
Synthesis and characterization of N-2-aryl-1,2,3-triazole based iridium complexes as photocatalysts with tunable photoredox potential†
Rong
Cai
a,
Wuming
Yan
a,
Matthew G.
Bologna
a,
Kaushalya
de Silva
a,
Zhao
Ma
b,
Harry O.
Finklea
a,
Jeffrey L.
Petersen
a,
Minyong
Li
 *b and
Xiaodong
Shi
*a
*b and
Xiaodong
Shi
*a
aC. Eugene Bennett Department of Chemistry, West Virginia University, Morgantown, WV 26506, USA. E-mail: Xiaodong.Shi@mail.wvu.edu; Fax: +1-304-293-4904; Tel: +1-304-293-0179
bDepartment of Medicinal Chemistry, School of Pharmacy, Shandong University, Jinan, Shandong 250012, China. E-mail: mli@sdu.edu.cn; Fax: +86-531-8838-2076
First published on 30th December 2014
Abstract
N-2-Aryl chelated 1,2,3-triazole-Ir(III) complexes with various substituents were prepared for the first time. These photoactive Ir(III) complexes were characterized by X-ray crystallography and their redox potentials were evaluated. This study revealed a new class of photocatalysts with tunable photoredox potentials.
The cationic Ir(III) polyimine complexes have been widely applied in various areas,1 including live cell imaging,2 electroluminescent materials,3 water oxidation etc.4 Recently, [Ir(ppy)2(bpy)]+ and [Ir(ppy)2(dtbbpy)]+ have gained tremendous attention as efficient photocatalysts in promoting organic transformations.5,6 The fast growth of photocatalytic research led to strong needs for new systems with structural novelty and potentially new reactivity. Herein, we report the application of N-2-aryl-1,2,3-triazoles as ligands for the synthesis of novel N-aryl chelated Ir(III) photocatalysts with tunable redox potentials.
After the discovery of “click chemistry”, 1,2,3-triazole (TA) became one of the most important heterocycles in chemical research.7 However, the studies regarding the binding ability of N-2-substituted triazole toward a metal cation are relatively rare. During the past several years, our group has been working on developing new synthetic methods toward triazole functionalization8 while investigating the coordination ability of various triazole derivatives.9 Inspired by recent success on Ir(III) promoted photocatalysis, we become interested in studying the TA binding ability with Ir(III) cations and hope to develop a new class of photoactive complexes with the potential to further extend the reaction scope of photocatalysis.
