
Selective monoalkylation of amines with light electrophiles using a flow microreactor system†
Thomas
Lebleu
,
Jacques
Maddaluno
and
Julien
Legros
*
Normandie Univ, COBRA UMR 6014, Univ Rouen, INSA Rouen and CNRS, 1 rue Lucien Tesnière, 76821 Mont-Saint-Aignan, France. E-mail: julien.legros@univ-rouen.fr
First published on 3rd February 2015
Abstract
The challenging direct monoalkylation of amines with light electrophilic reagents (C1 to C3) was performed through a flow microreactor approach. The efficiency of mixing coupled with short residence times (tR = 0.7–62 s) allows the transfer of a single alkyl group from R-OTf onto primary amines (R = Et, Pr) as well as on secondary amines (R = Me, Et, Pr, allyl, propargyl) with good selectivities.
The occurrence of several competitive transformations is a phenomenon frequently met in organic synthesis.1,2 In this line, the deceptively simple preparation of secondary – or even tertiary – amines by direct monoalkylation with electrophilic reagents, such as alkyl halides, often fails due to competitive consecutive overalkylation processes, even if a single equivalent of electrophile is used.3 While this phenomenon is less prominent with bulky electrophiles,3–7 monoalkylation with light primary alkyl chains (i.e. C1 to C3) remains extremely challenging and reports of this reaction with groups shorter than n-butyl are scarce.8,9 In this context, we recently disclosed that primary amines reacted with MeOTf to transfer a single CH3 group with good to high selectivity by simply using hexafluoroisopropanol (HFIP) as a solvent.10 The success of this method was ascribed to the specific H-bonding properties of HFIP11–14 that interacts with monoalkylated amines, thus reducing overalkylation. Unfortunately, HFIP is a rather expensive solvent and this alkylation process turns out to be less efficient with EtOTf, hampering wider applications and use on large scales. In this context, we now describe a flow microreactor approach to perform selective monoalkylation of primary and secondary amines with small alkyl triflates (Scheme 1).
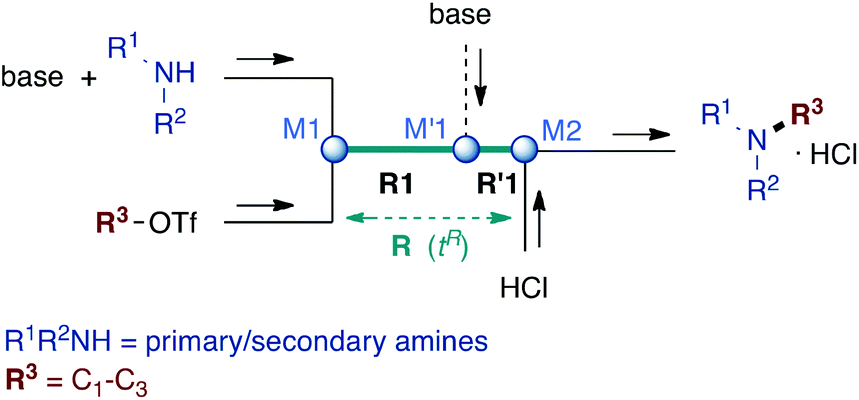 | ||
| Scheme 1 A flow microreactor system for selective monoalkylation of primary and secondary amines. | ||
Microflow technology has recently emerged as an outstanding tool to circumvent the problem of disguised chemical selectivity in competitive consecutive reactions.15–20 By virtue of excellent mixing and very precise control of reaction time in microreactors (residence time tR), highly reactive compounds can be generated in such systems and trapped before further adverse reaction occurs.21–31 Therefore, a flow microreactor approach to perform N-alkylation of primary amines with small alkyl chains is expected to ensure reaction control at the first alkylation step before overalkylation proceeds. For this purpose, we implemented a microflow system composed of two PEEK T-shaped micromixers M1 and M2 (V = 58 nL) and a microtube reactor R (inner diameter ∅ = 0.5 mm), the reagents being introduced via syringe pumps, as depicted in Scheme 1.
