and Pd(OPiv)2 were used as precatalysts. In addition, KOPiv, or a combination of K2CO3 and PivOH, were tested as the pivalate source (Table 2). We observed that the use of KOPiv tends to result in lower yields (entries 1 and 3). With a combination of K2CO3 and PivOH, the use of different precatalysts did not significantly affect the reaction outcome (entries 2 and 4).
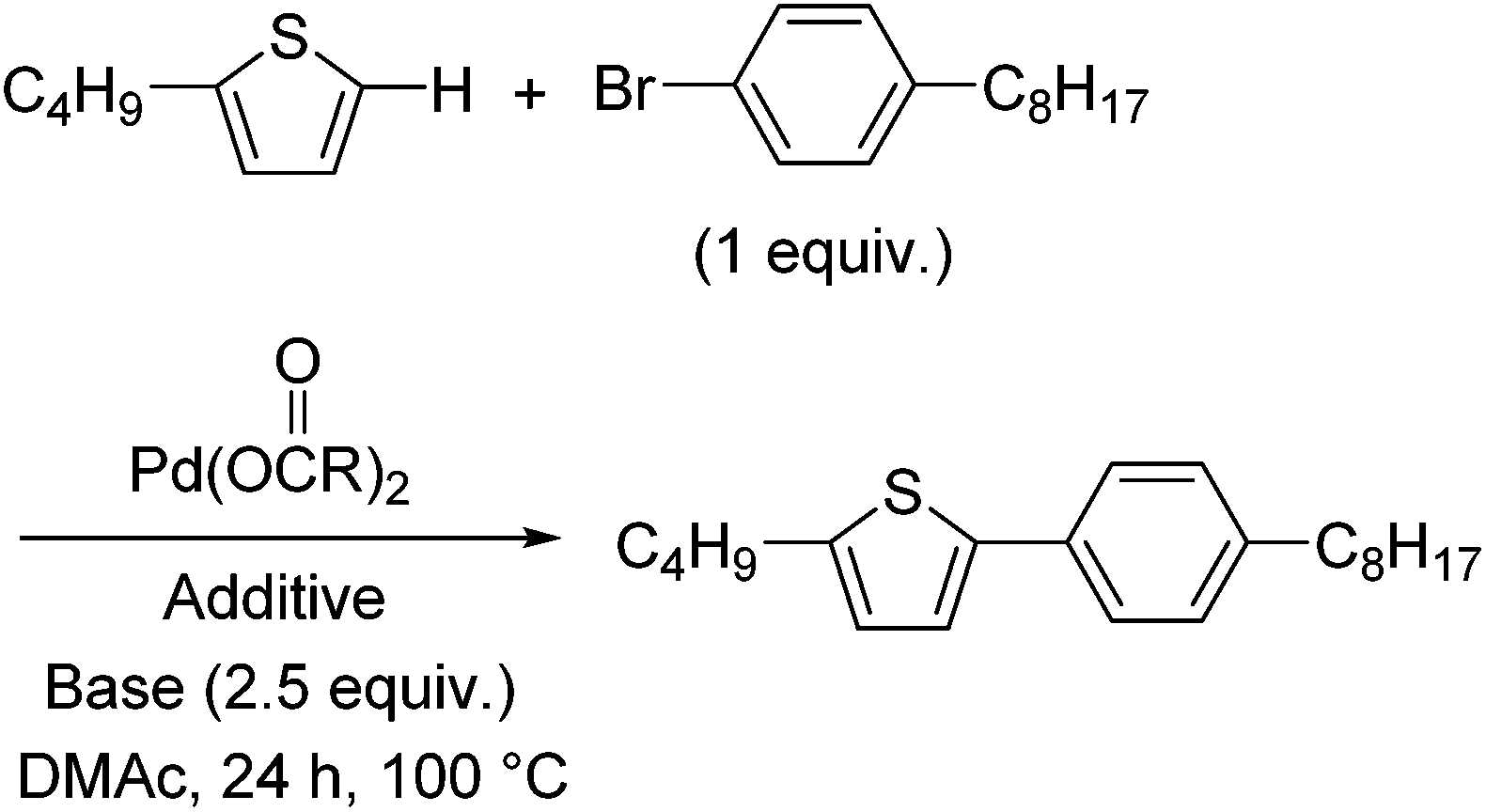
Fig. 1 shows a plot of reaction progress versus time for the four reactions, with the results taken from 1H NMR spectra recorded at various points during the reaction (Fig. S3†). In addition to the desired coupling product, 5,5′-dibutyl-2,2′-bithiophene and 4,4′-dioctyl-1,1′-biphenyl were also observed as side products in the crude mixture (Fig. 2), which was consistent with the result of the sub-stoichiometric reaction in Scheme 3. Since the mechanistic study showed a smooth Pd(0) formation from Pd(OPiv)2, the loading of Pd(OPiv)2 was expected to drop. Indeed, the reduced loading of the Pd catalyst was beneficial in suppressing the consumption of the thiophene substrate, and the disproportionation of the Pd species. Fig. 3 shows a plot of reaction progress vs. time for the reactions