DOI:
10.1039/C4QI00241E
(Research Article)
Inorg. Chem. Front., 2015,
2, 477-484
Amide-containing zinc(II) metal–organic layered networks: a structure–CO2 capture relationship†
Received
29th December 2014
, Accepted 11th March 2015
First published on 12th March 2015
Abstract
The self-assembly of zinc–organic coordination polymers [Zn2(1,3-bdc)2(bpda)2]·3DMF·0.5H2O (1, 1,3-bdc = 1,3-benzenedicarboxylate; bpda = N,N′-bis(pyridine-4-yl)-1,4-benzenedicarboxamide) and [Zn2(1,4-ndc)(dmc)2(bpda)2] (2, 1,4-ndc = 1,4-naphthalenedicarboxylate; dmc = dimethylcarbamate) through mixed-ligand coordination under hydro(solvo)thermal conditions is reported. Both compounds 1 and 2 are made up of a two-dimensional layered network with a decorated 44-sql topology from the associated dinuclear two-blade paddlewheel building units. The bis-amide groups of the bpda ligands in the layered structures of 1 and 2 are sheltered and participate in multiple hydrogen-bonding interactions. Compounds 1 and 2 are both highly thermally stable at temperatures over 300 °C and exhibit solid-state luminescence properties. In comparison with the CO2 adsorption behavior of the related compound {[Zn4(1,4-bdc)4(bpda)4]·5DMF·3H2O}n (3, 1,4-bdc = 1,4-benzenedicarboxylate), both the desolvated sample of 1 and the thermally-activated sample of 2 show a lower CO2 uptake capacity and a decreased Qst trace with increasing CO2 uptake. These results show that no meaningful cooperative binding occurs between the amide groups and CO2 molecules in the condensed 2D structures of 1 and 2. By varying the dicarboxylate ligand from 1,3-bdc, 1,4-ndc to 1,4-bdc, the amide-functionalized products 1–3 induce subtle changes in their structures. In particular, the intimate interrelationship between the structural characteristics of amide groups and the CO2 adsorption behavior of such compounds is clearly demonstrated.
Introduction
The design and construction of metal–organic frameworks (MOFs) has become a rapidly growing field in metal-based supramolecular chemistry and has triggered vigorous investigations in recent decades due to their potential in a range of achievable applications.1–10 Porous MOFs have currently been developed for use as materials in capturing CO2.7–11 Further modification of internal cavities of such frameworks and an understanding of the interrelationship between their structural characteristics and CO2 adsorption capability would be highly desirable.8–11