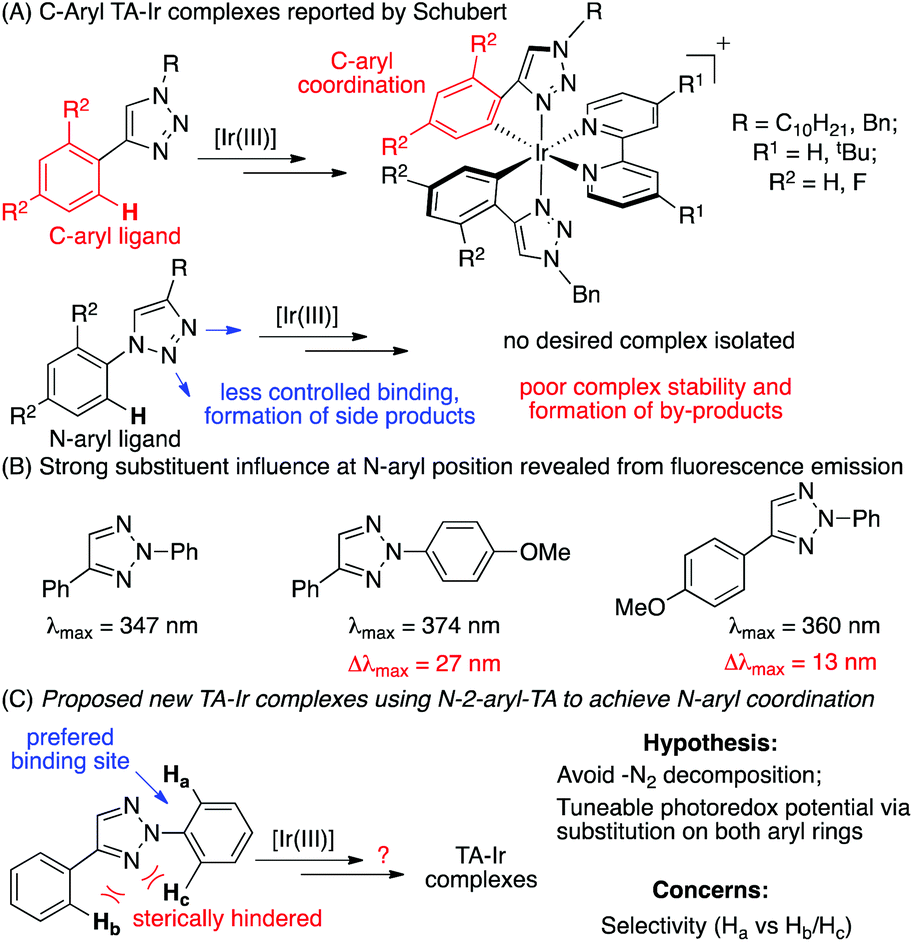
In 2009, Schubert and co-workers reported the first triazole coordinated Ir(III) complexes using N1-substituted 1,2,3-triazole as the C^N ligand.10 Later, De Cola and co-workers reported the electroluminescence properties of similar Ir(III) complexes (Scheme 1A).11 Although these and other pioneering studies12 revealed a good coordination ability of TA ligands with Ir(III), no photocatalytic reactivity has been reported. More importantly, as mentioned by Schubert, only C-aryl-TA chelated Ir(III) complexes could be produced. The attempts to form N-aryl-TA chelating complexes were unsuccessful due to either the poor complex stability or the formation of unidentified byproducts.
 | ||
| Scheme 1 New Ir(III) complexes with 1,2,3-triazole ligands. | ||
Among various triazole derivatives, one particularly interesting compound is the fluorescence active N-2-aryl triazoles (NATs).7b As revealed by the crystal structures, the N-2-aryl group holds a perfect co-planar conformation with the triazole ring, resulting in a strong fluorescence emission, whereas the N-1-isomer exhibits no emission.13 Moreover, substitution on the N-2-aryl group exhibited a strong influence on the fluorescence emission wavelength and intensity (Scheme 1B). Based on these results, we postulated that the N-2-aryl-triazole may be used as a ligand to prepare novel N-aryl-chelated Ir(III) complexes by avoiding the potential triazole decomposition (via loss of N2) and providing specific coordination sites.14 To test our hypothesis, N-2-aryl-triazoles were prepared and used for Ir(III) complex formation. The general synthetic route is summarized in Fig. 1.15
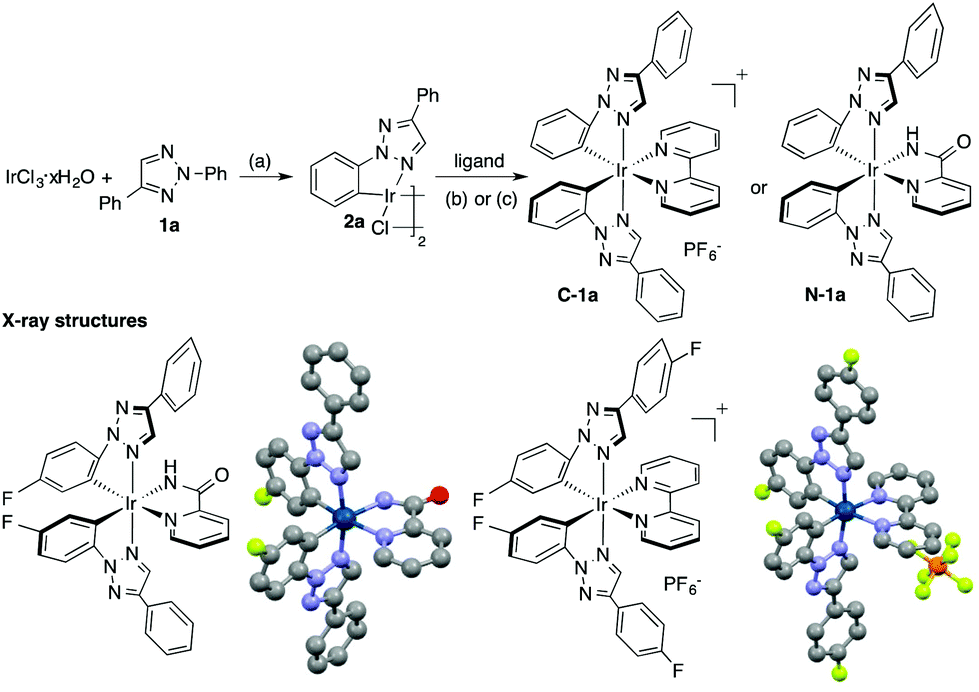 | ||
| Fig. 1 General synthetic route and X-ray crystal structures of N-aryl-chelated TA-Ir complexes: (a) 2-ethoxyethanol, 140 °C, N2, 24 h; (b) 2,2′-bispyridine (bpy, 2.5 eq.), 1,2-ethanediol, 120 °C, N2, 20 h, followed by anion exchange with NH4PF6; (c) 2-picolinamide (2.6 eq.), Na2CO3 (11 eq.), 2-ethoxyethanol, 140 °C, N2, 20 h. | ||