with either 2 mol% or 1 mol% of Pd(OPiv)2, where it can be seen that both reactions followed a similar course. These observations demonstrate that the use of 2 mol% of a Pd precatalyst was not required for the reaction to go to completion (i.e. the Pd precatalyst was present in excess).
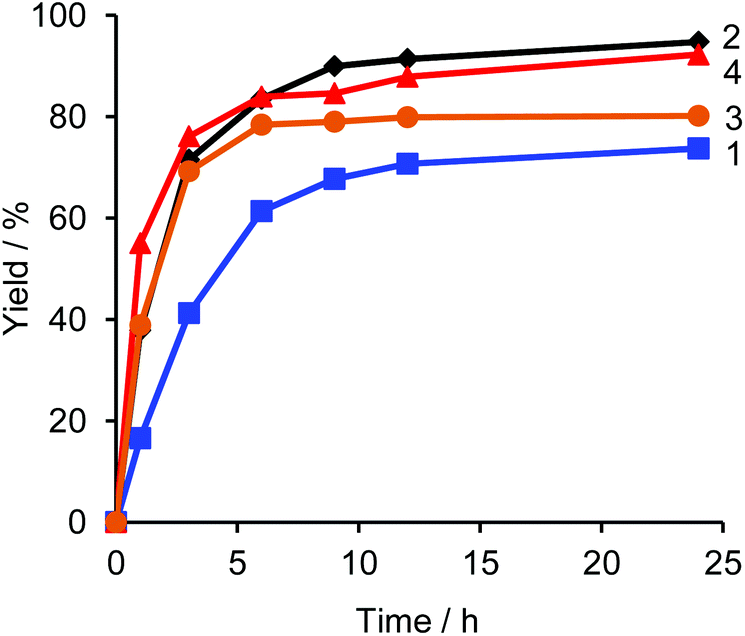 | ||
| Fig. 1 Time courses for the yields of catalytic reactions under the conditions in Table 2. | ||
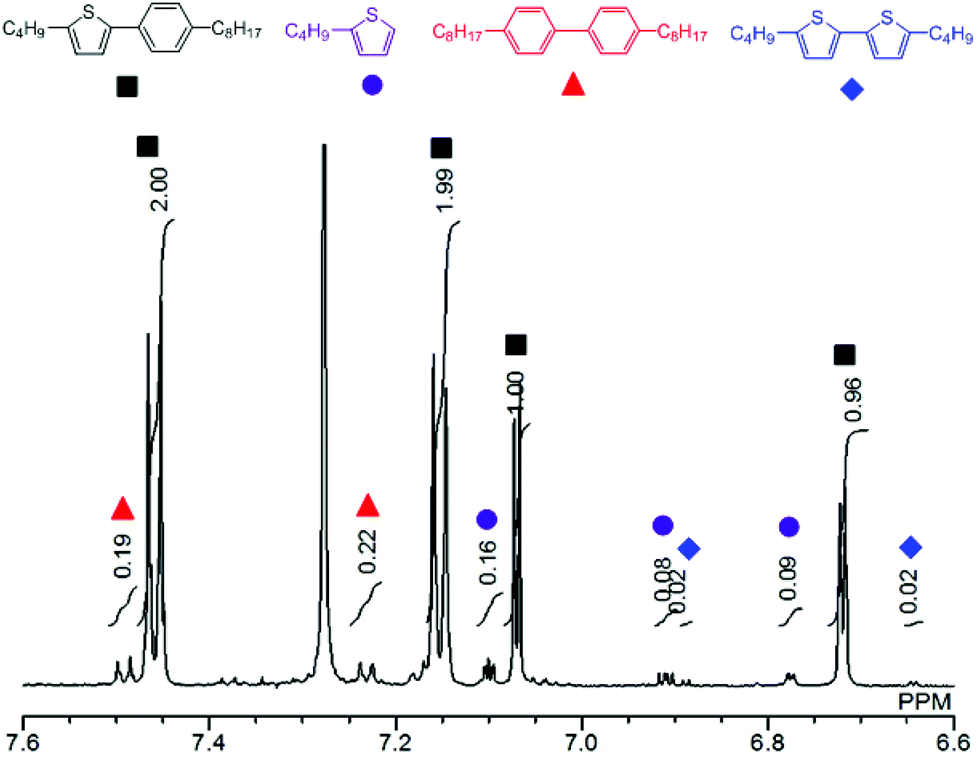 | ||
| Fig. 2 1H NMR spectrum of the reaction mixture at 24 h (Table 2, entry 4). | ||
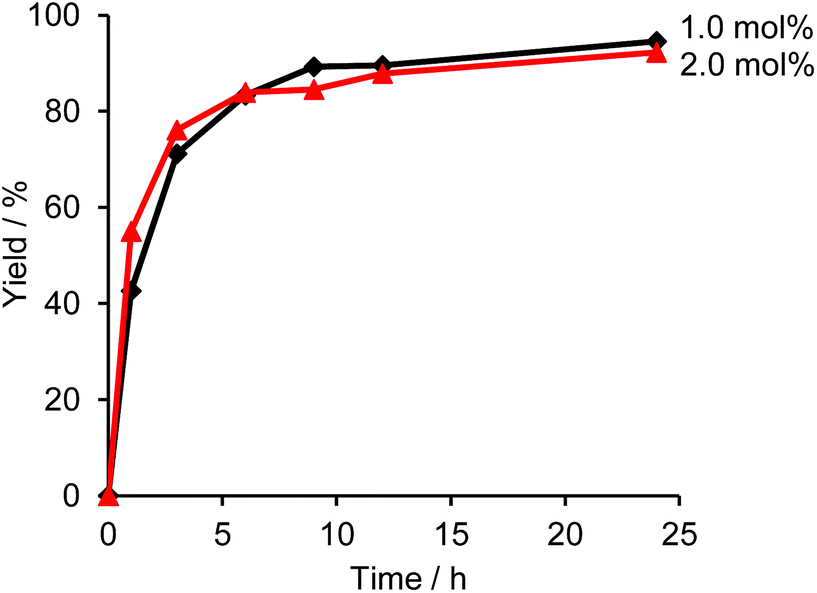 | ||
| Fig. 3 Time courses for the yields of the reactions with 2.0 mol% and 1.0 mol% of Pd(OPiv)2. The other reaction conditions are shown in Table 2. | ||
In addition, the reaction using 1 mol% of the Pd precatalyst resulted in the formation of a lower amount of 4,4′-dioctyl-1,1′-biphenyl by-product than when 2 mol% of the precatalyst was used (Fig. S4†). Thus, we propose that a lower concentration of the Pd catalyst in the reaction media is likely to suppress the bimolecular disproportionation reaction. We also attempted the reaction using lower amounts of the Pd precatalyst (0.5 mol% and 0.25 mol%) to