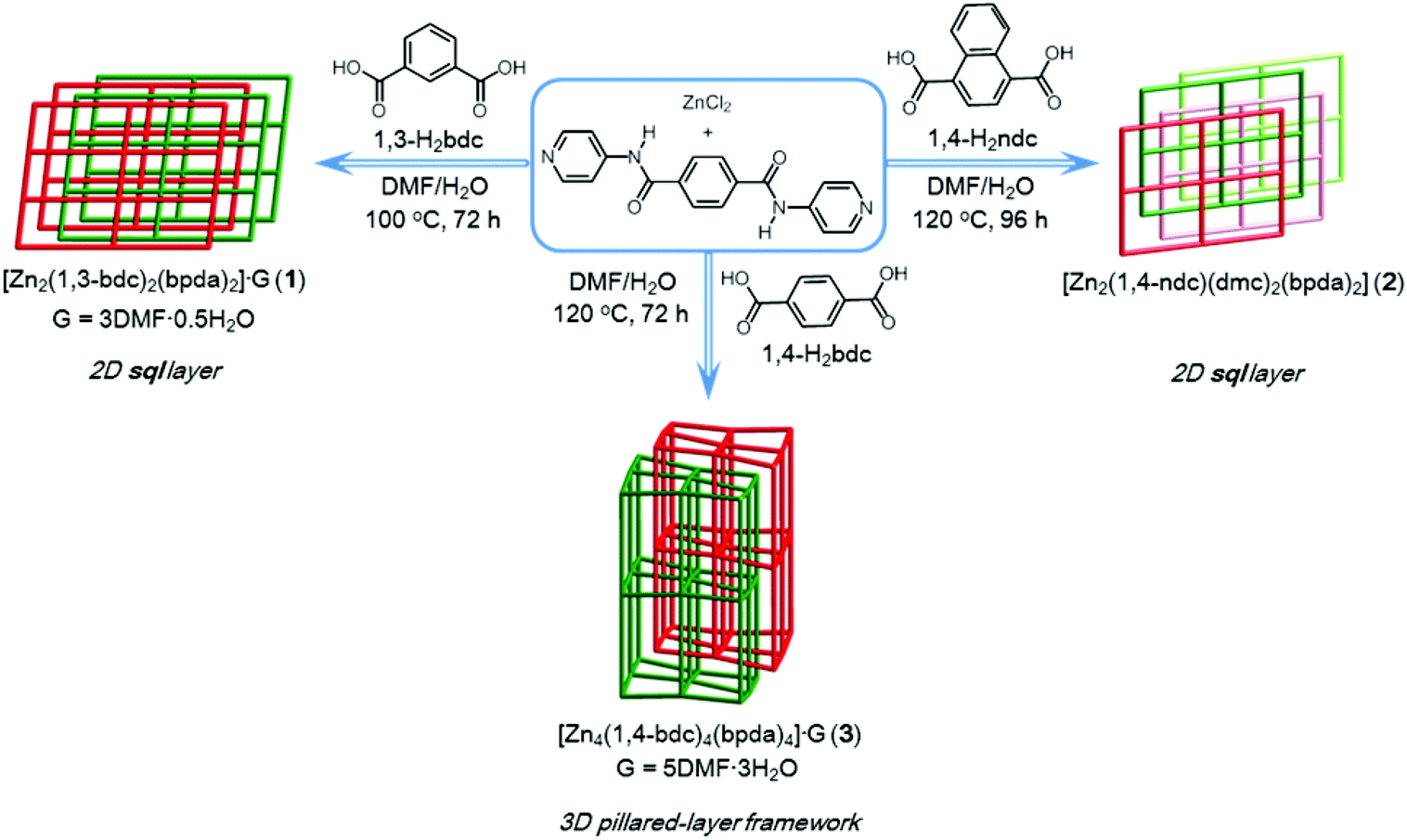
We recently reported on a unique CO2 adsorption behavior of an amide-containing MOF.11 A two-fold interpenetrated MOF {[Zn4(1,4-bdc)4(bpda)4]·5DMF·3H2O}n (3, where 1,4-bdc = 1,4-benzene dicarboxylate, bpda = N,N′-bis(pyridine-4-yl)-1,4-benzenedicarboxamide) showed interesting characteristics, in which the unsheltered amide groups inside the open-ended channels cooperatively bind CO2 molecules by not only amide:CO2 binding but also amide:CO2:CO2 binding. These interactions result in a high isosteric heat of CO2 adsorption (Qst), which increases significantly with increasing CO2 uptake. To further advance this ongoing project,11,12 we envisaged that the replacement of other similar dicarboxylate bridging ligands such as 1,3-bdc (1,3-benzenedicarboxylate) and 1,4-ndc (1,4-naphthalenedicarboxylate) might significantly alter the special arrangement of the amide groups, thereby affecting their CO2 adsorption behavior. Herein, we report on the structures and CO2 adsorption capabilities of a series of 1,3-bdc, 1,4-ndc and 1,4-bdc containing Zn(II) MOFs. An intimate interrelationship between the structural characteristics of their amide groups and CO2 sorption behavior is observed and described.
Experimental section
Materials and general characterization
The ligand N,N′-bis(pyridine-4-yl)-1,4-benzenedicarboxamide (bpda) was prepared according to the literature method.11 All other solvents and chemical reagents were purchased commercially and used as received without further purification. Infrared (IR) samples were prepared as KBr pellets, and spectra were obtained in the 4000–400 cm−1 range using a Perkin-Elmer Paragon 1000 FT-IR spectrophotometer; abbreviations used for the IR bands are w = weak, m = medium, s = strong. Elemental analyses were performed on a Perkin-Elmer 2400 CHN elemental analyzer. A Perkin-Elmer TGA-7 analyzer was used to carry out the thermogravimetric (TG) analysis under flowing nitrogen at a heating rate of 10 °C min−1. Powder X-ray diffraction (PXRD) measurements were performed on a Siemens D-5000 diffractometer at 40 kV, 30 mA for Cu Kα (λ = 1.5406 Å), with a step size of 0.02° in θ and a scan speed of 1 s per step size. The solid-state emission spectra were recorded on a Hitachi F-7000 Fluorescence Spectrophotometer at room temperature.
Synthesis of [Zn2(1,3-bdc)2(bpda)2]·3DMF·0.5H2O (1).
