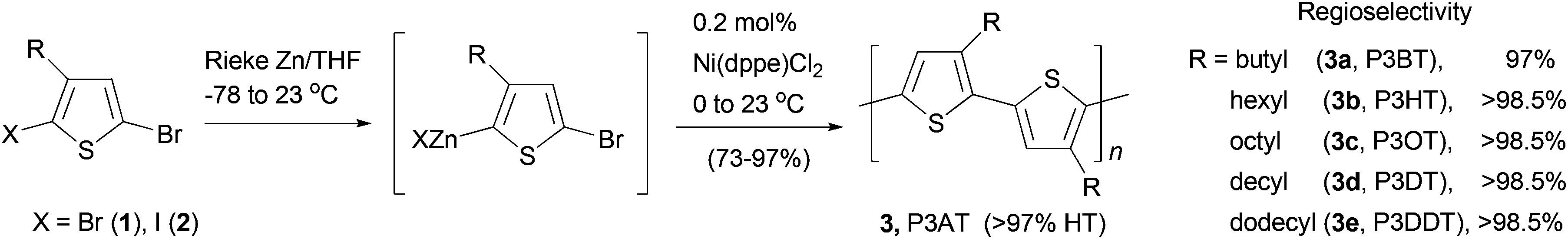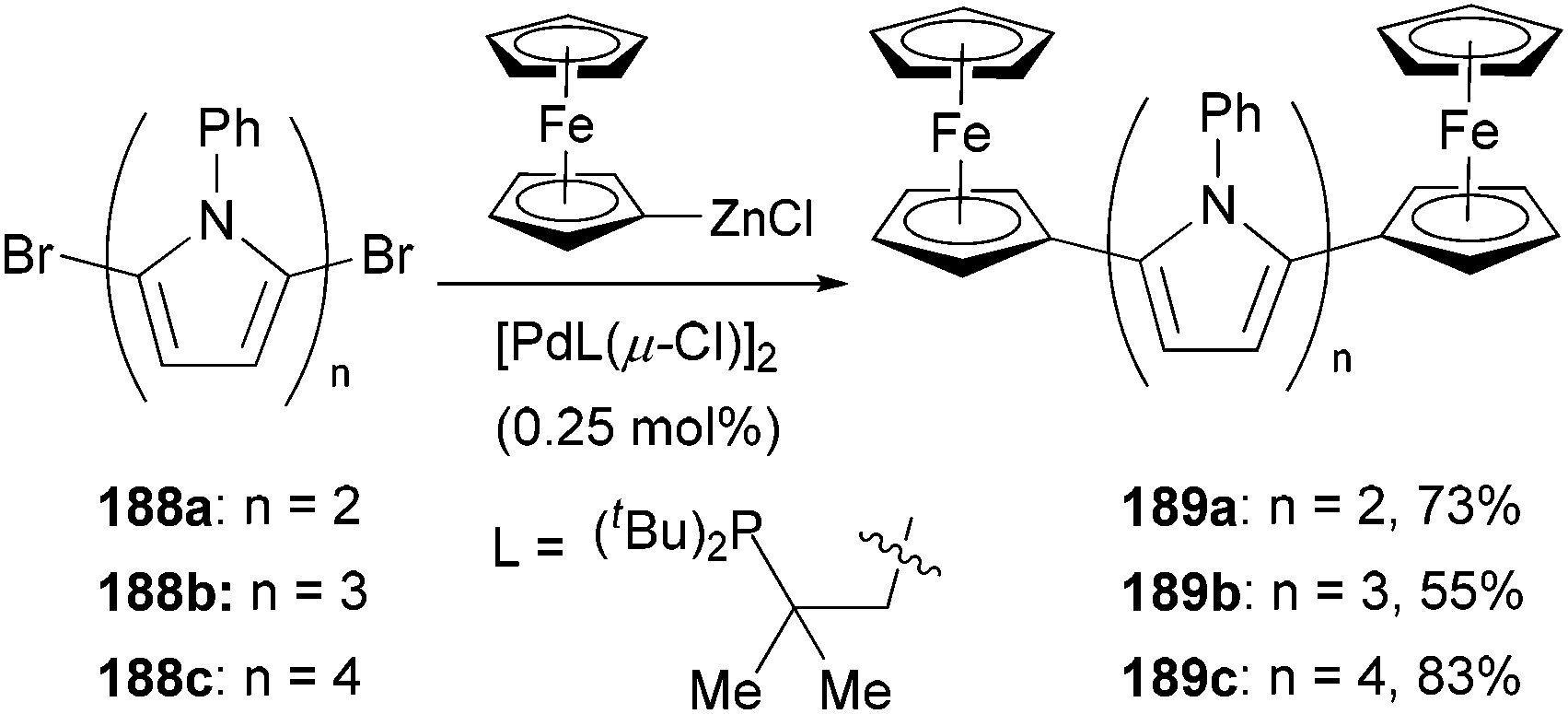DOI:
10.1039/C4QO00322E
(Review Article)
Org. Chem. Front., 2015,
2, 416-445
Negishi coupling in the synthesis of advanced electronic, optical, electrochemical, and magnetic materials†
Received
8th December 2014
, Accepted 4th February 2015
First published on 4th February 2015
Abstract
Negishi coupling is one of the most important modern organic synthetic methods for selective C–C bond formation, and has been extensively applied in the synthesis of organic electronic, optical, electrochemical, and magnetic materials. This report provides a critical overview of the efficiency and versatility of Negishi coupling as applied for synthesizing polymers, oligomers, and small molecules with a wide variety of structural features and various desirable functions for organic electronic, optoelectronic, and other advanced technologies.
 Shouquan Huo | Shouquan Huo received his B.S. and M.S. from Zhengzhou University and Ph.D. from Nanjing University under the supervision of Prof. Yangjie Wu. He has held a series of research positions at the Shanghai Institute of Organic Chemistry with Prof. Xiyan Lu, at the Institute for Molecular Sciences and Hokkaido University with Prof. Tamotsu Takahashi, and at Purdue University with Prof. Negishi. After spending one year with DSM Pharmaceuticals and four years with the Eastman Kodak company, he joined East Carolina University as an Assistant Professor in 2007 and was promoted to Associate Professor in 2013. His research interest is focused on organometallic chemistry. |
 Robert Mroz | Robert Mroz was born in New York, USA in 1991. He received his B.Sc. from the State University of New York at Oswego in 2014. He is currently a graduate student at East Carolina University under the advisement of Dr Shouquan Huo. His research focuses on the synthesis and design of phosphorescent organometallic complexes. |
 Jeffrey S. Carroll | Jeffrey S. Carroll was born in Massachusetts, USA in 1989. He received his Bachelor of Science degrees in Chemistry and Biology from East Carolina University in 2013, and is currently a graduate student at East Carolina University under the advisement of Dr Shouquan Huo. He will be attending the School of Dentistry at UNC-Chapel Hill in the fall of 2015. |
1. Introduction
Negishi coupling may be loosely defined as the palladium or nickel catalyzed cross-coupling reaction of organometals containing metals of intermediate electronegativity represented by Zn, Al, and Zr with organic electrophiles such as organic halides and sulfonates.1 Since its discovery in the middle to late 1970s,2–7 Negishi coupling has become a frequently used C–C bond formation method in modern organic synthesis.8–19 For his pioneering work in the field of palladium-catalyzed cross coupling reactions and the impact of Negishi coupling on organic synthesis, Professor Negishi, together with Professors Heck and Suzuki, was awarded the 2010 Nobel Prize in Chemistry.19 The methodological developments in Negishi coupling and its applications in the synthesis of organic molecules of chemical, biological, or medicinal importance have been extensively reviewed.1,9–23 However, its utility in the synthesis of molecules of electronic, optical, electrochemical, or magnetic importance has not been reviewed. Organic materials with excellent electronic24 and optical properties25 are the key components in organic electronic and optoelectronic technologies such as organic light-emitting diodes (OLEDs), organic photovoltaic cells, and organic field-effect transistors (OFETs). Organic electronic and optoelectronic technologies offer several advantages over conventional technologies based on inorganic semiconductors, two of which are the low cost of production and the flexibility of the devices. Some of the organics-based technologies, for instance, OLED display technology, have been developed to a considerably advanced stage, and have resulted in the marketing of commercial products. Material development has made a major contribution in advancing these technologies. A number of reviews have dealt with applications of palladium-catalyzed cross coupling reactions (Kumada coupling,26 Negishi coupling, Stille coupling,27 and Suzuki coupling28) in the synthesis of polymeric and oligomeric electronic conducting and semiconducting materials.29 Functional small molecules such as charge-transporting, redox-active, light-emitting and harvesting, and other optical materials have also played important roles in fabricating highly efficient organic electronic and optoelectronic devices. Negishi coupling, particularly with the use of organozinc reagents, has enabled cross coupling of all types of carbon atoms, namely sp, sp2, and sp3 carbons, and essentially all possible combinations of various types of organozincs and electrophiles to form carbon–carbon bonds.16 Therefore, Negishi coupling becomes a frequent choice in synthesizing these small functional molecules as well. This review will focus on the application of Negishi coupling in the synthesis of advanced materials including polymers, oligomers, and small functional molecules. The main objective of this review is to demonstrate the versatility and general applicability of Negishi coupling in the synthesis of diverse arrays of functional molecules.
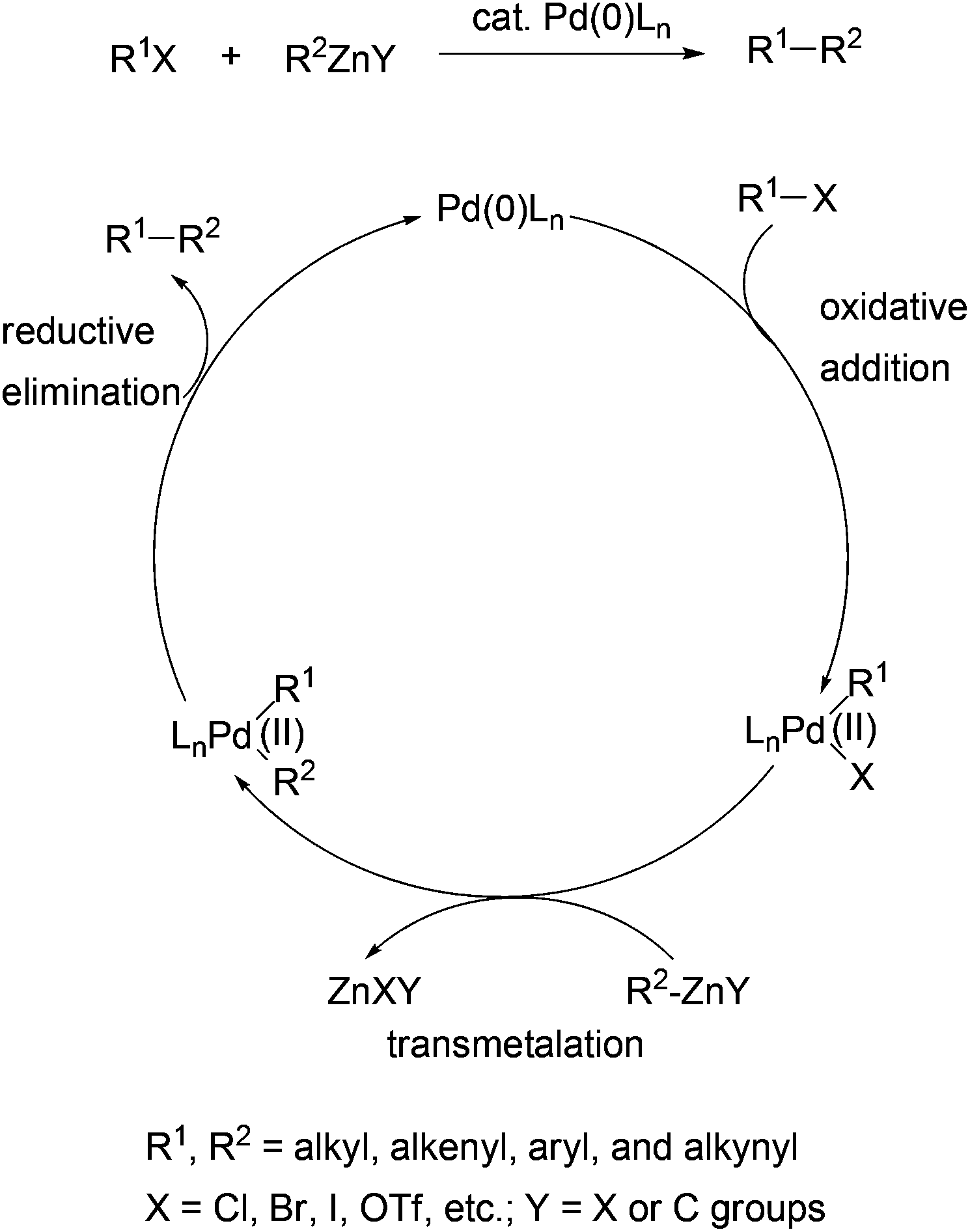
The generally accepted mechanism for Negishi coupling with organozinc reagents is shown in Scheme 1. The reaction involves the oxidative addition of an organic electrophile, typically a halide or a sulfonate ester, to palladium (0), transmetalation with an organozinc reagent, and reductive elimination to release the cross-coupling product and regenerate the catalyst. Negishi coupling becomes the choice of method mainly because of a few beneficial factors to organic synthesis which distinguish it from other cross couplings using organometals of Sn (Stille coupling), B (Suzuki coupling), and Mg (Kumada coupling). First, Negishi coupling proceeds with generally high efficiency, namely high yields and high selectivities. The selectivity could include the selectivity of the formation of the cross-coupled product and the stereoselectivity in the formation of structurally defined alkenes. Second, Negishi coupling has optimal balance between reactivity and chemoselectivity. Organozinc reagents are more reactive than their Sn and B counterparts, and can tolerate more functional groups than Grignard reagents. Owing to the higher reactivity of organozinc reagents, Negishi coupling can survive the presence of organic tin and boron functionalities, which permits the development of the sequential Negishi–Stille or Negishi–Suzuki coupling methodology and other useful synthetic strategies.30 Third, Negishi coupling often proceeds under mild conditions. Unlike the Suzuki and Stille couplings, the cross coupling with organozinc reagents typically does not require a base or other additives. Another feature of Negishi coupling is its operational simplicity. The organometals (Al, Zr, and Zn) used in Negishi coupling can be generated in situ and used directly in the subsequent cross couplings. Last but not least, there are multiple convenient and inexpensive accesses to organozinc reagents including transmetalation with organolithiums and Grignard reagents, and particularly, direct zinc insertion in organic halides.30–35 Unlike Stille coupling which uses toxic organostannes, Negishi coupling with organozincs is environmentally benign.
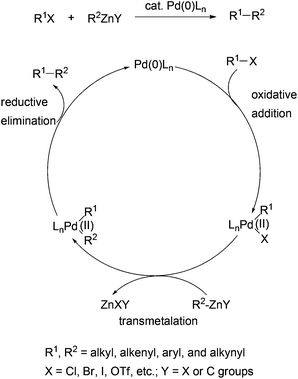 |
| | Scheme 1 Mechanism of Negishi coupling with organozinc reagents. | |
2. Organic electronic materials
2.1. Conducting polymers via Negishi polycondensation
Polythiophenes are a family of conjugated polymers with excellent conductivity, and in particular, those derived from 3-alkyl-substituted thiophenes36 have been used extensively in polymer-based electronic devices including OLEDs,37 OFETs,38 and organic solar cells.39 Regioselectivity (regioregularity) in the polymerization of 3-alkylthiophenes has profound influence on the conductivity of the formed polymers. Regioregular polymers usually display much higher conductivity than their regiorandom forms. Regioregular, head-to-tail poly(3-alkylthiophenes) (P3AT) can be prepared with excellent selectivity from the Ni-catalyzed cross coupling of the corresponding thiophenylzinc reagents.40 Organozinc reagents can be generated by the selective insertion of Rieke's zinc to either 3-alkyl-2,5-dibromothiophene (1) at −78 °C or 3-alkyl-2-bromo-5-iodothiophene (2) at 0 °C (Scheme 2). The polycondensation of these bifunctional organozinc reagents in the presence of catalyst NiCl2(dppe) (dppe = 1,2-bis(diphenylphosphino)ethane) produced polymers 3 in greater than 97% head-to-tail regioselectivity. Such polycondensation is termed as AB-type polycondensation since the bifunctional monomer bears both metal and halide groups required for the cross coupling. The regioselectivity was found to depend on both the metal and the ligand of the catalyst. The replacement of dppe with PPh3 or the switch from Ni to Pd results in much lower selectivity. The regioregular polymers show consistently lower band gaps and thus higher conductivities (103 S cm−1 for doped films) compared to their regiorandom forms (<10 S cm−1).41
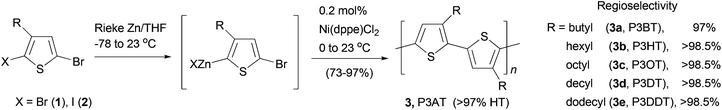 |
| | Scheme 2 Synthesis of P3ATs 3a–3e by Negishi polycondensation. | |
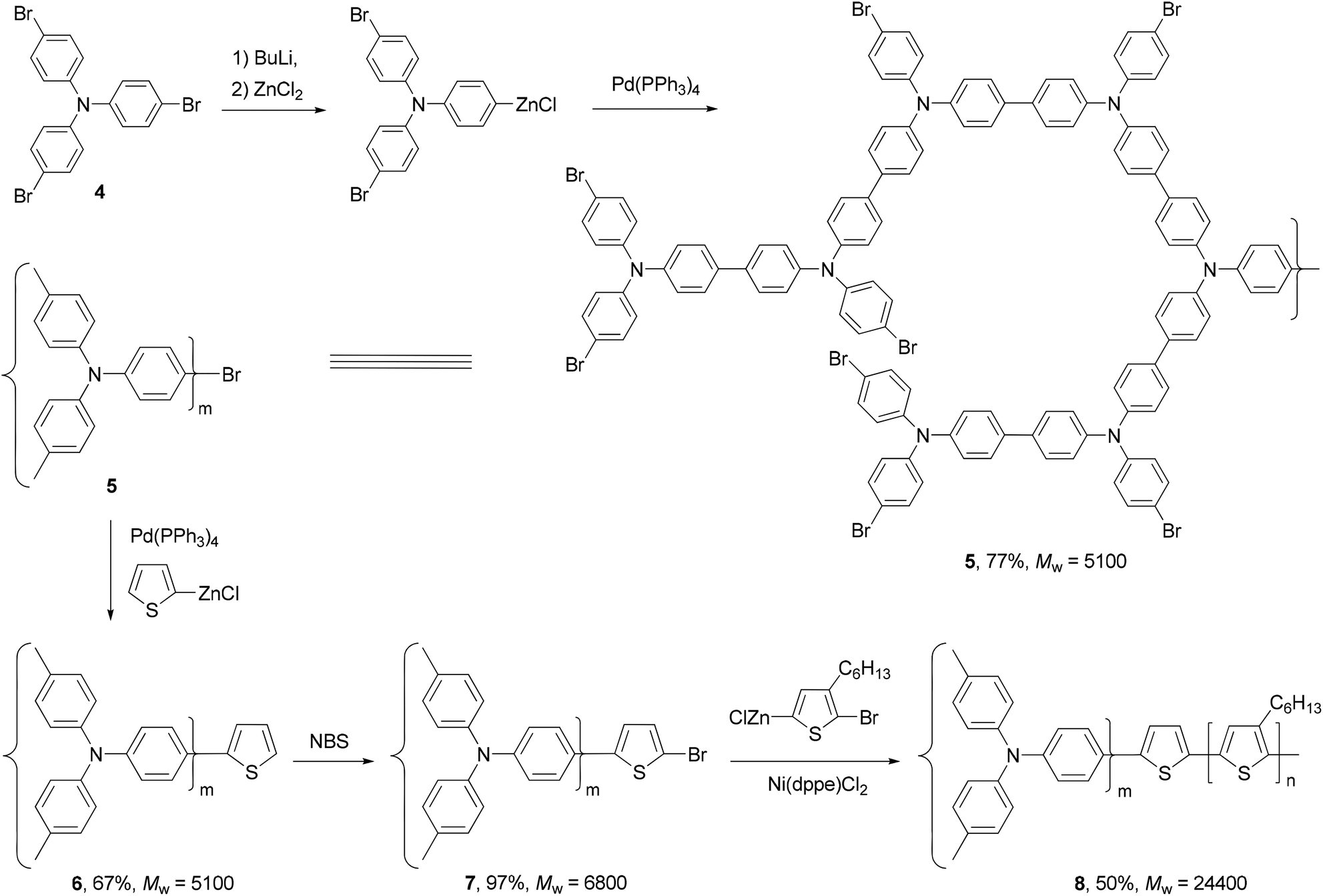
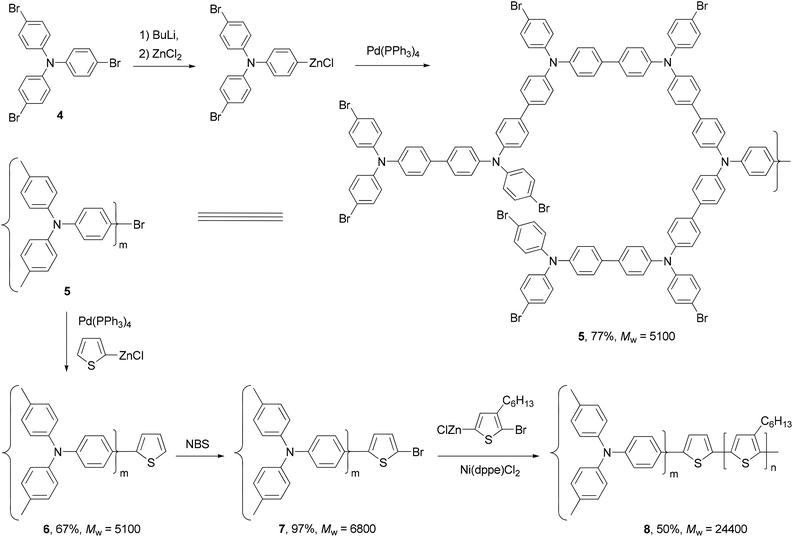
A star structured regioregular poly(3-hexylthiophene) with a conjugated hyperbranched poly(triphenylamine) was prepared by two consecutive Negishi polycondensations (Scheme 3).42 First, a hyperbranched poly(triphenylamine) 5 was synthesized by Negishi polycondensation starting from tris(4-bromophenyl)amine (4). The terminal bromo groups on the hyperbranched polymer 5 were subsequently converted to a thiophene by Negishi coupling with 2-thioenylzinc chloride to form 6, which was brominated with NBS to give 7. This functionalized hyperbranched polymer 7 was used as the core for the synthesis of the star structured polymer 8. The formation of regioregular P3HT was realized by the second Negishi polycondensation of 5-bromo-4-hexyl-2-thioenylzinc chloride. The star structured polymer has been shown to be electroactive and capable of attaining a high level of conductivity up to 100 S cm−1 in its doped form.
 |
| | Scheme 3 Synthesis of star structured polymer 8. | |
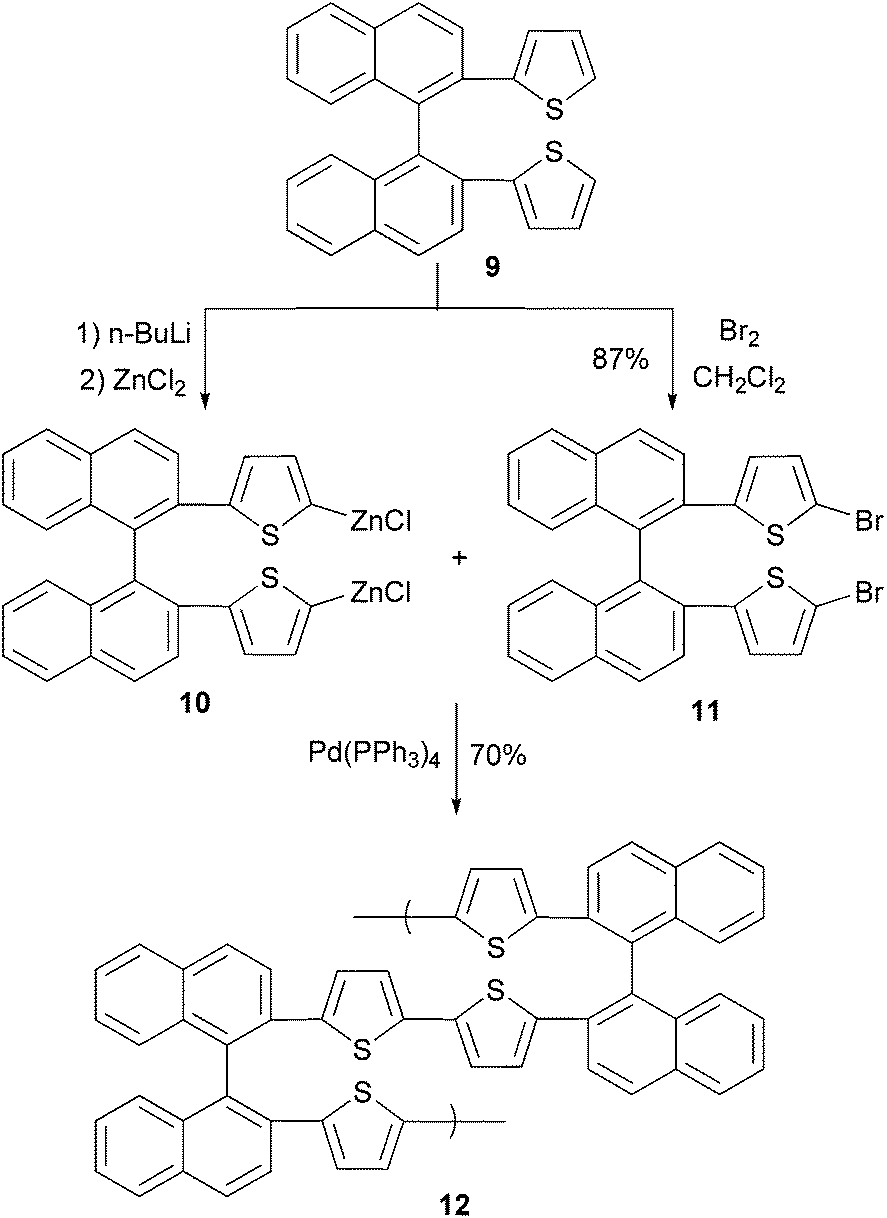
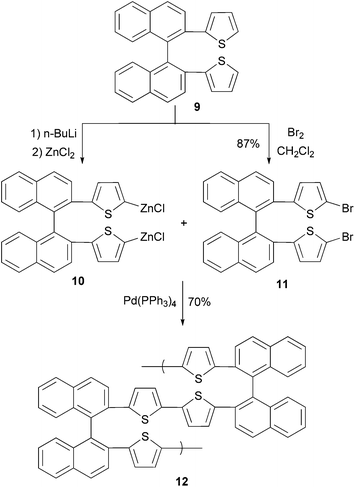
In a convergent synthesis shown in Scheme 4, 2,2′-di(2-thiophenyl)-1,1′-binaphthylene (9) is converted to its bifunctional organozinc monomer 10 and dibromide monomer 11. In the presence of Pd(PPh3)4, an AA/BB-type (two different bifunctional monomers AA and BB with each bearing two same functional groups) polycondensation produces binaphthylene–thiophene copolymer 12 where the oligothiophene is covalently bonded to the 2,2′-site of 1,1′-binaphthylene. The conductivity of the doped polymer was measured to be 3 × 10−5 S cm−1.43
 |
| | Scheme 4 Synthesis of binaphthylene–thiophene copolymer 12. | |
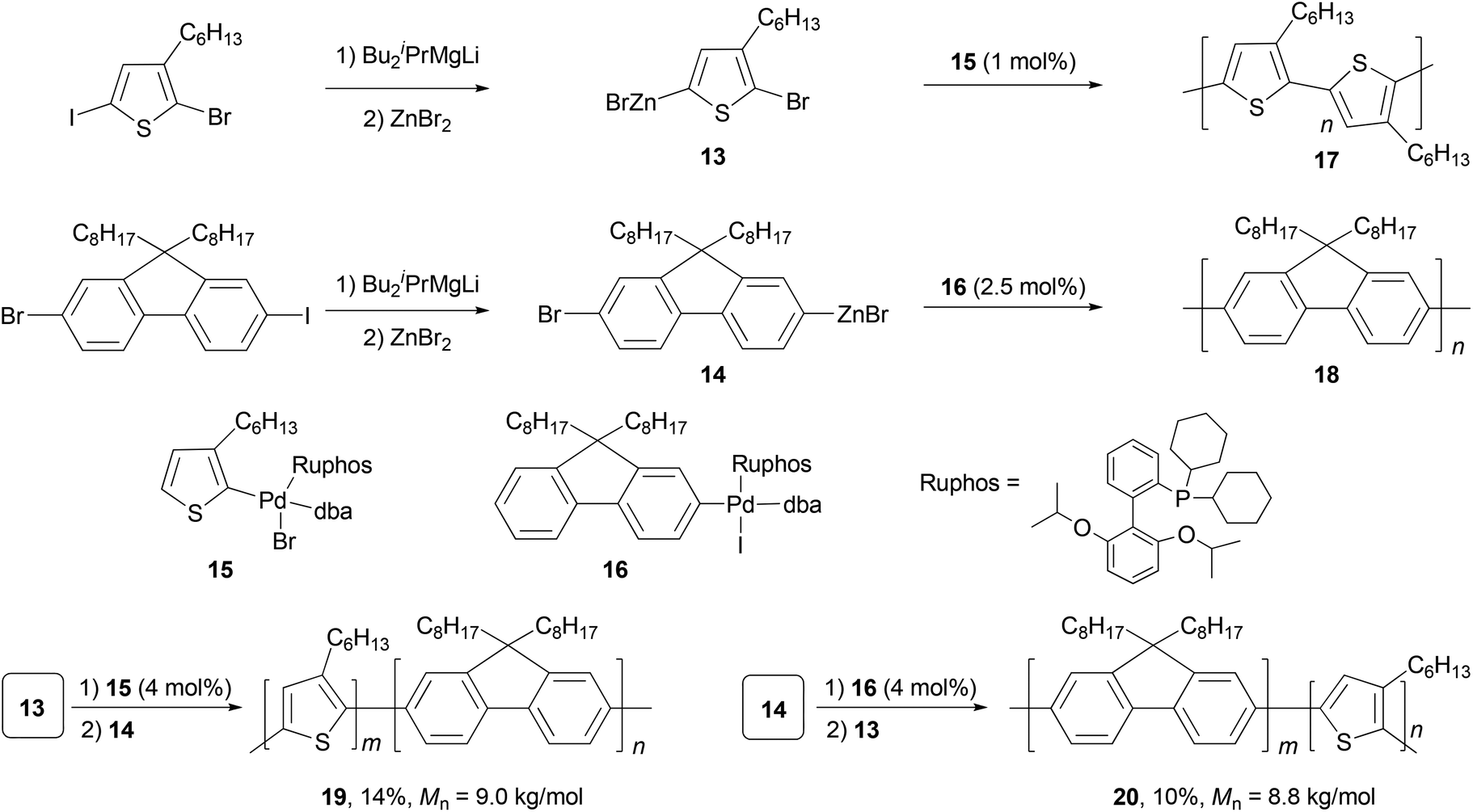
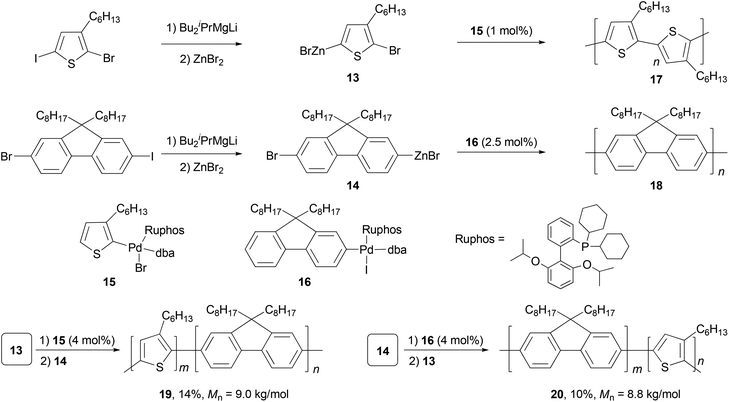
Polyfluorenes (PFs) represent another class of synthetic polymers that have the required electronic and very favorable optical properties for applications in organic electronic and optoelectronic technologies.44,45 Earlier, the synthesis of polyfluorenes was achieved with oxidative polymerization either chemically or electrochemically. A more controlled synthesis now relies on transition metal catalyzed cross coupling reactions. A universal chain-growth polymerization protocol was developed and demonstrated for the synthesis of both homopolymers, P3HT 17 and PF 18, and block-copolymers P3HT-b-PF (19) and PF-b-P3HT (20) (Scheme 5).46 The protocol is based on Negishi coupling using a stable palladium catalyst with electron-rich, sterically hindered Ruphos47 as the ligand. This method allows the one-pot synthesis of block-copolymers by successive monomer addition. The AB-type monomers 13 and 14 were generated by reacting 2-bromo-5-iodo-3-hexylthiophene and 2-bromo-7-iodo-9,9-dihexylfluorene with iPrMgCl-LiCl,48 followed by treatment with ZnBr2. The initiators 15 and 16 were prepared by oxidative addition of 2-bromo-3-hexylthiophene and 2-iodo-9,9-dioctylfluorene to Pd2(dba)3 (dba = dibenzylideneacetone) in the presence of Ruphos.
 |
| | Scheme 5 Synthesis of P3HT(17), PF(18), P3HT-b-PF(19), and PF-b-P3HT(20). | |
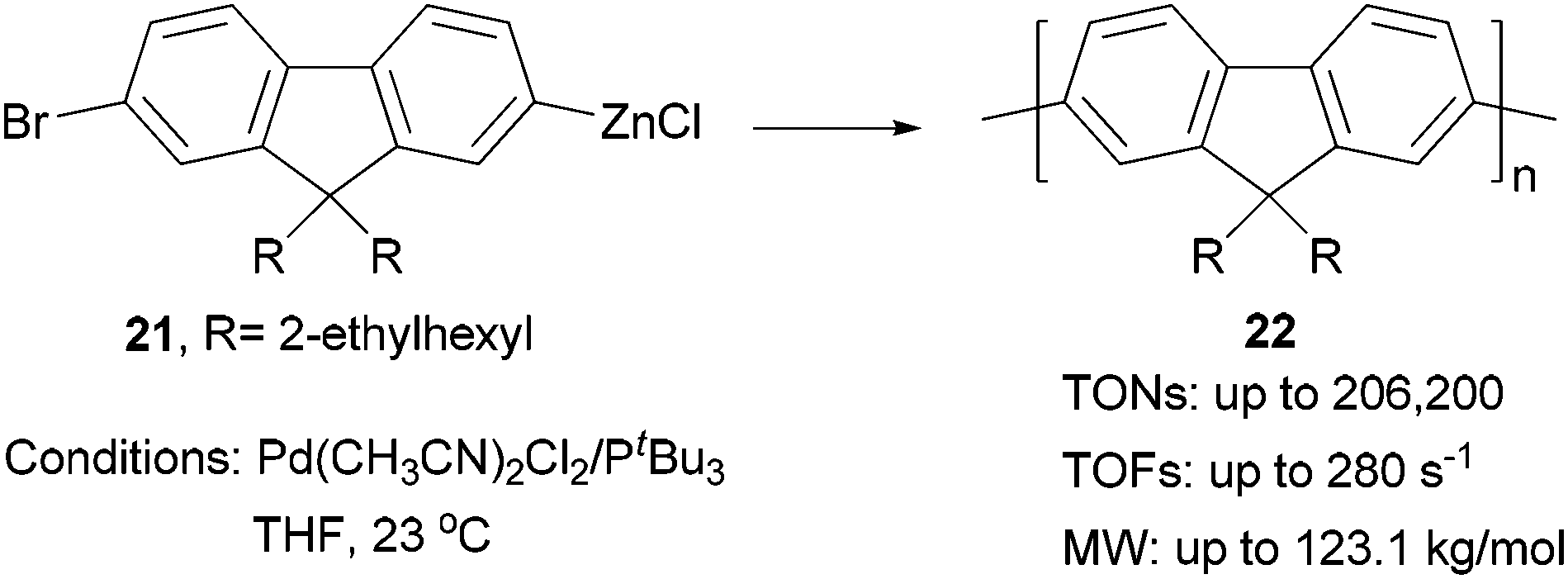
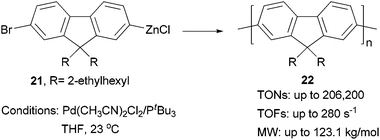
Although π-conjugated polymers are often synthesized by the Stille29c and Suzuki29a polycondensations, a recent study demonstrates that Negishi polycondensation can offer several critical advantages such as higher reactivity, lower catalyst loading, and higher molecular weight (MW).49 Kiriy and co-workers reported the Pd/PtBu3-catalyzed Negishi chain-growth polycondensation of AB-type monomer 21 to produce the polyfluorene 22, which proceeds with unprecedentedly high turnover numbers (TONs) of over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (highest TONs of 206200) and turnover frequencies (TOFs) of up to 280 s−1. The highest MW of 123.1 kg mol−1 was obtained at a monomer/catalyst ratio of 20000:1(Scheme 6). They also demonstrated that the related AA/BB-type step-growth polycondensation proceeds with two orders of magnitude lower TONs and TOFs under the same conditions.