As expected, treating triazole 1 with IrCl3·xH2O gave the corresponding chloro-bridged iridium(III) dimer 2 in excellent yields (generally >80%). Subsequent reactions with either a neutral N^N ligand (condition b) or an anionic N^N ligand (condition c) gave the corresponding cationic TA-Ir (C-1) or neutral TA-Ir (N-1) complexes. Both complexes were characterized by X-ray crystallography. Notably, unlike N-1-aryl triazoles, the N-2-aryl triazoles showed a strong coordination ability, forming stable Ir(III) complexes, which could be easily purified by column chromatography without decomposition. With this synthetic method, various TA-Ir complexes were prepared. Good to excellent yields were obtained in most cases, which warranted further applications of N-2-aryl triazoles as the C^N ligand for iridium photocatalysts.
As shown in Scheme 2A, the general mechanism of Ir-photocatalysis is initiated from the promotion of Ir(III) to the excited state Ir(III)* by visible light, followed by oxidative or reductive quenching through single-electron transfer. The resulting Ir(IV) or Ir(II) will be reduced or oxidized to regenerate the ground state Ir(III). The redox potential for each step is a crucial factor in catalyst design, simply because oxidation/reduction reactions require good matching of the redox potentials between catalysts and substrates. Compared to 2-phenylpyridine (ppy) ligands, N-2-aryl triazoles are more electron-deficient. Thus, higher oxidation potentials of the corresponding iridium complexes are expected.5c To evaluate the photophysical properties of these new TA-Ir complexes, UV-Vis absorption and fluorescence emission were examined.
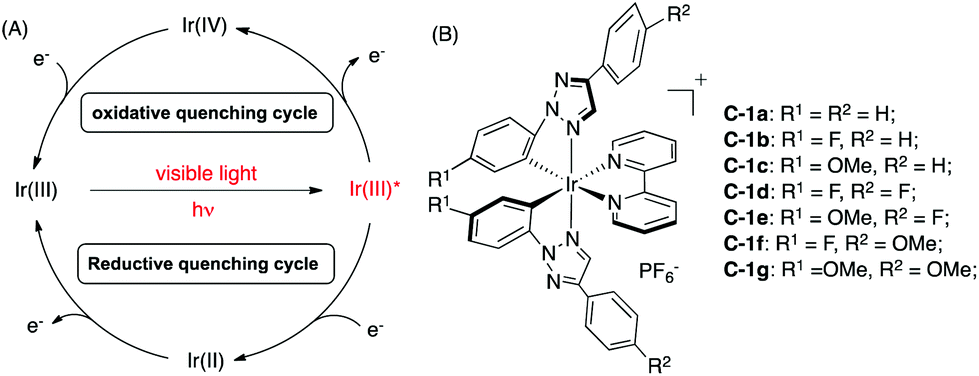 | ||
| Scheme 2 (A) The general mechanism for Ir(III) photocatalysis; (B) TA-Ir complexes prepared from N-2-aryl triazoles. | ||
Similar to other literature reported systems,15a the neutral complex N-1a (with the anionic N^N ligand) gave a very weak fluorescence emission and almost no photocatalytic reactivity. On the other hand, compared with [(ppy)2Ir(bpy)]PF6, the TA-Ir cationic complexes [(tapy)2Ir(bpy)]PF6 showed better absorption of the blue light and a stronger fluorescence emission in the visible light region (Fig. 2, see detailed spectral comparison in ESI†).