ZnCl2 (13.7 mg, 0.10 mmol), 1,3-benzenedicarboxylic acid (1,3-H2bdc, 16.8 mg, 0.10 mmol), bpda (31.9 mg, 0.10 mmol), DMF (7 mL), and H2O (0.5 mL) were sealed in a Teflon-lined stainless steel Parr acid digestion bomb and heated at 100 °C for 72 h, which was then allowed to slowly cool to 30 °C. Colorless block-shaped crystals were collected by filtration, washed with water, and dried in air. Yield: 51%. Elemental analysis calcd for 1 (C61H58N11O15.5Zn2): C, 55.34; H, 4.42; N, 11.64%. Found: C, 55.11; H, 4.61; N, 11.43%. IR (KBr pellet, cm−1): 3455 (w), 3259 (w), 3176 (w), 3085 (w), 2928 (w), 1682 (s), 1598 (s), 1562 (w), 1498 (m), 1442 (w), 1422 (m), 1391 (s), 1330 (m), 1297 (m), 1261 (w), 1211 (m), 1116 (w), 1098 (m), 1076 (w), 1019 (m), 956 (w), 835 (m), 750 (m), 715 (m), 658 (w), 604 (w), 575 (w), 537 (m).
Synthesis of [Zn2(1,4-ndc)(dmc)2(bpda)2] (2).
ZnCl2 (13.6 mg, 0.10 mmol), 1,4-naphthalenedicarboxylic acid (1,4-H2ndc, 21.9 mg, 0.10 mmol), bpda (32.0 mg, 0.10 mmol), DMF (7 mL), and H2O (0.5 mL) were sealed in a Teflon-lined stainless steel Parr acid digestion bomb and heated at 120 °C for 96 h, which was then allowed to slowly cool to 30 °C. Colorless block-shaped crystals were collected by filtration, washed with water, and dried in air. Yield: 50%. Elemental analysis calcd for 2 (C54H44N10O12Zn2): C, 56.12; H, 3.83; N, 12.12%. Found: C, 56.23; H, 3.87; N, 12.23%. IR (KBr pellet, cm−1): 3259 (w), 3174 (w), 3076 (w), 2926 (w), 2860 (w), 1686 (s), 1596 (s), 1568 (s), 1511 (s), 1422 (m), 1393 (w), 1370 (m), 1329 (s), 1299 (m), 1264 (m), 1210 (s), 1112 (m), 1098 (m), 1061 (w), 1019 (s), 835 (s), 797 (m), 770 (w), 736 (w), 711 (m), 661 (m), 603 (m), 565 (m), 537 (s).
Crystal data collection and refinement
Single-crystal X-ray diffraction analyses for 1 and 2 were performed on a Nonius Kappa CCD diffractometer, equipped with graphite monochromatized Mo Kα radiation (λ = 0.71073 Å). Intensity data were collected within the limits of 1.04° ≤ θ ≤ 27.50° at 150(2) K for 1 and of 1.87° ≤ θ ≤ 26.37° at 100(2) K for 2. The crystal structures were solved with direct methods and refined with a full-matrix least-squares procedure on F2 using the SHELX-97 program package.13 Nonhydrogen atoms were found from the difference Fourier maps and most of them were treated anisotropically except where noted. In 1, one pyridine ring of the bpda ligand was treated disorderedly over two occupied sites with equal refined site occupancy factors. All of the lattice DMF and H2O molecules were refined isotropically, where some of them were treated disorderedly. Whenever possible, the hydrogen atoms were located on a difference Fourier map and refined. In other cases, the hydrogen atoms were geometrically fixed. Experimental details for X-ray data collection and the refinements are summarized in Table S1.† CCDC 1029622 and 1029623 contain the supplementary crystallographic data for this paper.
Low-pressure adsorption measurements
An ASAP 2020 Accelerated Surface Area and Porosimetry analyzer from Micrometrics was used to perform low-pressure carbon dioxide (CO2) adsorption measurements. Research grade CO2 (99.9995% purity) was used in all adsorption measurements. The materials were degassed prior to analysis. Fresh samples were loaded into a sample tube of known weight and activated at 120 °C under a high vacuum for about 24 h to completely remove guest solvent molecules. After activation, the sample and tube were re-weighed to determine the precise mass of the evacuated sample. CO2 adsorption isotherms were measured at 195 K, 273 K, and 298 K.