determine if the Pd catalyst concentration could be lowered further. However, we observed that both reactions using less than 1 mol% of the catalyst afforded lower reaction yields, as can be seen in Fig. S5.† We could therefore conclude from these results that the optimal amount of Pd precatalysts for the reaction is 1 mol%, which is also consistent with the previously reported results.18,19
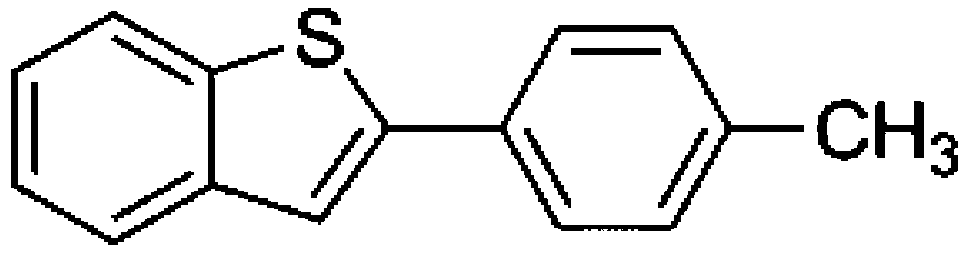
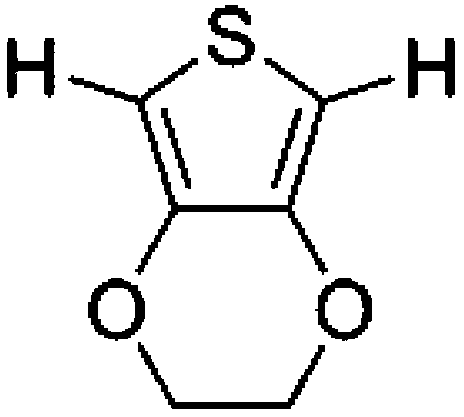
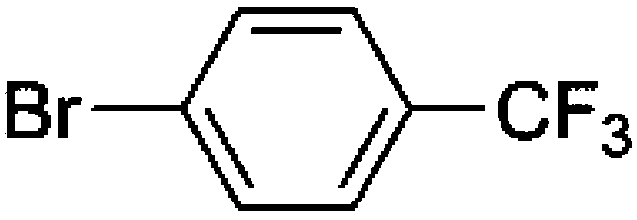
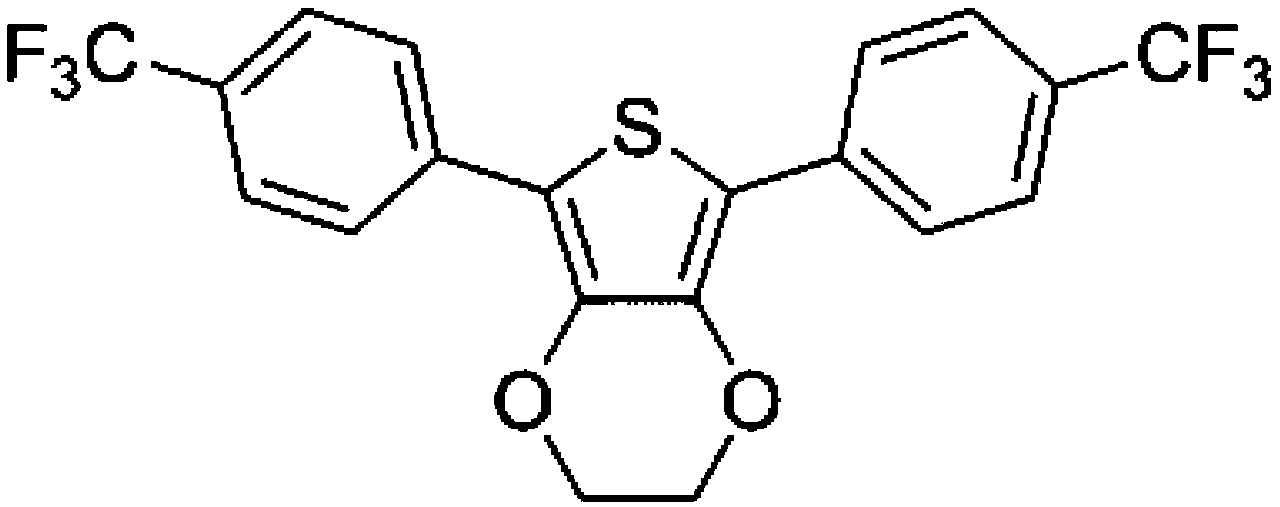
To confirm usability of the optimised conditions, direct arylation reactions of heteroarenes were conducted using 1.0 mol% of Pd(OPiv)2 (Table 3). The reaction of a thiazole derivative afforded the coupling product in 92% isolated yield (entry 1). It should be noted that the high yield was achieved from the reaction with a 1:1 ratio of the substrates; an excess amount of the substrate was not needed because consumption of the substrates was minimized by decreasing the catalyst loading. The reaction of benzothiophene also afforded a good yield (entry 2). In the reaction of 3,4-ethylenedioxythiophene with two equivalents of 