First experiments consisted of reacting benzylamine and EtOTf in nitromethane7 in the microreactor R and stopping the reaction with 6 N HCl in M2 at various residence times (tR) and temperatures to determine optimal conditions in terms of conversion/selectivity towards monoethylated product BnNHEt (Table 1). First reactions were carried out at 20 °C (entries 1 and 2). Thus, at tR = 1 s less than 50% conversion was reached but with a promising 84% selectivity toward the desired BnNHEt (BnNHEt = 41%, BnNEt2 = 8% yield; entry 1). At longer reaction time (tR = 20 s), transformation reaches its maximum (80% conversion) albeit with lower selectivity (76%; entry 2). Performing the reaction at higher temperatures allowed faster conversion and higher selectivity: at 80 °C, 62% of BnNHEt was obtained along with 15% of BnNEt2 (81% selectivity) within 0.5 s (entry 3). In contrast, halving the flow (tR = 1 s) produced the same quantity of undesired BnNEt2 (15%) whereas only 55% of the desired product was afforded, highlighting the importance of fast mixing for high selectivity (entry 4). Raising the temperature to 95 °C afforded slightly lower results, probably due to mixing disruption at this temperature close to the boiling point of the solvent (bp(MeNO2) = 100 °C; entry 5).
| Entry | Base | Temp. (°C) | t R (s) | Ratioc (%) | |
|---|---|---|---|---|---|
| BnNHEt | BnNEt2 | ||||
| a Microflow system according to Scheme 1: micromixers M (V = 58 nL), microtube reactor R (ID = 0.5 mm); see ESI for details. b Conditions: BnNH2 (0.8 mmol), EtOTf (1.2 mmol), base (0.8 mmol) in MeNO2, 6 N aq. HCl (1 mL). c Measured by 1H NMR spectroscopy. d Base (0.2 mmol) at M1 and additional base (0.6 mmol) added at M′1. | |||||
| 1 | — | 20 | 1 | 41 | 8 |
| 2 | — | 20 | 20 | 61 | 19 |
| 3 | — | 80 | 0.5 | 62 | 15 |
| 4 | — | 80 | 1 | 55 | 15 |
| 5 | — | 95 | 0.2 | 61 | 17 |
| 6 | DIPEA | 80 | 0.7 | 54 | 33 |
| 7 | DBU | 80 | 0.9 | 55 | 25 |
| 8 | DABCO | 80 | 1.1 | 57 | 22 |
| 9 | 2,6-Lutidine | 80 | 0.7 | 56 | 16 |
| 10 | 2,6-Lutidine | 80 | 0.7 | 67 | 16 |
| 11d | 2,6-Di-tert-butyl-4-methylpyridine | 80 | 0.9 | 63 | 16 |
| 12d | Proton sponge® | 80 | 0.7 | 41 | 54 |
In order to reach higher conversions, the effect of an additional base mixed with BnNH2 was assessed under the above optimized conditions (tR = 80 °C). The choice of bases depends on their solubility in the solvent as well as on their basicity/nucleophilicity in order to avoid proton transfer toward benzylamine products and competitive trapping of the alkylating agent. For these reasons, various tertiary amines have been used, and best results were obtained at tR ≤ 1.1 s (entries 6–12). Thus, with 1 equivalent of Hünig's base conversion improved (87%) but at the expense of selectivity (62%; entry 6). The use of tertiary alicyclic amines, DBU and DABCO, did not afford better results (ca. 55% BnNHEt along with >22% BnNEt2; entries 7 and 8). Sterically hindered pyridines were then evaluated (entries 9–12). Results with 2,6-lutidine were disappointing since only moderate yield and selectivity for the monoethyl amine were obtained (56% yield; entry 9). However, using an additional micromixer M′1 to introduce a part of 2,6-lutidine in a separate step (0.25 eq. with BnNH2 at M1 and 0.75 eq. through M′1 as depicted in Scheme 1) led to significant improvements: 67% of BnNHEt were obtained within 0.7 s, accompanied by 16% of BnNEt2 (81% selectivity; entry 10). The evaluation of other bases under these two-step addition conditions did not bring improvements: 2,6-di-tert-butyl-4-methylpyridine gave slightly lower results whereas Proton sponge® was shown to favour the overalkylated product (95% conversion yielding 54% of BnNEt2; entry 12). It is worth noting that all these experiments are associated with short residence times that completely avoid the formation of quaternary ammonium salts.