Results and discussion
Synthesis and general characterization
Compound 1 was prepared by reacting ZnCl2, bpda, and 1,3-H2bdc in a DMF–H2O solution under hydro(solvo)thermal conditions through a single-step self-assembly process, whereas compound 2 was prepared by a synthetic procedure similar to that used for 1, using 1,4-H2ndc instead of 1,3-H2bdc (Scheme 1). Single-crystal X-ray diffraction analyses were carried out to determine the crystal structures of 1 and 2. Powder X-ray diffraction (PXRD) patterns measured with the bulk materials of 1 and 2 confirm the phase purity of both materials (Fig. S1 and S2†).
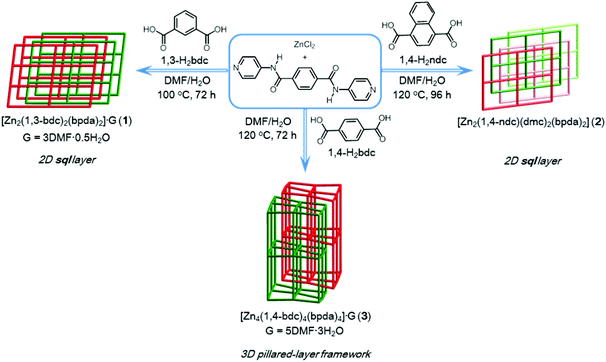 |
| | Scheme 1 | |
Crystal structure description
[Zn2(1,3-bdc)2(bpda)2]·3DMF·0.5H2O (1).
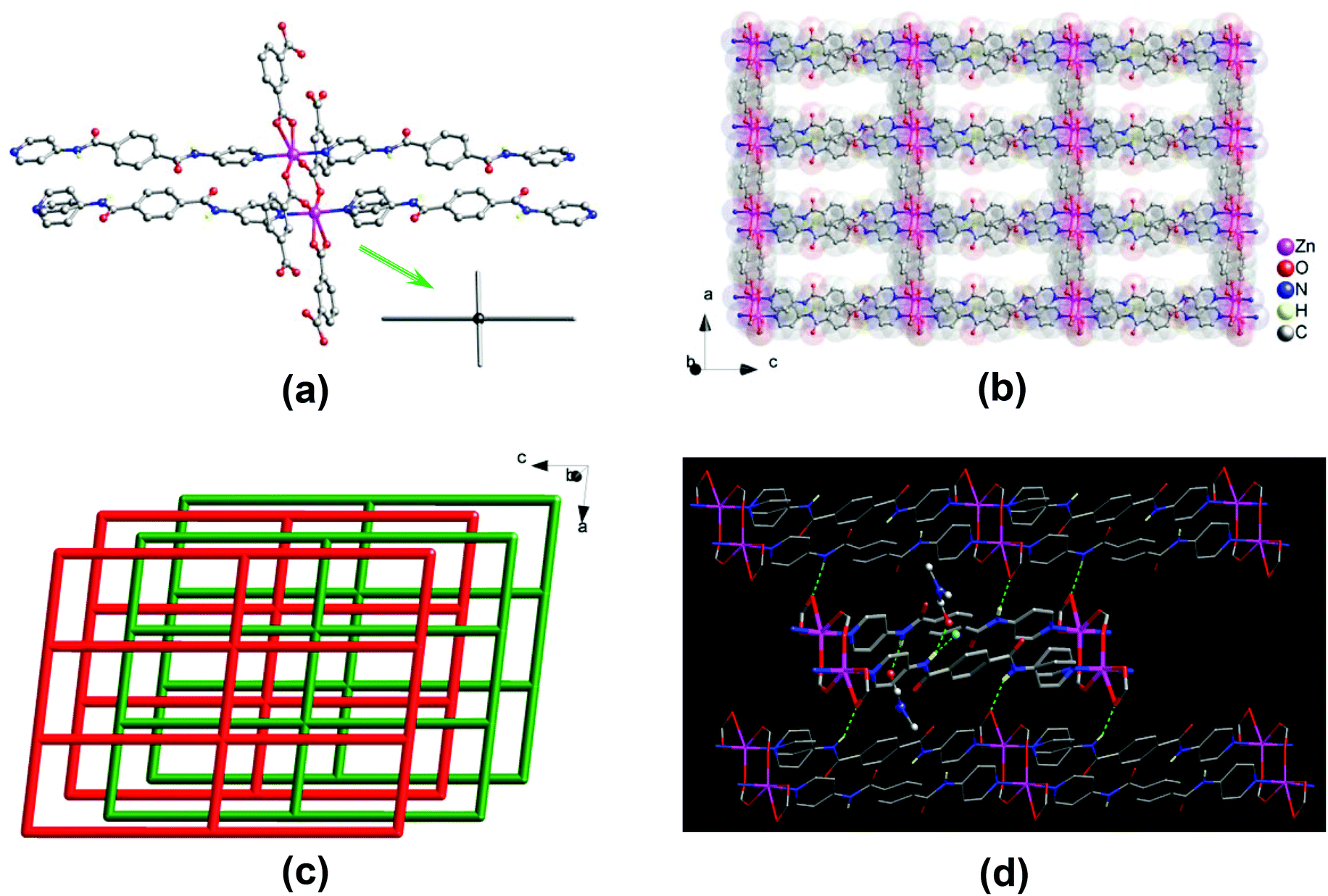
A single-crystal X-ray diffraction analysis revealed that compound 1 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit consists of two Zn(II) centers, two 1,3-bdc2− and two bpda ligands, and a total of three lattice DMF and half of lattice water molecules. Two crystallographically distinct Zn(II) centers adopt a five-coordinated {ZnN2O3} trigonal bipyramidal geometry (Fig. 1a), with τ values of 0.89 and 0.87,14 surrounded by two pyridine nitrogens of two different bpda ligands at the axial positions and three carboxyl oxygens of three different 1,3-bdc2− ligands at the equatorial plane. The Zn–N bond lengths are in the range of 2.142(3)–2.165(3) Å, and the Zn–O bond lengths are in the range of 2.008(3)–2.045(3) Å. Each 1,3-bdc2− ligand bridges three Zn(II) centers with one carboxyl group in a μ2–η1:η1-bridging bidentate coordination mode and another in a monodentate mode. It should be noted that the remaining oxygen of the monodentate carboxylate group also interacts weakly with the Zn(II) center (Zn⋯O, 2.59 and 2.65 Å).
. The asymmetric unit consists of two Zn(II) centers, two 1,3-bdc2− and two bpda ligands, and a total of three lattice DMF and half of lattice water molecules. Two crystallographically distinct Zn(II) centers adopt a five-coordinated {ZnN2O3} trigonal bipyramidal geometry (Fig. 1a), with τ values of 0.89 and 0.87,14 surrounded by two pyridine nitrogens of two different bpda ligands at the axial positions and three carboxyl oxygens of three different 1,3-bdc2− ligands at the equatorial plane. The Zn–N bond lengths are in the range of 2.142(3)–2.165(3) Å, and the Zn–O bond lengths are in the range of 2.008(3)–2.045(3) Å. Each 1,3-bdc2− ligand bridges three Zn(II) centers with one carboxyl group in a μ2–η1:η1-bridging bidentate coordination mode and another in a monodentate mode. It should be noted that the remaining oxygen of the monodentate carboxylate group also interacts weakly with the Zn(II) center (Zn⋯O, 2.59 and 2.65 Å).