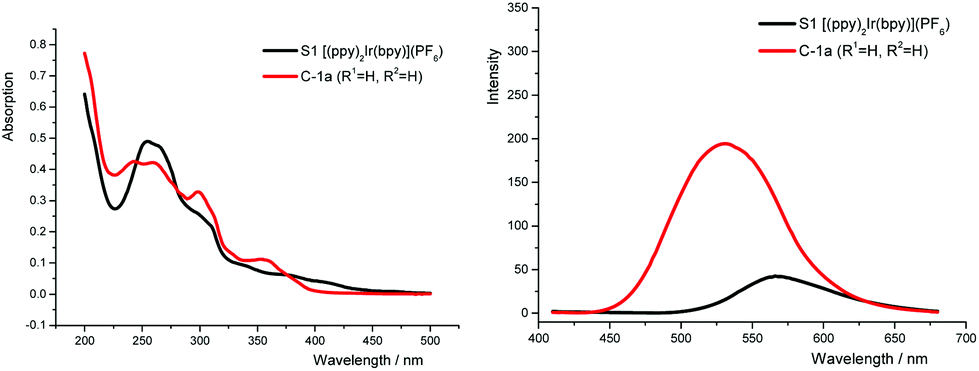 | ||
| Fig. 2 UV-Vis (left) and fluorescence (right) spectral comparison. | ||
The fluorescence emission and redox potentials of some representative TA-Ir complexes are summarized in Table 1.
| R1 | R2 | λ em (nm) | Φ | τ (ns) | E 1/2 (V) | ||||
|---|---|---|---|---|---|---|---|---|---|
| (Ir4+/3+) | (Ir4+/3+*) | (Ir3+/2+) | (Ir3+*/2+) | ||||||
| a S1 = [(ppy)2Ir(bpy)]PF6. All potentials are given versus the saturated calomel electrode (SCE). Measurements were performed at room temperature in acetonitrile using an internal standard Fc/Fc+ redox couple (0.40 V vs. SCE). b PF6− is used as the counter anion. c The quantum yields were calculated relative to Ru(bpy)3(PF6)2 (Φ = 0.062 in ACN). d Excited-state lifetime. | |||||||||
| S1 | — | — | 566 | 1.28 | −0.91 | −1.38 | 0.81 | ||
| C-1a | H | H | 530 | 0.32 | 266 | 1.54 | −0.80 | −1.34 | 1.00 |
| C-1b | F | H | 503 | 0.39 | 358 | 1.67 | −0.80 | −1.33 | 1.14 |
| C-1c | OMe | H | 550 | 0.02 | 30 | 1.37 | −0.88 | −1.35 | 0.90 |
| C-1d | F | F | 501 | 0.29 | 270 | 1.66 | −0.82 | −1.33 | 1.15 |
| C-1e | OMe | F | 554 | 0.02 | 32 | 1.37 | −0.87 | −1.35 | 0.89 |
| C-1f | F | OMe | 508 | 0.21 | 172 | 1.61 | −0.83 | −1.32 | 1.12 |
| C-1g | OMe | OMe | 558 | 0.01 | 18 | 1.25 | −0.90 | −1.37 | 0.85 |
The data in Table 1 revealed a clear substituent effect on the Ir4+/3+ redox potential. First, with more electron-deficient phenyl-triazole (pta) ligand, C-1a gave a higher Ir4+/3+ redox potential (1.54 V) than [Ir(ppy)2(bpy)]PF6 (S1, 1.28 V). The introduction of electron withdrawing groups (such as F, C-1b) at the R1 position (N-2 aryl) further increased the E1/2 (Ir4+/3+) to 1.67 V. In contrast, complex C-1c (with the electron donating OMe) gave a lower Ir4+/3+ redox potential (1.37 V), which was still higher than ppy complex S1. A similar trend was also observed in the Ir3+*/2+ redox potential, though with a smaller variation. However, substitution at the R2 position indicated little influence on the redox potential (e.g.C-1b, 1.67 V vs.C-1d, 1.66 V), suggesting that the electronic effect influence from the ring that is directly touching the metal center is much more important. Similarly, introducing EWG at the R1 position (C-1b) helped to increase the excited-state lifetime and quantum yield while introducing EDG (C-1c) impaired the excited-state lifetime and quantum yield.