1-bromo-4-trifluoromethylbenzene, the diarylated product was obtained in 97% yield (entry 3). The yield was higher than that of the reported reaction using 5 mol% of a Pd precatalyst with 10 mol% of a phosphine ligand.30 These results show that the decreased amount of the Pd precatalyst without the addition of a phosphine ligand can effectively promote the direct arylation reaction of heteroarenes under the optimised conditions.
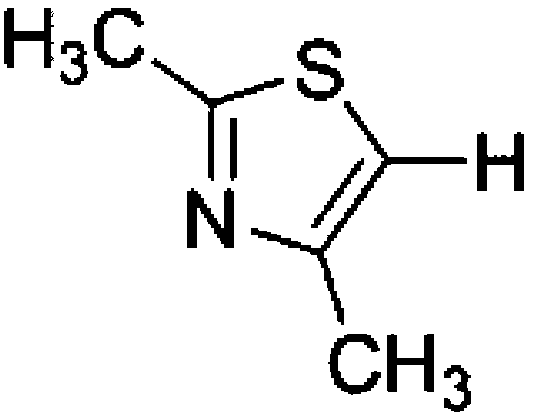
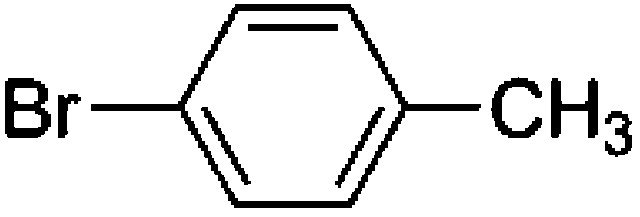
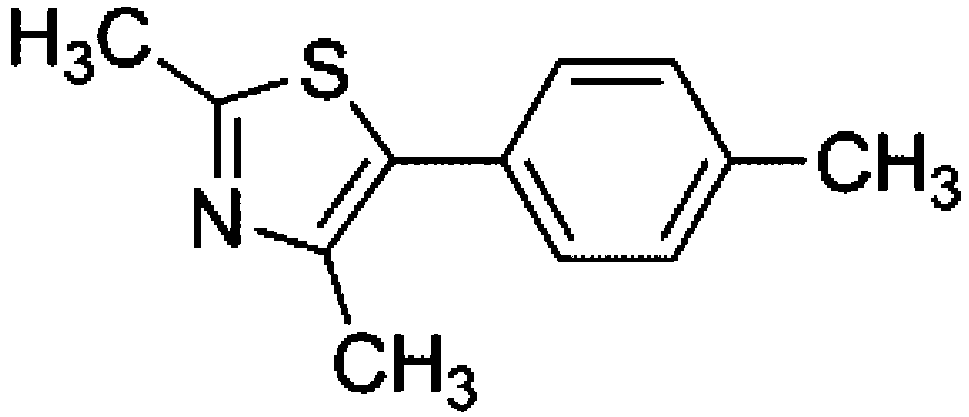
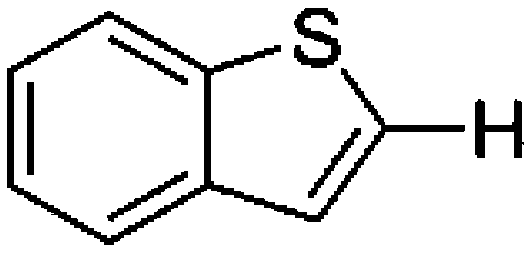
Conclusions
In the Pd-catalysed direct arylation reaction, in the absence of a phosphine ligand, an oxidative homocoupling reaction of a thiophene substrate contributes to the formation of a catalytically active Pd(0) species from a Pd(II) precatalyst. A biaryl compound was observed to form as a side product via disproportionation of the Pd intermediate. As the consumption of the thiophene substrate and disproportionation of the Pd intermediate depends on the amount of a Pd(II) precatalyst present in the reaction mixture, the use of a minimal amount of the Pd precatalyst is the key to achieving a high selectivity and yield