000 (highest TONs of 206200) and turnover frequencies (TOFs) of up to 280 s−1. The highest MW of 123.1 kg mol−1 was obtained at a monomer/catalyst ratio of 20000:1(Scheme 6). They also demonstrated that the related AA/BB-type step-growth polycondensation proceeds with two orders of magnitude lower TONs and TOFs under the same conditions.
 |
| | Scheme 6 Highly efficient synthesis of polyfluorene by Negishi polycondensation. | |
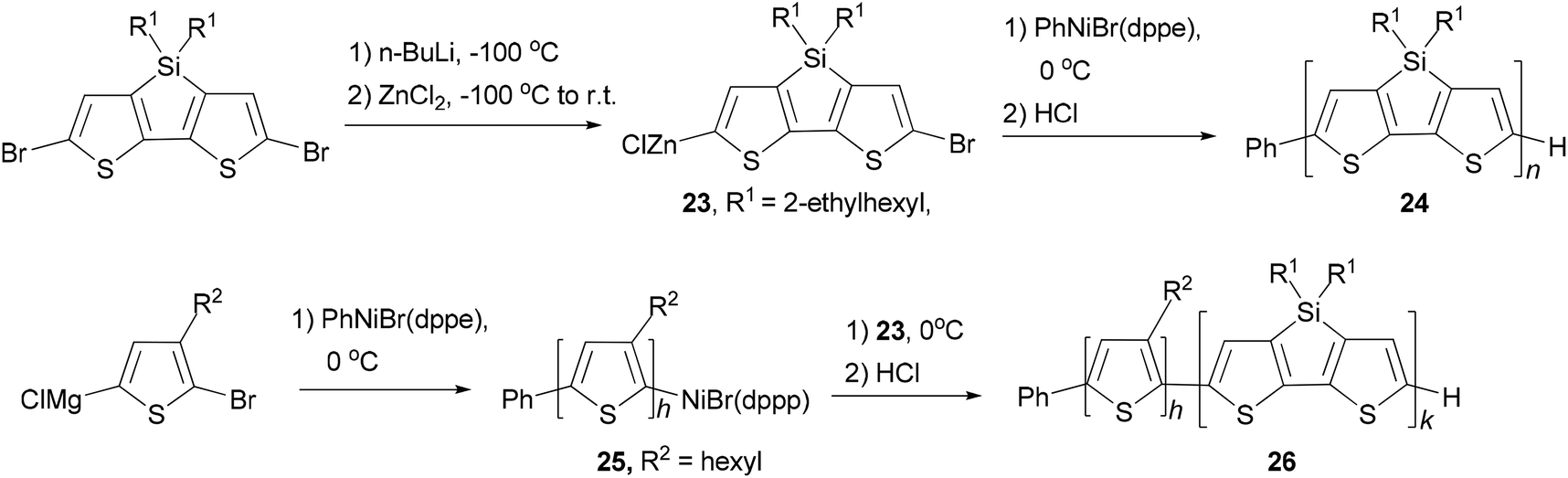
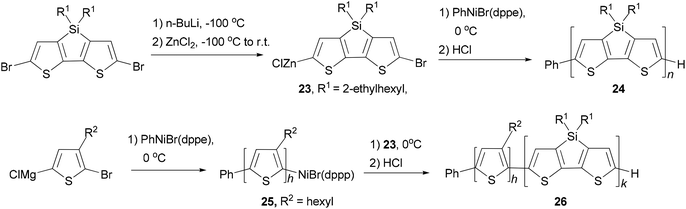
A quasi-living Negishi-type catalyst-transfer polycondensation of a zinc–organic DTS-based monomer 23 provides access to narrowly distributed poly(4,4-bis(2-ethylhexyl)dithieno[3,2-b:2′,3′-d]silole) (PDTS) (24) with controlled molecular weight (Scheme 7).50 The synthesis of well-defined all-conjugated diblock copolymers 26 containing a PDTS block is accomplished by a combination of Kumada and Negishi catalyst-transfer polycondensations (KCTP and NCTP, respectively). Notably, it was shown that living P3HT chains 25 obtained by the KCTP of magnesium–organic thiophene-based monomer efficiently initiate the NCTP of zinc–organic DTS-based monomer 23. The purity of the DTS-based monomer was found to be a crucial factor for achieving a clean chain-growth polymerization process.
 |
| | Scheme 7 Synthesis of diblock copolymer 26 using a combined KCTP and NCTP. | |
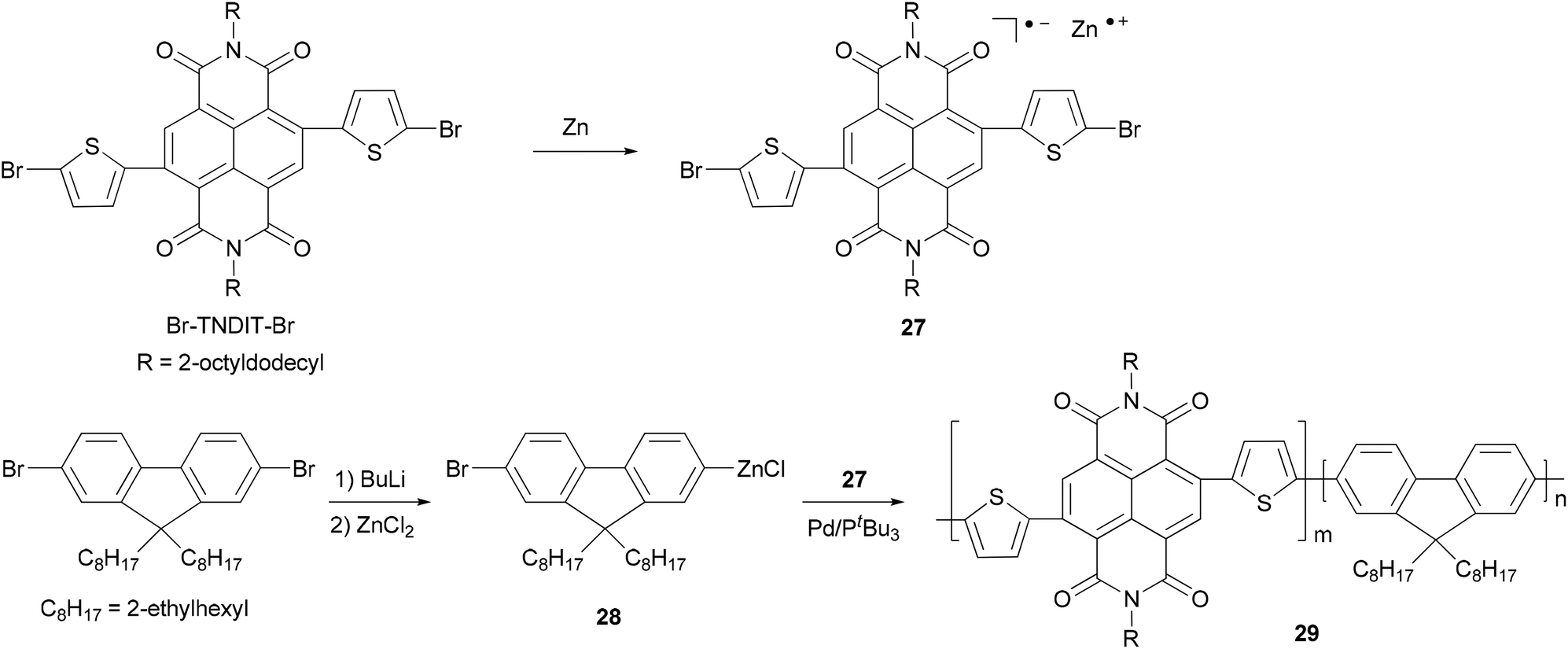
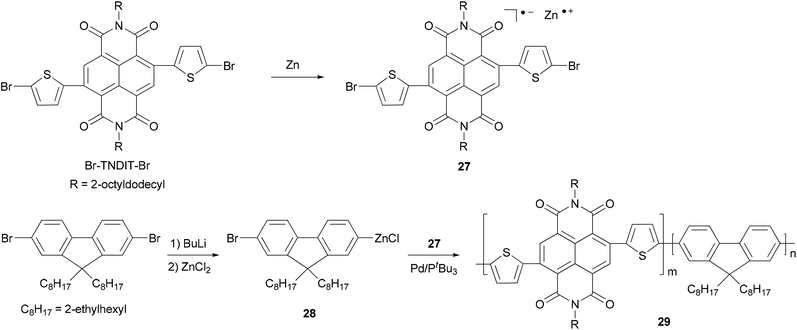
Chain-growth catalyst-transfer Negishi polycondensation has recently been successfully applied for the copolymerization of electron-rich and electron-deficient monomers,51 anion-radical 27 formed upon the reaction of activated Zn powder and Br–TNDIT–Br (Br–TNDIT–Br = 2,6-bis(2-bromothien-5-yl)naphthalene-1,4,5,8-tetracarboxylic-N,N′-bis(2-octyldodecyl)diimide) and AB-type fluorine-based monomer 28 generated from monolithiation of 2,7-dibromo-9,9-bis(2-ethylhexyl)-9H-fluorene followed by transmetalation with ZnCl2. The copolymerization proceeds rapidly in the presence of Pd catalyst having a bulky and electron-rich tri-tert-butylphosphine ligand. Even though the two monomers are added simultaneously, the polymerization gives a sharp gradient copolymer or even block-like copolymer 29, due to the much faster Negishi condensation of the fluorenic monomer 28 (Scheme 8).49
 |
| | Scheme 8 Synthesis of block copolymer 29. | |
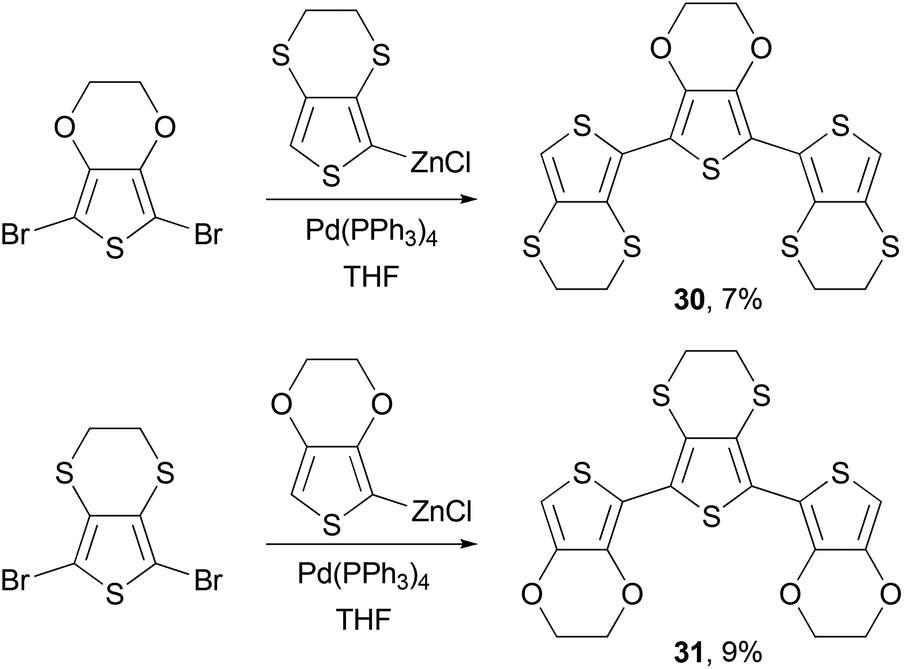
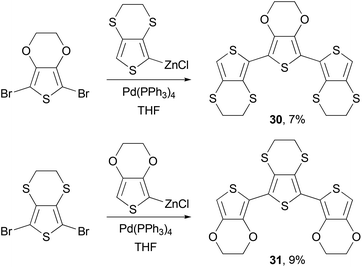
Negishi coupling is also frequently used to synthesize monomers for other polymerization reactions. Poly(ethylene-3,4-dioxythiophene) (PEDOT) has become one of the most popular polythiophene derivatives used for device applications, due to its exceptional hole injection properties, high conductivity and stability.52,53 PEDOT-PSS (PEDOT-polystyrene sulfonic acid) is now a standard hole injection material used in solution-processed OLED devices and solar cells. The p-doped PEDOTs have demonstrated conductivity of several hundred S cm−1, whereas the sulfur analog of PEDOT, poly(ethylene-3,4-dithiathiophene) (PEDTT) has showed electrical conductivity of 0.1 and 0.4 S cm−1 in its chemically doped (with FeCl4−) and electrochemically doped (with ClO4−) states, respectively.54 The large difference in the band gap between PEDOT and PEDTT promoted Skabara and co-workers to synthesize hybrid copolymers PSOS and POSO and compare their electronic and electrochemical properties.55 The monomers SOS (30) and OSO (31) were synthesized as shown in Scheme 9.
 |
| | Scheme 9 Synthesis of monomers SOS (30) and OSO (31). | |
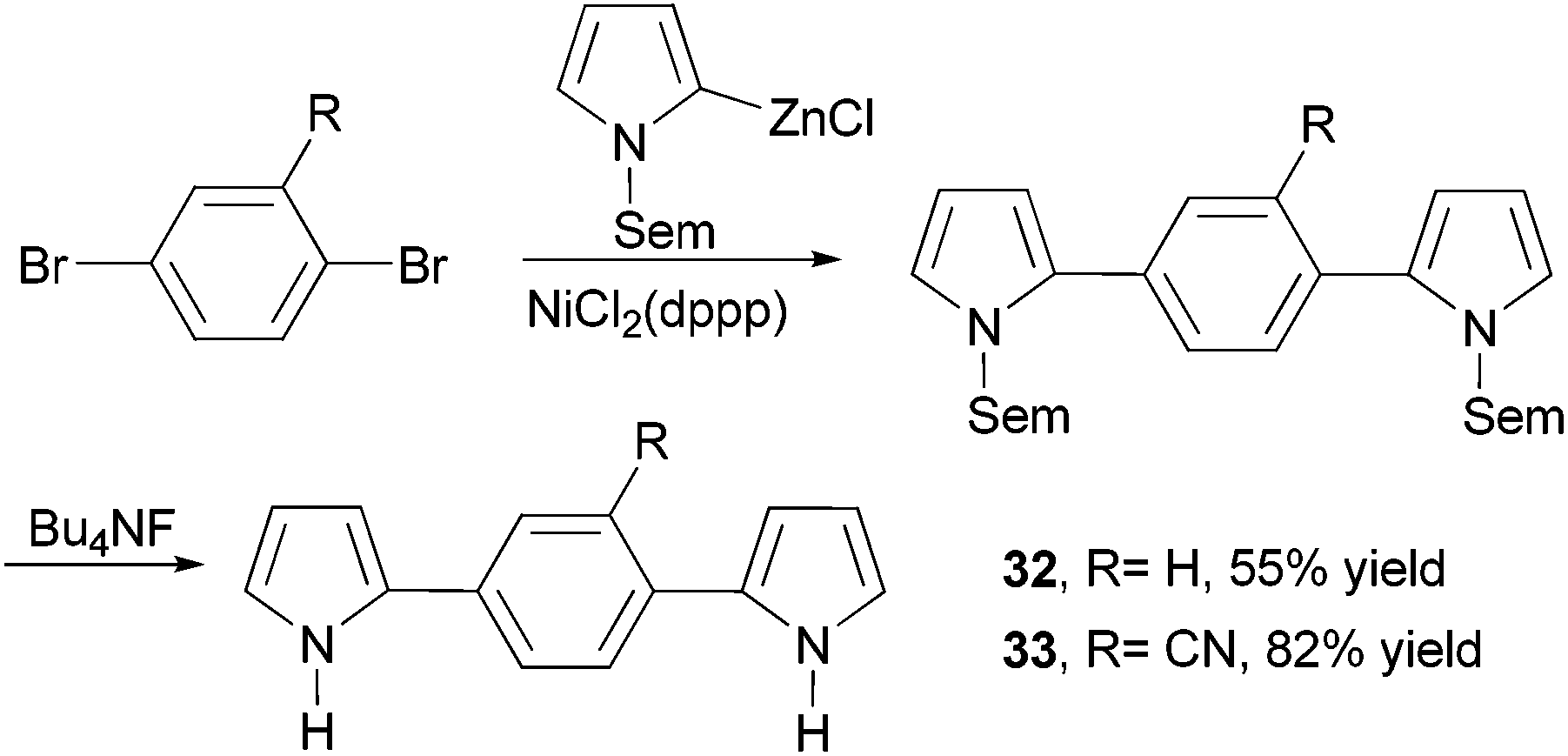
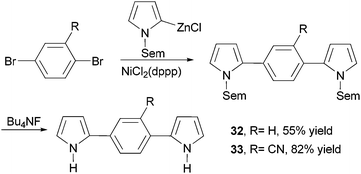
Dipyrrolylphenylenes 32 and 33 were prepared by the nickel-catalyzed cross coupling of N-Sem-pyrrol-2-ylzinc chloride (Sem = [2-(trimethylsilyl)ethoxy]methyl) and the appropriate dibromo derivatives (Scheme 10).56 The Sem protecting group can be easily removed by reacting with Bu4NF (TBAF). Anodic coupling of monomers 32 and 33 produced conducting conjugated polymers poly-32 (σ = 0.3 S cm−1) and poly-33 (σ = 0.1 S cm−1), respectively.
 |
| | Scheme 10 Synthesis of dipyrrolylphenylenes 32 and 33. | |
2.2 Small molecule semiconducting materials
Since the discovery of vapor-deposited OLED57 and photovoltaic58 devices with a p–n junction structure, small molecules have attracted a great deal of attention in order to improve the efficiency and stability of the devices. In general, these small molecules can be divided into electron-transporting and hole-transporting materials. Electron transporting materials should have a relatively low-lying LUMO, and typically contain electron-deficient heteroaromatic structures, while hole-transporting materials are dominated by triarylamines that can form a stable radical cation. A material capable of transporting/trapping both electrons and holes may be used as the host in the emissive layer of an OLED device to enhance the device performance.59
Electron transporting materials.
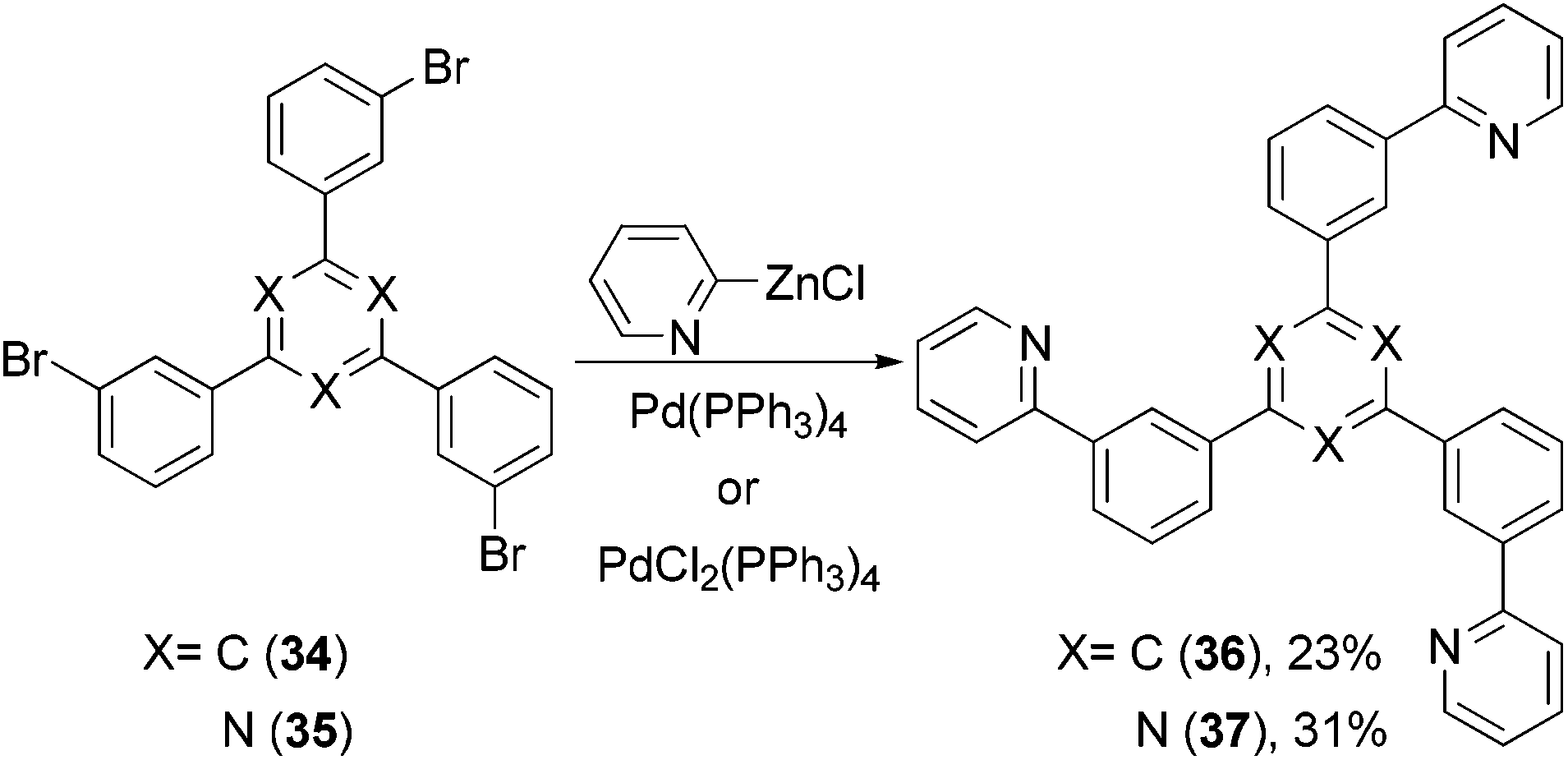
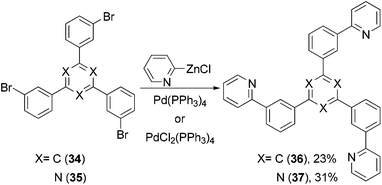
1,3,5-Tri(m-pyrid-2-yl-phenyl)benzene (36) was synthesized by the Negishi coupling of 1,3,5-tri(m-bromophenyl)-benzene (34) with 2-pyridylzinc bromide60 and evaluated as an electron transporting material in OLED devices (Scheme 11), which was compared with isomeric 1,3,5-tri(m-pyrid-3-yl-phenyl)benzene and 1,3,5-tri(m-pyrid-4-yl-phenyl)benzene prepared by Suzuki coupling. Similar Negishi coupling was applied to the synthesis of a 1,3,5-triazine-core-containing electron transporting material 37.61
 |
| | Scheme 11 Synthesis of 36 and 37. | |
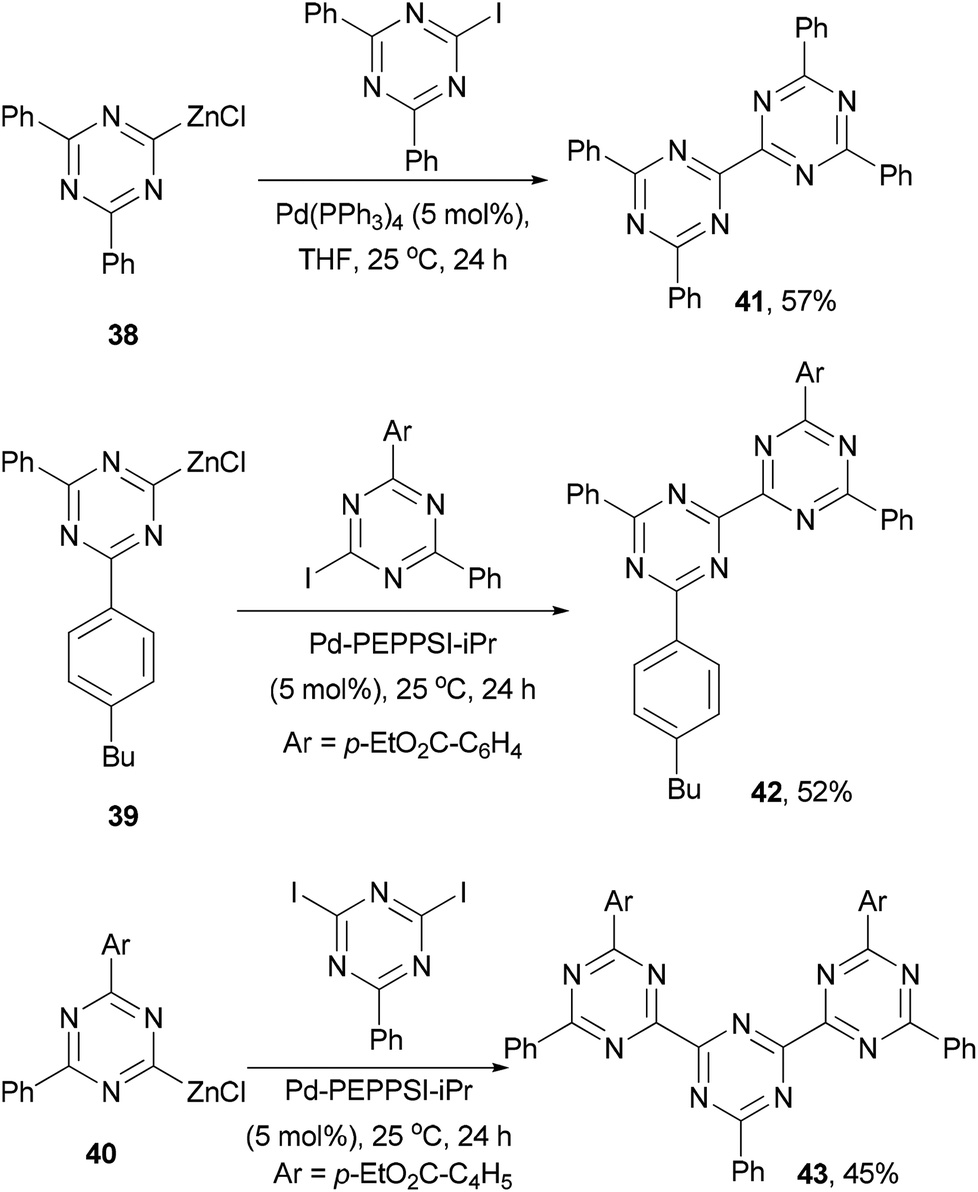
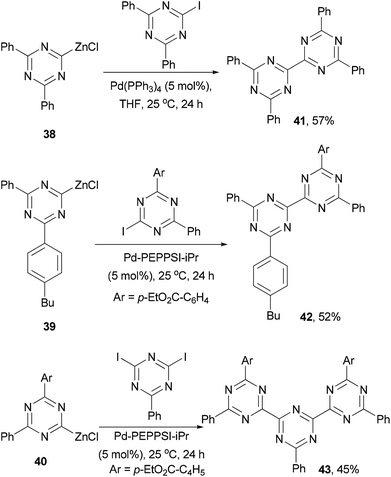
Knochel and co-workers developed a method of generating 2-zincated 1,3,5-triazines via an iodine–magnesium exchange of iodo- and diiodo-1,3,5-triazines with alkyl Grignard reagents followed by transmetalation with ZnCl2.62 The zinc reagents 38–40 undergo palladium catalyzed cross coupling with iodo- or diiodo-1,3,5-triazines to give dimeric and trimeric derivatives 41–43, respectively, in moderate yields (Scheme 12).62 These materials could be used as electron-transporting materials in optoelectronic devices.63
 |
| | Scheme 12 Synthesis of oligomeric 1,3,5-triazines 41–43. | |
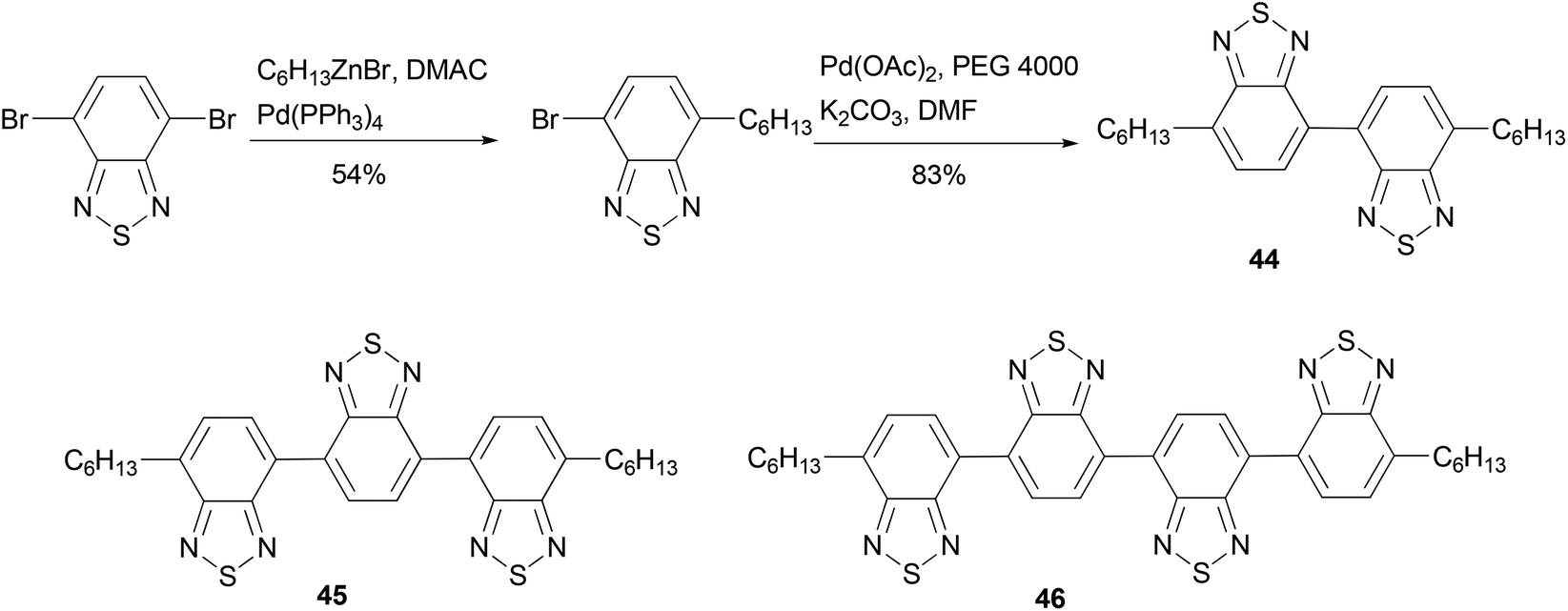
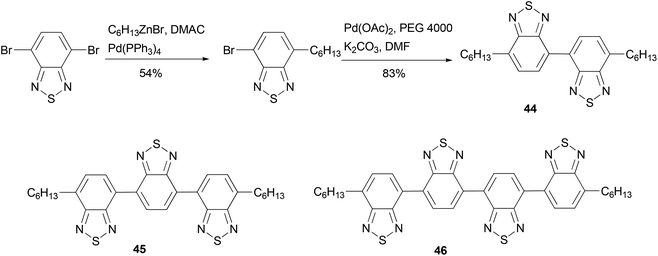
Electron-deficient 2,1,3-benzothiadiazole oligomeric assemblies 44–46 were prepared through palladium-mediated homo-coupling or cross coupling (Scheme 13).64 To improve the solubility of the oligomers, an alkyl group is introduced by Negishi coupling using the alkylzinc reagent conveniently generated using Huo's protocol.33 Compared with a commercial electron-transporting material 2-(4-biphenyl)-5-(4-tert-butylphenyl)-1,3,4-oxadiazole, compounds 45 and 46 show much lower LUMO values, suggesting that the energy barriers between the oligomers and the cathode would be greatly reduced when they are used in OLEDs. These optoelectronic properties show that oligomers are promising electron-transporting materials. The introduction of an alkyl group improves the solubility of the material not only for easy isolation and purification but also for fabricating devices using low-cost solution-processes.
 |
| | Scheme 13 Synthesis of alkylated oligomeric 2,1,3-benzothiadiazoles 44–46. | |
Hole transporting materials.
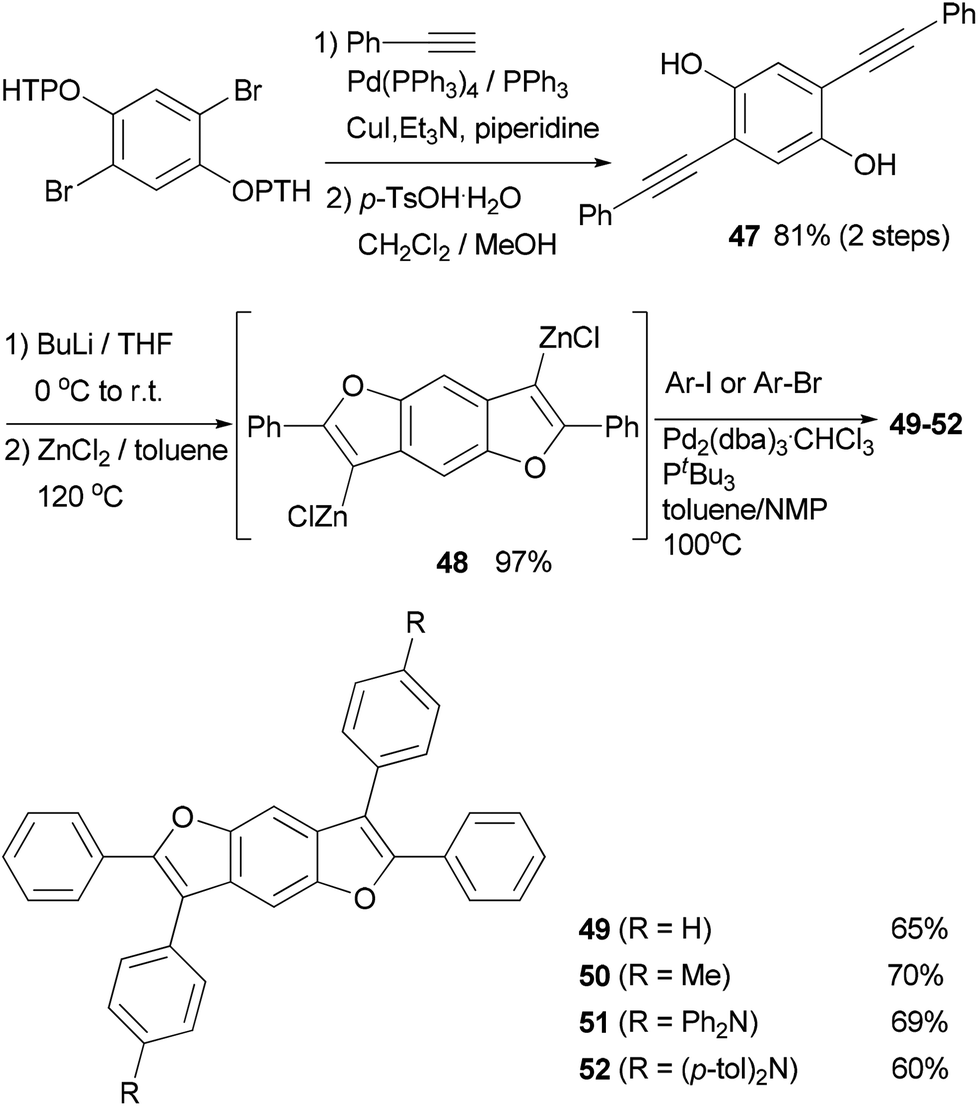
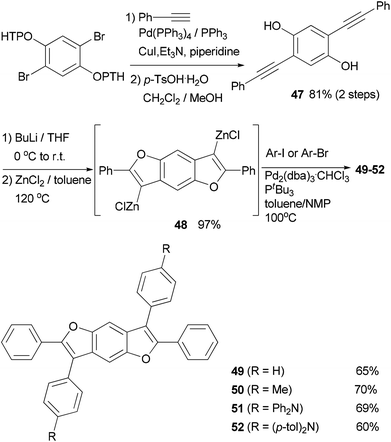
A series of 2,3,6,7-tetraarylbenzo-[1,2-b:4,5-b′]difurans (BDFs) were synthesized based on a zinc-mediated annulation–Negishi coupling synthetic methodology.65 The precursor 47 for the annulation was prepared by the Sonogashira coupling66 of protected 2,5-dibromohydroqinone with phenylacetylene. Zinc-mediated annulation produced the 3,7-dizinciobenzodifuran 48 in quantitative yield, which underwent Negishi coupling with arylzinc halides to give 2,3,6,7-tetraarylbenzo-[1,2-b:4,5-b′]difurans 49–52 in good yields65a (Scheme 14). A toluene/N-methylpyrrolidone (NMP) mixture was used as the solvent to improve the yields.67 These compounds were evaluated as efficient hole transporting materials in OLED devices showing a performance comparable to or better than that of a device made with the commonly used hole transporting materials α-NPD (N,N′-di(1-naphthyl)-N,N′-diphenyl-(1,1′-biphenyl)-4,4′-diamine).68 Compounds 49, 51, and 52 have a higher hole mobility (μ = 5.6 × 10−4 to 2.8 × 10−3 cm2 V s−1) than α-NPD (μ = 3.6 × 10−4 cm2 V s−1).65a
 |
| | Scheme 14 Synthesis of 49–52 by the zinc-mediated annulation–Negishi coupling tandem process. | |
Bipolar host materials.