The reduction potential E1/2 (Ir3+/2+) of all tested C-1 TA-Ir complexes were almost the same even with different C^N ligands. This is likely due to the fact that Ir(III) reduction is more related to the metal–ligand charge transfer (MLCT) through the π* orbital of the N^N ligand (bpy).1 To fully elucidate the ligand effect on the redox potential, we prepared triazole-pyridine (tapy)16 as a new type of the N^N ligand to coordinate with Ir cations. The reaction between the chloro-bridged iridium dimer 2a and N-2 tapy gave messy mixtures with no desired complex isolated. Interestingly, a much cleaner reaction was obtained with N-1-tapy. Although growing a single crystal is unsuccessful at this moment, the TA-Ir complexes with N-1-tapy as the N^N ligand have been successfully prepared and characterized by 1H, 13C, 19F NMR and HRMS. The fluorescence emission and redox potentials of these complexes were then determined as shown in Table 2.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| R1 | λ em (nm) | Φ c | τ d (ns) | E 1/2 (V) | ||||
| (Ir4+/3+) | (Ir4+/3+*) | (Ir3+/2+) | (Ir3+*/2+) | |||||
| a S1 = [(ppy)2Ir(bpy)]PF6; S2 = [(ppy)2Ir(dtbbpy)]PF6. b ,c,dSame conditions as in Table 1 applied. | ||||||||
| S1 | — | 566 | 1.28 | −0.91 | −1.38 | 0.81 | ||
| S2 | — | 560 | 0.20 | 175d | 1.25 | −0.96 | −1.48 | 0.73 |
| C-2 | — | 546 | 0.12 | 110 | 1.37 | −0.90 | −1.48 | 0.79 |
| C-3a | H | 462, 486 | 0.06 | 217 | 1.61 | −0.94 | −1.45 | 1.10 |
| C-3b | F | 458, 482 | 0.07 | 375 | 1.75 | −0.82 | −1.41 | 1.16 |
| C-3c | OMe | 508 | 0.01 | 108 | 1.37 | −1.07 | −1.43 | 1.01 |
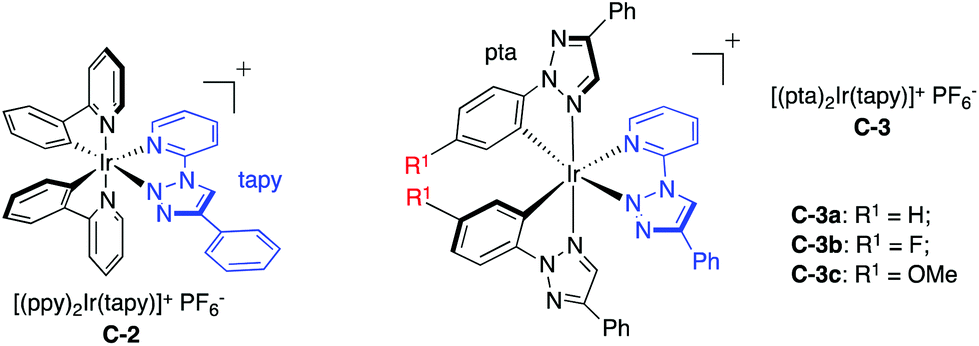
Interestingly, compared to complex S1 (with the ppy N^N ligand), complex C-2 (with the tapy N^N ligand) resulted in a clear increase of the Ir3+/2+ reduction potential, from 1.38 V to 1.48 V, similar to complex S2 (with the dtbbpy N^N ligand). Notably, the Ir3+/2+ reduction potential remained almost the same when changing the C^N ligand to pta. These results highlighted the excellent viability of these new triazole-based Ir photocatalysts: using pta as the C^N ligand to adjust the Ir4+/3+ oxidation potential while using tapy as the N^N ligand to tune the Ir3+/2+ reduction potential.
Two typical photocatalytic reactions6c,17 were performed to prove the feasibility of these new TA-Ir complexes and good catalytic reactivities were observed as shown in Fig. 3.