of the desired coupling product. These findings show that a lager amount of a catalyst does not always afford better results. Optimisation of the reaction conditions using a reduced amount of the Pd precatalyst (1 mol%) resulted in a successful reaction affording the desired product in good yield and good selectivity. These insights for high conversion and selectivity are particularly important when the direct arylation reaction was utilized in polycondensation to afford high-molecular-weight polymers without structural defects.31,32Experimental
Synthesis of 2-butyl-5-(4-octylphenyl)thiophene
A mixture of Pd(OAc)2 (4.4 mg, 0.020 mmol), pivalic acid (34 μL, 0.30 mmol), 2-butylthiophene (147 μL, 1.0 mmol), 1-bromo-4-octylbenzene (235 μL, 1.0 mmol), and K2CO3 (345 mg, 2.5 mmol) was stirred in anhydrous DMAc (4.0 mL) for 24 h at 100 °C under a nitrogen atmosphere. The reaction mixture was cooled to room temperature and diluted with CHCl3. The organic phase was washed with water and brine, and dried over Na2SO4. The product was isolated by column chromatography on silica gel using a mixture of CHCl3 and hexane (1:7) as an eluent. The solvents were removed in vacuo to give 2-butyl-5-(4-octylphenyl)thiophene (261 mg, 80%). 