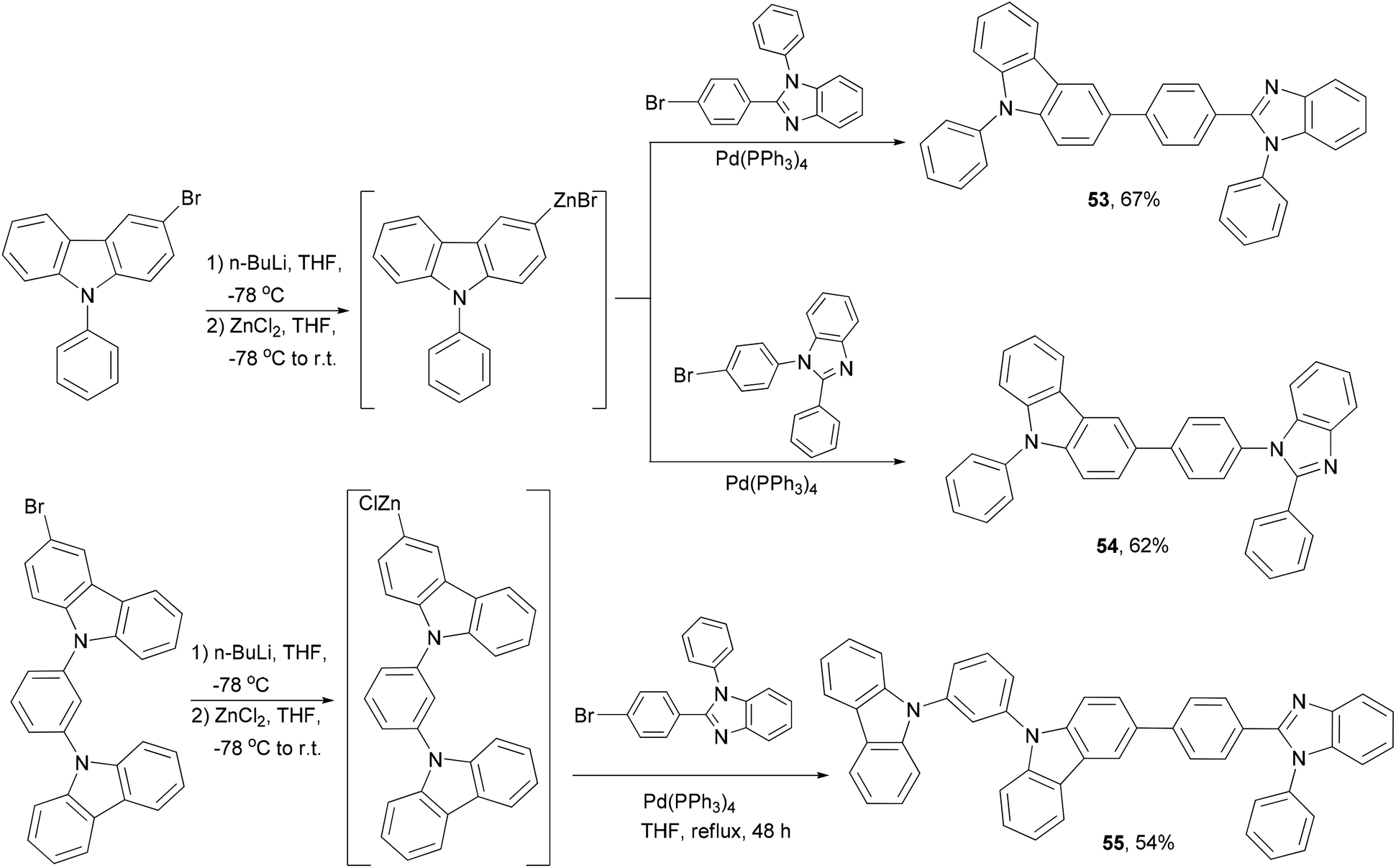
Host materials refer to the host used in the emissive layer of OLED devices. Bipolar hosts are able to transport both electrons and holes and have been used to improve the performance of OLED devices,59 in particular, to reduce the drive voltage and increase the efficiency. A series of hybrid bipolar host materials 53–55 were synthesized by linking a carbazole motif and a benzimidazole motif using the Negishi coupling method (Scheme 15).69 These materials were successfully incorporated into phosphorescent green and blue OLEDs as the host materials showing high external quantum efficiency. A carbazolyl-substituted BDF derivative, 2,6-diphenyl-3,7-bis[4-(N-carbazolyl)phenyl]benzo[1,2-b:4,5-b′] difuran, which was synthesized by using the same method as shown in Scheme 14, also displays bipolar properties and has been used to fabricate efficient OLED devices with a simple homojunction structure.65c
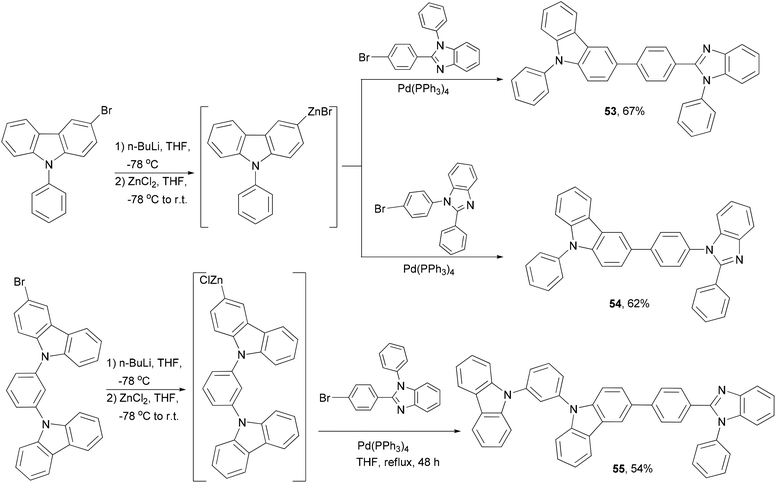 |
| | Scheme 15 Synthesis of hybrid bipolar host materials 53–55. | |
3. Organic optical materials
Organic optical/photonic materials refer to the molecules that can either interact with light, such as absorbing, reflecting, refracting, and rotating light, or emit light under stimulations. Organic optical materials that are important to organic electronic and optoelectronic devices include but are not limited to nonlinear optical (NLO) materials, liquid crystals, light emitters, and light harvesting materials. Transparency, i.e. not absorbing visible light, is also an important optical property of organic electronic materials that can be an important component in optoelectronic devices such as capsulation.
3.1 Nonlinear optical materials
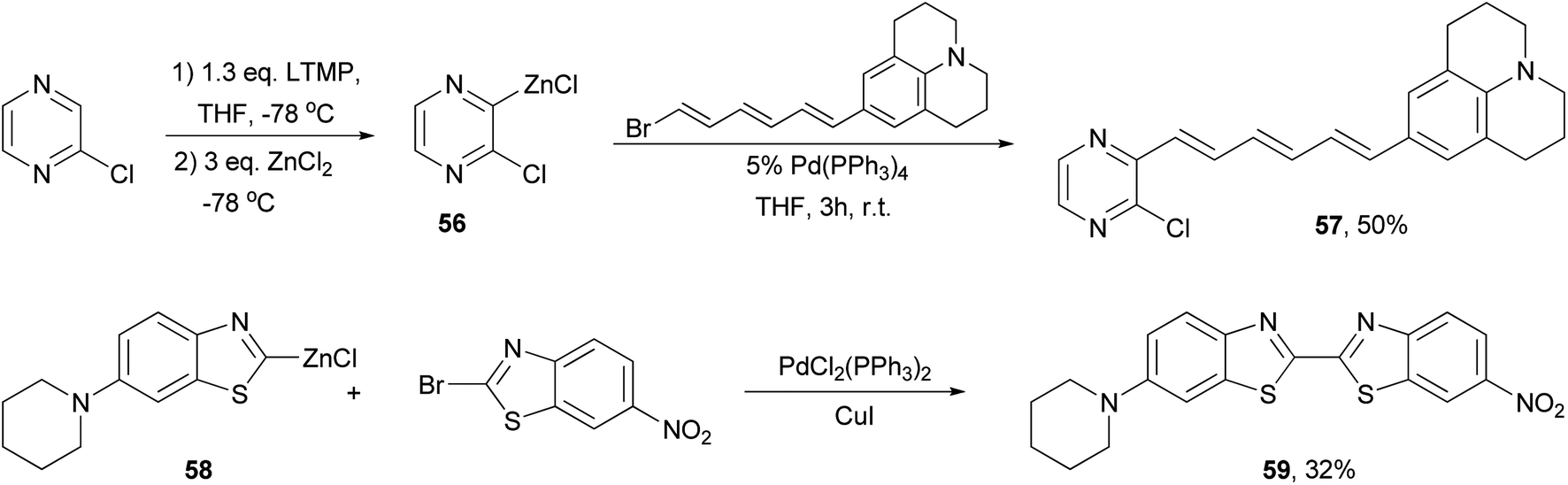
A push–pull structure with a pyrazine core and hexatriene chain 57 was efficiently constructed by the reaction of the pyrazinyl zinc reagent 56 with 6-julolidinyl-1-bromohexatriene in the presence of Pd(PPh3)4 (Scheme 16).70 The organozinc reagent was generated in situ by deprotonating 2-chloropyrazine with LTMP (lithium 2,2,6,6-tetramethylpiperidide) followed by treatment with zinc chloride. The molecule displayed promising NLO properties with a dipole moment of μ = 8.0 D and a scalar product μβ of 1040 × 10−48 esu as compared to DANS71 (dimethylaminonitrostilbene) (μ = 6.6 D; μβ = 350 × 10−48 esu). In addition, the compounds also emitted intense red light (653 nm) with a large Stokes shift (5783 cm−1). A similar strategy was also applied for functionalizing 2,6-dichloropyrazine.70 Another push–pull molecule 6-nitro-6′-piperidyl-2,2′-bisbenzothiazole (59) was prepared by the palladium-catalyzed cross coupling of the organozinc derivative 58 and 2-bromo-6-nitrobenzothiazole (Scheme 16).72 The zinc reagent was prepared by the deprotonation of 6-(N-piperidyl)benzothiazole with n-butyllithium at −100 °C followed by treatment with zinc chloride. Compound 59 exhibited good NLO properties (μβ = 375 × 10−48 esu) and a dual, oxidative and reductive, three-stage single-electron redox character.
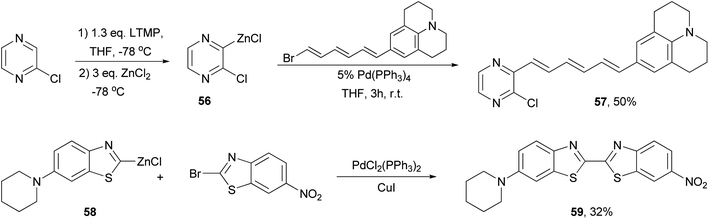 |
| | Scheme 16 Synthesis of push–pull NLO materials 57 and 59. | |
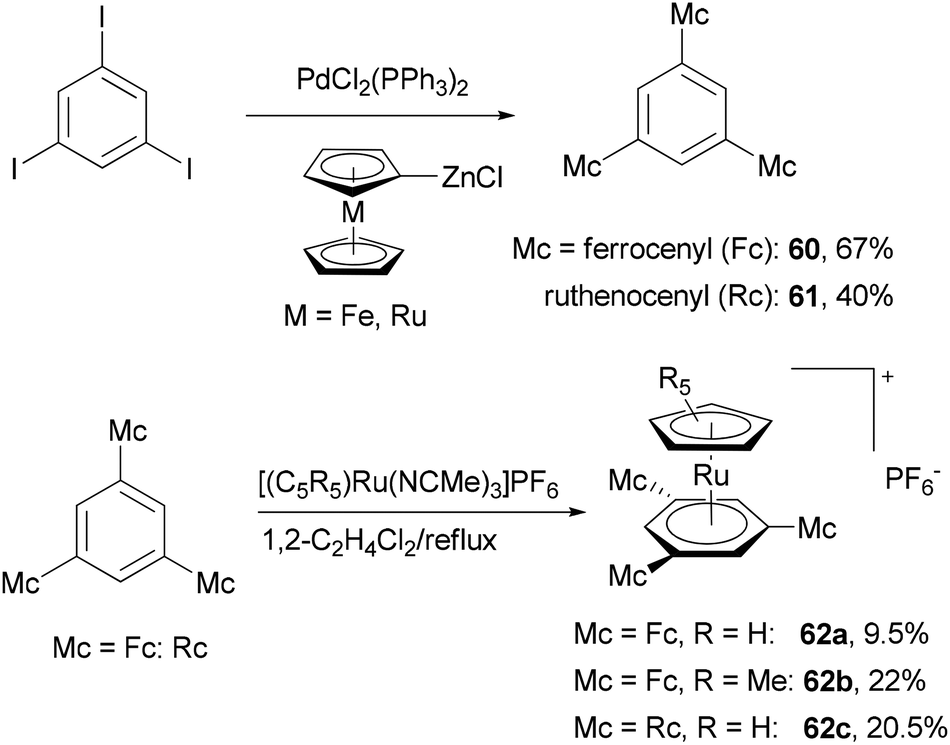
The Negishi cross-coupling reaction of 1,3,5-triiodobenzene with ferrocenyl- and ruthenocenylzinc chlorides yields the corresponding 1,3,5-trimetallocenylated benzene derivatives 60 and 61 in good yields (Scheme 17).73 When 1,3,5-tribromobenzene was used, the yields were much lower, being 35% and 13% for 60 and 61, respectively. The metallocenylzinc reagents were prepared by deprotonating the metallocenes with butyl lithium followed by treatment with zinc chloride.74 The resulting 60 and 61 were used as ligands to synthesize the trigonal-pyramidal four-sandwich NLO materials 62a–c. Compound 62a demonstrates a larger first hyperpolarizability than the all-ruthenium congener 62c due to the better electron-donating properties of the ferrocene substituents.
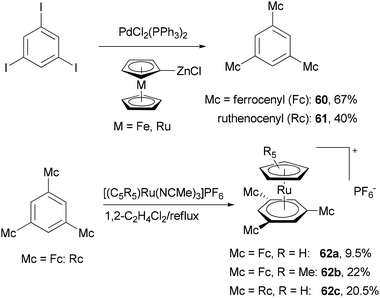 |
| | Scheme 17 Synthesis of the trigonal-pyramidal four-sandwich NLOphores 62a–c. | |
3.2 Liquid crystals
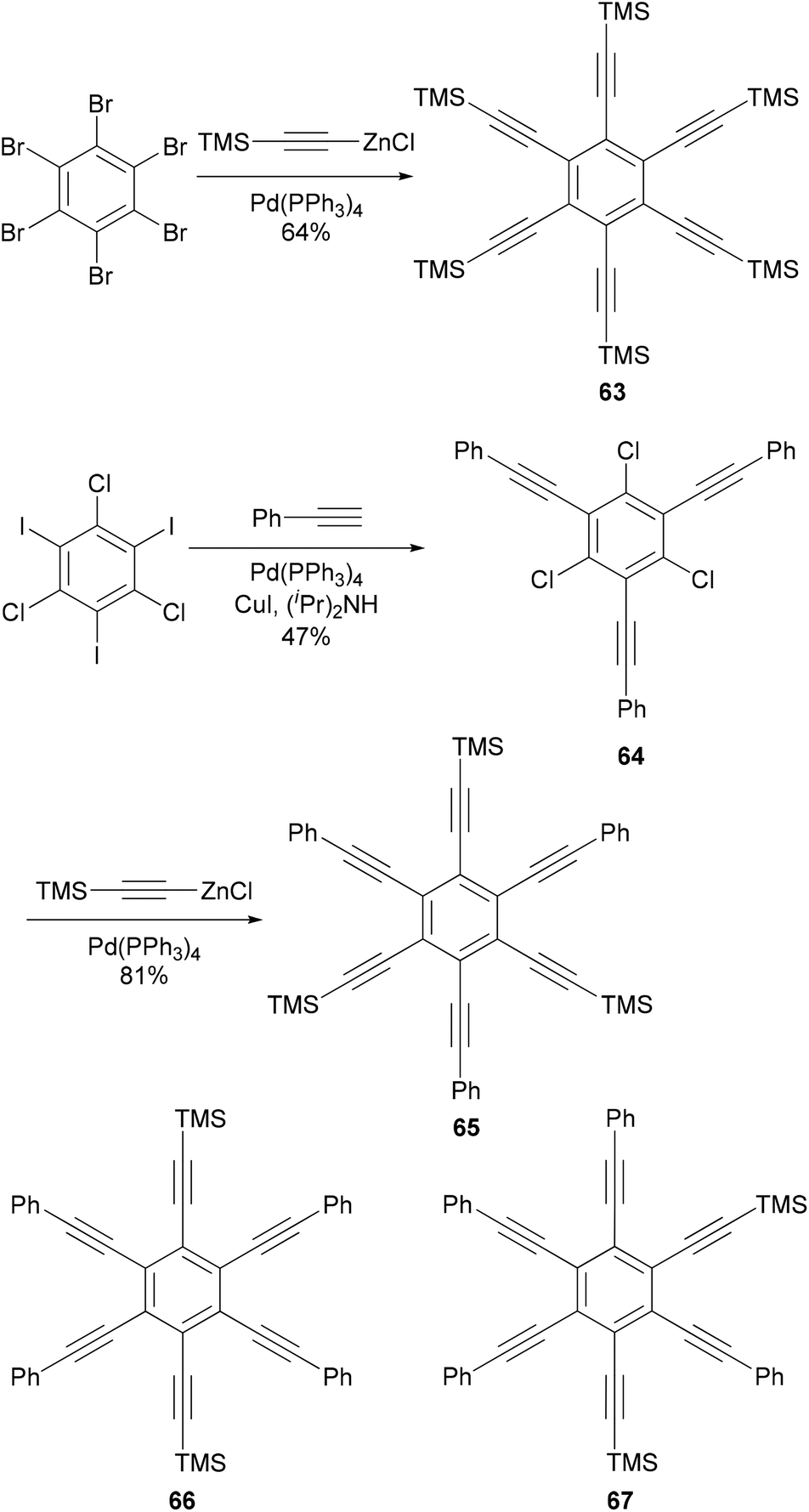
Hexaethynylbenzene derivatives are attractive molecules because of their potential applications as liquid crystals, nonlinear optical materials, core structures for dendritic materials, and building blocks for two-dimensional carbon network graphenes. Tobe and co-workers reported an efficient synthesis of hexaethynylbenzene 6375 (Scheme 18) using the Negishi alkynylation of hexabromobenzene. Toluene was added as the co-solvent to raise the refluxing temperature. The yields of 63 were much better than those reported previously using Sonogashira coupling.76,77 On the other hand, the reaction of 2,4,6-trichloro-1,3,5-triazine with [(trimethylsilyl)ethynyl]zinc chloride in the presence of Pd(PPh3)4 catalyst proceeded smoothly at room temperature to give 2,4,6-tris[(trimethylsilyl)ethynyl]triazine in 80% yield, reflecting the higher reactivity of electron-deficient heteroaryl halides.75 A stepwise Sonogashira and Negishi coupling reaction of chloroiodobenzene derivatives with appropriate ethynyl reagents efficiently produced differentially substituted hexaethynylbenzenes. For instance, the Sonogashira coupling of 1,3,5-trichloro-2,4,6-triiodobenzene with phenylacetylene produced 64 in 47% yield, which was further cross coupled with [(trimethylsilyl)ethynyl]zinc chloride in the presence of Pd(PPh3)4 to introduce three more ethynyl groups and give the hexasubstituted benzene 65 in 81% yield (Scheme 18).75 The other two compounds 66 and 67 were prepared similarly from appropriate hexahalobenzenes. Negishi coupling gives generally satisfactory yields. The success of this differential substitution method clearly benefits from the higher reactivity of organozinc reagents.
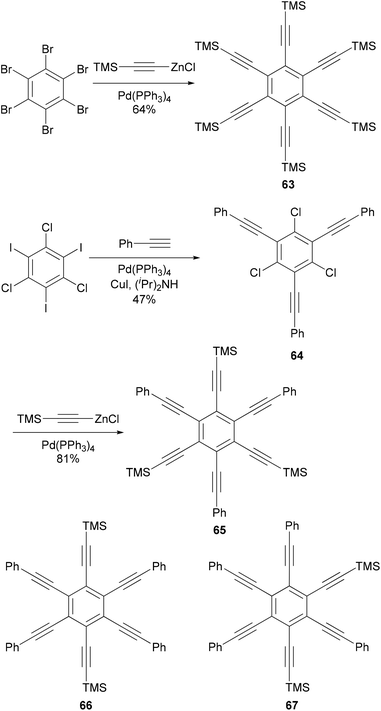 |
| | Scheme 18 Synthesis of hexaalkynylbenzenes 63 and 65. | |
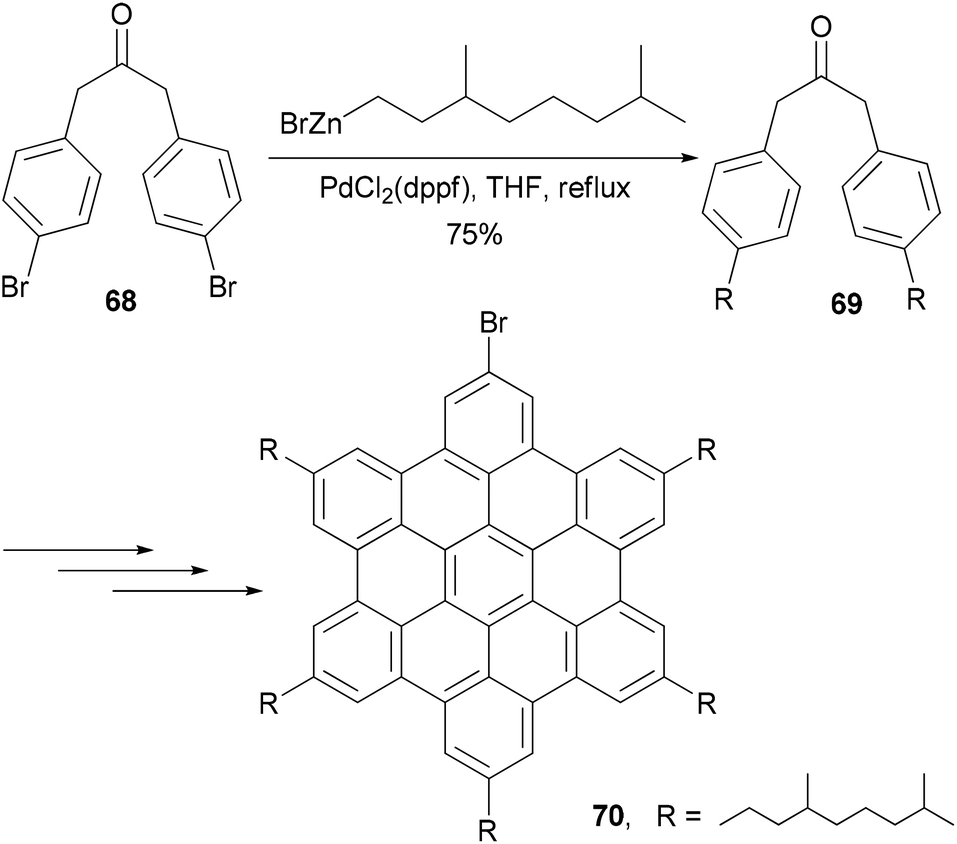
Alkyl- or phenyl-alkyl substituted hexabenzocoronenes (HBCs) are discotic liquid crystalline materials with extremely large phase widths (up to 250 °C)78 and charge carrier mobilities along the columnar axis (1.13 cm2 V s−1).79 In the synthesis of thermotropic liquid crystalline derivatives of hexabenzocoronene (HBC) substituted at the periphery by one bromo and five alkyl groups, palladium catalyzed cross coupling reactions were used to improve the synthesis of the HBC precursors.80 Specifically, Negishi coupling was used to introduce the flexible alkyl chain in the synthesis of a precursor 69 that is used in the synthesis of the mono bromo HBC 70 (Scheme 19). The cross coupling of the dibromide 68 with 3,7-dimethyloctanylzinc bromide in the presence of PdCl2(dppf) afforded 69 in 75% yield. Kumada coupling with the Grignard reagent was also successful, but involves tedious steps of protecting and deprotecting the carbonyl group. It should be noted that in the absence of functional groups, Kumuda coupling works very well as reported in this paper.
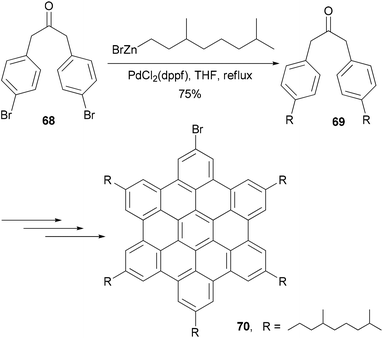 |
| | Scheme 19 Synthesis of alkylated hexabenzocoronene 70. | |
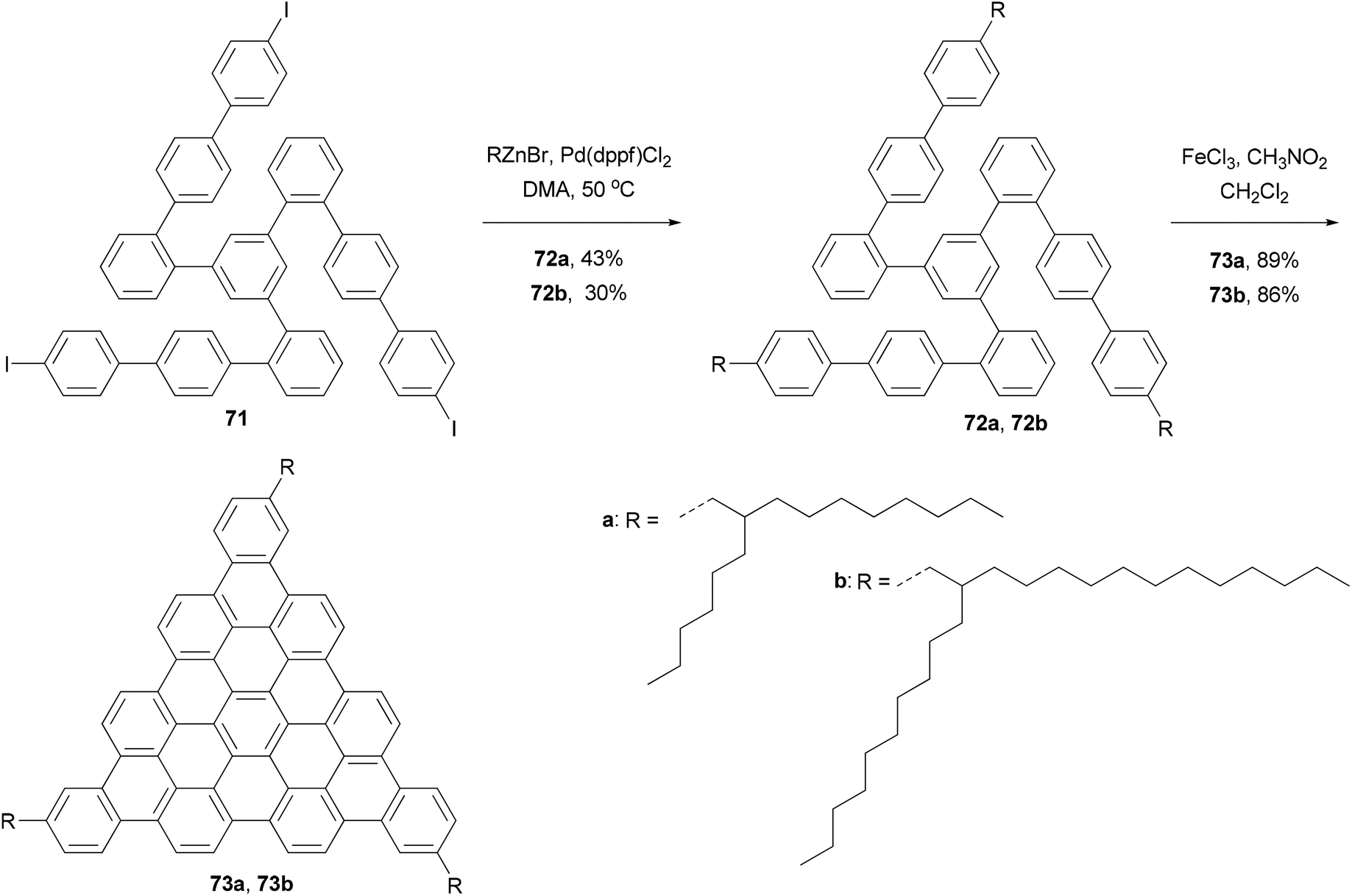
In the synthesis of triangle-shaped discotic graphenes with three swallow-tailed alkyl substituents 73a and 73b,81 a three-fold cross coupling is required to introduce the alkyl substituents. Kumada coupling was unsuccessful because of the inactivity and server homo-coupling of the magnesium reagents. The author then turned to Negishi coupling using the corresponding zinc reagents easily prepared using Huo's protocol,33 and the desired products 72a and 72b were obtained in reasonable yields (Scheme 20). Both 73a and 73b exhibit a characteristic discotic liquid crystalline phase with an extremely high stability; they did not have an isotropic phase up to 500 °C. The introduction of the swallow-tailed substituents allows a facile purification, control over the thermotropic properties, and solution fabrication of highly efficient photovoltaic devices. When 73b was blended with N,N′-bis(1-ethylpropyl)-3,4,9,10-perylenetetracarboxy-diimide (PDI) in a ratio of 4:6, and was spin-coated to a photovoltaic device with the structure of indium tin oxide (ITO)/(73b + PDI)/Ag, an external quantum efficiency (EQE) of 19% at 490 nm of illumination was achieved.81
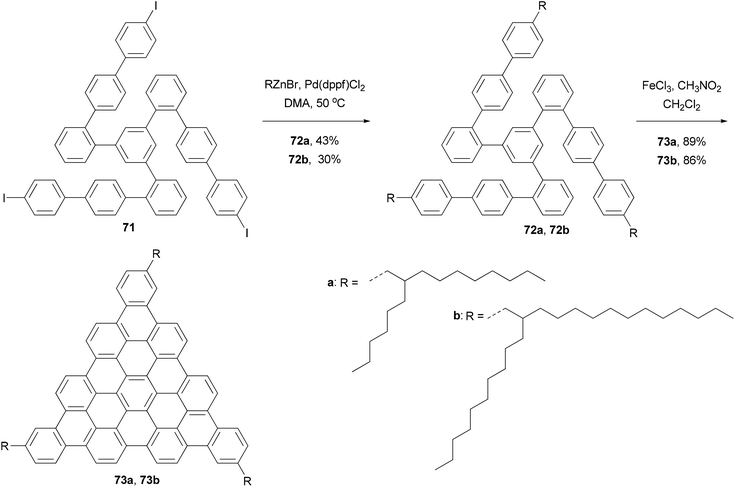 |
| | Scheme 20 Synthesis of alkylated triangle-shaped graphenes 73a and 73b. | |
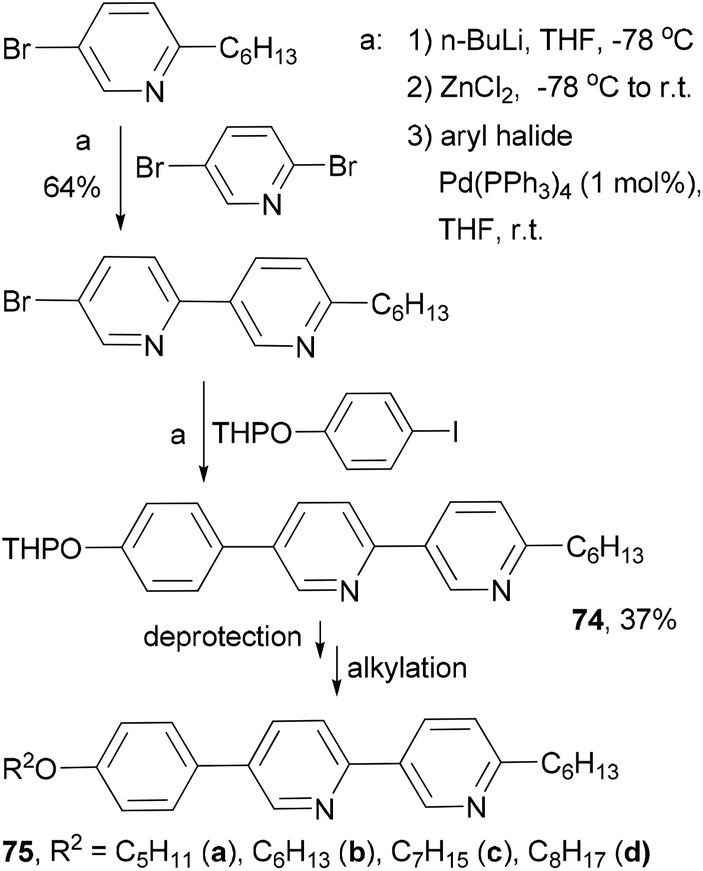
Fifteen 2,5-disubstituted pyridine-based liquid crystals are synthesized by exploiting the different reactivities of the bromine atoms in 2,5-dibromopyridine under Negishi coupling conditions.82 The use of consecutive Negishi couplings to prepare the precursor 74 is illustrated in Scheme 21.