In order to determine the scope and limitation of these conditions (amine and R-OTf in MeNO2 reacted in a microreactor at 80 °C, with 2,6-lutidine added in two steps) a variety of primary and secondary amines were reacted with various triflates as electrophilic partners. However, according to the nature of the substrate/triflate, erosion and/or clogging of the microreactor occurred. Therefore, the composition and size of the micro-mixers and -reactors were modified so as to provide a system as universal as possible. Thus, larger stainless steel micromixers M1, M′1 and M2 (V = 570 nL) and tubing (∅ = 0.762 mm) were retained (Table 2). Under these new conditions, N-ethylbenzylamine was obtained with the same 67% yield within 9.3 s (entry 1).‡ These conditions were successfully applied to other benzylamines as well as to anilines (62–69%; entries 2–5). Propylation was also achieved with the highest yield in monoadduct at longer tR = 15 s and 31 s, to afford respectively N-propyl-benzylamine (64%; entry 6) and N-propyl-2,6-aniline (62%; entry 7). Then, alkylation of secondary amines was also studied (entries 8–16). The above flow microreactor conditions allowed successful N-ethylation and N-propylation of dibenzylamine and N-benzylaniline to afford the corresponding tertiary amines (64–95%; entries 8–11). The easy introduction of the versatile allyl and propargyl groups was also evidenced (entries 12–14): allylation gave good yields of the expected products (81% and 85%; entries 12 and 13) whereas propargylation was not as successful due to the instability of propargyl triflate (34%, entry 14). Although the very challenging N-methylation of primary amines was ineffective in a microreactor, the transfer of a single methyl group to secondary amines could however be successfully achieved.§ Thus, N-methyl-dibenzylaniline and N-methyl-N-benzylaniline were obtained in good yields (64% and 75%, respectively; entries 15 and 16).
| Entry | t R (s) | Product | % Ratioc (% yield) |
|---|---|---|---|