1H NMR (400 MHz, CDCl3): 7.50 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 7.10 (d, J = 3.6 Hz, 1H), 6.75 (d, J = 3.2 Hz, 1H), 2.84 (t, J = 7.6 Hz, 2H), 2.63 (t, J = 7.6 Hz, 2H), 1.76–1.69 (m, 2H), 1.68–1.60 (m, 2H), 1.50–1.40 (m, 2H), 1.38–1.28 (m, 10H), 0.98 (t, J = 7.6 Hz, 3H), 0.92 (t, J = 6.8 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): 145.5, 142.3, 142.3, 132.7, 129.3, 125.9, 125.3, 122.6, 36.1, 34.2, 32.4, 31.9, 30.4, 30.0, 29.8, 29.7, 23.1, 22.7, 14.6, 14.3. GC-MS: m/z = 328 (Calcd for [M]+: 328). Elemental analysis: calculated for C22H32S: C, 80.42%; H, 9.81%; Found: C: 80.14%, H, 9.88%.
Acknowledgements
This work was supported by Industrial Technology Research Grant Program in 2011 from New Energy and Industrial Technology Development Organization (NEDO) of Japan, and partly supported by Grant-in-Aid for Young Scientists (B) (25810070), Challenging Exploratory Research (25620094) and Scientific Research (B) (25288052). The authors thank the Chemical Analysis Center of University of Tsukuba for measurements of NMR spectroscopy and elemental analysis.Notes and references
- T. Satoh and M. Miura, Chem. Lett., 2007, 36, 200 CrossRef CAS.