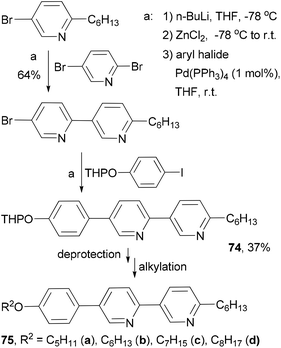 |
| | Scheme 21 Synthesis of liquid crystals 75a–d. | |
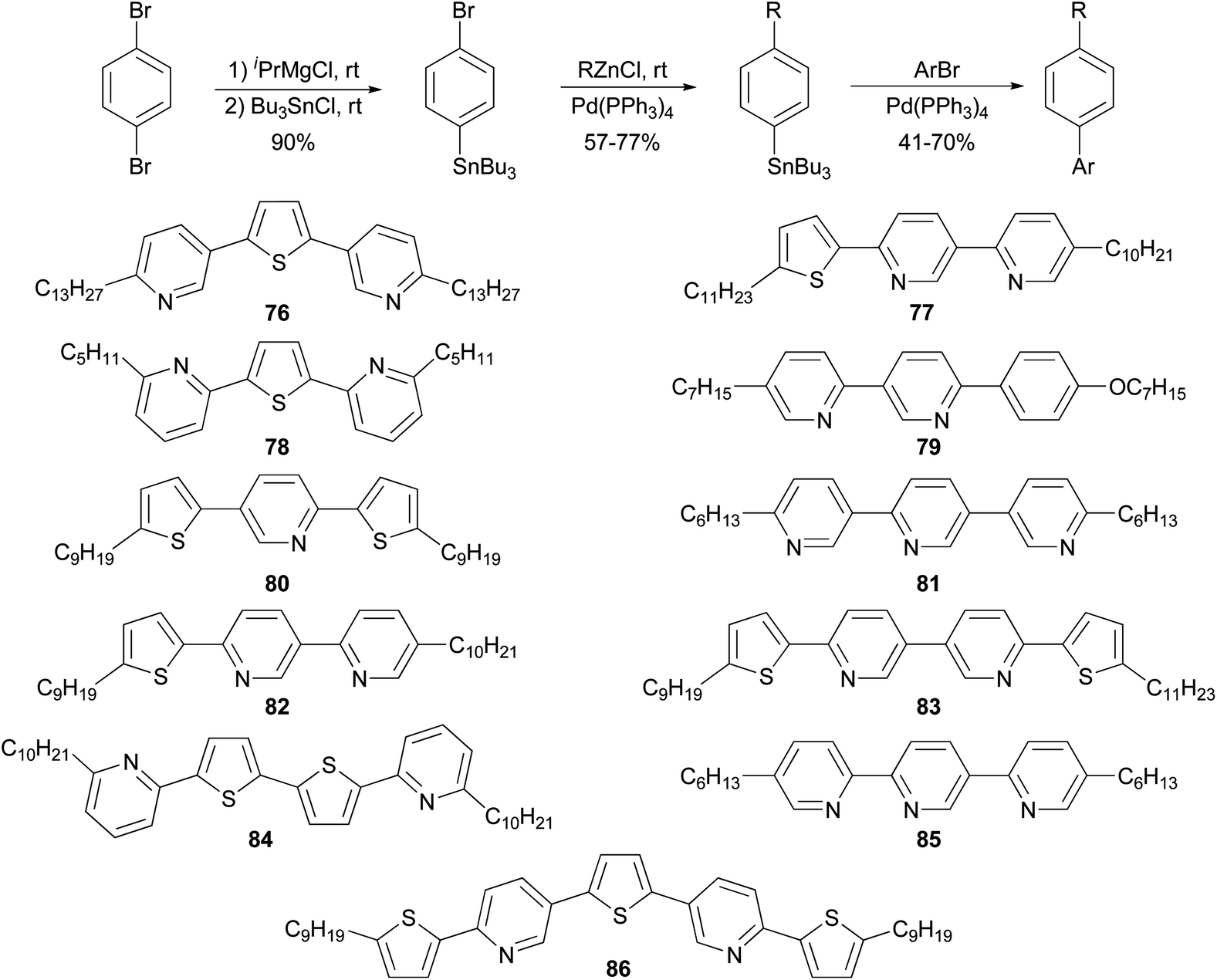
The high reactivity of organozinc reagents in Negishi coupling has been often exploited in selective cross coupling in the presence of less reactive stannyl and boron groups. For example, the 2-bromo-5(or 6)-tributylstannylpyridines, prepared from dibromopyridines and i-PrMgCl at room temperature, undergo Negishi coupling with either alkyl or arylzinc chlorides without affecting the tributylstannyl group (Scheme 22).83 The newly produced alkyl- and arylsubstituted pyridylstannanes are suitable for further functionalization by Stille coupling, which results in the synthesis of a group of 11 liquid crystalline materials 76–86 with aromatic cores comprised of pyridine and thiophene rings.
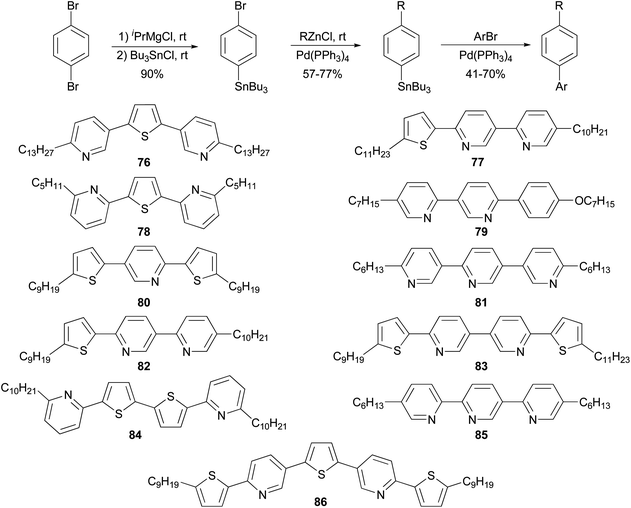 |
| | Scheme 22 Negishi coupling in the presence of the organostannyl functional group. | |
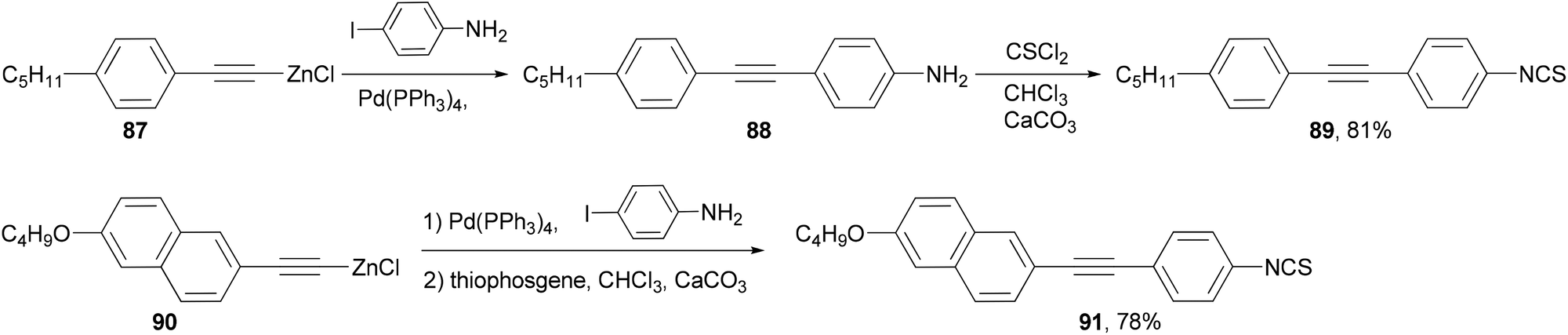
A number of structural moieties including core units (such as phenyl, naphthyl and thiophenyl), linking groups (such as ethynyl), terminal substituents (such as cyano, isothiocyanato and fluoro), and lateral fluoro substituents have been incorporated into materials designed to confer a high birefringence on nematic mixtures.84 The materials are all prepared through convergent syntheses involving palladium-catalyzed cross-coupling reactions. In the demanding case of introducing an aniline group, the Negishi coupling of alkynylzinc reagents 87 and 90, which can be readily prepared from the terminal alkyne by deprotonation with butyllithium and transmetalation with zinc chloride, had to be used to synthesize liquid crystal molecules 89 and 91 (Scheme 23).
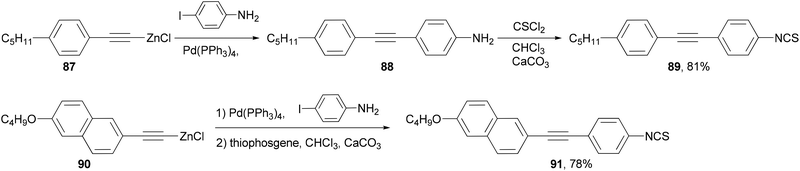 |
| | Scheme 23 Synthesis of 89 and 91. | |
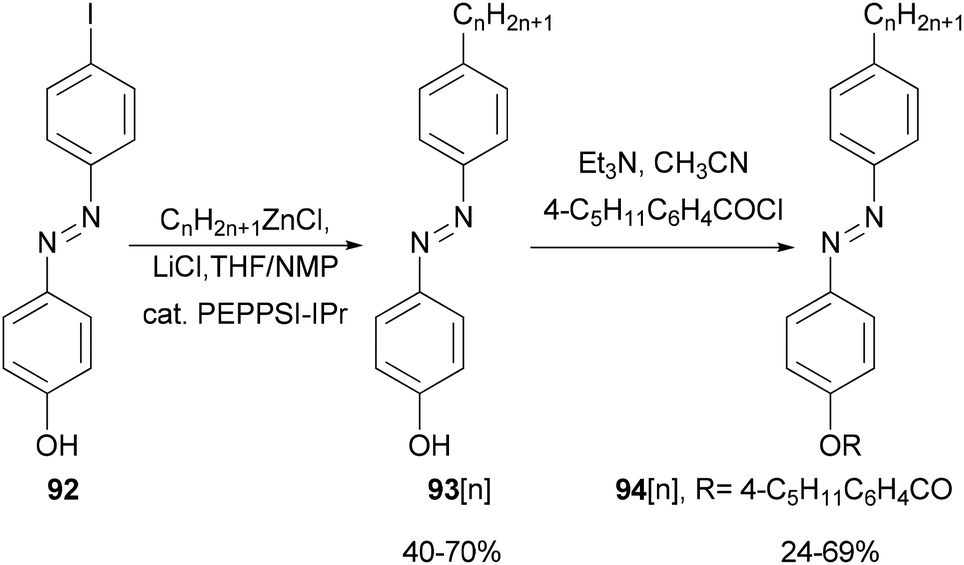
Functionalized azobenzenes were among the first successful nematic liquid crystals used in the display industry. A homologous series of 4-(4-alkylphenylazo)phenols 93[n] (n = 2–22, even number of carbon atoms in the alkyl chains) were prepared in yields of 40–70% (Scheme 24)85 by Negishi coupling of alkylzinc chlorides with 4-(4-iodophenylazo)phenol (92) under Organ's conditions.86 The phenols were converted to the corresponding 4-pentylbenzoates 94[n], which exhibit enantiotropic nematic phases even for 94[22].
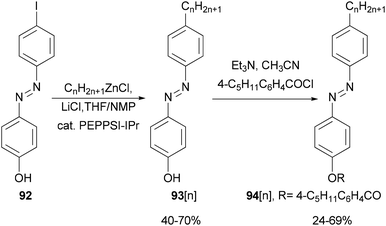 |
| | Scheme 24 Synthesis of 94[n]. | |
3.3 Light-emitting materials
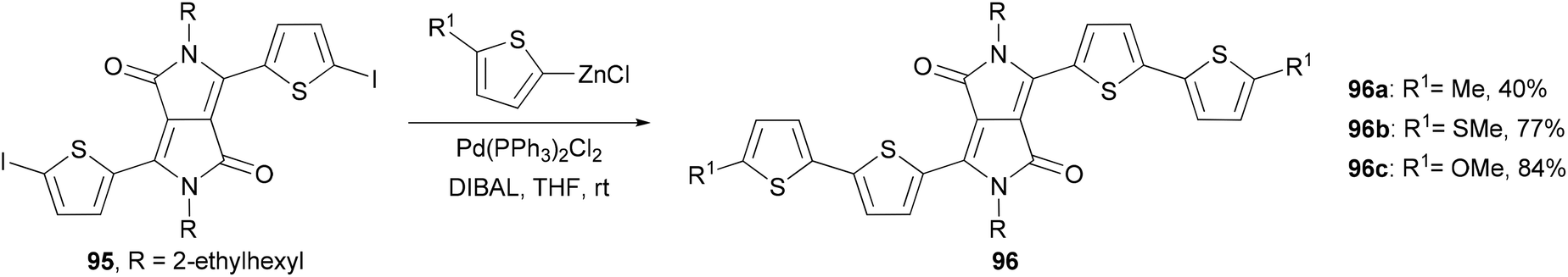
A series of 2,2′-bithiophene-functionalized diketopyrrolopyrrole (DPP) fluorescent dyes bearing different electron-donating and electron-withdrawing substituents at the terminal thiophene units were synthesized by palladium-catalyzed cross-coupling reactions including Negishi coupling (Scheme 25).87 The diiodide 95 was prepared for the first time in 67% yield by deprotonating the corresponding thiophene-functionalized diketopyrrolopyrrole with LDA (lithium diisopropylamide) followed by iodinolysis. The cross coupling of 95 with 5-substituted 2-thioenylzinc chlorides gives 96a–c, which display intense red emissions upon photoexcitation in a solution of dichloromethane.
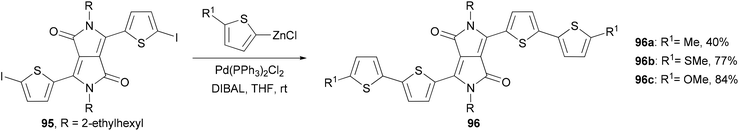 |
| | Scheme 25 Synthesis of fluorescent dyes 96a–c. | |
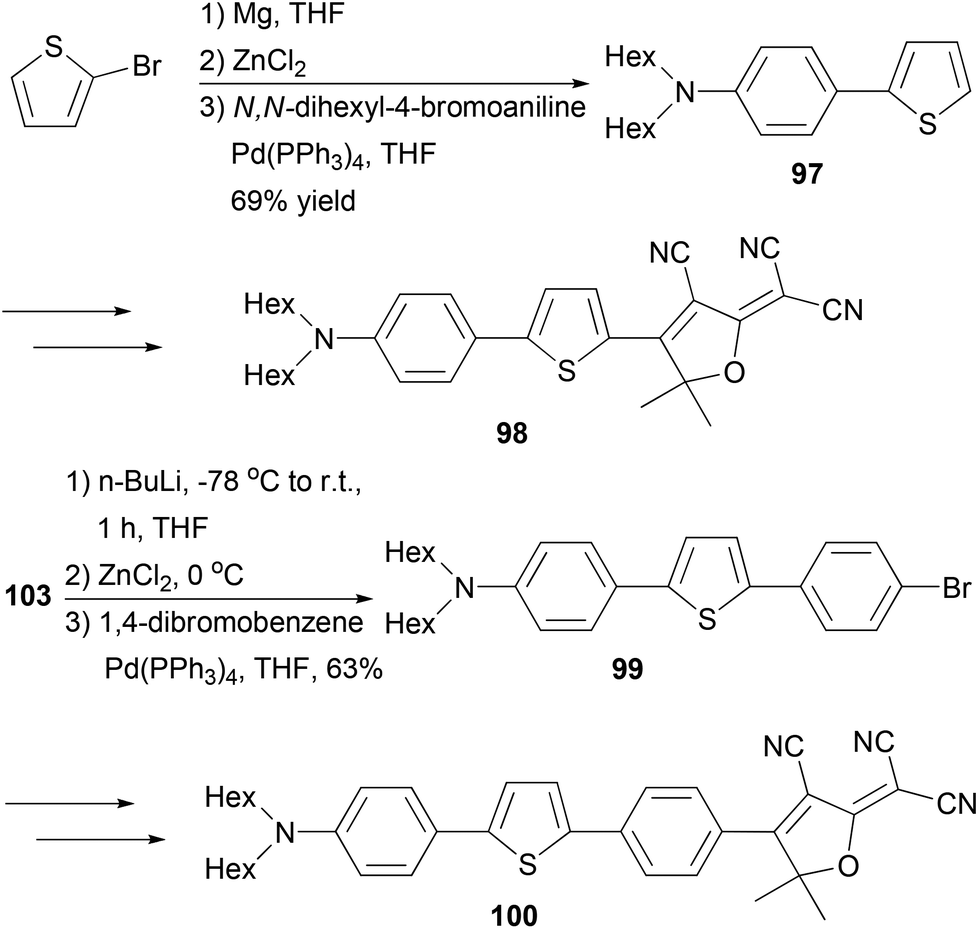
Fluorescent dye materials 98 and 100 comprising of an amine donor, a conjugated system, and a dicyanomethylenedihydrofuran acceptor group were synthesized (Scheme 26). The conjugated system was constructed by Negishi coupling.88 The dye 97 emits in red (λmax = 631 nm) with a high quantum yield of 74%, while compound 100 with extended conjugation emits in deep red (λmax = 709 nm) with an impressive quantum yield of 34% in toluene.
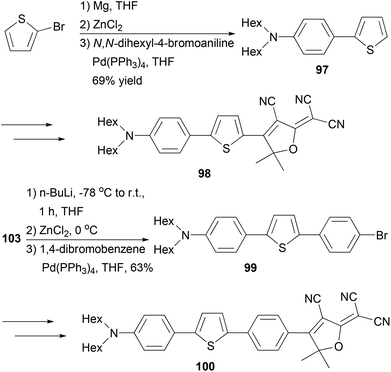 |
| | Scheme 26 Synthesis of fluorescent dyes 97 and 100. | |
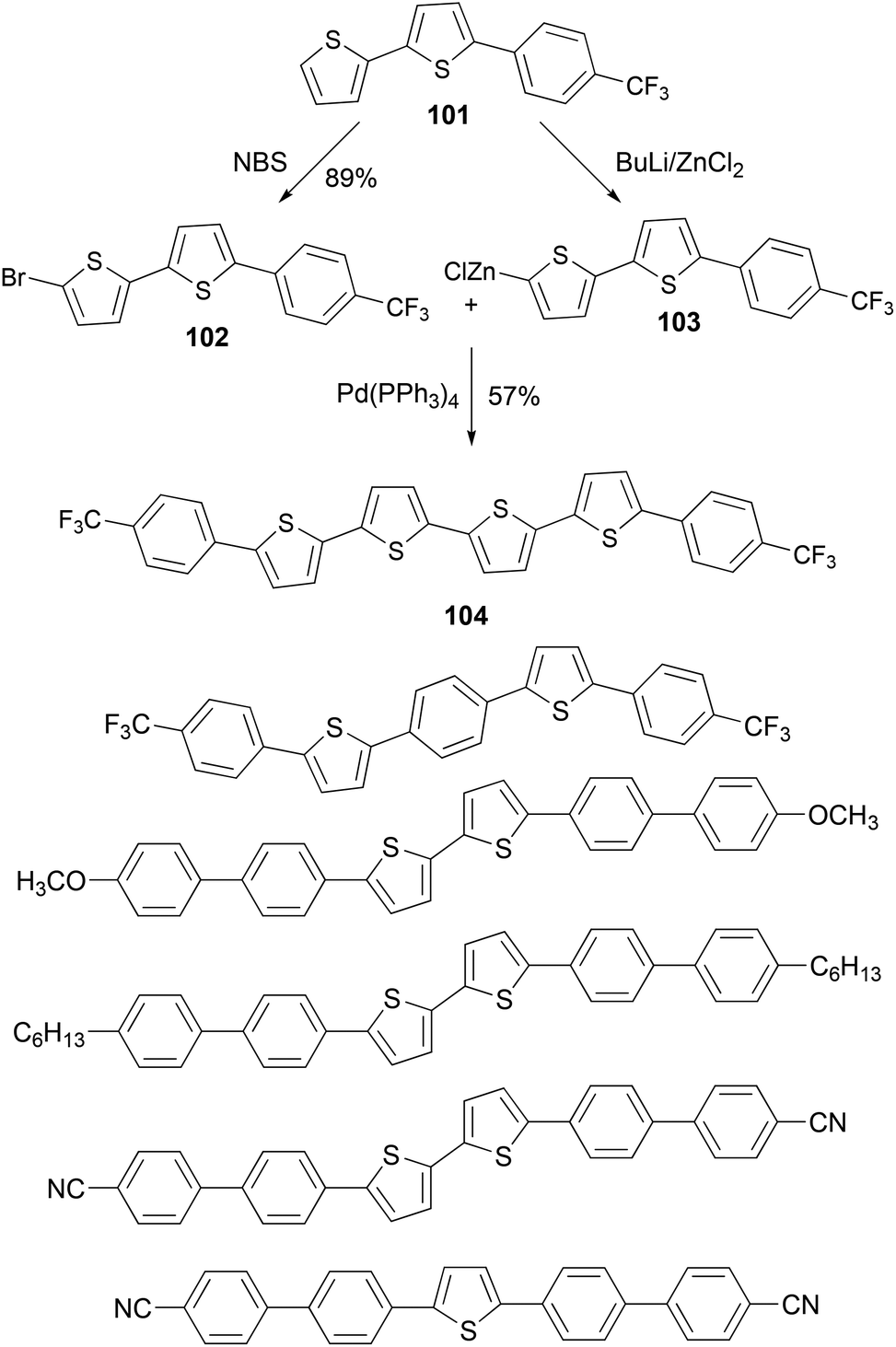
A series of thiophene–phenylene co-oligomers have been synthesized using the Negishi coupling approach,89 as listed in Scheme 27. Some of the syntheses are carried out in a convergent manner. For instance, the bithiophene derivative 101 was brominated to form bromide 102 and was converted to the organozinc reagent 103, and the cross coupling of 102 and 103 produced the thiophene–phenylene conjugated oligomer 104. These oligomers can be used in light-emitting transistors.90
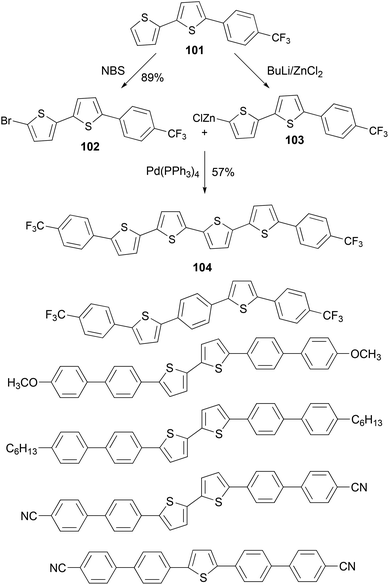 |
| | Scheme 27 Convergent synthesis of 104. | |
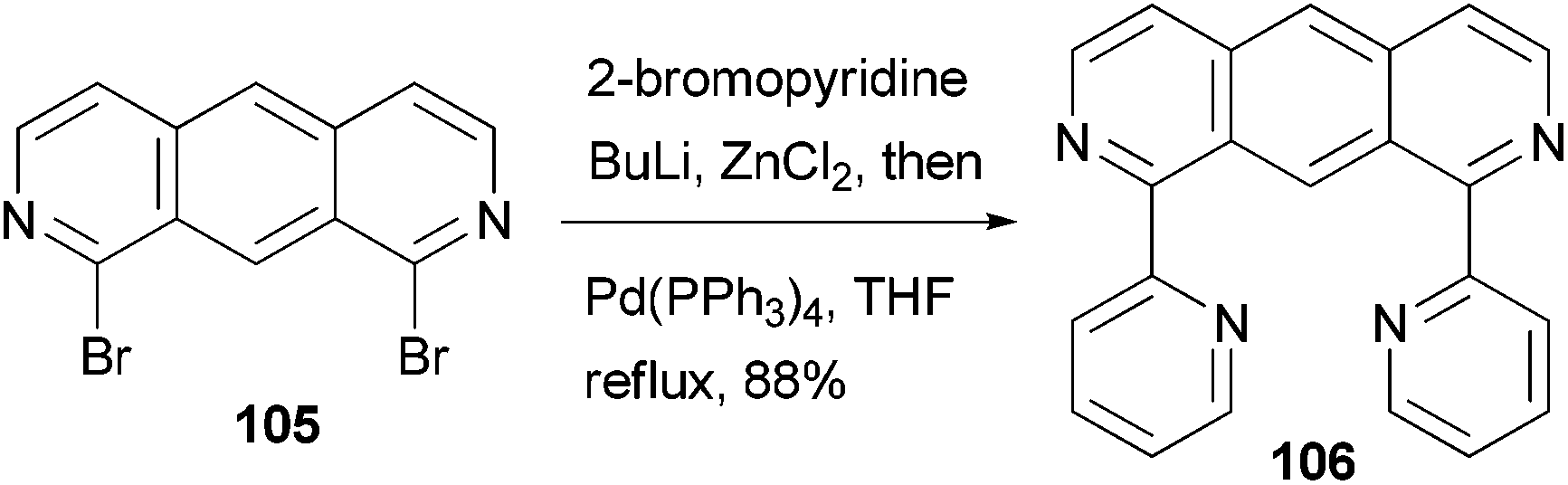
A fluorescent ditopic ligand 106 was synthesized by the cross coupling of 1,8-dibromo-2,7-diazaanthracene 105 with 2-pyridylzinc chloride, which is generated in situ from 2-bromopyridine (Scheme 28).91 A successful double cross-coupling reaction requires thoroughly dried zinc chloride (24 h at 55 °C under reduced pressure). Upon irradiation at 366 nm, ligand 106 dimerizes at the 9 and 10 positions to form the head-to-tail tetra-bpy ligand. This thermally stable photodimer can be dissociated back to 106 using high energy irradiation (254 nm). The photodimerization can be easily monitored by the decrease in the photoluminescence of 106.
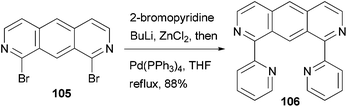 |
| | Scheme 28 Synthesis of photodimerizable ditopic ligand 106. | |
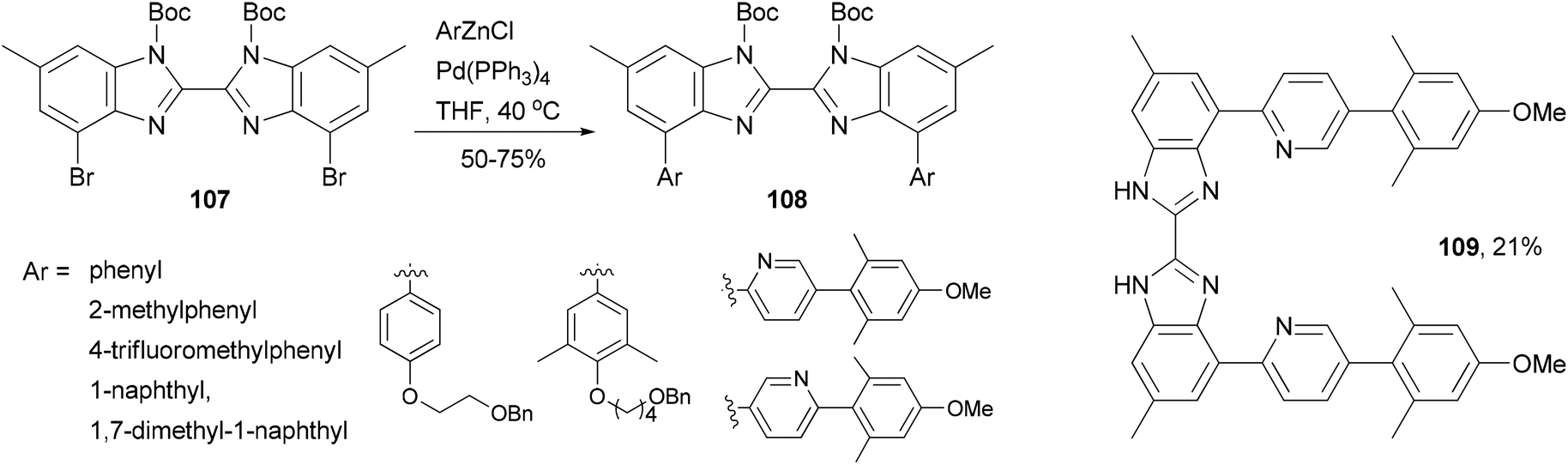
A series of 4,4′-bisaryl-2,2′-bisbenzimidazoles 108 have been synthesized from the corresponding 4,4′-dibromo-2,2′-bisbenzimidazoles 107 by Negishi coupling reactions (Scheme 29).92 This procedure affords highly substituted bisbenzimidazoles. In the case of the cross coupling of 107 with a 2-pyridylzincate, the deprotected form 109 was obtained under the reaction conditions. These bisbenzimidazole-based organic dyes may also serve as multi-dentate ligands for metal complexes.
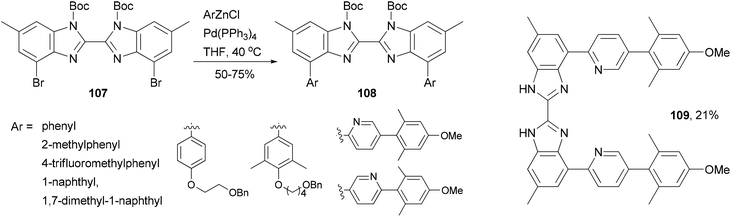 |
| | Scheme 29 Synthesis of 4,4′-bisaryl-2,2′-bisbenzimidazoles 108 and 109. | |
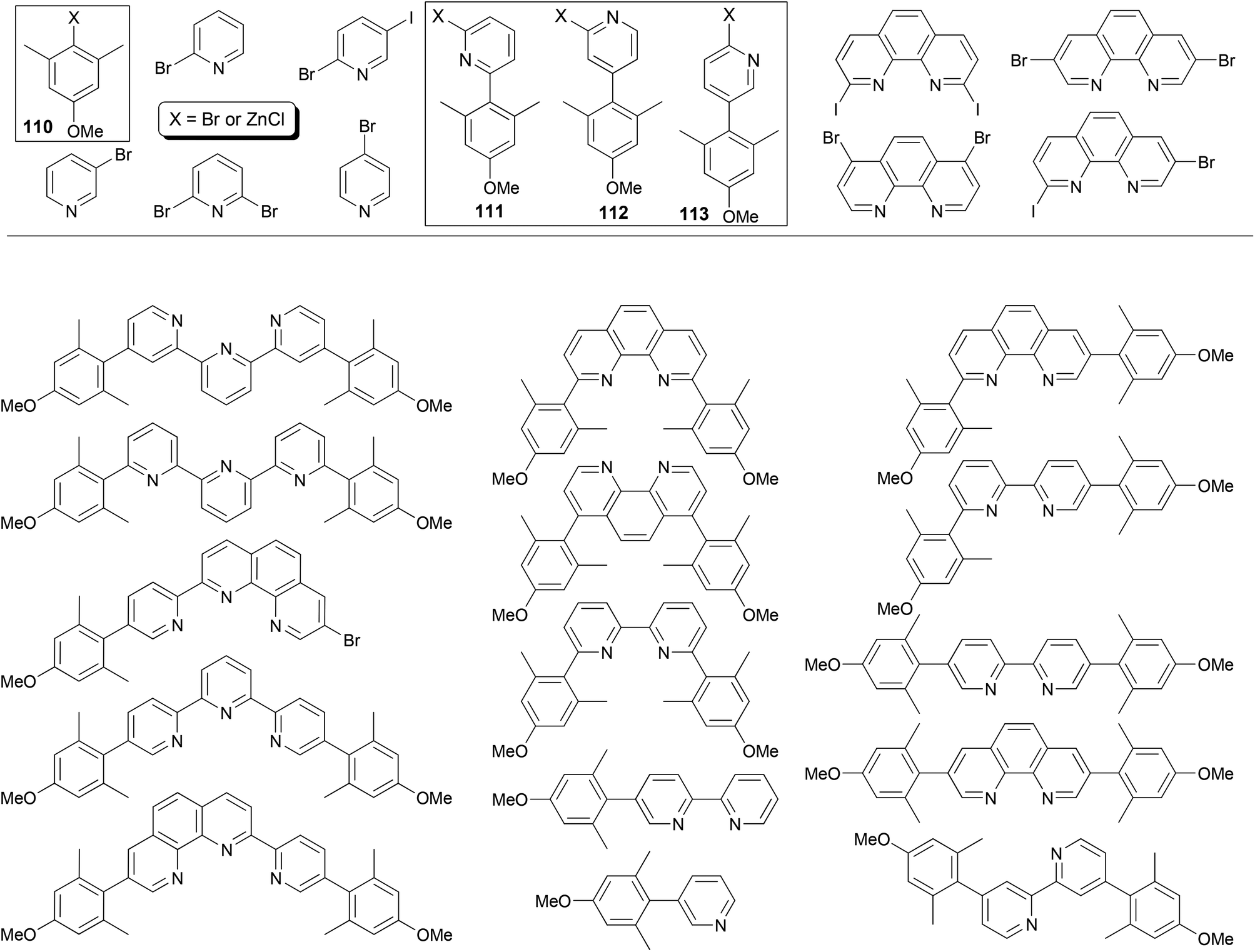
The power of Negishi coupling may be best illustrated by a simple, general procedure developed for the synthesis of a series of fluorescent α-, β-, and γ-substituted bipyridines, terpyridines, phenanthrolines, and pyridylphenanthrolines.93 The procedure is based on a series of manipulations of palladium-catalyzed cross coupling reactions of organozinc reagents and heteroaryl halides. The method has been showcased with the “manisyl” (4-methoxyl-2,6-dimethylphenyl) group as the aryl prototype (Fig. 1). Starting from commercially available 3,5-dimethylanisole, 4-methoxy-2,6-dimethylbromobenzene 110-Br (110, X = Br) was prepared according to a literature procedure94 and then converted into the organozinc reagent 110-ZnCl. The Negishi coupling of 110-ZnCl with 2,6-dibromopyridine and 2-bromo-5-iodopyridine gave 111-Br and 112-Br, respectively. The cross coupling of 110-ZnCl with 4-bromopyridine (in the hydrochloride form) gave 113-H (113, X = H), which was converted to 113-NH2 (113, X = NH2) under Chichibabin conditions95 then 113-I (113, X = I) under non-aqueous Sandmeyer conditions.96 Compounds 111-Br, 112-Br, and 113-I can be converted into their organozinc reagents 111-ZnCl, 112-ZnCl, and 113-ZnCl, respectively.97 Pairing for Negishi coupling by choosing one from the four organozinc reagents 110–113-ZnCl and the other from the aryl halides listed in Fig. 1 including 110–112-Br and 113-I creates a large array of ligand patterns (Fig. 1), which were synthesized in generally satisfactory yields (60–80% in most cases). The compounds emit from ultraviolet to blue light and exhibit quantum yields as high as 87% in acetonitrile. The simplicity and efficiency of Negishi coupling can hardly be matched by other cross coupling methods in this remarkable approach, especially with Negishi coupling as essentially the sole player.
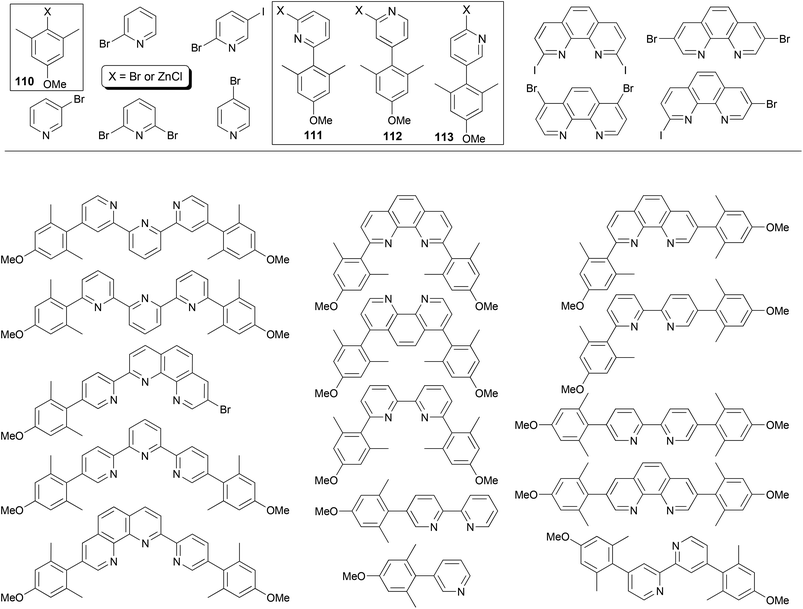 |
| | Fig. 1 An array of fluorescent ligands made by Negishi coupling. | |
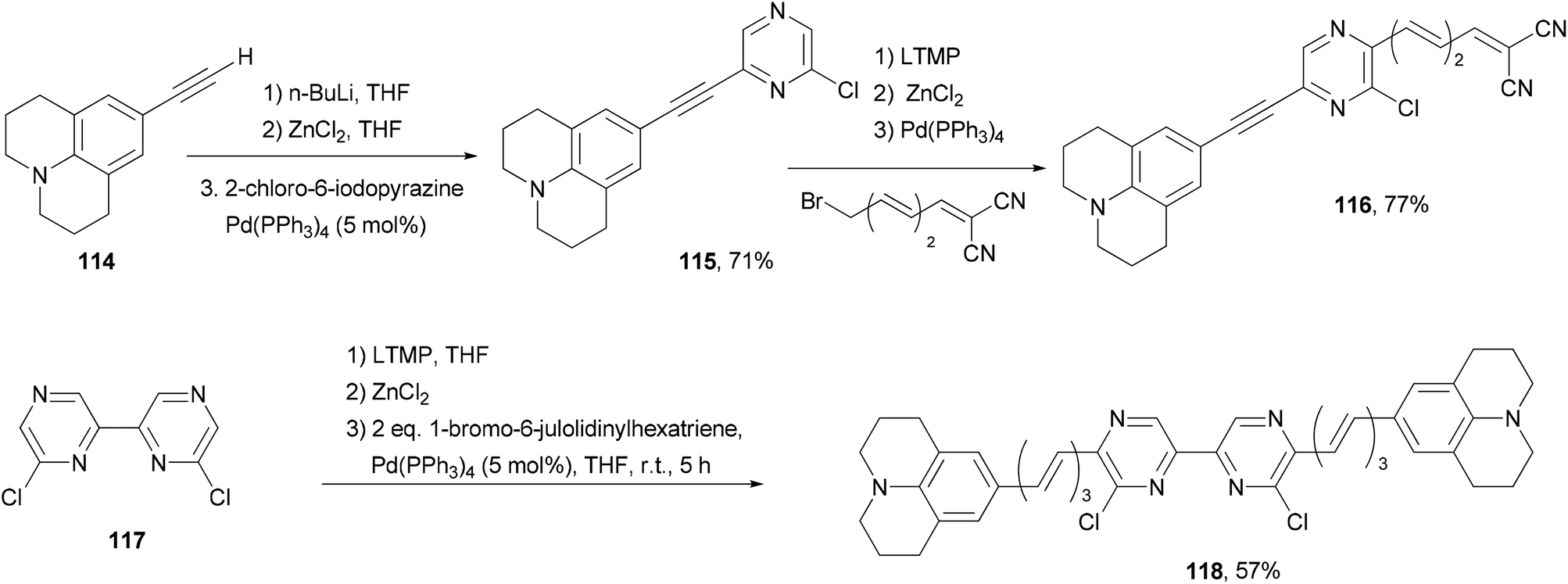
Back-to-back Negishi couplings have been used to prepare push–pull molecule 116 with extended conjugation (Scheme 30).98 Terminal alkyne 114 was first converted to the corresponding zinc reagent and then cross coupled with 2-chloro-6-iodopyrazine chemoselectively to produce 115 in good yield. Compound 115 was selectively deprotonated with LTMP and converted to a zinc reagent, which reacted with (3E,5E)-6-bromo-1,1-dicyanohexa-1,3,5-triene to give 116 in good yield. Two-fold Negishi coupling is required for the synthesis of 118 from 6,6′-dichloro-2,2′-bipyrazine (117). These molecules exhibit emissions in the near infrared region (707–744 nm). They also absorb strongly blue to orange visible light.