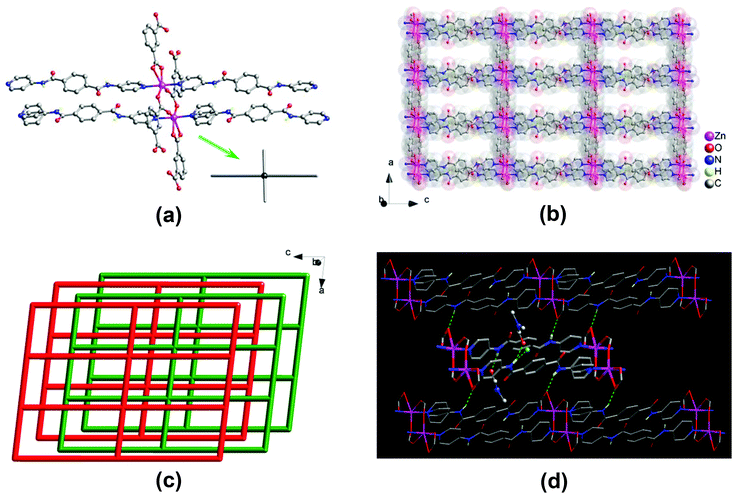 |
| | Fig. 1 Crystal structure of 1: (a) view of a two-blade paddlewheel-like [Zn2(CO2)4N4] cluster as a 4-connected node; (b) view of the 2D double-edged layered structure; (c) topologic view of the packing of the 44-sql layers along slight off the crystallographic [010] direction, showing an ABAB offset-stacking fashion; and (d) highlighting the N–H⋯O hydrogen-bonding interactions. | |
Two Zn(II) centers are held together by two carboxyl groups from two 1,3-bdc2− ligands, thus forming a dinuclear [Zn2(CO2)4N4] secondary building unit (SBU) with a Zn⋯Zn separation of 4.03 Å, which resembles a two-bladed paddlewheel (Fig. 1a). The SBUs are connected to each other by 1,3-bdc2− ligands to generate a neutral one-dimensional (1D) Zn–1,3-bdc chain array, which is then linked by pairs of bpda ligands with amide and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups oriented in an antiparallel manner (trans-conformation) to yield a neutral two-dimensional (2D) double-edged zinc(II)–organic 44-sql layered network (Fig. 1b),15 with a nanosized mesh of 10.06 × 20.11 Å2. These 2D layers are stacked offset in an ABAB fashion along the crystallographic [010] direction (Fig. 1c). The open channels thus formed in two different window sizes with effective dimensions of approximately 2 × 4 and 4 × 9 Å2, respectively. The lattice DMF and H2O molecules are located within the open channels (Fig. S3a†), occupying an extra-framework volume of approximately 28%.16 The bis-amide groups of the coordinated bpda ligands participate in multiple hydrogen-bonding interactions. The whole crystal structure is stabilized by moderate N–H⋯O hydrogen-bonding interactions among the offset-stacked layers (Fig. 1d and S3b†); these net-to-net hydrogen bonds are located between the amide hydrogens of the bpda ligands in one layer and the carboxyl oxygens of the 1,3-bdc2− ligands in neighbouring layers (N⋯O = 2.88 and 2.90 Å, Table S2†). In addition, the amide groups of the bpda ligands also interact with the lattice DMF and H2O molecules through net-to-guest N–H⋯O hydrogen bonds (N⋯O = 2.90–3.02 Å, Table S2†).