- L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed.
- R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, 17 CrossRef CAS.
- Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66 CrossRef CAS PubMed.
- L.-C. Campeau, D. R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35 CAS.
- D. Zhao, J. You and C. Hu, Chem. – Eur. J., 2011, 17, 5466 CrossRef CAS PubMed.
- S. Xu, E. H. Kim, A. Wei and E. Negishi, Sci. Technol. Adv. Mater., 2014, 15, 044201 CrossRef.
- J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, 2010 Search PubMed.
- E. Negishi, Bull. Chem. Soc. Jpn., 2007, 80, 233 CrossRef CAS.
- A. Suzuki and Y. Yamamoto, Chem. Lett., 2011, 40, 894 CrossRef CAS.
- F. Ozawa, A. Kubo and T. Hayashi, Chem. Lett., 1992, 21, 2177 CrossRef.
- C. Amatore, A. Jutand and M. A. M′Barki, Organometallics, 1992, 11, 3009 CrossRef CAS.
- T. Mandai, T. Matsumoto and J. Tsuji, Tetrahedron Lett., 1993, 34, 2513 CrossRef CAS.
- C. S. Wei, G. H. M. Davies, O. Soltani, J. Albrecht, Q. Gao, C. Pathirana, Y. Hsiao, S. Tummala and M. D. Eastgate, Angew. Chem., Int. Ed., 2013, 52, 5822 CrossRef CAS PubMed.
- F. Požgan, J. Roger and H. Doucet, ChemSusChem, 2008, 1, 404 CrossRef PubMed.
- Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 3308 CrossRef CAS PubMed.
- Y. Fujinami, J. Kuwabara, W. Lu, H. Hayashi and T. Kanbara, ACS Macro Lett., 2012, 1, 67 CrossRef CAS.
- K. Yamazaki, J. Kuwabara and T. Kanbara, Macromol. Rapid Commun., 2012, 34, 69 CrossRef PubMed.
- S. J. Choi, J. Kuwabara and T. Kanbara, ACS Sustainable Chem. Eng., 2013, 1, 878 CrossRef CAS.
- T. Itahara, M. Hashimoto and H. Yumisashi, Synthesis, 1984, 255 CrossRef CAS.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef.
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- B. Biswas, M. Sugimoto and S. Sakaki, Organometallics, 2000, 19, 3895 CrossRef CAS.
- B. Liégault, D. Lapointe, L. Caron, A. Vlassova and K. Fagnou, J. Org. Chem., 2009, 74, 1826 CrossRef PubMed.
- J. A. Molina de la Torre, P. Espinet and A. C. Albéniz, Organometallics, 2013, 32, 5428 CrossRef CAS.
- M. Wakioka, Y. Nakamura, Y. Hihara, F. Ozawa and S. Sakaki, Organometallics, 2013, 32, 4423 CrossRef CAS.
- M. Wakioka, Y. Nakamura, Q. Wang and F. Ozawa, Organometallics, 2012, 31, 4810 CrossRef CAS.
- K. Osakada and T. Yamamoto, Coord. Chem. Rev., 2000, 198, 379 CrossRef CAS.
- Y. Suzaki, T. Yagyu and K. Osakada, J. Organomet. Chem., 2007, 692, 326 CrossRef CAS PubMed.
- C.-Y. Liu, H. Zhao and H. Yu, Org. Lett., 2011, 13, 4068 CrossRef CAS PubMed.
- F. Lombeck, H. Komber, S. I. Gorelsky and M. Sommer, ACS Macro Lett., 2014, 3, 819 CrossRef CAS.
- S. Kowalski, S. Allard and U. Scherf, Macromol. Rapid Commun., 2014 DOI:10.1002/marc.201400557.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00351a |
| This journal is © the Partner Organisations 2015 |