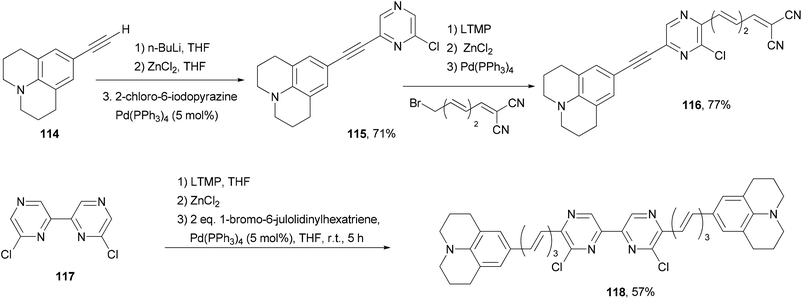 |
| | Scheme 30 Synthesis of near infrared fluorescent dyes 116 and 118. | |
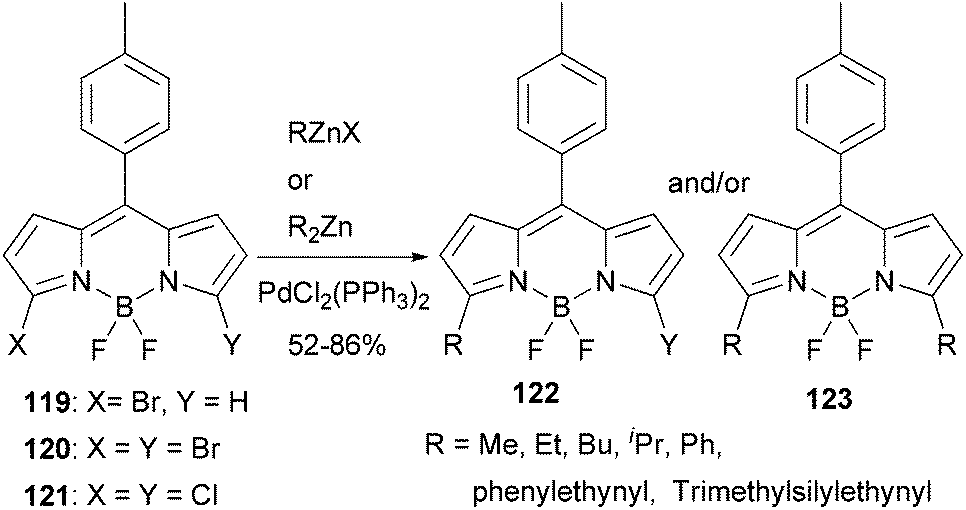
BODIPY (boron dipyrrin or boron dipyrromethene) dyes constitute one of the most important families of luminophores, due to their easily tunable absorption and emission properties.99 BODIPY dyes have very broad photonic applications such as sensors, laser dyes, photodynamic therapy, OLEDs and solar cells. A recent study shows that Negishi coupling is a promising tool for modifying BODIPY dyes.100 3-Bromo, 3,5-dibromo, and 3,5-dichloroBODIPYs, 119, 120, and 121, respectively, reacted smoothly with various organozinc reagents in the presence of PdCl2(PPh3)2 to give the 3,5-substituted BODIPY derivatives (Scheme 31). Control of the reaction to obtain monosubstituted products from both 120 and 121 is possible, which allows the introduction of two different groups at the 3- and 5-positions. Most of the reactions gave satisfactorily high yields except for benzylation, which took place with a low yield of 20%. A modified ligand may be required in this demanding case.
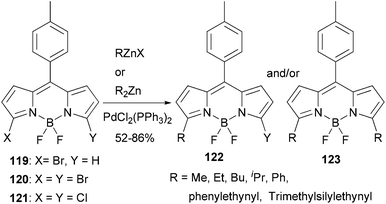 |
| | Scheme 31 Modification of BODIPYs using Negishi coupling. | |
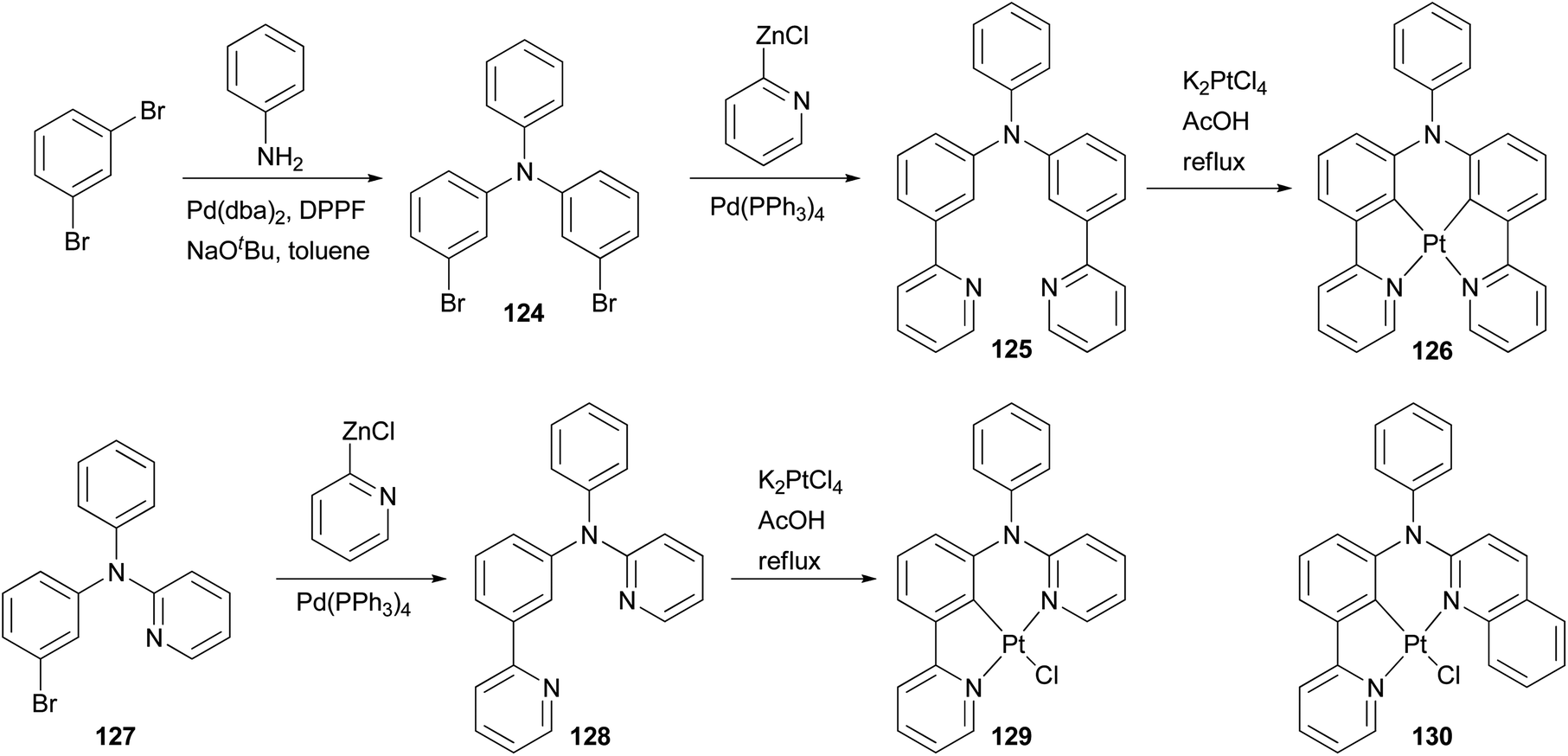
Phosphorescent materials based on transition metal complexes have recently attracted a great deal of attention because of their various applications in chemical and biological fields, and in particular as triplet emitters in OLED devices.101 The ligands for these complexes are typically conjugated aromatic and/or heteroaromatic assemblies that can be easily constructed by palladium-catalyzed cross coupling reactions. Huo and coworkers have designed and synthesized several types of tridentate and tetradentate cyclometalating ligands. The platinum complexes based on these ligands possess a less common five–six-membered metallacycle, and display high photoluminescence quantum yields in dichloromethane at room temperature.102–106 Their application in OLED devices has been demonstrated.102 An efficient synthetic strategy based on the combination of palladium catalyzed C–C and C–N bond cross coupling reactions was developed to synthesize these cyclometalating ligands. As illustrated in Scheme 32, the Negishi coupling of 2-pyridylzinc chloride with dibromide 124 efficiently installs two pyridyl groups to give 125, which reacts with K2PtCl4 as a tetradentate cyclometalating ligand to form a red emitting complex 126.102 Ligand 128 for a highly luminescent platinum complex 129 was also prepared from 127 using Negishi coupling, and a similar approach was applied to the synthesis of orange emitter130.104
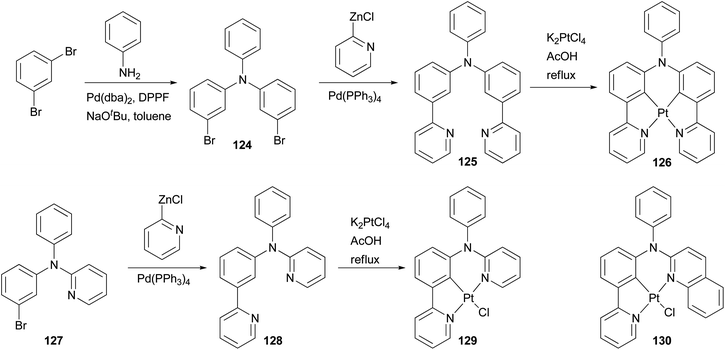 |
| | Scheme 32 Synthesis of cyclometalating ligands 125 and 128. | |
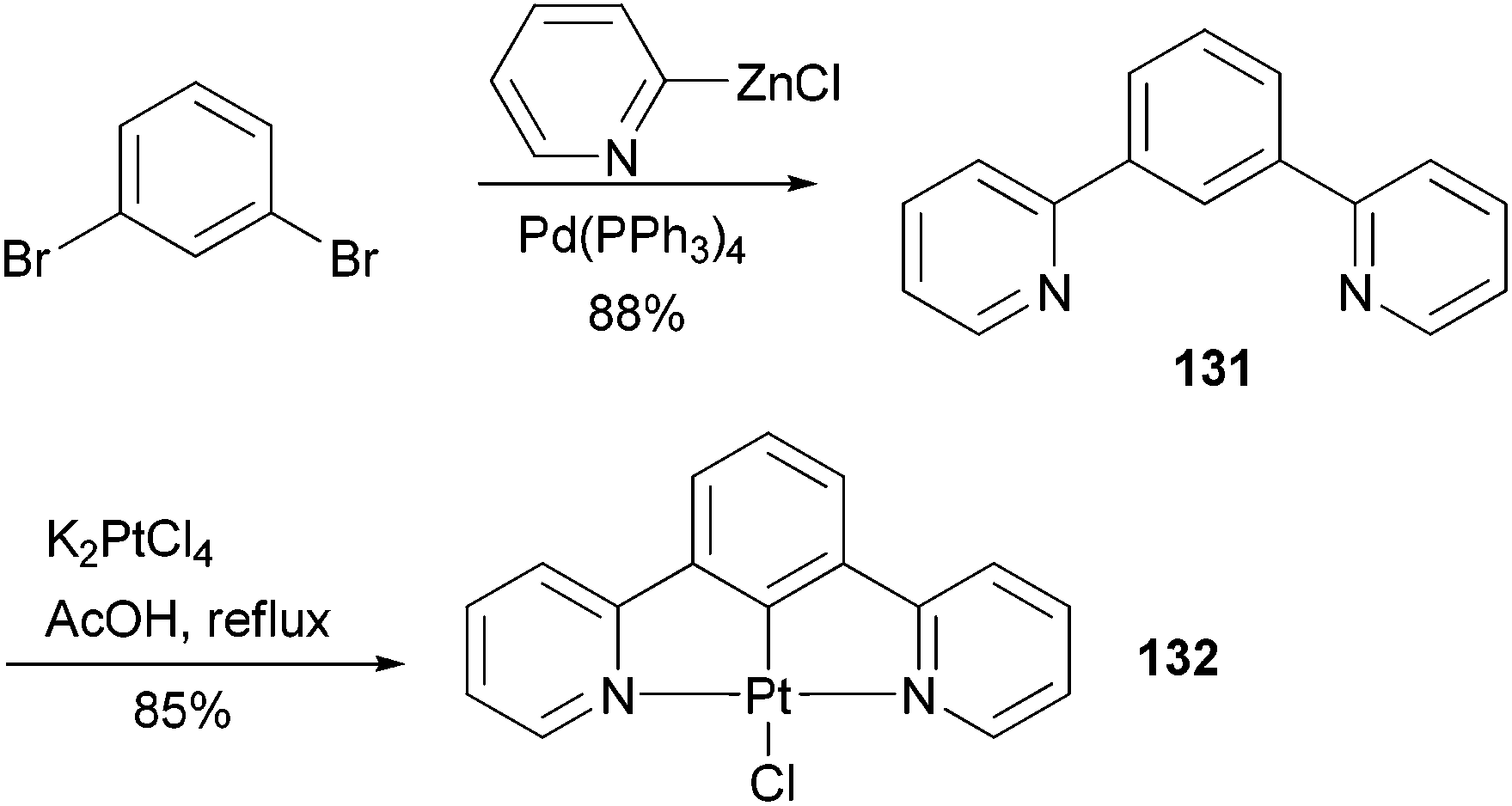
Complex 132107 is a prototype in the family of highly luminescent N^C^N-coordinated platinum complexes and has been used as a triplet emitter in OLED devices showing high quantum efficiency.107c The synthesis of ligand 131 originally relied upon the Stille coupling of 1,3-dibromobenzene with a 2-pyridylstannane reagent,107a however, recent reports demonstrated that Negishi coupling is a much better choice to construct this ligand and most likely its analogues as well (Scheme 33).107c,108
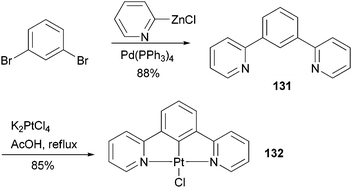 |
| | Scheme 33 Synthesis of 131. | |
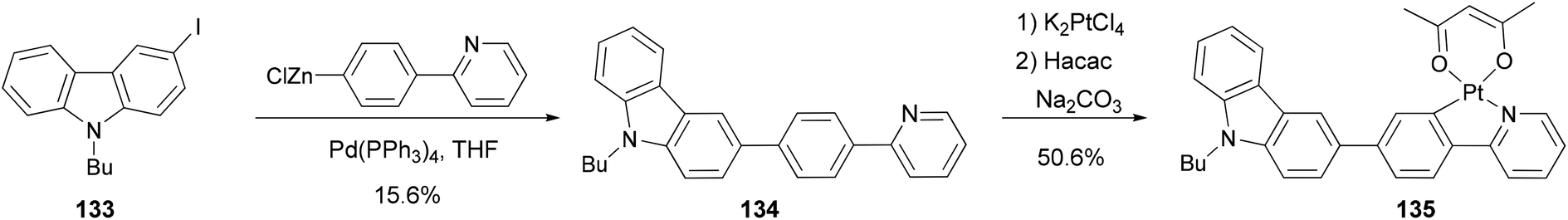
Zhao and coworkers109 synthesized a carbazole-capped cyclometalated platinum(II) complex 135 used in an oxygen sensor. The bidentate ligand 134 is synthesized by the Negishi coupling of 133 with the 4-(2-pyridyl)phenylzinc chloride generated from the corresponding bromide as shown in Scheme 34.
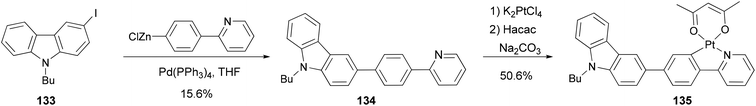 |
| | Scheme 34 Synthesis of bidentate ligand 134. | |
3.4 Light-harvesting materials
Porphyrins are important macrocyclic heteroaromatic compounds and play various important roles in nature, particularly in photosynthesis. They are commonly used as building blocks in supramolecular chemistry and recently as light harvesting components in dye-sensitized solar cells (DSSCs).110 One of the easiest ways to modify porphyrins is to make substitutions at their peripheral positions and Negishi coupling has proved to be an efficient tool for such transformation.111–114
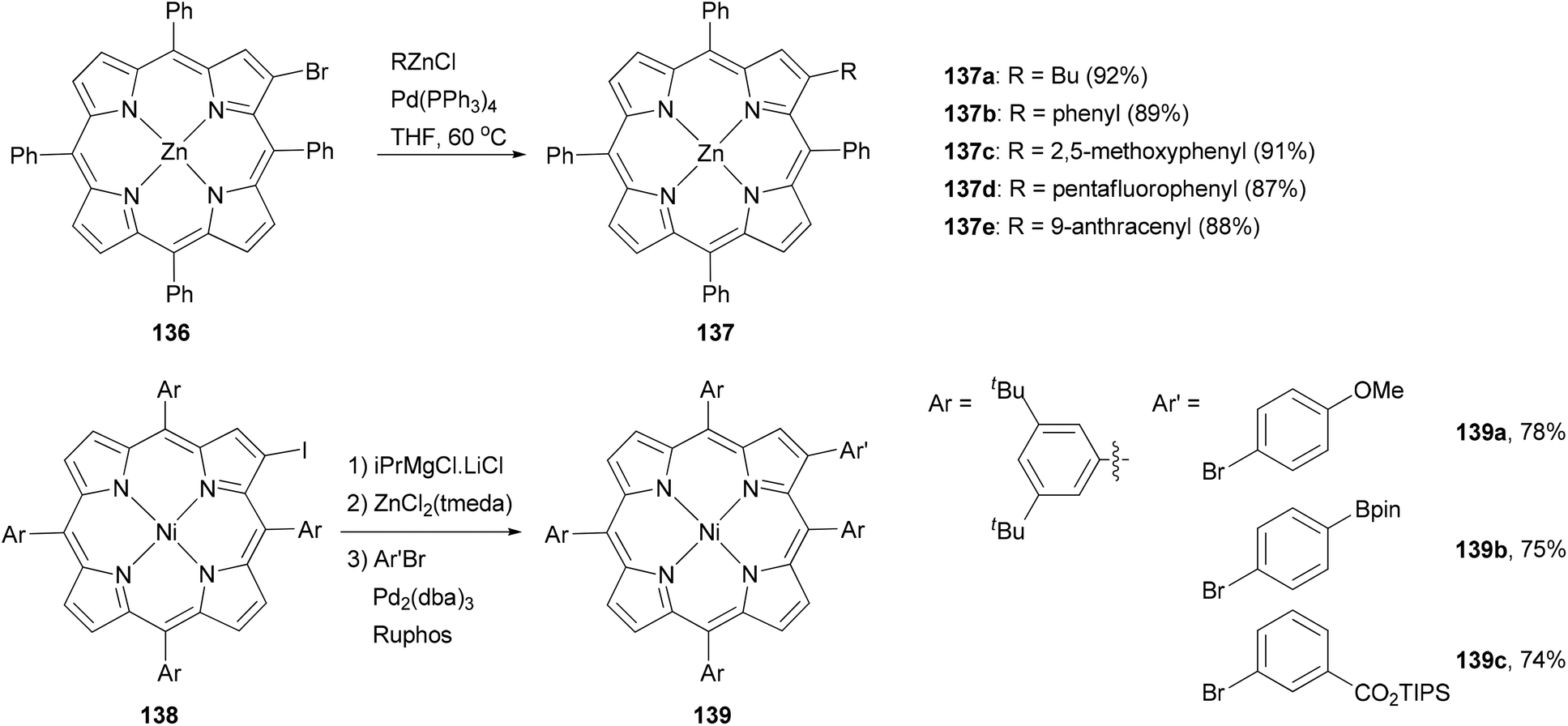
Functionalization at the β-position of porphyrins using Negishi coupling was first reported by Therien and co-workers.111 The β-bromoporphyrin 136 reacted with either butyl or arylzinc chloride in the presence of Pd(PPh3)4 to give the β-substituted product 137 in excellent yields (Scheme 35). More recently, a different approach was adopted by generating the zinc reagent from iodoporphyrin 138, which cross coupled with aryl bromides to give 139 in high yields.114
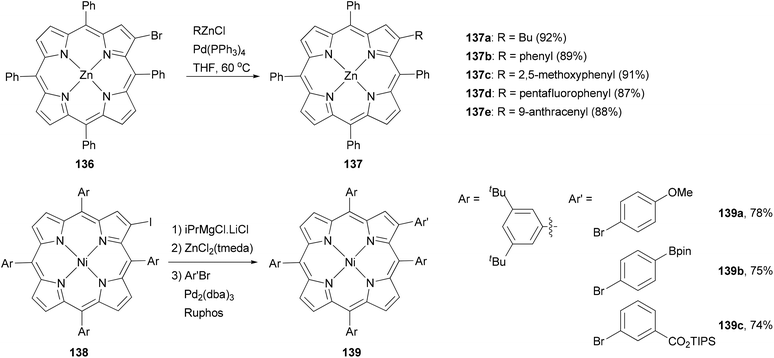 |
| | Scheme 35 Negishi coupling at the β position of porphyrins. | |
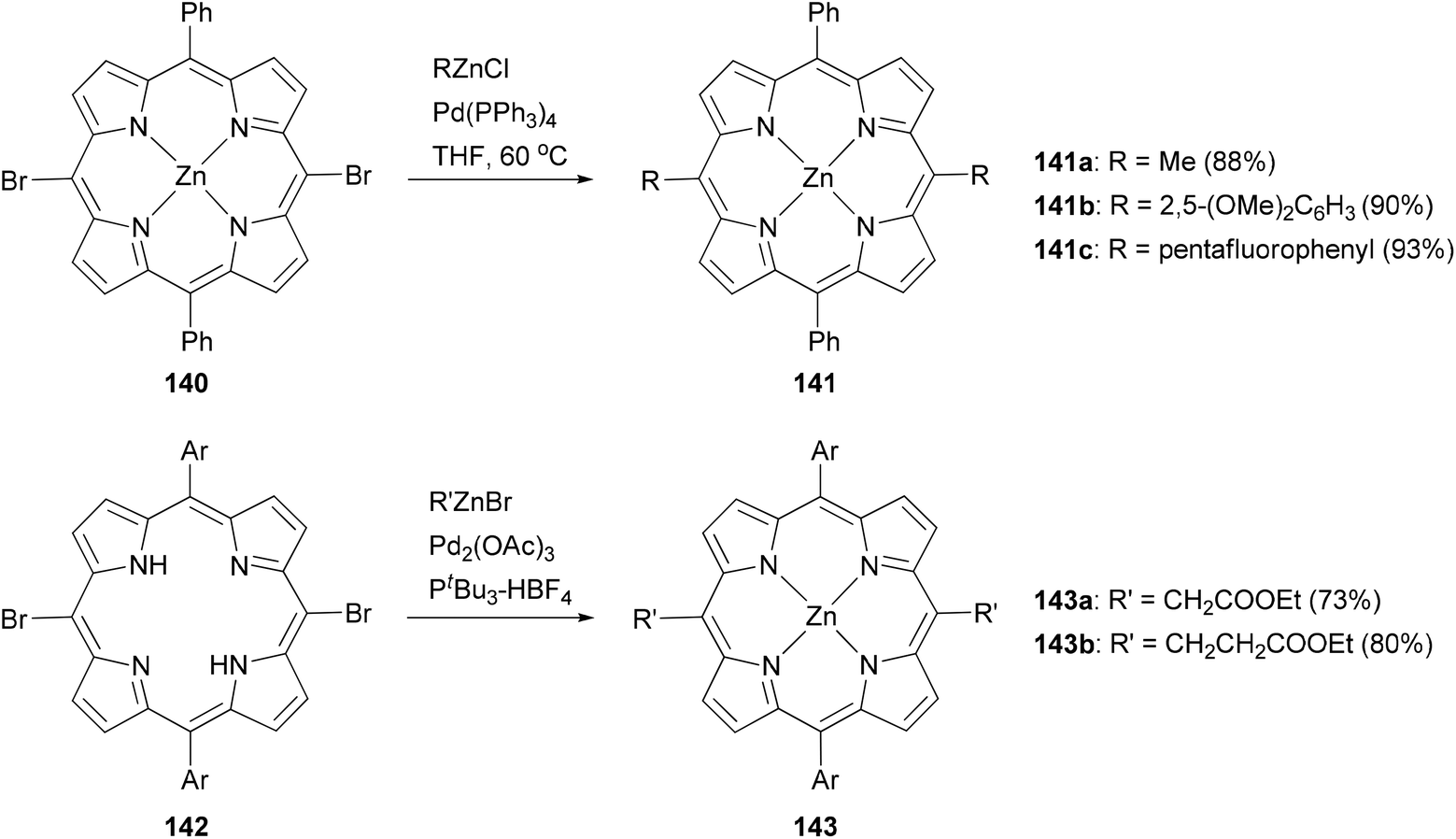
Modification at the meso-position by Negishi coupling has also been demonstrated. The reaction of meso-dibromo-substituted porphyrin 140 with methyl, 2,5-dimethoxyphenyl, and pentafluorophenylzinc chlorides produced 141a–c, respectively, in excellent yields.111b Takanami and co-workers introduced various functionalized alkyl and aryl groups to the meso positions of free porphyrin ligands.112a For instance, the reaction of 142 with functionalized alkylzinc bromides gives 143 in high yields (Scheme 36). In addition, they also reported an interesting cyanation at the β or meso position of porphyrins via the palladium catalyzed cross coupling of 2-cyanoethylzinc bromide with β or meso-bromoporphyrin. 2-Cyanoethylzinc bromide serves as the cyanide source.112b
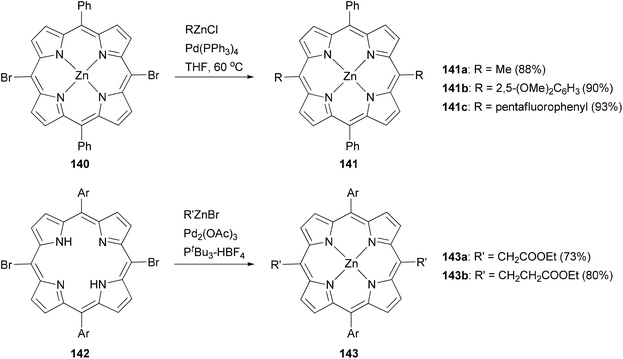 |
| | Scheme 36 Negishi coupling at the meso positions of porphyrin. | |
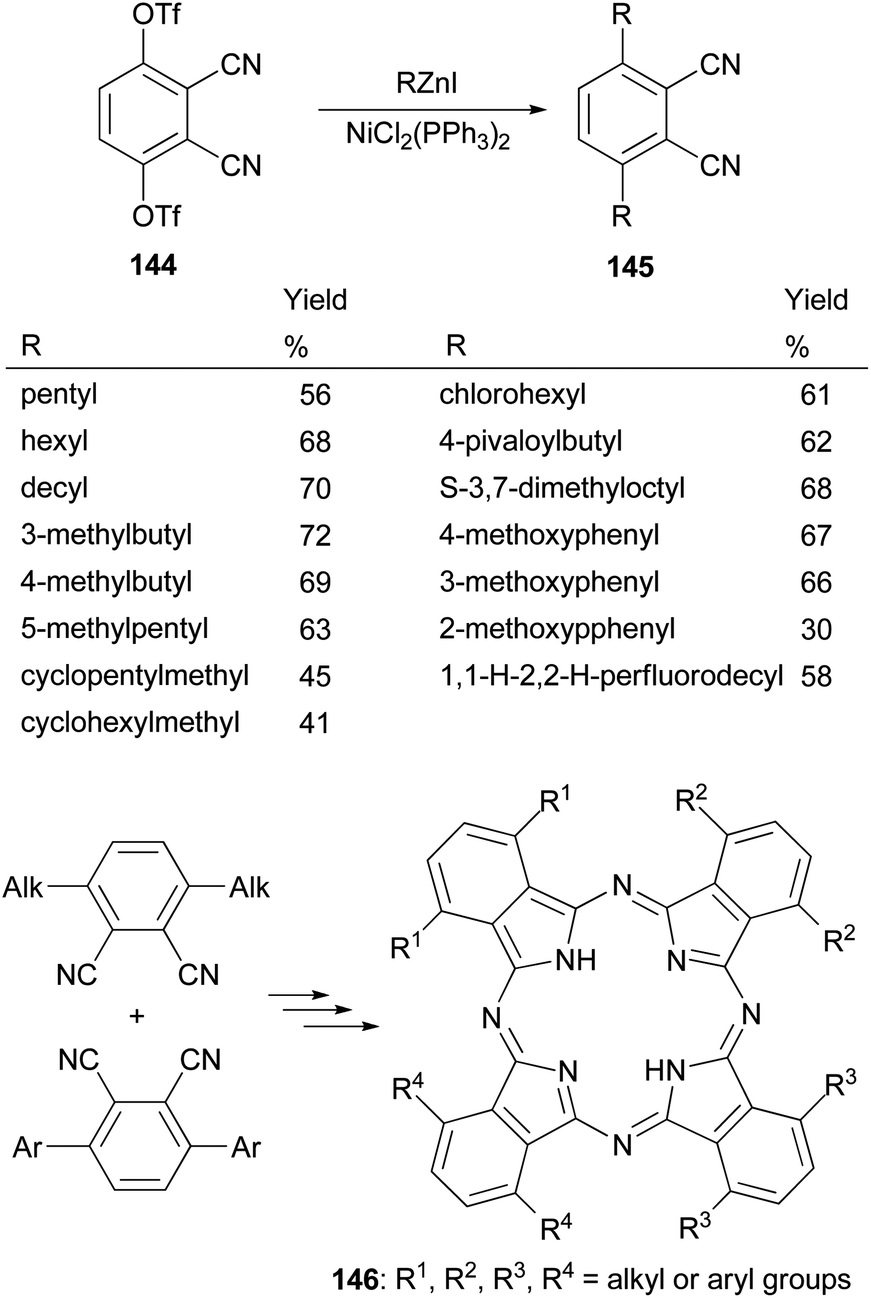
Phthalocyanines, structurally similar to porphyrins, are another family of optically important compounds which have found applications in solar cells, photodynamic therapy, optical data storage and are being used as nonlinear optical and optical limiting materials. With Negishi coupling, a series of precursors 145 for the synthesis of phthalocyanines 146 can be prepared from the bis-triflate derivative 144.115–117 The inexpensive NiCl2(PPh3)2 was used as an effective catalyst for this transformation. Suzuki coupling with the in situ generated tridecylborane to introduce the alkyl group was reported to give the desired product in much lower yields (∼30%) under various conditions including using PdCl2(dppf) as the catalyst and K3PO4 or K2CO3 as the bases (Scheme 37).117
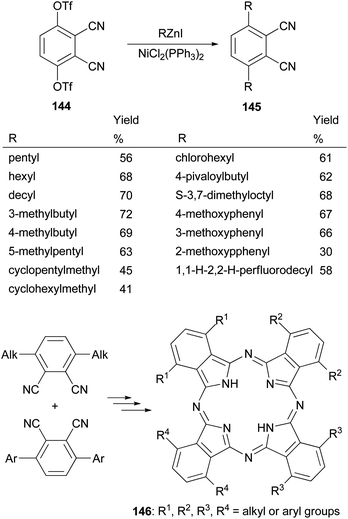 |
| | Scheme 37 Synthesis of phthalocyanines’ precursors 145. | |
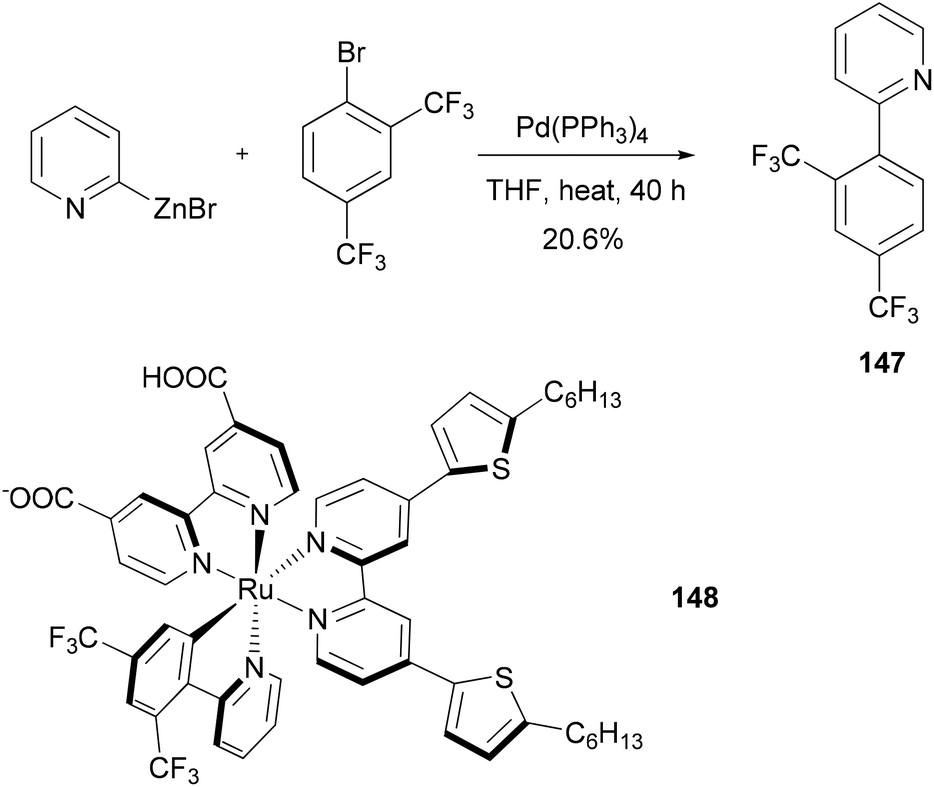
Dye sensitized solar cells (DSSCs) offer a promising alternative to conventional photovoltaic technologies. A tris-heteroleptic cyclometalated Ru(II) sensitizer 148 is prepared, which demonstrates high power output in a DSSC with a performance of 7.3% (η) under AM1.5 irradiation and 8.3% at half of that light intensity. The cyclometalating ligand 147, was prepared by a Negishi coupling of 2-pyridylzinc bromide and 1-bromo-2,4-bis(trifluoromethyl)benzene (Scheme 38).118
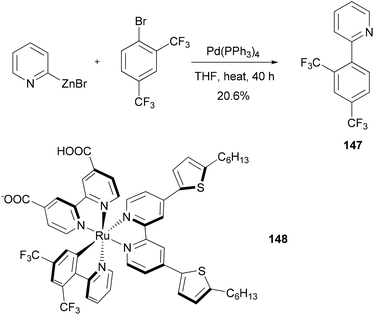 |
| | Scheme 38 Synthesis of cyclometalating ligand 147. | |
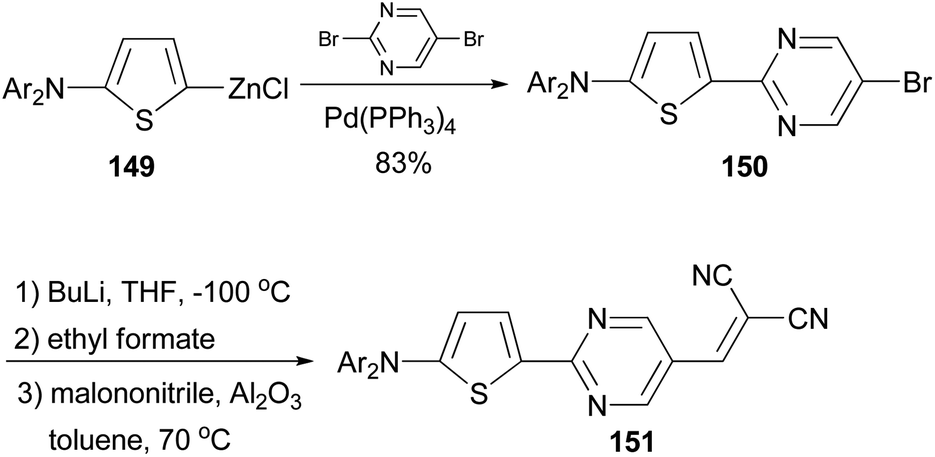
A donor–acceptor–acceptor molecule suitable for vacuum-processing was synthesized by a three-step reaction as shown in Scheme 39.119 First, the palladium-catalyzed coupling reaction of the organozinc reagent 149 derived from 2-(N,N-di-(p-tolyl)amino)thiophene with 5-bromo-2-iodopyrimidine afforded 150, which was then converted to the corresponding carbaldehyde by lithiation with butyllithium and subsequently quenched with ethyl formate. Finally, the aldehyde was condensed with malononitrile to yield the target compound 151via the Knöevenagel reaction in the presence of basic aluminium oxide. When compound 151 was used as the donor material in vacuum-deposited planar-mixed heterojunction solar cells with C70 as the acceptor, a power conversion efficiency as high as 6.4% was achieved.