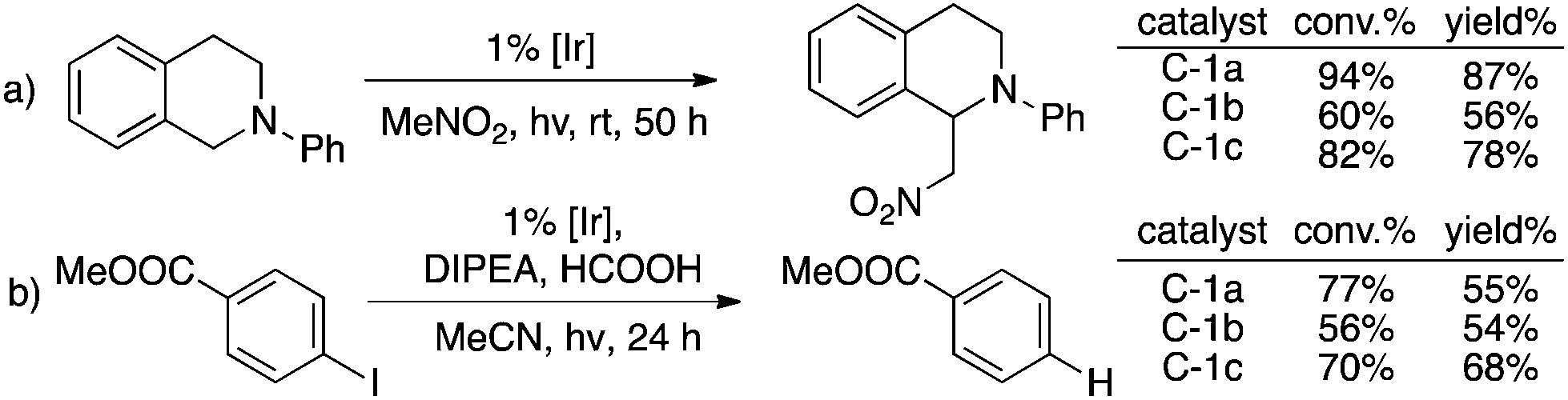 | ||
| Fig. 3 TA-Ir as effective new photocatalysts. | ||
In conclusion, we herein report the synthesis and characterization of N-2-aryl-1,2,3-triazole-Ir(III) complexes (TA-Ir). Various complexes have been prepared and their photophysical properties were evaluated. Tunable redox potentials were achieved by varying substituents on either the pta C^N ligand or the tapy N^N ligand, which indicated a promising future for these new photocatalysts. Investigation on challenging photocatalytic reactions using this new class of photocatalysts is currently ongoing in our lab.
We thank the NSF (CHE-1362057) and NSFC (21228204) for the financial supports.
Notes and references
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS.
- (a) K. K.-W. Lo, W.-K. Hui, C.-K. Chung, K. H.-K. Tsang, T. K.-M. Lee, C.-K. Li, J. S.-Y. Lau and D. C.-M. Ng, Coord. Chem. Rev., 2006, 250, 1724–1736 CrossRef CAS PubMed; (b) K. Y. Zhang and K. K.-W. Lo, Inorg. Chem., 2009, 48, 6011–6025 CrossRef CAS PubMed; (c) Q. Zhao, M. Yu, L. Shi, S. Liu, C. Li, M. Shi, Z. Zhou, C. Huang and F. Li, Organometallics, 2010, 29, 1085–1091 CrossRef CAS; (d) G. Zhang, H. Zhang, Y. Gao, R. Tao, L. Xin, J. Yi, F. Li, W. Liu and J. Qiao, Organometallics, 2013, 33, 61–68 CrossRef.
- (a) V. V. Grushin, N. Herron, D. D. LeCloux, W. J. Marshall, V. A. Petrov and Y. Wang, Chem. Commun., 2001, 1494–1495 RSC; (b) A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377–7387 CrossRef CAS PubMed; (c) J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS PubMed; (d) A. B. Tamayo, S. Garon, T. Sajoto, P. I. Djurovich, I. M. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 8723–8732 CrossRef CAS PubMed.