| a Microflow system: micromixers M1, M′1 and M2 (V = 570 nL), microtube reactor R (ID = 0.762 mm); total flow rate at M2: 2.83 mL min−1. b Conditions: BnNH2 (0.8 mmol), R-OTf (1.2 mmol), base (0.2 then 0.6 mmol) in MeNO2, 6 N aq. HCl (1 mL). c Measured by 1H NMR spectroscopy. d Microflow system: micromixers M1, M′1 and M2 (V = 58 nL), microtube reactor R (ID = 0.5 mm); total flow rate at M2: 5.66 mL min−1. | |||
| 1 | 9.3 |
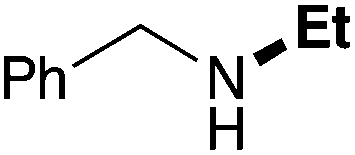
|
67 |
| 2 | 9.3 |
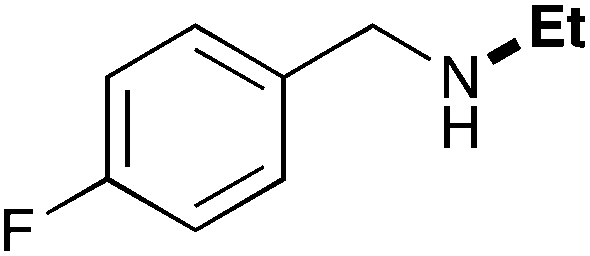
|
65 |
| 3 | 9.3 |
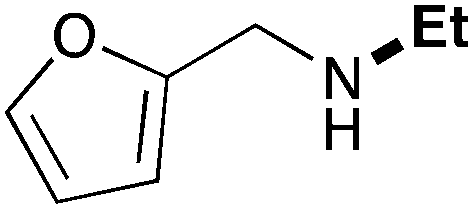
|
62 |
| 4 | 9.3 |
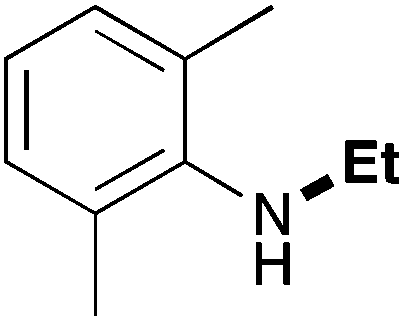
|
69 |
| 5 | 9.3 |
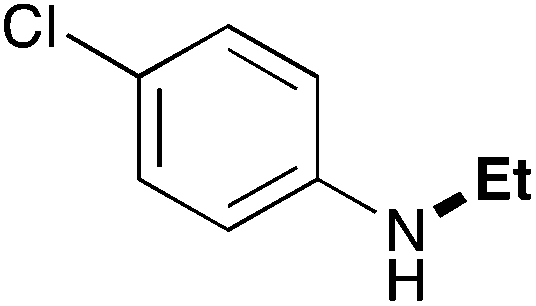
|
64 |
| 6 | 15 |
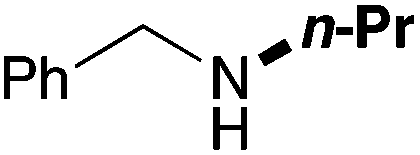
|
64 |
| 7 | 31 |

|
60 |
| 8 | 18.7 |
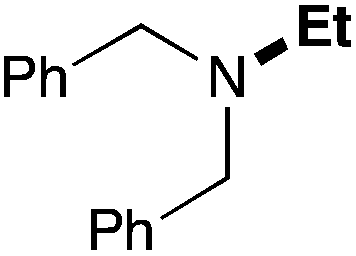
|
95 (84) |
| 9 | 38.8 |

|
89 (79) |
| 10 | 18.7 |
|
84 (80) |
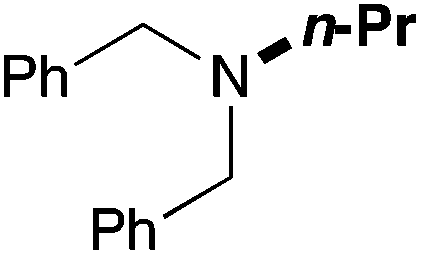
| 11 | 62 |
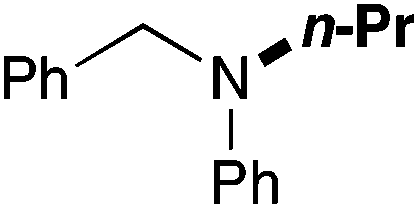
|
75 (60) |
| 12 | 18.7 |
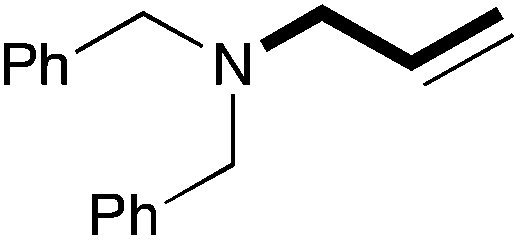
|
81 (70) |
| 13 | 62 |
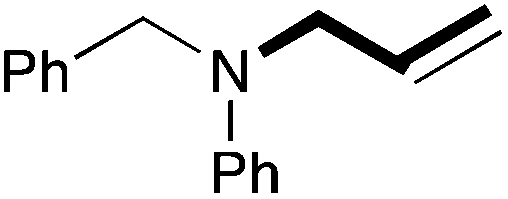
|
85 (76) |
| 14 | 18.7 |
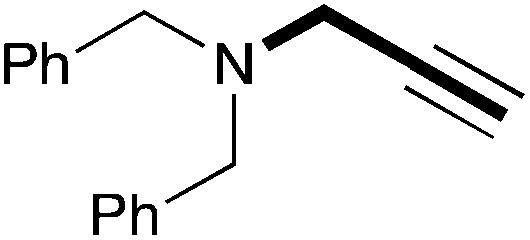
|
(34) |
| 15 | 9.6 |
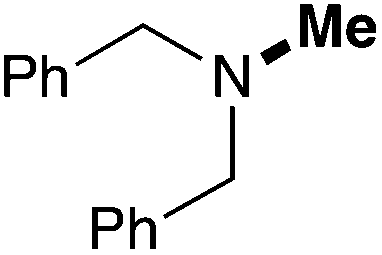
|
64 (52) |
| 16d | 1 |
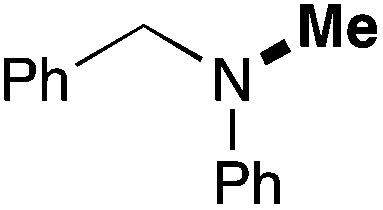
|
75 (69) |
In conclusion, we have developed an effective and reactant-efficient method to perform the challenging direct mono-N-alkylation of primary and secondary amines with small alkyl groups (C1–C3) by virtue of flow microreactor features (fast mixing and precise reaction time control). Thus, ethyl and propyl chains were efficiently transferred in a monoselective fashion onto primary amines whereas the reaction from secondary amine substrates could be extended to allyl and even methyl groups.
Acknowledgements
Prof. J.-i. Yoshida (Kyoto University) is gratefully acknowledged for helpful discussions. This project was funded by the European France (Manche) – England cross-border cooperation program INTERREG IV A “AI-CHEM CHANNEL” and co-financed by ERDF. The authors also thank the Labex SYNORG and the Région Haute-Normandie (MicroChim Project) for financial support.Notes and references
- P. Rys, Angew. Chem., Int. Ed. Engl., 1977, 16, 807–817 CrossRef
.
- P. Rys, Acc. Chem. Res., 1976, 9, 345–351 CrossRef CAS