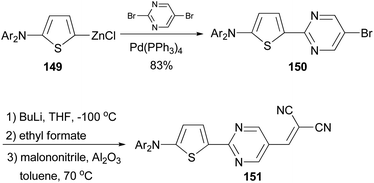 |
| | Scheme 39 Synthesis of 151. | |
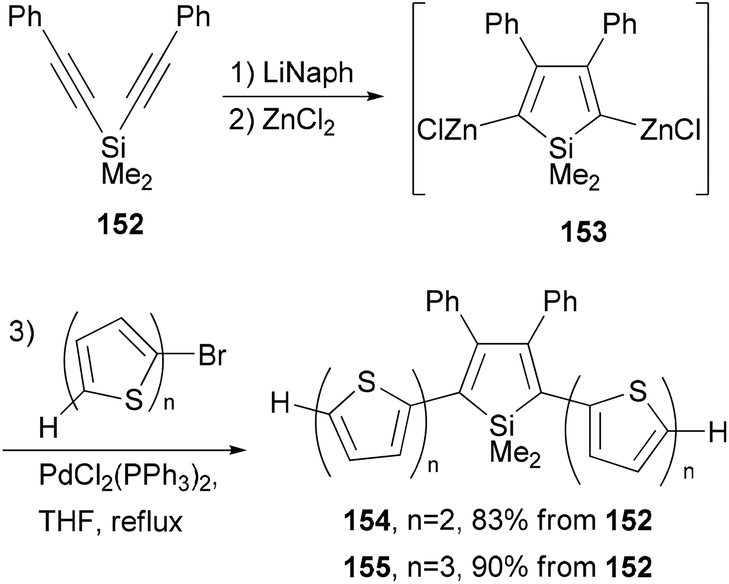
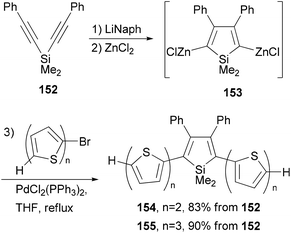
A series of donor–acceptor materials based on polythiophene modified with silole moieties were prepared by the electrochemical anodic polymerization of 2,5-bis([2,2′-bithiophen]-5-yl)-1,1-dimethyl-3,4-diphenylsilole (154) and 2,5-bis([2,2′-terthiophen]-5-yl)-1,1-dimethyl-3,4-diphenylsilole (155), as well as the copolymerization of these monomers with 2,2′-bithiophene, and their electrochemical and photovoltaic properties were investigated.120 The required electrochemically active monomers 154 and 155 are prepared by the one-pot, three-step methodology involving Tamao's reductive cyclization121 of 152 with lithium naphthalenide (LiNaph), transmetalation with ZnCl2, and Negishi cross coupling of the resulting organozinc reagent 153 (Scheme 40).
 |
| | Scheme 40 Synthesis of 154 and 155. | |
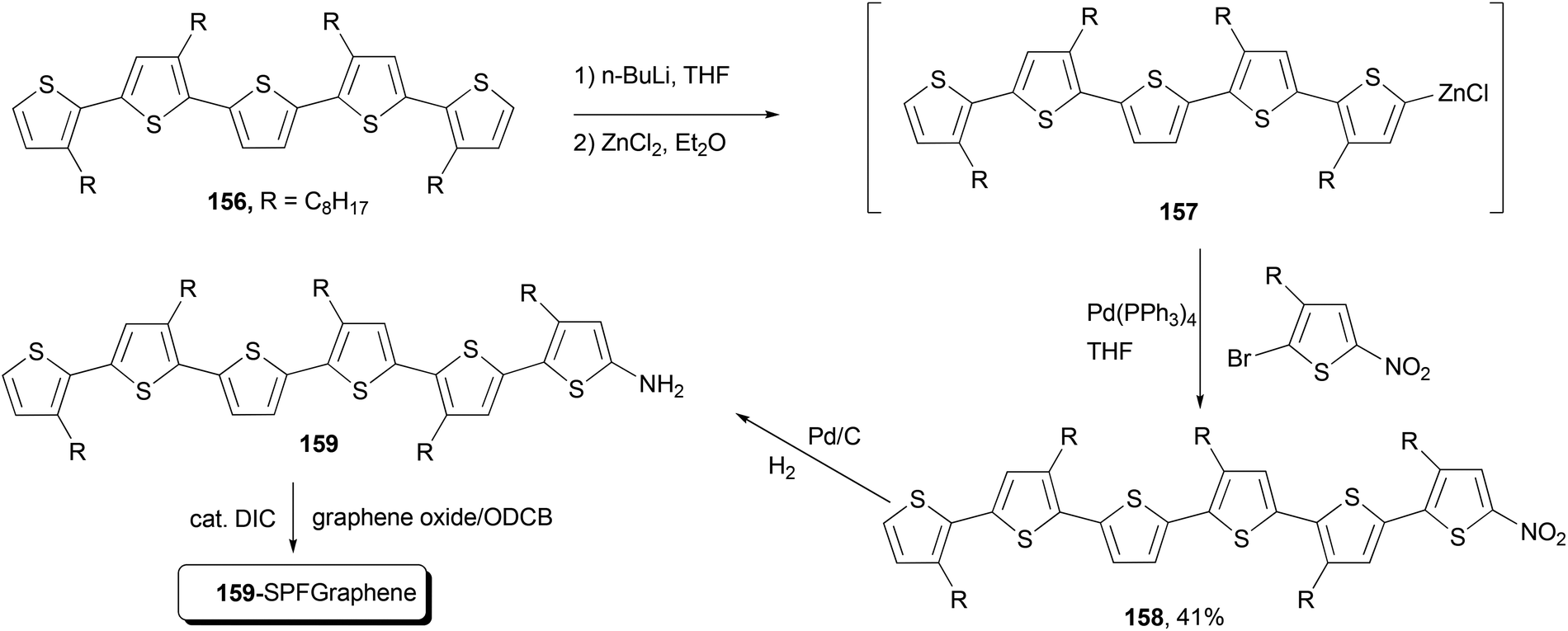
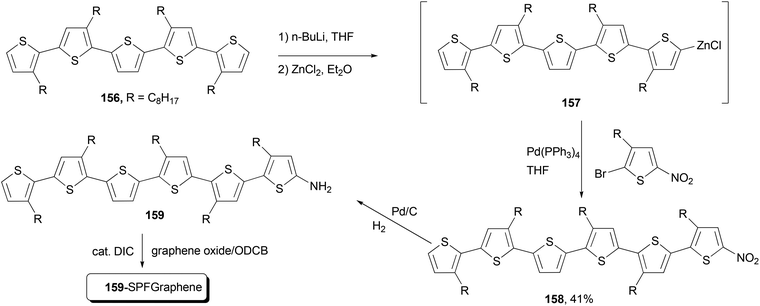
Functional oligothiophenes are frequently used π-conjugated materials that serve as active components in organic electronic devices and molecular electronics.122 Oligothiophene 158 was prepared by utilizing the Negishi cross-coupling reaction of 157 with 2-bromo-5-nitro-3-octylthiophene, which, after hydrogenation reduction of the nitro group, was covalently bonded to the graphene to form the oligothiophene–graphene nanohybrid 159-SPFGraphene (Scheme 41).123 The attachment of the electron-acceptor group (graphene oxide sheet) onto the oligothiophene molecules results in an improved absorption compared with its parent compound in the whole spectral region and an efficient quenching of photoluminescence. This modified graphene shows superior solution processability and better optical limiting effect than the benchmark optical limiting material C60.123
 |
| | Scheme 41 Synthesis of 159-SPFGraphene. | |
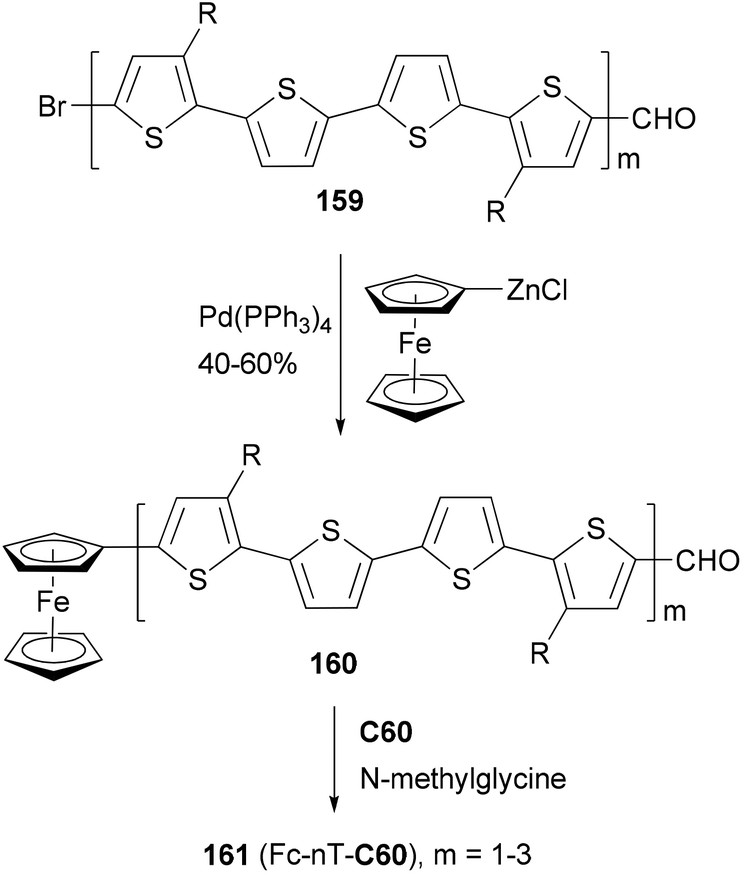
A strongly electron-donating ferrocenyl group is introduced into the oligothiophenes 159 (m = 1–3) by a Negishi coupling of ferrocenylzinc chloride. The ferrocene substituted derivatives 160 (m = 1–3) are covalently bonded to C60 to form the ferrocene–oligothiophene–fullerene triads 161(Fc-nT-C60, m = 1–3) (Scheme 42).124 In the triads, the conjugation between the ferrocene and the oligothiophene serves to promote electron transfer either from the excited oligothiophene to the fullerene or from the oligothiophene to the excited fullerene. Negishi coupling was also used to attach the 3,4-ethylenedioxythiophene (EDOT) moiety to a core structure of heptacyclic polyarene 10,15-dihydro-5H-di-indeno[1,2-a; 1′,2′-c]fluorene (truxene, Tr) and elongate the thiophene moiety in the synthesis of star-shaped D–π-B–A derivatives end-capped with pyrrolidino fullerene.125 These compounds can be used as potential light-harvesting materials in solar cell devices.
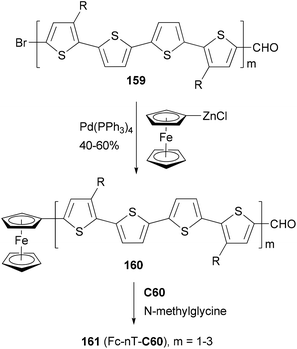 |
| | Scheme 42 Synthesis of triads 161 (Fc-nT-C60). | |
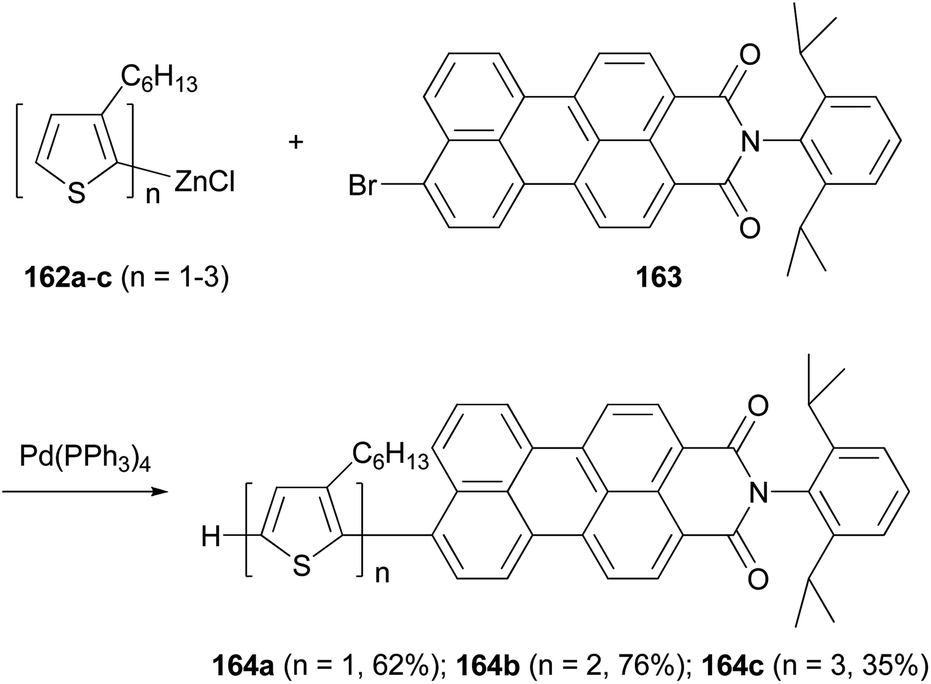
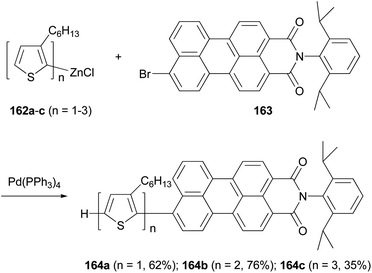
A series of donor–acceptor systems, consisting of head-to-tail coupled oligo(3-hexylthiophene)s covalently linked to perylenemonoimide are created via effective palladium-catalyzed cross-coupling reactions of 162a–c with 163 in good to excellent yields (Scheme 43).126 The synthesized perylenyl–oligothiophenes 164a–c display a strong absorption between 300 and 550 nm and a nearly complete fluorescence quenching of the perylene acceptor. Furthermore, the HOMOs of these hybrid compounds are lower than the work function of a PEDOT-coated ITO electrode, and their LUMOs are higher than both the LUMO of the fullerene derivative [6,6]-phenyl-C61-butyric acid methyl ester (PCBM) and the work function of aluminum cathode, which makes them suitable light-harvesting materials for applications in photovoltaic devices.126
 |
| | Scheme 43 Synthesis of 164a–c. | |
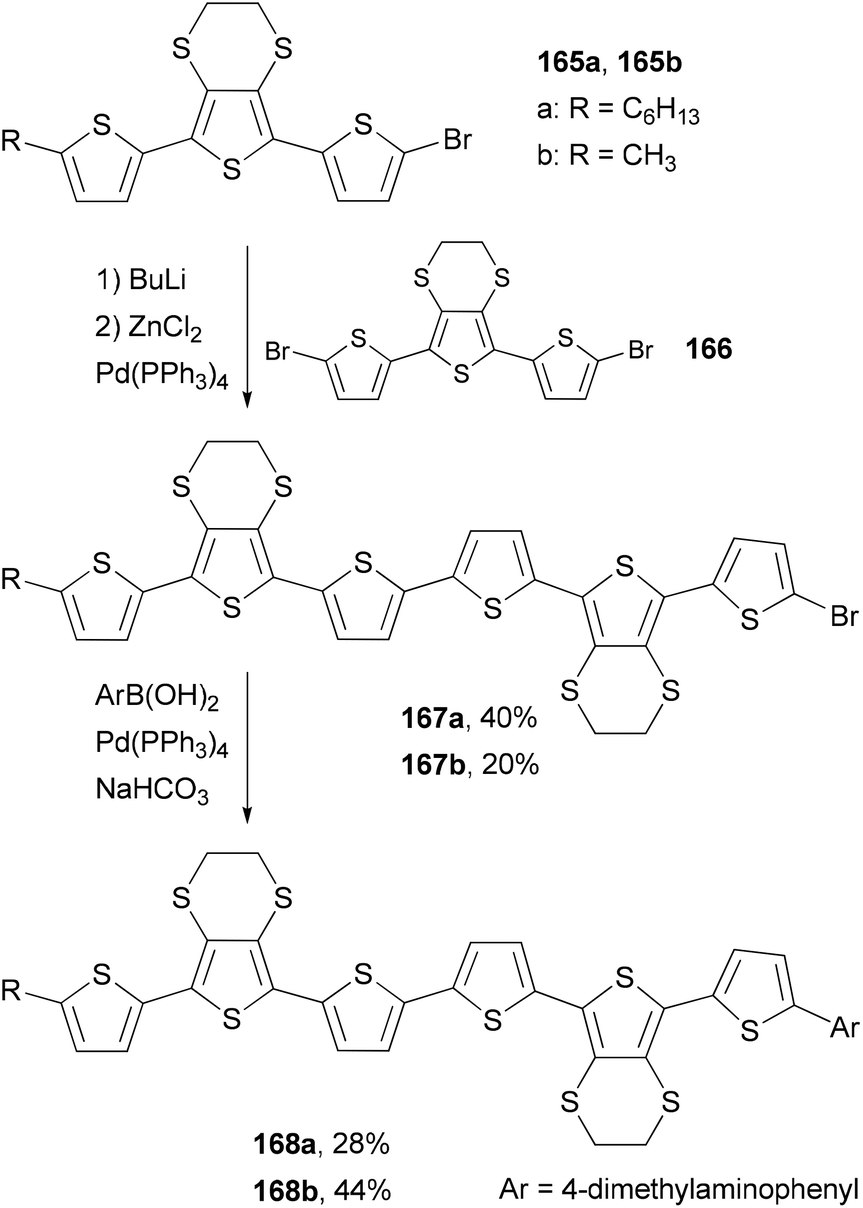
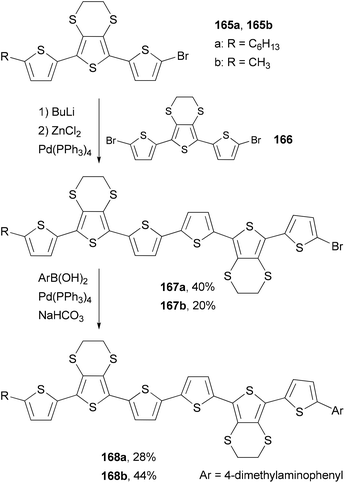
Skabara and co-workers prepared sexithiophenes 168a and 168b with a 4-(dimethylamino)phenyl end-capping group by using a combination of the Negishi and Suzuki couplings.127a The mono brominated terthiophene intermediate 165 is converted to the corresponding organozinc reagent, which cross-couples with the dibrominated terthiophene 166 to give 167. The second step involves the Suzuki coupling of 167 with 4-(dimethylamino)phenylboronic acid, giving sexithiophenes 168. Compared with similar sexithiophenes with both ends capped symmetrically with a hexyl or methyl group, which have been used as electron donor materials in a bilayer photovoltaic device,127b the introduction of the end-capping 4-(dimethylamino)phenyl group in 168a and 168b lowers the band gap and shifts the absorption to a longer wavelength by about 10 nm. Sexithiophenes are also efficient luminescence quenchers of quantum dots (Scheme 44).127a
 |
| | Scheme 44 Synthesis of 168a and 168b. | |
4. Electrochemical materials
Electrochemical materials discussed here refer to materials displaying unique electrochemical properties such as reversible and stable multiple electron redox processes, which may be used in advanced organic electronic and optical devices, energy storage devices, redox-responsive sensors, and catalysis. Multinuclear metallocene complexes have various applications from molecular electronics to homogeneous catalysis. Owing to its electron-richness, ferrocene (Fc) undergoes a one-electron oxidation to form a stable ferrocenium ion (Fc+) at a low potential. This process is reversible, making ferrocene a commonly used standard in electrochemistry. Molecules with multiple ferrocenyl groups are particularly attractive, mainly because of their unique redox properties and electrochemical stability. They can be used as multi-electron redox catalysts, electron storage devices, and as surface modifiers of electrodes.
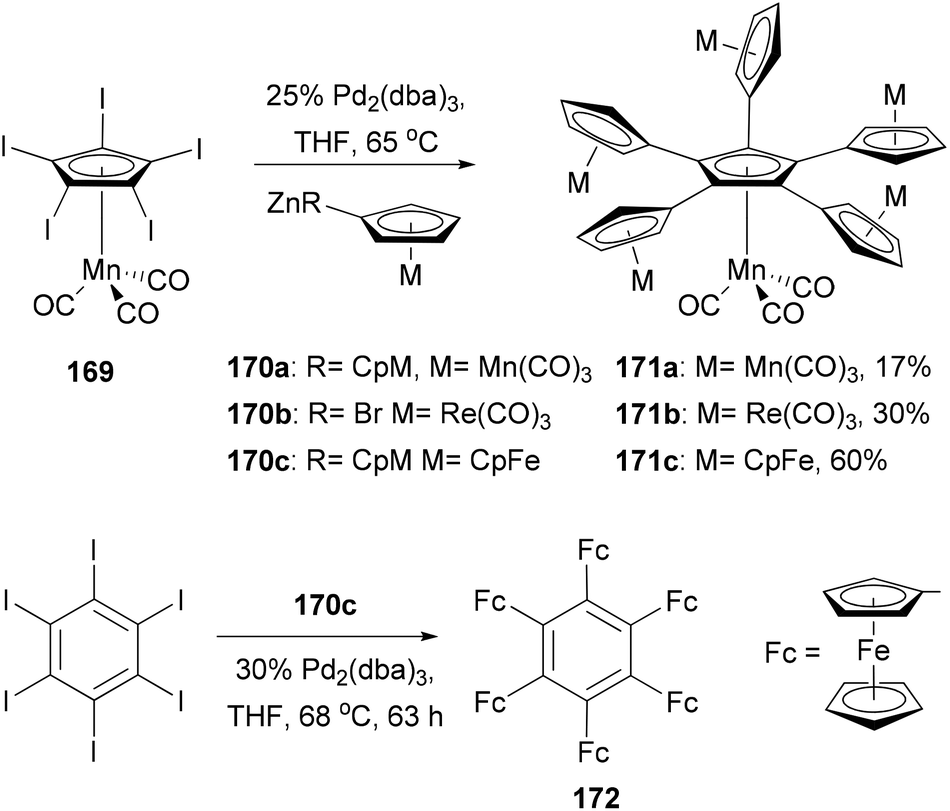
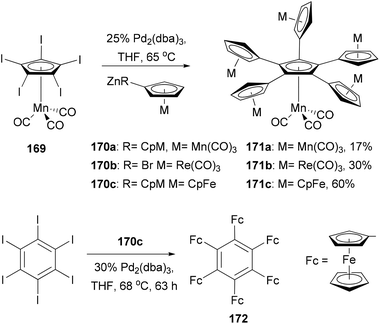
There is a tremendous effort to construct a π-conjugated system with multiple ferrocenyl motifs, so that the electronic communication between ferrocenyl centers can be assessed. Star-shaped two-dimensional oligoferrocene derivatives with directly connected metallocene fragments are expected to have optimal electronic communication between the metal centers as found in the linear poly(ferrocenes).128 However, the synthesis of such an oligoferrocene was a challenge until the first synthesis of radial oligocyclopentadienyl metal complexes 171a–c in 2006 by Vollhardt and co-workers129 (Scheme 45). After extensive experimentation with various cross-coupling reagents,130 they found that a five-fold Negishi coupling turned out to be the most efficient tool to introduce multiple metallocenyl groups to an aromatic core. As shown in Scheme 45, the reaction of metallocenyl zinc reagents 170a–c with [Mn(C5I5)(CO)3] 169 gave the sexicyclopentadienyl metal complexes 171a–c, respectively, in satisfactory yields. Electrochemical investigation of the ferrocene derivative 171c revealed three separate redox waves corresponding to a single one-electron transition (half-wave potential E1/2 = 5 mV vs. Fc/Fc+) and a pair of two-electron transitions (E1/2 = 169, 282 mV), indicating some intramolecular electronic communication between the metal centers. Sterically more congested hexaferrocenylbenzene 172 was also prepared, for the first time, by the Negishi coupling of 170c with hexaiodobenzene in 4% yield, while pentaferrocenylbenzene was isolated in 56% yield.131 The use of hexabromobenzene is less effective in forming highly ferrocenylated benzene. Three clearly separated redox waves, a single one-electron (E1/2 = −162.8 mV vs. Fc/Fc+), a two-electron (−32.3 mV), and a three-electron transition wave (222.4 mV), were observed from the voltammogram of 172.
 |
| | Scheme 45 Synthesis of sexicyclopentadienyl metal complexes 171a–c and hexaferrocenylbenzene 172. | |
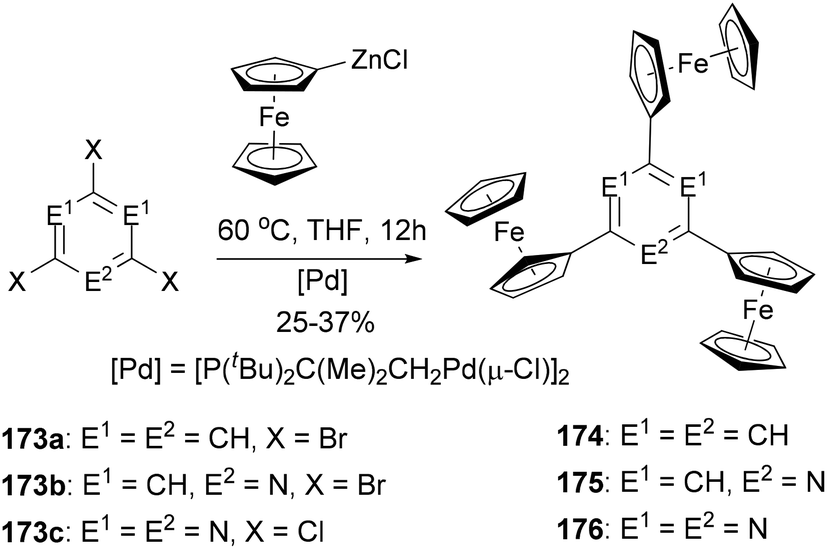
A series of di- and triferrocenyl (hetero)aromatics were prepared using the Negishi C–C cross-coupling of ferrocenylzinc chloride with multi halogenated heteroarenes.132 1,3,5-Triferrocenylbenzene (174), 2,4,6-triferrocenylpyridine (175), and 2,4,6-triferrocenyl-1,3,5-triazine (176) were synthesized by the reaction of ferrocenylzinc chloride with 173a–c, respectively (Scheme 46). Three well-defined, separated by 140–185 mV (ΔE1/2, difference in the individual half-wave potential), and reversible one-electron processes were observed for 174–176, pointing to weak intermetallic interactions.
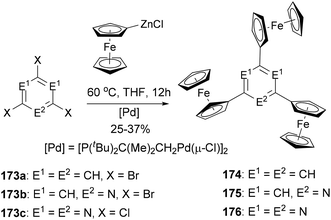 |
| | Scheme 46 Synthesis of triferrocenyl(hetero)arenes 174–176. | |
The redox activity of the ferrocenyl group has been incorporated into the concept of a redox-driven single molecular motor.133 The rotor 178 composed of five terminal electroactive groups has been prepared and connected to a tripodal ligand (stator) by means of a ruthenium(II) center (Scheme 47). The five ferrocenyl groups were introduced by the coupling of ferrocenylethynylzinc chloride with the ruthenium(II) complex 177 as shown in Scheme 47. Attempts under Sonogashira conditions with various catalysts and phosphines, including even the very bulky and electron-rich tris(tert-butyl)phosphine, failed. This system can be considered as the heart of a future molecular motor, since it is particularly well-suited for the preparation of a family of molecules through variations of the hydrotris(indazolyl) borate ligand. The stator can be functionalized to allow its deposition on various surfaces.
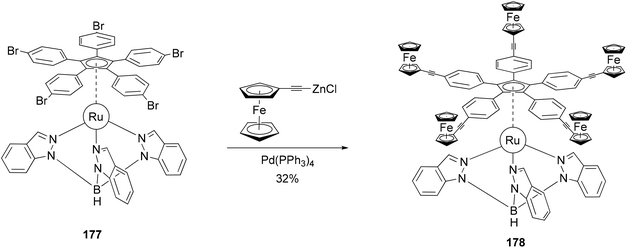 |
| | Scheme 47 Synthesis of molecular motor 178. | |
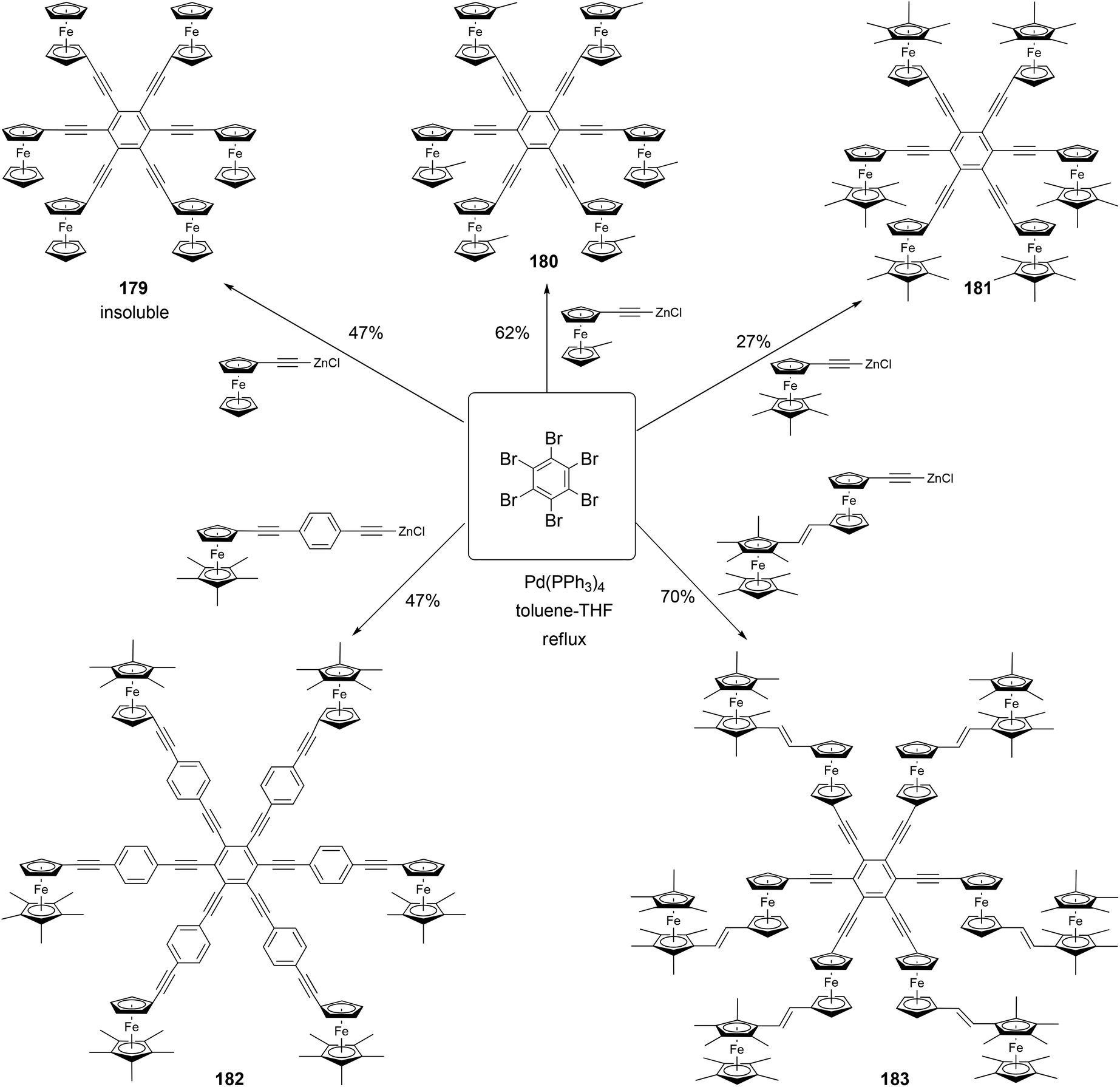
Astruc and coworkers have synthesized a family of rigid redox stars 117–183 by the Negishi coupling of hexabromobenzene with various alkynylzinc reagents (Scheme 48).134,135 Once again, attempts to use Sonogashira coupling failed to produce the desired hexasubstituted products. Cyclic voltammetry studies show that these compounds display a single wave for the six-electron oxidation when using NBu4PF6 as the supporting electrolyte, whereas splitted multiple electron transfer processes are observed when NBu4BAr4 (Ar = 3,5-bistrifluoromethylphenyl) is used as the supporting electrolyte. This, in combination with the study of other closely related family members of rigid ferrocenyl-terminated redox stars, confirms the lack of electronic communication between the redox centers and a significant through-space electrostatic effect among the oxidized ferrocenyl groups.134,135
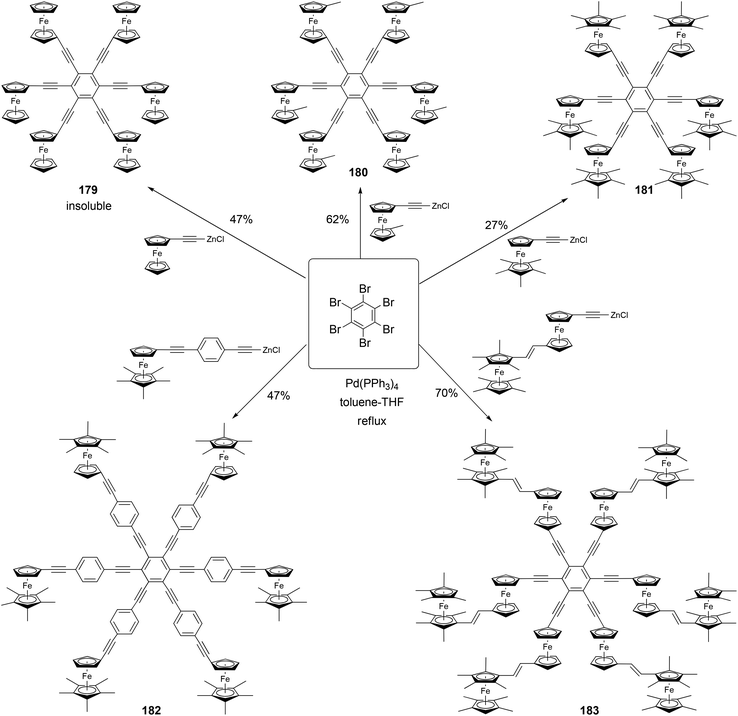 |
| | Scheme 48 Synthesis of redox stars 179–183 by six-fold Negishi coupling. | |
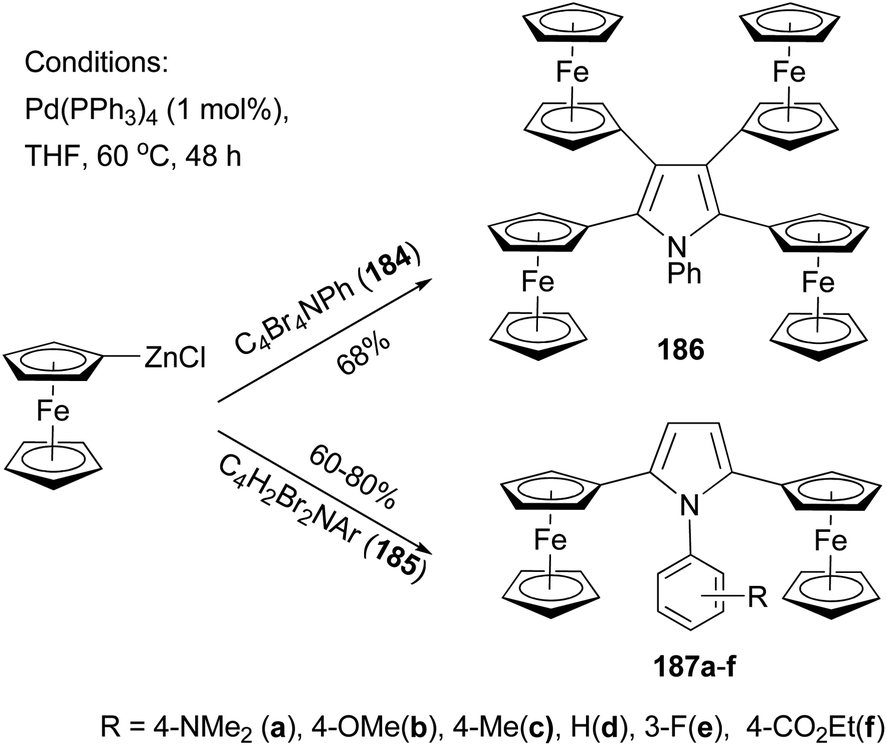
Recently, Lang and co-workers have used Negishi coupling to introduce ferrocenyl groups to the peripheries of pyrrole, furan, and thiophene cores.136–140 Tetraferrocenyl pyrrole 186 was synthesized by the palladium catalyzed cross coupling of ferrocenylzinc chloride with 2,3,4,5-tetrabromo-1-phenyl-1H-pyrrole (184) in 68% percent yield.136 Under the same conditions, di-ferrocenylpyrrole 187a–f with various substituents on the N-phenyl ring were prepared from 2,5-dibromo-1-aryl-1H-pyrrole (185) in high yields (Scheme 49).137 Similar compounds with a furan or thiophene core were also prepared using similar Negishi coupling.138 Compound 186 displays four reversible one-electron transfer processes with remarkably high ΔE1/2 values and reduction potentials of E0f = −280, 51, 323, and 550 mV (ΔE1/2 = 322, 264, and 233 mV) using [NBu4][B(C6F5)4] as the supporting electrolyte.136 Compounds 187c displays two electrochemically reversible one-electron transfer processes with even higher ΔE1/2 value and reduction potentials of E0f = −238 and E0f = 212 mV (ΔE1/2 = 450 mV). These results, compared with those from other ferrocenyl substituted aromatic and heteroaromatic compounds described previously, suggest a strong intermetallic communication between ferrocenyl and ferrocenium termini when using a pyrrole as the connecting unit. The substituents on the phenyl ring of compounds 187a–f have an influence on both the redox potentials and the separation of the redox potentials, with electron-donating groups (187a–c) increasing the ΔE1/2 values, while electron-withdrawing groups (187e an 187f) decreasing the redox separation.137 A total of 13 thiophene derivatives with one to five ferrocenyl groups substituted at the periphery positions of thiophene are prepared by Negishi coupling using ferrocenylzinc chloride in generally satisfactory yields.139 Ferrocenyl-substituted thiophenes also display redox separations, but not as large as those displayed by their pyrrole counterparts.