- (a) M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS; (b) N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2007, 130, 210–217 CrossRef PubMed; (c) D. R. Whang, D. R. Whang, K. Sakai, K. Sakai and S. Y. Park, Angew. Chem., Int. Ed., 2013, 52, 11612–11615 CrossRef CAS PubMed.
- Recent reviews on visible light photocatalysis: (a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2010, 40, 102 RSC; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed; (b) J. Du and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 14604–14605 CrossRef CAS PubMed; (c) A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed; (d) S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350–19353 CrossRef CAS PubMed; (e) M. Neumann, S. Fueldner, B. Koenig and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed; (f) S. Maity and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 9562–9566 CrossRef CAS PubMed; (g) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed; (h) Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567–9571 CrossRef CAS PubMed; (i) S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheelhouse, G. Rassias, M. Médebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505–2508 CrossRef CAS PubMed; (j) S. Zhu, A. Das, L. Bui, H. Zhou, D. P. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823–1829 CrossRef CAS PubMed.
- (a) J. Crowley and D. McMorran, in Click Triazoles, ed. J. Košmrlj, Springer, Berlin, Heidelberg, 2012, pp. 31–83 Search PubMed; (b) B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
- (a) Y. Chen, Y. Liu, J. L. Petersen and X. Shi, Chem. Commun., 2008, 3254–3256 RSC; (b) Y. Liu, W. Yan, Y. Chen, J. L. Petersen and X. Shi, Org. Lett., 2008, 10, 5389–5392 CrossRef CAS PubMed; (c) W. Yan, Q. Wang, Y. Chen, J. L. Petersen and X. Shi, Org. Lett., 2010, 12, 3308–3311 CrossRef CAS PubMed.
- (a) H. Duan, S. Sengupta, J. L. Petersen, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2009, 131, 12100–12102 CrossRef CAS PubMed; (b) H. Duan, S. Sengupta, J. L. Petersen and X. Shi, Organometallics, 2009, 28, 2352–2355 CrossRef CAS; (c) X. Y., Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Petersen and X. Shi, Chem. Sci., 2013, 4, 3712–3716 RSC; (d) Q. Wang, S. E. Motika, N. G. Akhmedov, J. L. Petersen and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 5418–5422 CrossRef CAS PubMed.
- B. Beyer, C. Ulbricht, D. Escudero, C. Friebe, A. Winter, L. González and U. S. Schubert, Organometallics, 2009, 28, 5478–5488 CrossRef CAS.
- J. M. Fernández-Hernández, C.-H. Yang, J. I. Beltrán, V. Lemaur, F. Polo, R. Fröhlich, J. R. M. Cornil and L. De Cola, J. Am. Chem. Soc., 2011, 133, 10543–10558 CrossRef PubMed.
- (a) S. Ladouceur, D. Fortin and E. Zysman-Colman, Inorg. Chem., 2011, 50, 11514–11526 CrossRef CAS PubMed; (b) K. N. Swanick, S. Ladouceur, E. Zysman-Colman and Z. Ding, Chem. Commun., 2012, 48, 3179 RSC.
- W. Yan, Q. Wang, Q. Lin, M. Li, J. L. Petersen and X. Shi, Chem. – Eur. J., 2011, 17, 5011–5018 CrossRef CAS PubMed.
- There is one reported example of the N-2-phenyl-1,2,3-triazole-Ir(III) complex, though with no studies on the substituent influence: N. M. Shavaleev, R. Scopelliti, M. Grätzel and M. K. Nazeeruddin, Inorg. Chim. Acta, 2012, 388, 84–87 CrossRef CAS PubMed.
- (a) Y. You and S. Y. Park, J. Am. Chem. Soc., 2005, 127, 12438–12439 CrossRef CAS PubMed; (b) S. Ladouceur, D. Fortin and E. Zysman-Colman, Inorg. Chem., 2011, 50, 11514–11526 CrossRef CAS PubMed.
- Previously reported tapy compounds: I. Stengel, A. Mishra, N. Pootrakulchote, S.-J. Moon, S. M. Zakeeruddin, M. Gratzel and P. Bauerle, J. Mater. Chem., 2011, 21, 3726–3734 RSC.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, UV, fluorescence spectra, cyclic voltammetry and X-ray data with CCDC numbers. CCDC 1012360 and 1010939. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00281d |
| This journal is © the Partner Organisations 2015 |