- R. N. Salvatore, C. H. Yoon and K. W. Jung, Tetrahedron, 2001, 57, 7785–7811 CrossRef CAS
- R. N. Salvatore, A. S. Nagle, S. E. Schmidt and K. W. Jung, Org. Lett., 1999, 1, 1893–1896 CrossRef CAS
- R. N. Salvatore, S. E. Schmidt, S. I. Shin, A. S. Nagle, J. H. Worrell and K. W. Jung, Tetrahedron Lett., 2000, 41, 9705–9708 CrossRef CAS
- R. N. Salvatore, A. S. Nagle and K. W. Jung, J. Org. Chem., 2002, 67, 674–683 CrossRef CAS PubMed
- S. Bhattacharyya, U. Pathak, S. Mathur, S. Vishnoi and R. Jain, RSC Adv., 2014, 4, 18229–18233 RSC
- R. M. Yebeutchou and E. Dalcanale, J. Am. Chem. Soc., 2009, 131, 2452–2453 CrossRef CAS PubMed
- M. Selva, P. Tundo and A. Perosa, J. Org. Chem., 2001, 66, 677–680 CrossRef CAS PubMed
- T. Lebleu, X. Ma, J. Maddaluno and J. Legros, Chem. Commun., 2014, 50, 1836–1838 RSC
- C. Laurence, J. Legros, P. Nicolet, D. Vuluga, A. Chantzis and D. Jacquemin, J. Phys. Chem. B, 2014, 118, 7594–7608 CrossRef CAS PubMed
- D. Vuluga, J. Legros, B. Crousse, A. M. Z. Slawin, C. Laurence, P. Nicolet and D. Bonnet-Delpon, J. Org. Chem., 2011, 76, 1126–1133 CrossRef CAS PubMed
- A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS PubMed
- C. Laurence, J. Legros, A. Chantzis, A. Planchat and D. Jacquemin, J. Phys. Chem. B, 2015 DOI:10.1021/jp512372c
-
J. Yoshida, Flash chemistry: fast organic synthesis in microsystems, Wiley, Hoboken, N.J., 2008 Search PubMed
- K. S. Elvira, X. C. i. Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed
- J. Yoshida, A. Nagaki and T. Yamada, Chem. – Eur. J., 2008, 14, 7450–7459 CrossRef CAS PubMed
- J. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896–9904 RSC
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed
- J. Yoshida, A. Nagaki, T. Iwasaki and S. Suga, Chem. Eng. Technol., 2005, 28, 259–266 CrossRef CAS
- T. Fukuyama, M. Kobayashi, M. T. Rahman, N. Kamata and I. Ryu, Org. Lett., 2008, 10, 533–536 CrossRef CAS PubMed
- I. C. Wienhöfer, A. Studer, M. T. Rahman, T. Fukuyama and I. Ryu, Org. Lett., 2009, 11, 2457–2460 CrossRef PubMed
- F. Lévesque and P. H. Seeberger, Org. Lett., 2011, 13, 5008–5011 CrossRef PubMed
- D. Webb and T. F. Jamison, Org. Lett., 2012, 14, 568–571 CrossRef CAS PubMed