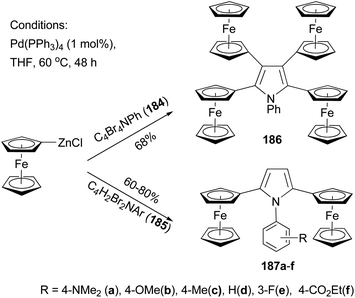 |
| | Scheme 49 Synthesis of 186 and 187a–f. | |
The large redox separation displayed by compounds 187a–f suggests that pyrrole is an excellent connecting unit for two ferrocenyl centers allowing efficient electronic interaction between the two redox centers, which implies a possible model for molecular wires containing oligo(pyrrole) units. To this end, a series of oligopyrroles 188a–c were prepared by the iterative alternation of bromination and Negishi coupling of the corresponding pyrrolyl derivatives, which were capped by two ferrocenyl groups through Negishi coupling again with ferrocenylzinc chloride to form molecular wires 189a–c (Scheme 50).140 The redox splitting is decreased as the number of pyrrole units increases, and the ferrocenyl moieties in quarterpyrrole 189c are oxidized simultaneously.
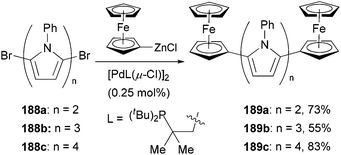 |
| | Scheme 50 Synthesis of ferrocene capped oligopyrroles 189a–c. | |
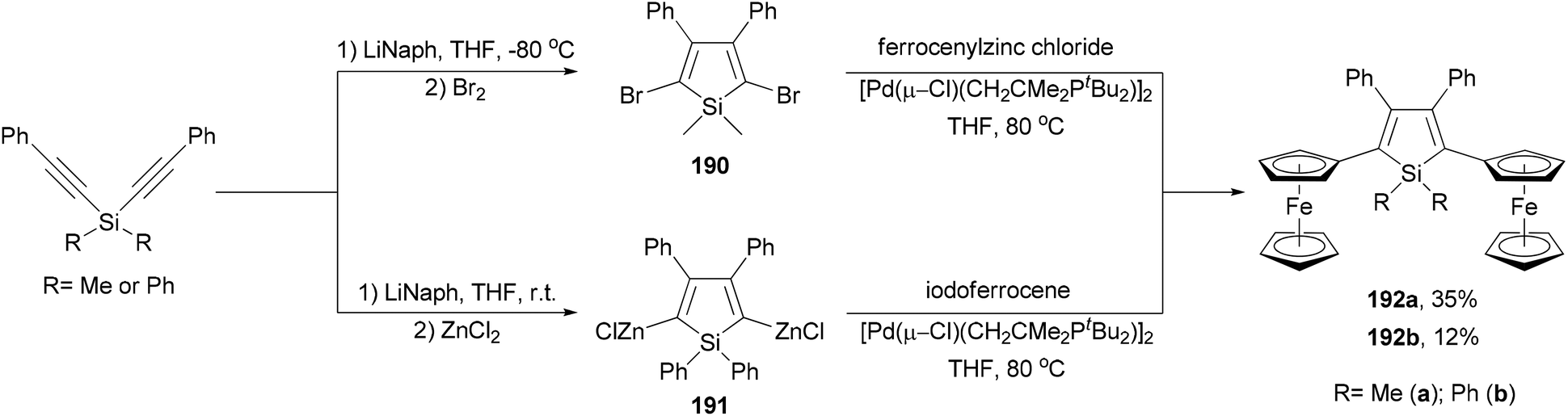
Ferrocenyl-substituted siloles 192a and 192b are both prepared by the reductive cyclization of dialkynylsilanes and subsequent Negishi coupling but with different strategies (Scheme 51).141 Silacyclopentadiene 190 was prepared by the reductive cyclization of dimethylbis(phenylethynyl)silane followed by bromination. Cross coupling of the dibromide with ferrocenylzinc chloride gave 192a in 35% yield. For the synthesis of 192b, the organozinc reagent 191 had to be used to couple with iodoferrocene since the other combination did not work. Interestingly, compounds 192a and 192b underwent two sequential ferrocenyl-based redox processes with separations of 300 and 280 mV (ΔE1/2), respectively, which are similar to that displayed by diferrocenylfuran (ΔE1/2 = 290 mV).141
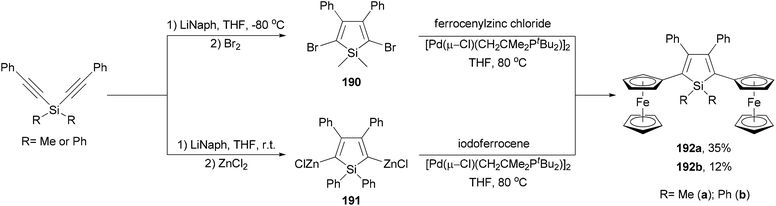 |
| | Scheme 51 Synthesis of ferrocenyl-substituted siloles 192a and 192b. | |
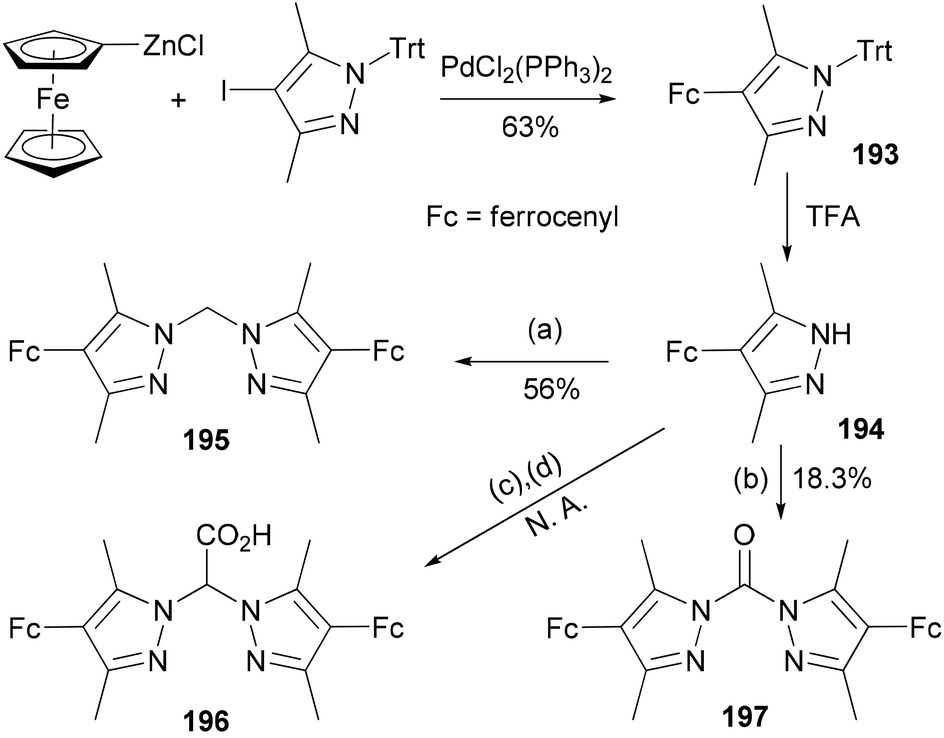
The compound 4-ferrocenyl-3,5-dimethylpyrazole (194) is synthesized from 4-iodo-3,5-dimethyl-1-tritylpyrazole via a Negishi cross-coupling reaction142 and the subsequent deprotection of the intermediate 193.143 From 194, chelating ligands 195–197 are prepared under various conditions (Scheme 52). The redox properties and complexation of 196 with iron were studied. Ligands 195 and 196 were suggested for future model complexes by mimicking the redox processes of Rieske dioxygenases.
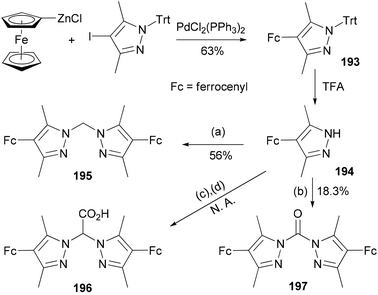 |
| | Scheme 52 Synthesis of 193. Conditions: (a) KOH, K2CO3, CH2Cl2, TEBAC; (b) NEt3, triphosgene, (c) Br2CO2H, KOtBu, TEBAC; (d) H2O, HCl. | |
Negishi coupling has been used to create a new binding site in a complex resulting in a heterodimetallic ruthenium–osmium complex (Scheme 53).144 Ligand 198 was prepared in high yield by the cross coupling of 2,2′-dibromo-4,4′-bipyridine with 2-pyridylzinc bromide (2 equivalents) at room temperature in THF with Pd(PPh3)4 as the catalyst. The high yield of the mono cross-coupled product 198 is due to the precipitation of its ZnBr2 complex from the reaction mixture. Zinc bromide can be sequestered by treating the complex with EDTA. The reaction of 198 with RuCl2(bpy)2 (bpy = 2,2′-bipyridine) gives 199. Complex 199 reacted with excess 2-pyridylzinc bromide in the presence of palladium catalyst to form 200 almost quantitatively, which was converted to the heterodinuclear complex 201 by reacting with OsCl2(bpy)2. The oxidation of the osmium metal center does not alter the oxidation potential of the ruthenium metal center, indicating weak metal–metal interactions in these complexes.
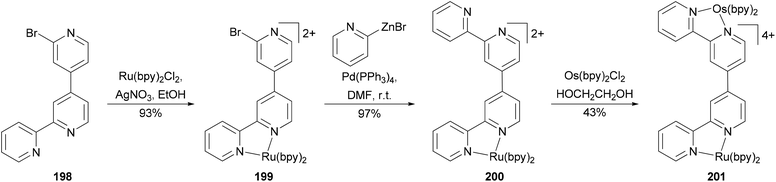 |
| | Scheme 53 Synthesis of heterodimetallic Ru-Os complex 201. | |
Ren and co-workers demonstrate that Negishi coupling is a facile, mild, and high-yield approach, superior to the Suzuki method, for biphenyl formation at the periphery of diruthenium coordination and organometallic compounds.145 As shown in Scheme 54, coordination compounds 202a–c and organometallic compounds 203a–c were prepared in 75–92% yields (47% for 203a) by using Negishi coupling, and the reactions proceed at room temperature.145a In contrast, 202a was previously prepared using Suzuki coupling only in 41% yield.145b
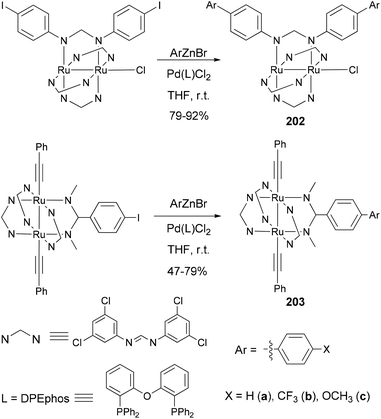 |
| | Scheme 54 Synthesis of 202 and 203. | |
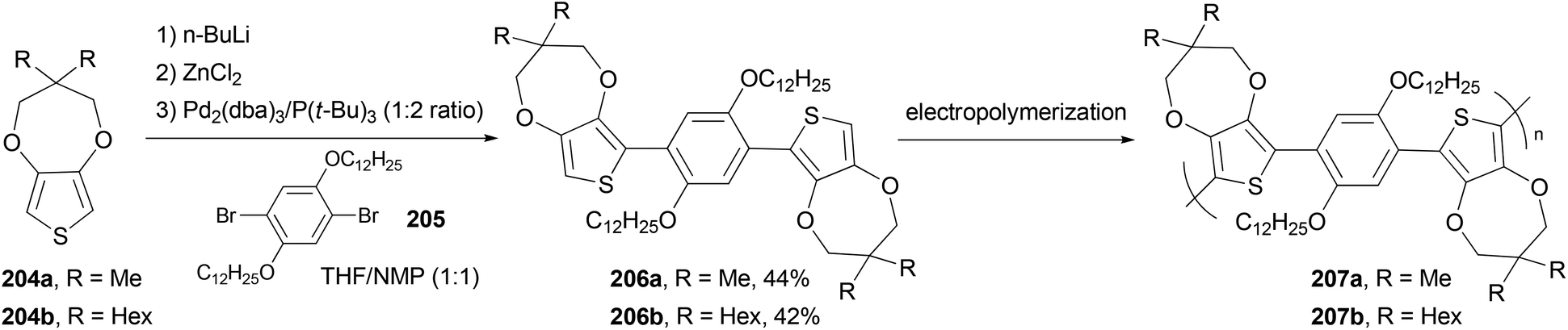
The monomers 1,4-bis(3,3-dimethyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepin-6-yl)-2,5-didodecyloxybenzene (206a) and 1,4-bis(3,3-dihexyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepin-6-yl)-2,5-didodecyloxybenzene (206b) were synthesized via Negishi coupling of the alkyl-substituted ProDOT 204a and 204b and the didodecyloxyphenylene unit 205 in ca. 40% yields (Scheme 55).146 Both the monomers were efficiently electropolymerized to form electroactive films exhibiting redox switching at fairly low potentials (ca. +0.1 V vs. Fc/Fc+). Polymer 207b electrochemically switches between orange, blue, and highly transmissive-gray colors, making it potentially useful in large area electrochromic displays.
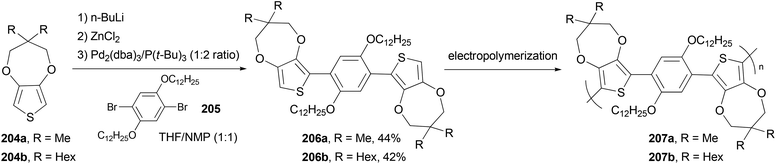 |
| | Scheme 55 Synthesis of 207a and 207b. | |
5. Magnetic materials and high spin organic polyradicals
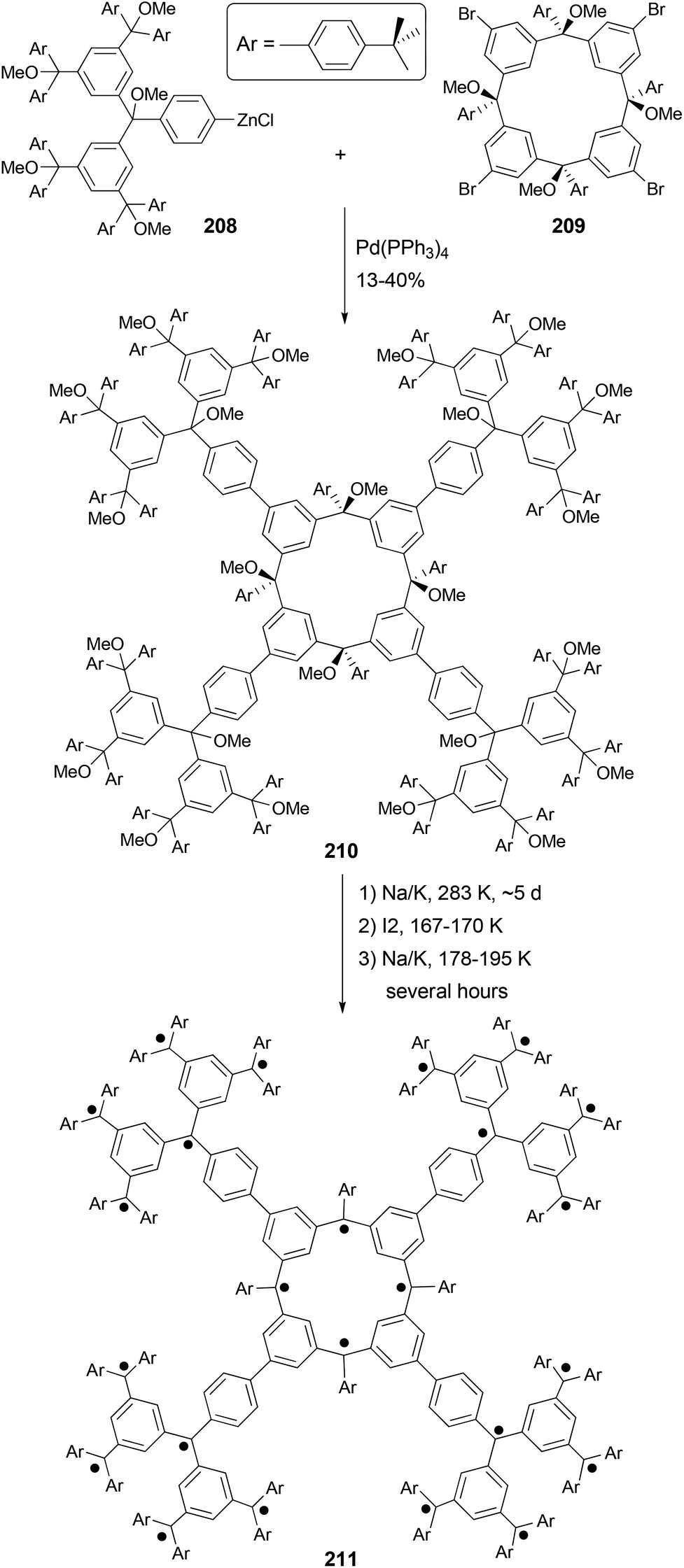
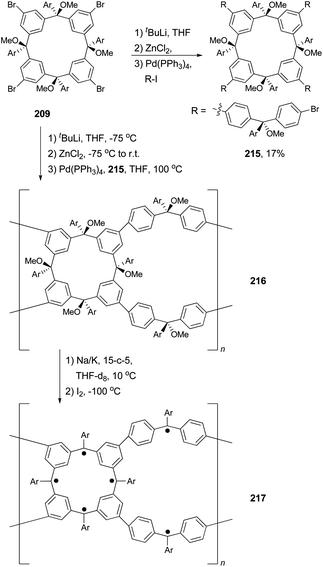
Organic molecules with a very high spin possess a large number of ferromagnetically coupled unpaired electrons (parallel spins), and such paramagnetic organic polyradicals are not only the building blocks for organic magnets147 but may also exhibit other interesting electronic, optical, and electrochemical properties.148 Achieving a high spin quantum number S (high-spin ground state) becomes a major task for the researchers in this field. A unique dendritic macrocyclic organic spin cluster 211 with a very high spin of S = 10 has been synthesized.149 The synthesis of the precursor 210 relies on the attachment of four dendritic branches to the macrocyclic core by the four-fold Negishi coupling of the zincated pentaether 208, generated in situ from the corresponding bromopentaether by a bromine–lithium exchange using tBuLi followed by treatment with ZnCl2, with macrocyclic tetrabromide 209 (Scheme 56). Yields ranging from 10–40% were reported for this highly demanding synthesis.
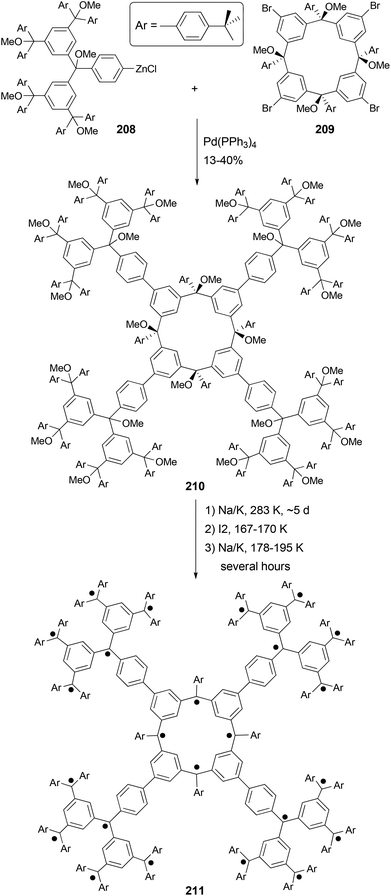 |
| | Scheme 56 Synthesis of organic spin cluster 211. | |
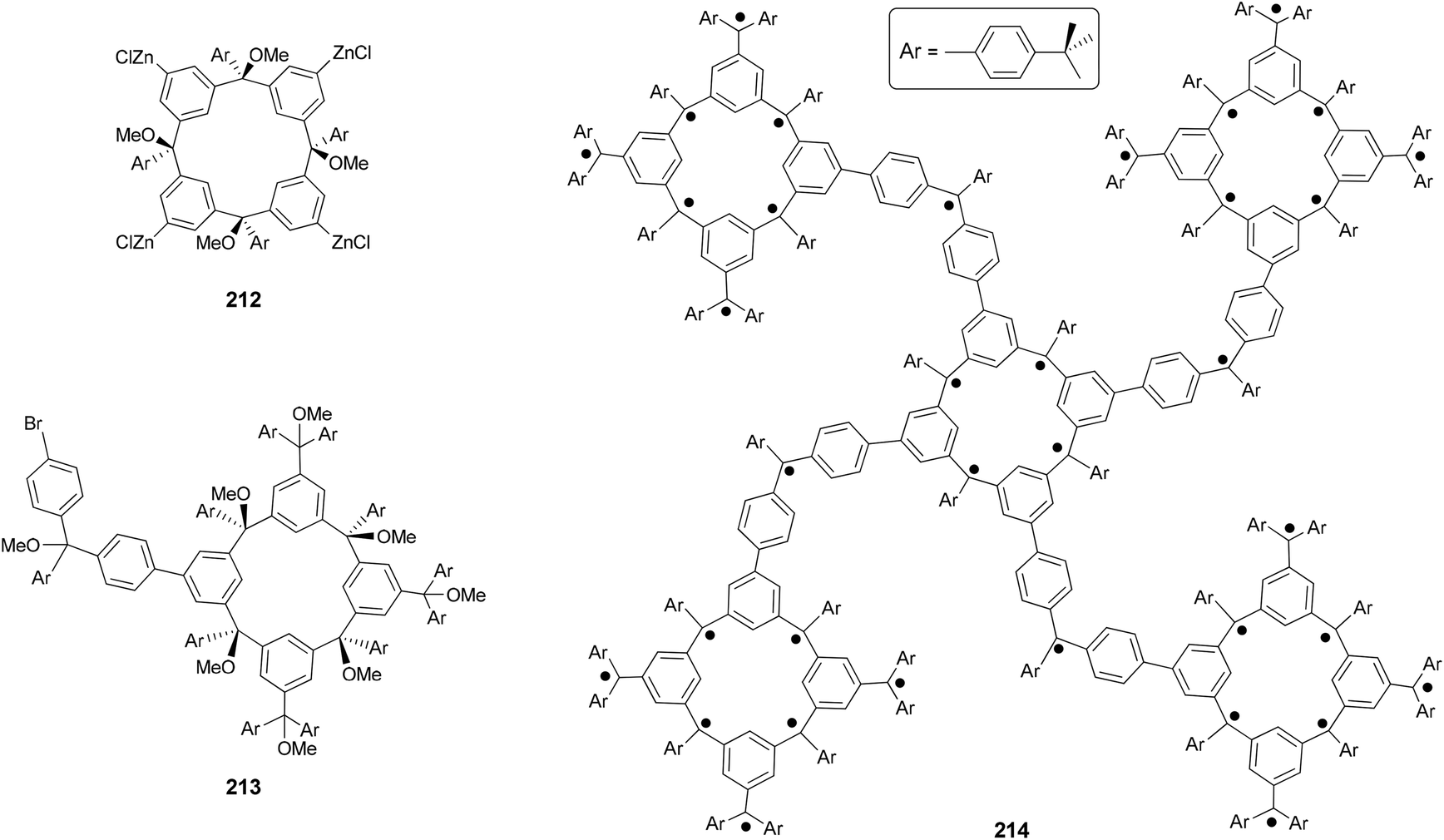
The Negishi protocol was further employed in a macrocycle–macrocycle coupling to make a series of macrcycle assemblies by linking two to five calix[4]arene-based polyarylmethyl polyether macrocycles.150 Polyarylmethyl polyradicals derived from these assemblies display a very high spin S = 5–13. Remarkably, polyarylmethyl polyradical 214 (Fig. 2) has the highest spin quantum number of S = 13 for an organic molecule reported so far. The precursor to the high spin macrocycle assembly 214 was prepared by the four-fold Negishi coupling of tetrazincated tetraether macrocycle 212 (from 209) with monobrominated polyether macrocycle 213 in 7–32% yields under the same conditions described in Scheme 56.
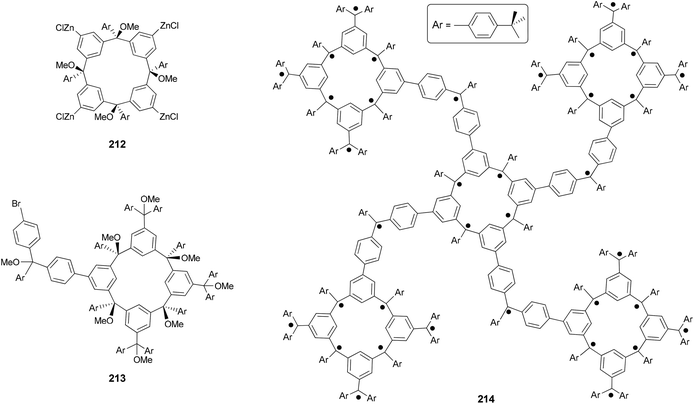 |
| | Fig. 2 Structure of high spin polyarylmethyl polyradical macrocycle assembly 214. | |
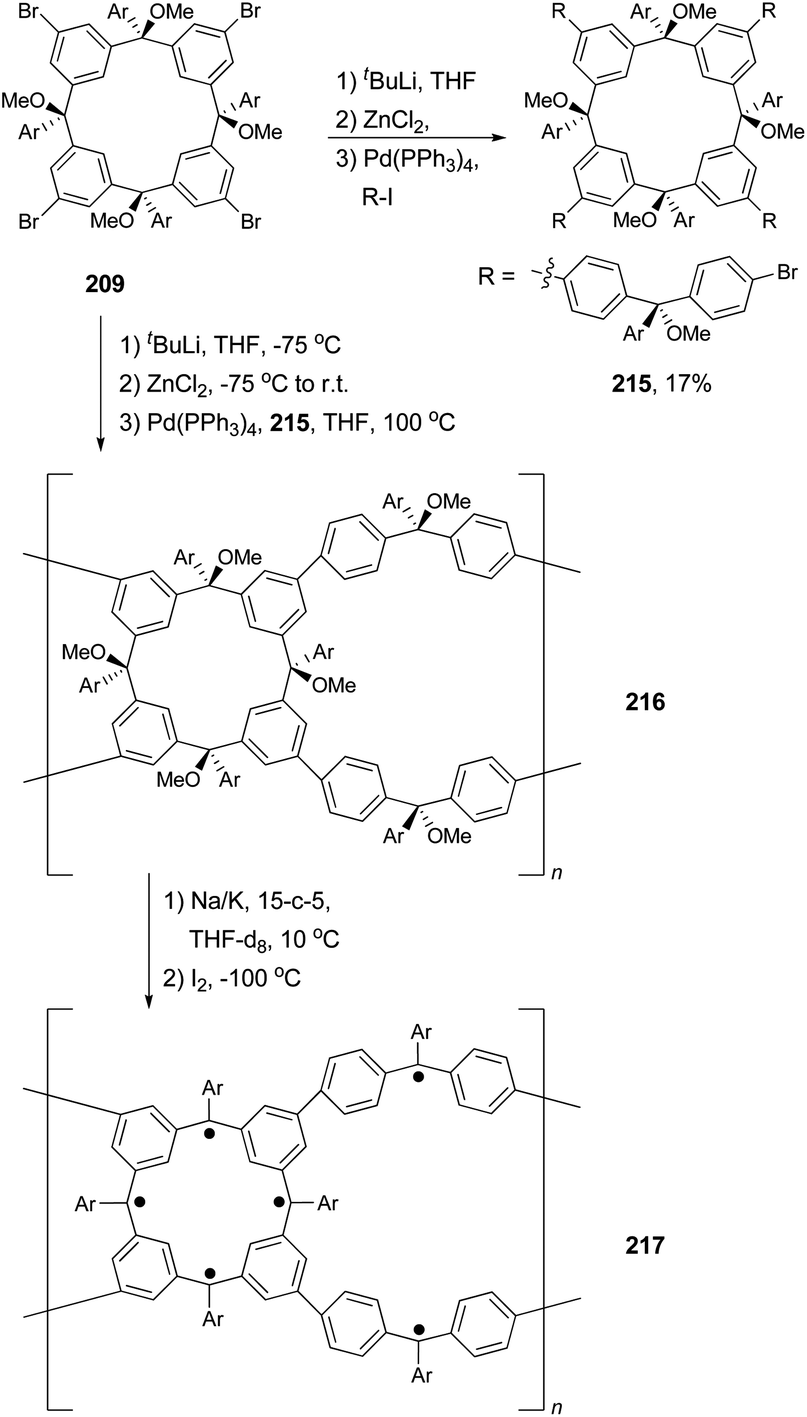
Polymers with a high spin quantum number (S) were very much limited to S < 5 until Rajca's novel design and synthesis of polymer networks with cross-linked macrocyclic modules possessing an average of S ≥ 40.151 The cross-linking was realized by Negishi coupling as shown in Scheme 57. First, tetrabromide 209 was converted to a new tetrabromide 215via a one-pot lithium–bromine exchange/zincation/four-fold cross coupling process. In the second step, 209 was again converted to its zincated derivative (212) and cross coupled with the tetrabromide 215 in the presence of catalyst Pd(PPh3)4 to give the cross-linked polymer 216, the precursor to 217. In a later study, the highly cross-linked polymer is produced using the same synthetic strategy, and displays an effective magnetic moment corresponding to an average S of about 5000 and slow reorientation of the magnetization by a small magnetic field (less than or equal to 1 oersted) below a temperature of about 10 K.152 The magnetic behavior falls between insulating spin glasses and blocked super-paramagnets.
 |
| | Scheme 57 Synthesis of cross-linked magnetic polymer 217. | |
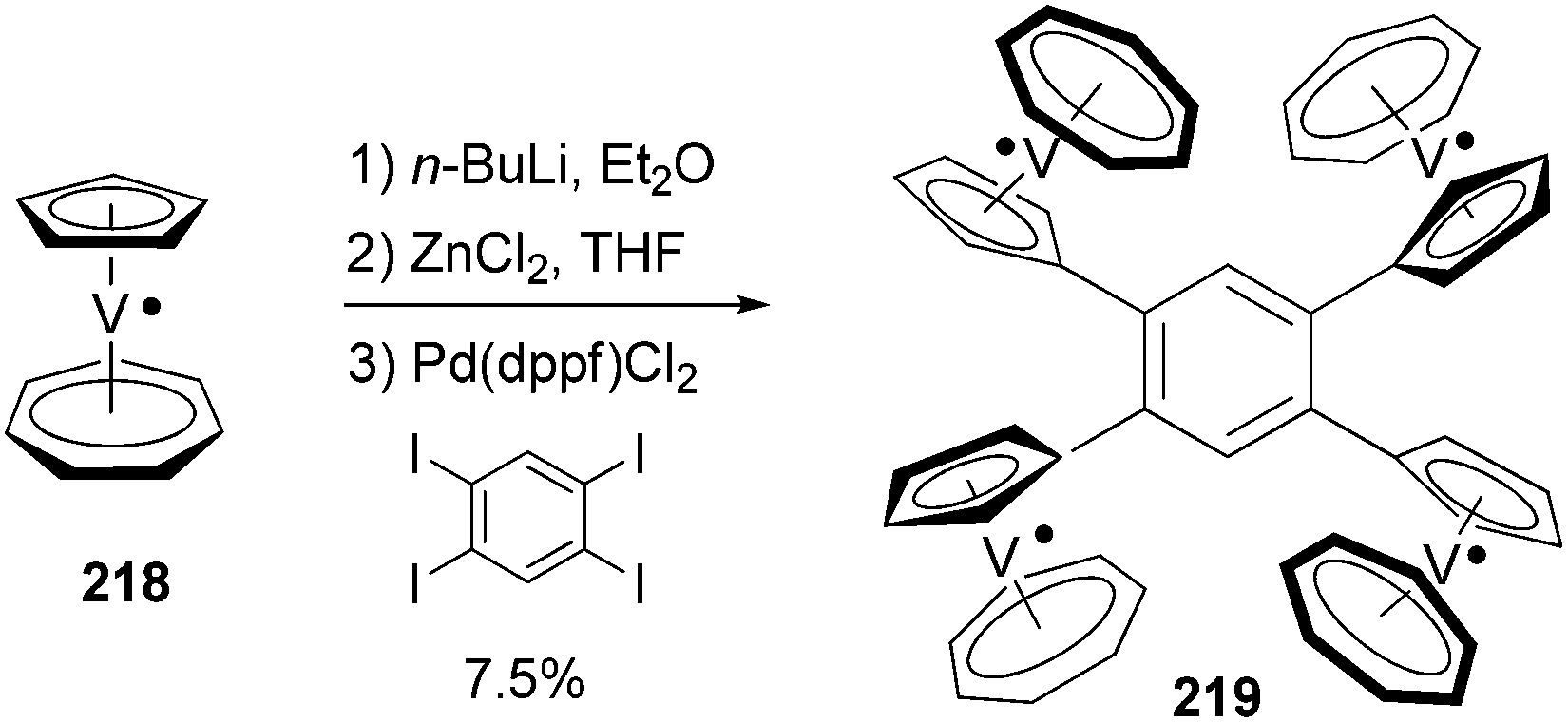
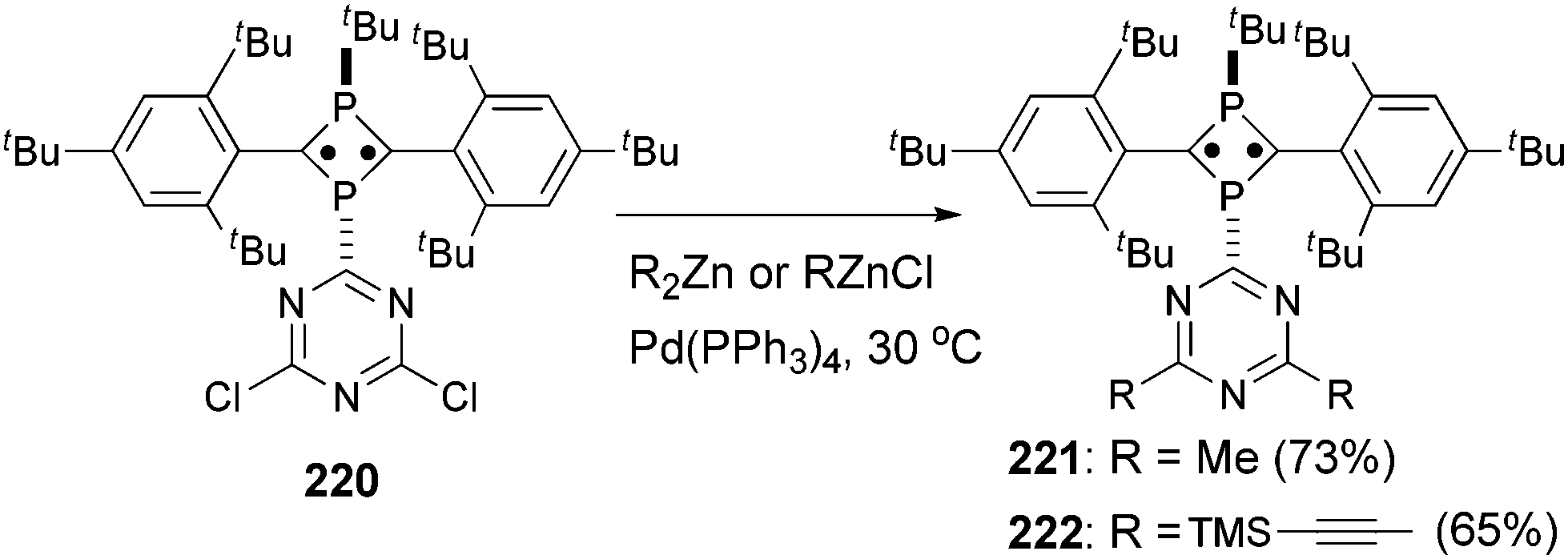
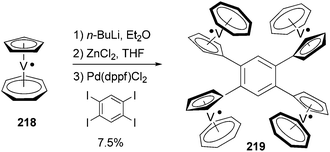
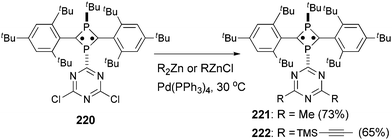
Tetra- and octaradicals based on calix[4]arene nitroxides were prepared recently by Rajca and co-workers, and Negishi coupling was once again used to introduce nitroxide fragments to the calix[4]arene core.153 Elschenbroich and co-workers reported an organometallic tetraradical displaying pronounced electro- and magnetocommunication. The organometallic tetraradical 219 was prepared via a four-fold Negishi coupling of (C7H7)V(C5H4ZnCl) (218) with 1,2,4,5-tetraiodobenzene (Scheme 58).154 More recently, Negishi coupling was applied to the modification of a triazine-substituted biradical 220 to obtain more stable derivatives 221 and 222 (Scheme 59).155 The Sonogashira coupling conditions were not suitable for preparing 222 because of an undesirable Hoffmann reaction with the amine.
 |
| | Scheme 58 Synthesis of tetraradical 219. | |
 |
| | Scheme 59 Negishi coupling of biradical 220. | |
6. Conclusions and perspectives
Negishi coupling has played an important role in the development of advanced organic materials, allowing the synthesis of a wide variety of molecules exhibiting different functions including electronic conducting, light-emitting and other optical functions, redox activities, and magnetic properties. Many of the syntheses are very challenging but can be tackled with Negishi coupling. For example, reactions involving multifold Negishi cross couplings often give satisfactory results that cannot be achieved by using other methods. Negishi coupling is also extremely efficient in introducing an aromatic or heteroaromatic unit because many of the aromatic/heteroaromatic zinc reagents can be generated in situ from inexpensive starting materials and used directly in the cross coupling. More importantly, the reaction usually proceeds under mild conditions and gives a high yield of the desired product, and in most cases, palladium catalysts with simple triphenylphosphine ligands can serve the purpose of the synthesis. Although polycondensation using Suzuki coupling predominates the synthesis of conducting polymers, a recent study demonstrates that Negishi polycondensation can have several clear advantages such as a lower catalyst loading (higher TONs) and a faster reaction rate (higher TOFs).49 Moisture and oxygen-free conditions seem to be the limitation of Negishi coupling compared to Suzuki coupling, however, such conditions, in many cases, are beneficial to the synthesis itself in terms of the efficacy of reaction, for example, producing high-quality polymers and increasing the lifetime of the catalyst. As more of its unique merits are discovered, we believe that Negishi coupling will gain increasing popularity in organic synthesis. Advanced technologies based on organic electronic, optical, electrochemical, and magnetic materials can have a profound influence on improving the quality of human life while, in the meantime, also address associated global issues such as energy crisis and environmental conservation. Negishi coupling will continue to make its unique contribution to the development of new materials with multiple functions to improve these technologies.
References
-
E. Negishi, in Handbook of Organopalladium Chemistry, ed. E. Negishi, John Wiley & Sons, New York, 2002, vol. 1, ch. 3, pp. 229–247 Search PubMed.
- E. Negishi and S. Baba, J. Chem. Soc., Chem. Commun., 1976, 596–597 RSC.
- E. Negishi and S. Baba, J. Am. Chem. Soc., 1976, 98, 6729–6731 CrossRef.
- E. Negishi, A. O. King and N. Okukado, J. Org. Chem., 1977, 42, 1821–1823 CrossRef CAS.
- E. Negishi and D. E. Van Horn, J. Am. Chem. Soc., 1977, 99, 3168–3170 CrossRef CAS.
- A. O. King, N. Okukado and E. Negishi, J. Chem. Soc., Chem. Commun., 1977, 683–684 RSC.
- A. O. King, E. Negishi, F. J. Villani Jr. and A. Silveira Jr., J. Org. Chem., 1978, 43, 358–360 CrossRef CAS.
- E. Negishi, N. Okukado, A. O. King, D. E. Van Horn and B. I. Spiegel, J. Am. Chem. Soc., 1978, 100, 2254–2256 CrossRef CAS.
-
E. Negishi, in Aspects of Mechanism and Organometallic Chemistry, ed. J. H. Brewster, Plenum, New York, 1978, pp. 285–317 Search PubMed.
- E. Negishi, T. Takahashi, S. Baba, D. E. Van Horn and N. Okukado, J. Am. Chem. Soc., 1987, 109, 2393–2401 CrossRef CAS.
- E. Negishi, Acc. Chem. Res., 1982, 15, 340–348 CrossRef CAS.
-
E. Negishi, X. Zeng, Z. Tan, M. Qian, Q. Hu and Z. Huang, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. De Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004, vol. 2, pp. 815–889 Search PubMed.
- E. Negishi, Q. Hu, Z. Huang, M. Qian and G. Wang, Aldrichimica Acta, 2005, 38, 71–88 CAS.
- E. Negishi, Bull. Chem. Soc. Jpn., 2007, 80, 233–257 CrossRef CAS.
- E. Negishi, Z. Huang, G. Wang, S. Mohan, C. Wang and H. Hattori, Acc. Chem. Res., 2008, 41, 1474–1485 CrossRef CAS PubMed.
- V. B. Phapale and D. J. Cardenas, Chem. Soc. Rev., 2009, 38, 1598–1607 RSC.
- E. Negishi, G. Wang, H. Rao and Z. Xu, J. Org. Chem., 2010, 75, 3151–3182 CrossRef CAS PubMed.
- C. Valente, M. E. Belowich, N. Hadei and M. G. Organ, Eur. J. Org. Chem., 2010, 4343–4354 CAS.
- E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed.
-
E. Colacino, J. Martinez and F. Lamaty, in Modern Tools for the Synthesis of Complex Bioactive Molecules, ed. J. Cossy and S. Arseniyadis, Wiley & Sons, Hoboken, 2012, pp. 33–75 Search PubMed.
- M. M. Heravi, E. Hashemi and N. Nazari, Mol. Diversity, 2014, 18, 441–472 CrossRef CAS PubMed.
- Other recent reviews on cross coupling reactions:
(a) G. C. Fu, Acc. Chem. Res., 2008, 41, 1555–1564 CrossRef CAS PubMed;
(b) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC;
(c) C. Valente, S. Ç Alimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed.
- For selected recent reviews:
(a) O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546–2555 CrossRef CAS PubMed;
(b) Y. Lin, H. Fan, Y. Li and X. Zhan, Adv. Mater., 2012, 24, 3087–3106 CrossRef CAS PubMed;
(c) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed;
(d) T. M. Figueira-Duarte and K. Müellen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS PubMed;
(e) X. Zhao and X. Zhan, Chem. Soc. Rev., 2011, 40, 3728–3743 RSC;
(f) A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS;
(g) X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed;
(h) A. L. Kanibolotsky, I. F. Perepichka and P. J. Skabara, Chem. Soc. Rev., 2010, 39, 2695–2728 RSC.
-
(a) T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38–68 CrossRef CAS PubMed;
(b) M. O'Neill and S. M. Kelly, Adv. Mater., 2011, 23, 566–584 CrossRef PubMed;
(c) H. Dong, H. Zhu, Q. Meng, X. Gong and W. Hu, Chem. Soc. Rev., 2012, 41, 1754–1808 RSC;
(d) H. Mori, T. Tanaka and A. Osuka, J. Mater. Chem. C, 2013, 1, 2500–2519 RSC;
(e) D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310 CrossRef CAS PubMed;
(f) W. Lee, J. Park, V. Subramanian and H. Takezoe, Opt. Mater. Exp., 2014, 4, 1088–1091 CrossRef.
- K. Tamao, J. Organomet. Chem., 2002, 653, 23–26 CrossRef CAS.
- V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1–652 CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- For selected reviews, see:
(a) J. Sakamoto, M. Rehahn, G. Wegner and A. D. Schlüter, Macromol. Rapid Commun., 2009, 30, 653–687 CrossRef CAS PubMed;
(b) M. Hoyos, M. L. Turner and O. Navarro, Curr. Org. Chem., 2011, 15, 3263–3290 CrossRef CAS;
(c) B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed;
(d) F. P. V. Koch and M. Heeney, Syn. Polym., 2012, 1, 155–198 CAS;
(e) Y. H. Geng, L. Huang, S. P. Wu and F. S. Wang, Sci. China: Chem., 2010, 53, 1620–1633 CrossRef CAS;
(f) J. Soloducho, J. Cabaj, K. Olech, P. Data and M. Lapkowski, Curr. Org. Chem., 2013, 17, 283–295 CrossRef CAS;
(g) T. Higashihara and E. Goto, Polym. J., 2014, 46, 381–390 CrossRef CAS;
(h) S. Xu, E. H. Kim, A. Wei and E. Negishi, Sci. Technol. Adv. Mater., 2014, 15, 044201 CrossRef.
- C. Wang, T. Tobrman, Z. Xu and E. Negishi, Org. Lett., 2009, 11, 4092–4095 CrossRef CAS PubMed.
-
(a) L. Zhu, R. M. Wehmeyer and R. D. Rieke, J. Org. Chem., 1991, 56, 1445–1453 CrossRef CAS;
(b) R. D. Rieke, M. V. Hanson, J. D. Brown and Q. J. Niu, J. Org. Chem., 1996, 61, 2726–2730 CrossRef CAS PubMed.
- C. Jubert and P. Knochel, J. Org. Chem., 1992, 57, 5425–5431 CrossRef CAS.
- S. Huo, Org. Lett., 2003, 5, 423–425 CrossRef CAS PubMed.
- A. Krasovskiy, V. Malakhov, A. Gavryushin and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 6040–6044 CrossRef CAS PubMed.
-
(a) A. Metzger, M. A. Schade and P. Knochel, Org. Lett., 2008, 10, 1107–1110 CrossRef CAS PubMed;
(b) A. Metzger, F. M. Piller and P. Knochel, Chem. Commun., 2008, 5824–5826 RSC.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
- I. F. Perepichka, D. F. Perepichka, H. Meng and F. Wudl, Adv. Mater., 2005, 17, 2281–2305 CrossRef CAS.
- I. McCulloch, M. Heeney, M. L. Chabinyc, D. De Longchamp, R. J. Kline, M. Colle, W. Duffy, D. Fischer, D. Gundlach, B. Hamadani, R. Hamilton, L. Richter, A. Salleo, M. Shkunov, D. Sparrowe, S. Tierney and W. Zhang, Adv. Mater., 2009, 21, 1091–1109 CrossRef CAS.
- Y. Li and Y. Zou, Adv. Mater., 2008, 20, 2952–2958 CrossRef CAS.
-
(a) T.-A. Chen and R. D. Rieke, J. Am. Chem. Soc., 1992, 114, l0087–10088 Search PubMed;
(b) T.-A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233–244 CrossRef CAS.
- T.-C. Chen and R. D. Rieke, Synth. Met., 1993, 60, 175–177 CrossRef CAS.
- F. Wang, M. S. Wilson, R. D. Rauh, P. Schottland and J. R. Reynolds, Macromolecules, 1999, 32, 4272–4278 CrossRef CAS.
- J. Li, A. Rajca and S. Rajca, Synth. Met., 2003, 137, 1507–1508 CrossRef CAS.
- L.-H. Xie, C.-R. Yin, W.-Y. Lai, Q.-L. Fan and W. Huang, Prog. Polym. Sci., 2012, 37, 1192–1264 CrossRef CAS PubMed.
- M. Leclerc, J. Polym. Sci., A, 2001, 39, 2867–2873 CrossRef CAS.
- M. Verswyvel, P. Verstappen, L. De Cremer, T. Verbiest and G. Koeckelberghs, J. Polym. Sci. A, 2011, 49, 5339–5349 CrossRef CAS.
- J. E. Milne and S. L. Buchwald, J. Am. Chem. Soc., 2004, 126, 13028–13032 CrossRef CAS PubMed.
- A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333–3336 CrossRef CAS PubMed.
- R. Tkachov, V. Senkovskyy, T. Beryozkina, K. Boyko, V. Bakulev, A. Lederer, K. Sahre, B. Voit and A. Kiriy, Angew. Chem., Int. Ed., 2014, 53, 2402–2407 CrossRef CAS PubMed.
- T. Erdmann, J. Back, R. Tkachov, A. Ruff, B. Voit, S. Ludwigs and A. Kiriy, Polym. Chem., 2014, 5, 5383–5390 RSC.
- R. Tkachov, H. Komber, S. Rauch, A. Lederer, U. Oertel, L. Haüßler, B. Voit and A. Kiriy, Macromolecules, 2014, 47, 4994–5001 CrossRef CAS.
- C. Winder and N. S. Sariciftci, J. Mater. Chem., 2004, 14, 1077–1086 RSC.
- S. Kirchmeyer and K. Reuter, J. Mater. Chem., 2005, 15, 2077–2088 RSC.
- C. Wang, J. L. Schindler, C. R. Kannewurf and M. G. Kanatzidis, Chem. Mater., 1995, 7, 58–68 CrossRef CAS.
- H. J. Spencer, P. J. Skabara, M. Giles, I. McCulloch, S. J. Coles and M. B. Hursthouse, J. Mater. Chem., 2005, 15, 4783–4792 RSC.
- G. Zotti, S. Zecchin, G. Schiavon, A. Berlin, G. Pagani, M. Borgonovo and R. Lazzaroni, Chem. Mater., 1997, 9, 2876–2886 CrossRef CAS.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS PubMed.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS PubMed.
-
(a) D. A. K. Vezzu, J. C. Deaton, M. Shayeghi, Y. Li and S. Huo, Org. Lett., 2009, 11, 4310–4313 CrossRef CAS PubMed;
(b) A. Chaskar, H. Chen and K.-T. Wong, Adv. Mater., 2011, 23, 3876–3895 CrossRef CAS PubMed.
- S.-J. Su, Y. Takahashi, T. Chiba, T. Takeda and J. Kido, Adv. Funct. Mater., 2009, 19, 1260–1267 CrossRef CAS.
- S.-J. Su, H. Sasabe, Y.-J. Pu, K. Nakayama and J. Kido, Adv. Mater., 2010, 22, 3311–3316 CrossRef CAS PubMed.
- Z. Peng, B. A. Haag and P. Knochel, Org. Lett., 2010, 12, 5398–5401 CrossRef CAS PubMed.
- H. Zhong, E. Xu, D. Zeng, J. Du, J. Sun, S. Ren, B. Jiang and Q. Fang, Org. Lett., 2008, 10, 709–712 CrossRef CAS PubMed.
- E. Xu, H. Zhong, J. Du, D. Zeng, S. Ren, J. Sun and Q. Fang, Dyes Pigm., 2009, 80, 194–198 CrossRef CAS PubMed.
-
(a) H. Tsuji, C. Mitsui, L. Ilies, Y. Sato and E. Nakamura, J. Am. Chem. Soc., 2007, 129, 11902–11903 CrossRef CAS PubMed;
(b) M. Nakamura, L. Ilies, S. Otsubo and E. Nakamura, Angew. Chem., Int. Ed., 2006, 45, 944–947 CrossRef CAS PubMed;
(c) H. Tsuji, C. Mitsui, Y. Sato and E. Nakamura, Adv. Mater., 2009, 21, 3776–3779 CrossRef CAS.
-
(a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef;
(b)
K. Sonogashira, in Metal-Catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, NewYork, 1998, pp. 203–229 Search PubMed.
- C. Dai and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 2719–2724 CrossRef CAS PubMed.
- S. A. Van Slyke, C. H. Chen and C. W. Tang, Appl. Phys. Lett., 1996, 69, 2160–2162 CrossRef CAS PubMed.
- Y.-M. Chen, W.-Y. Hung, H.-W. You, A. Chaskar, H.-C. Ting, H.-F. Chen, K.-T. Wong and Y.-H. Liu, J. Mater. Chem., 2011, 21, 14971–14978 RSC.
- N. Hebbar, Y. Ramondenc, G. Plé, G. Dupas and N. Plé, Tetrahedron, 2009, 65, 4190–4200 CrossRef CAS PubMed.
- L. Chen, W. Tam, S. H. Stevenson, G. R. Meredith, G. Rikken and S. R. Marder, J. Phys. Chem., 1991, 95, 10631–10643 CrossRef.
- F. López-Calahorra, M. Martínez-Rubio, D. Velasco, E. Brillas and L. Julia, Tetrahedron, 2004, 60, 285–289 CrossRef PubMed.
- S. Steffens, M. H. Prosenc, J. Heck, I. Asselberghs and K. Clays, Eur. J. Inorg. Chem., 2008, 1999–2006 CrossRef CAS.
-
(a) F. Rebiere, O. Samuel and H. B. Kagan, Tetrahedron Lett., 1990, 31, 3121–3124 CrossRef CAS;
(b) U. T. Müller-Westerhoff, Z. Yuan and G. Ingram, J. Organomet. Chem., 1993, 463, 163–167 CrossRef.
- M. Sonoda, A. Inaba, K. Itahashi and Y. Tobe, Org. Lett., 2001, 3, 2419–2421 CrossRef CAS PubMed.
- R. Diercks, J. C. Armstrong, R. Boese and K. P. C. Vollhardt, Angew. Chem., Int. Ed. Engl., 1986, 25, 268–269 CrossRef.
- T. X. Neenan and G. M. Whitesides, J. Org. Chem., 1988, 53, 2489–2496 CrossRef CAS.
- P. Herwig, C. W. Kayser, K. Müllen and H. W. Spiess, Adv. Mater., 1996, 8, 510–513 CrossRef CAS.
-
(a) A. M. van de Craats, J. M. Warman, K. Müllen, Y. Geets and J. D. Brand, Adv. Mater., 1998, 10, 36–38 CrossRef CAS;
(b) A. M. van de Craats, J. M. Warman, A. Fechtenkötter, J. D. Brand, M. A. Harbison and K. Müllen, Adv. Mater., 1999, 11, 1469–1472 CrossRef CAS.
- A. Fechtenkotter, N. Tchebotareva, M. Waston and K. Müllen, Tetrahedron, 2001, 57, 3769–3783 CrossRef CAS.
- X. Feng, M. Liu, W. Pisula, M. Takase, J. Li and K. Müllen, Adv. Mater., 2008, 20, 2684–2689 CrossRef CAS PubMed.
- Y. A. Getmanenko, R. J. Twieg and B. D. Ellman, Liq. Cryst., 2006, 33, 267–288 CrossRef CAS.
- Y. A. Getmanenko and R. J. Twieg, J. Org. Chem., 2008, 73, 830–839 CrossRef CAS PubMed.
- M. Hird, K. J. Toyne, J. W. Goodby, G. W. Gray, V. Minter, R. P. Tuffin and D. G. McDonnell, J. Mater. Chem., 2004, 14, 1731–1743 RSC.
- L. Johnson, B. Ringstrand and P. Kaszynski, Liq. Cryst., 2009, 36, 179–185 CrossRef CAS.
- M. G. Organ, S. Avola, I. Dubovyk, N. Hadei, E. A. B. Kantchev, C. J. O'Brien and C. Valente, Chem. – Eur. J., 2006, 12, 4749–4755 CrossRef CAS PubMed.
- H. Bürckstümmer, A. Weissenstein, D. Bialas and F. Würthner, J. Org. Chem., 2011, 76, 2426–2432 CrossRef PubMed.
- Z. Lu, N. Liu, S. J. Lord, S. D. Bunge, W. E. Moerner and R. J. Twieg, Chem. Mater., 2009, 21, 797–810 CrossRef CAS PubMed.
- T. Katagiri, S. Ota, T. Ohira, T. Yamao and S. Hotta, J. Heterocycl. Chem., 2007, 44, 853–862 CrossRef CAS.
- S. Hotta and T. Yamao, J. Mater. Chem., 2011, 21, 1295–1304 RSC.
- D. Jouvenot, E. C. Glazer and Y. Tor, Org. Lett., 2006, 8, 1987–1990 CrossRef CAS PubMed.
- Y. Yasui, D. K. Frantz and J. S. Siegel, Org. Lett., 2006, 8, 4989–4992 CrossRef CAS PubMed.
- J. C. Loren and J. S. Siegel, Angew. Chem., Int. Ed., 2001, 40, 754–757 CrossRef CAS.
- A. Pelter and R. Drake, Tetrahedron, 1994, 50, 13775–13800 CrossRef CAS.
- J.-C. Chambron and J.-P. Sauvage, Tetrahedron, 1987, 43, 895–904 CrossRef CAS.
- L. Friedman and J. F. Chlebowski, J. Org. Chem., 1968, 33, 1636–1638 CrossRef CAS.
- I. Klement, M. Rottlander, C. E. Tucker, T. N. Majid, P. Knochel, P. Venegas and G. Cahiez, Tetrahedron, 1996, 52, 7201–7220 CrossRef CAS.
- N. Hebbar, C. Fiol-Petit, Y. Ramondenc, G. Plé and N. Plé, Tetrahedron, 2011, 67, 2287–2298 CrossRef CAS PubMed.
-
(a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed;
(b) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- G. Duran-Sampedro, E. Palao, A. R. Agarrabeitia, S. de la Moya, N. Boensb and M. J. Ortiz, RSC Adv., 2014, 4, 19210–19213 RSC.
-
(a) M. A. Baldo, M. E. Thompson and S. R. Forrest, Pure Appl. Chem., 1999, 71, 2095–2106 CrossRef CAS;
(b) H. Sasabe and J. Kido, Eur. J. Org. Chem., 2013, 7653–7663 CrossRef CAS.
- D. A. K. Vezzu, J. C. Deaton, J. S. Jones, L. Bartolotti, C. F. Harris, A. P. Marchetti, M. Kondakova, R. D. Pike and S. Huo, Inorg. Chem., 2010, 49, 5107–5119 CrossRef CAS PubMed.
- D. Ravindranathan, D. A. K. Vezzu, L. Bartolotti, P. D. Boyle and S. Huo, Inorg. Chem., 2010, 49, 8922–8928 CrossRef CAS PubMed.
- D. A. K. Vezzu, D. Ravindranathan, A. W. Garner, L. Bartolotti, M. E. Smith, P. D. Boyle and S. Huo, Inorg. Chem., 2011, 50, 8261–8273 CrossRef CAS PubMed.
- S. Huo, C. F. Harris, D. A. K. Vezzu, J. P. Gagnier, M. E. Smith, R. D. Pike and Y. Li, Polyhedron, 2013, 52, 1030–1040 CrossRef CAS PubMed.
- C. F. Harris, D. A. K. Vezzu, L. Bartolotti, P. D. Boyle and S. Huo, Inorg. Chem., 2013, 52, 11711–11722 CrossRef CAS PubMed.
-
(a) D. J. Cárdenas, A. M. Echavarren and M. C. Ramírez de Arellano, Organometallics, 1999, 18, 3337–3341 CrossRef;
(b) J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS PubMed;
(c)
S. Huo, J. C. Deaton and A. F. Sowinski, U.S. Patent 7,029,766, 24, 2006 Search PubMed.
-
(a) P. G. Bomben, K. C. D. Robson, P. A. Sedach and C. P. Berlinguette, Inorg. Chem., 2009, 48, 9631–9643 CrossRef CAS PubMed;
(b) D. A. K. Vezzu, Q. Lu, Y.-H. Chen and S. Huo, J. Inorg. Biochem., 2014, 134, 49–56 CrossRef CAS PubMed.
- W. Wu, W. Wu, S. Ji, H. Guo and J. Zhao, J. Organomet. Chem., 2011, 696, 2388–2398 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
-
(a) S. G. DiMagno, V. S.-Y. Lin and M. J. Therien, J. Am. Chem. Soc., 1993, 115, 2513–2515 CrossRef CAS;
(b) S. G. DiMagno, V. S.-Y. Lin and M. J. Therien, J. Org. Chem., 1993, 58, 5983–5993 CrossRef CAS.
-
(a) T. Takanami, M. Yotsukura, W. Inoue, N. Inoue, F. Hino and K. Suda, Heterocycles, 2008, 76, 439–453 CrossRef CAS PubMed;
(b) T. Takanami, M. Hayashi, H. Chijimatsu, W. Inoue and K. Suda, Org. Lett., 2005, 7, 3937–3940 CrossRef CAS PubMed.
- M. C. Balaban, A. Eichhöfer, G. Buth, R. Hauschild, J. Szmytkowski, H. Kalt and T. S. Balaban, J. Phys. Chem. B, 2008, 112, 5512–5521 CrossRef CAS PubMed.
- K. Fujimoto, H. Yorimitsu and A. Osuka, Eur. J. Org. Chem., 2014, 4327–4334 CrossRef CAS.
- A. N. Cammidge, C.-H. Tseng, I. Chambrier, D. L. Hughes and M. J. Cook, Tetrahedron Lett., 2009, 50, 5254–5256 CrossRef CAS PubMed.
- D. J. Tate, R. Anémian, R. J. Bushby, S. Nanan, S. L. Warriner and B. J. Whitaker, Beilstein J. Org. Chem., 2012, 8, 120–128 CrossRef CAS PubMed.
- M. J. Heeney, S. A. Al-Raqa, A. Auger, P. M. Burnham, A. N. Cammidge, I. Chambrier and M. J. Cook, J. Porphyrins Phthalocyanines, 2013, 17, 650–664 CrossRef.
- P. G. Bomben, T. J. Gordon, E. Schott and C. P. Berlinguette, Angew. Chem., Int. Ed., 2011, 50, 10682–10685 CrossRef CAS PubMed.
- S.-W. Chiu, L.-Y. Lin, H.-W. Lin, Y.-H. Chen, Z.-Y. Huang, Y.-T. Lin, F. Lin, Y.-H. Liu and K.-T. Wong, Chem. Commun., 2012, 48, 1857–1859 RSC.
-
(a) P. M. DiCarmine, X. Wang, B. L. Pagenkopf and O. A. Semenikhin, Electrochem. Commun., 2008, 10, 229–232 CrossRef CAS PubMed;
(b) J. C. Byers, P. M. DiCarmine, M. M. A. R. Moustafa, X. Wang, B. L. Pagenkopf and O. A. Semenikhin, J. Phys. Chem. B, 2009, 113, 15715–15723 CrossRef CAS PubMed.
- S. Yamaguchi, T. Endo, M. Uchida, T. Izumizawa, K. Furukawa and K. Tamao, Chem. – Eur. J., 2000, 6, 1683–1692 CAS.
- A. Mishra, C.-Q. Ma and P. Baüerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS PubMed.
- Y. Liu, J. Zhou, X. Zhang, Z. Liu, X. Wan, J. Tianb, T. Wanga and Y. Chen, Carbon, 2009, 47, 3113–3121 CrossRef CAS PubMed.
- H. Kanato, K. Takimiya, T. Otsubo, Y. Aso, T. Nakamura, Y. Araki and O. Ito, J. Org. Chem., 2004, 69, 7183–7189 CrossRef CAS PubMed.
- J.-L. Wang, X.-F. Duan, B. Jiang, L.-B. Gan, J. Pei, C. He and Y.-F. Li, J. Org. Chem., 2006, 71, 4400–4410 CrossRef CAS PubMed.
- J. Cremer, E. Mena-Osteritz, N. G. Pschierer, K. Müllen and P. Bäuerle, Org. Biomol. Chem., 2005, 3, 985–995 CAS.
-
(a) C. R. Mason, Y. Li, P. O'Brien, N. J. Findley and P. J. Skabara, Beilstein J. Org. Chem., 2011, 7, 1722–1731 CrossRef CAS PubMed;
(b) C. R. Mason, P. J. Skabara, D. Cupertino, J. Schofield, F. Meghdadi, B. Ebner and N. Serdar Saricifti, J. Mater. Chem., 2005, 15, 1446–1453 RSC.
- M. Wagner, Angew. Chem., Int. Ed., 2006, 45, 5916–5918 CrossRef CAS PubMed.
- Y. Yu, A. D. Bond, P. W. Leonard, K. P. C. Vollhardt and G. D. Whitener, Angew. Chem., Int. Ed., 2006, 45, 1794–1799 CrossRef CAS PubMed.
-
Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- Y. Yu, A. D. Bond, P. W. Leonard, U. J. Lorenz, T. V. Timofeeva, K. P. C. Vollhardt, G. D. Whitener and A. A. Yakovenko, Chem. Commun., 2006, 2572–2574 RSC.
- U. Pfaff, A. Hildebrandt, D. Schaarschmidt, T. Hahn, S. Liebing, J. Kortus and H. Lang, Organometallics, 2012, 31, 6761–6771 CrossRef CAS.
- A. Carella, G. Rapenne and J.-P. Launay, New J. Chem., 2005, 29, 288–290 RSC.
- A. K. Diallo, J.-C. Daran, F. Varret, J. Ruiz and D. Astruc, Angew. Chem., Int. Ed., 2009, 48, 3141–3145 CrossRef CAS PubMed.
- A. K. Diallo, C. Absalon, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2011, 133, 629–641 CrossRef CAS PubMed.
- A. Hildebrandt, D. Schaarschmidt and H. Lang, Organometallics, 2011, 30, 556–563 CrossRef CAS.
- A. Hildebrandt and H. Lang, Dalton Trans., 2011, 40, 11831–11837 RSC.
- A. Hildebrandt, D. Schaarschmidt, R. Claus and H. Lang, Inorg. Chem., 2011, 50, 10623–10632 CrossRef CAS PubMed.
- J. M. Speck, R. Claus, A. Hildebrandt, T. Rüffer, E. Erasmus, L. van As, J. C. Swarts and H. Lang, Organometallics, 2012, 31, 6373–6380 CrossRef CAS.
- U. Pfaff, A. Hildebrandt, D. Schaarschmidt, T. Rüffer, P. J. Low and H. Lang, Organometallics, 2013, 32, 6106–6117 CrossRef CAS.
- S. W. Lehrich, A. Hildebrandt, T. Rüffer, M. Korb, P. J. Low and H. Lang, Organometallics, 2014, 33, 4836–4845 CrossRef CAS.
- T. Mochida, F. Shimizu, H. Shimizu, K. Okazawa, F. Sato and D. Kuwahara, J. Organomet. Chem., 2007, 692, 1834–1844 CrossRef CAS PubMed.
- S. Tampier, S. M. Bleifuss, M. M. Abd-Elzaher, J. Sutter, F. W. Heinemann and N. Burzlaff, Organometallics, 2013, 32, 5935–5945 CrossRef CAS.
- Y.-Q. Fang, M. I. J. Polson and G. S. Hanan, Inorg. Chem., 2003, 42, 5–7 CrossRef CAS PubMed.
-
(a) L. Zhang, Z. Huang, W.-Z. Chen, E. Negishi, P. E. Fanwick, J. B. Updegraff, J. D. Protasiewicz and T. Ren, Organometallics, 2007, 26, 6526–6528 CrossRef CAS;
(b) G.-L. Xu and T. Ren, Inorg. Chem., 2006, 45, 10449–10456 CrossRef PubMed.
- E. M. Galand, J. K. Mwaura, A. A. Argun, K. A. Abboud, T. D. McCarley and J. R. Reynolds, Macromolecules, 2006, 39, 7286–7294 CrossRef CAS.
-
(a) H. Iwamura and N. Koga, Acc. Chem. Res., 1993, 26, 346–351 CrossRef CAS;
(b) A. Rajca, Chem. Rev., 1994, 94, 871–893 CrossRef CAS.
-
(a) I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC;
(b) E. P. Tomlinson, M. E. Hay and B. W. Boudouris, Macromolecules, 2014, 47, 6145–6158 CrossRef CAS.
-
(a) A. Rajca, J. Wongsriratanakul, S. Rajca and R. Cerny, Angew. Chem., Int. Ed., 1998, 37, 1229–1232 CrossRef CAS;
(b) S. Rajca, A. Rajca, J. Wongsriratanakul, P. Butler and S. Choi, J. Am. Chem. Soc., 2004, 126, 6972–6986 CrossRef CAS PubMed.
- A. Rajca, J. Wongsriratanakul and S. Rajca, J. Am. Chem. Soc., 2004, 126, 6608–6626 CrossRef CAS PubMed.
- A. Rajca, S. Rajca and J. Wongsriratanakul, J. Am. Chem. Soc., 1999, 121, 6308–6309 CrossRef CAS.
- A. Rajca, J. Wongsriratanakul and S. Rajca, Science, 2001, 294, 1503–1505 CrossRef CAS PubMed.
- A. Olankitwanit, V. Kathirvelu, S. Rajca, G. R. Eaton, S. S. Eaton and A. Rajca, Chem. Commun., 2011, 47, 6443–6445 RSC.
- C. Elschenbroich, O. Schiemann, O. Burghaus and K. Harms, Chem. Commun., 2005, 2149–2151 RSC.
- S. Ito, Y. Ueta, T. T. T. Ngo, M. Kobayashi, D. Hashizume, J. Nishida, Y. Yamashita and K. Mikami, J. Am. Chem. Soc., 2013, 135, 17610–17616 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Professor Ei-ichi Negishi on the occasion of his 80th birthday. |
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.