- H. Usutani, Y. Tomida, A. Nagaki, H. Okamoto, T. Nokami and J. Yoshida, J. Am. Chem. Soc., 2007, 129, 3046–3047 CrossRef CAS PubMed
- A. Nagaki, H. Kim and J. Yoshida, Angew. Chem., Int. Ed., 2009, 48, 8063–8065 CrossRef CAS PubMed
- H. Kim, A. Nagaki and J. Yoshida, Nat. Commun., 2011, 2, 264 CrossRef PubMed
- A. Nagaki, C. Matsuo, S. Kim, K. Saito, A. Miyazaki and J. Yoshida, Angew. Chem., Int. Ed., 2012, 51, 3245–3248 CrossRef CAS PubMed
- J. Wu, X. Yang, Z. He, X. Mao, T. A. Hatton and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 8416–8420 CrossRef CAS PubMed
- A. Nagaki, D. Ichinari and J. Yoshida, J. Am. Chem. Soc., 2014, 136, 12245–12248 CrossRef CAS PubMed
- A. Nagaki, D. Ichinari and J. Yoshida, Chem. Commun., 2013, 49, 3242 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and analytical data. See DOI: 10.1039/c4qo00342j |
| ‡ Typical procedure for the ethylation of benzylamine (all syringes were filled with the reagents and the corresponding quantity of MeNO2 to obtain a total volume of 1 mL). Syringe 1 (S1, 1 mL) was filled with benzylamine (88 μL, 0.8 mmol), 2,6-lutidine (18.5 μL, 0.2 mmol) in MeNO2. Syringe 2 (S2, 1 mL) was filled with EtOTf (164 μL, 1.2 mmol) in MeNO2. Syringe 3 (S3, 1 mL) was filled with 2,6-lutidine (74.1 μL, 0.6 mmol) and MeNO2. Syringe 4 (S4, 1 mL) contained a solution of 6 N aq. HCl. Micromixers (M) and microreactors (R) were immersed in a hot bath at 80 °C. Solutions in S1 and S2 were introduced into M1 (V = 570 nL) (flow rate = 707 μL min−1) and passed through R1 (V = 220 μL). The resulting solution was reacted with 2,6-lutidine (S3) in M′1 (V = 570 nL) (flow rate = 707 μL min−1) and passed through R′1 (V = 23 μL). Finally the reaction was quenched with HCl (S4) in M2 (flow rate = 707 μL min−1) and collected in a flask. Volatiles were evaporated under vacuum and some drops of 2 N aq. NaOH were added until pH > 9 was reached. The solution was extracted with CH2Cl2 (×3) and the combined organic layers were dried on MgSO4, filtered and evaporated under vacuum. |
| § N-methylation of BnNH2 without a base afforded BnNHMe (41%) along with BnNMe2 (17%) and BnN+Me3 (8%). |
| This journal is © the Partner Organisations 2015 |