O groups oriented in an antiparallel manner (trans-conformation) to yield a neutral two-dimensional (2D) double-edged zinc(II)–organic 44-sql layered network (Fig. 1b),15 with a nanosized mesh of 10.06 × 20.11 Å2. These 2D layers are stacked offset in an ABAB fashion along the crystallographic [010] direction (Fig. 1c). The open channels thus formed in two different window sizes with effective dimensions of approximately 2 × 4 and 4 × 9 Å2, respectively. The lattice DMF and H2O molecules are located within the open channels (Fig. S3a†), occupying an extra-framework volume of approximately 28%.16 The bis-amide groups of the coordinated bpda ligands participate in multiple hydrogen-bonding interactions. The whole crystal structure is stabilized by moderate N–H⋯O hydrogen-bonding interactions among the offset-stacked layers (Fig. 1d and S3b†); these net-to-net hydrogen bonds are located between the amide hydrogens of the bpda ligands in one layer and the carboxyl oxygens of the 1,3-bdc2− ligands in neighbouring layers (N⋯O = 2.88 and 2.90 Å, Table S2†). In addition, the amide groups of the bpda ligands also interact with the lattice DMF and H2O molecules through net-to-guest N–H⋯O hydrogen bonds (N⋯O = 2.90–3.02 Å, Table S2†).
[Zn2(1,4-ndc)(dmc)2(bpda)2] (2).
A single-crystal X-ray diffraction analysis revealed that 2 crystallizes in the monoclinic space group P21/c. The asymmetric unit consists of one Zn(II) center and half of one 1,4-ndc2−, one dmc−, and one bpda ligands, where Hdmc = dimethylcarbamic acid. The dmc− ligand is in situ generated via the hydrolysis of the DMF solvent under the hydro(solvo)thermal conditions used. The Zn(II) center adopts a five-coordinated {ZnN2O3} trigonal bipyramidal geometry (Fig. 2a), surrounded by two pyridine nitrogens of two different bpda ligands at the axial positions and three carboxyl oxygens of one 1,4-ndc2− and two different dmc− ligands at the equatorial plane (τ = 0.78).14 The Zn–N bond lengths are in the range of 2.151(2)–2.154(2) Å, and the Zn–O bond lengths are in the range of 1.981(2)–2.108(2) Å. Each 1,4-ndc2− ligand bridges two Zn(II) centers with the carboxyl group in a monodentate coordination mode. The remaining carboxylic oxygen is also in weak contact with the Zn(II) center (Zn⋯O, 2.723 Å). Each dmc− ligand adopts a μ2–η1:η1-bridging bidentate coordination mode to bridge two Zn(II) centers.
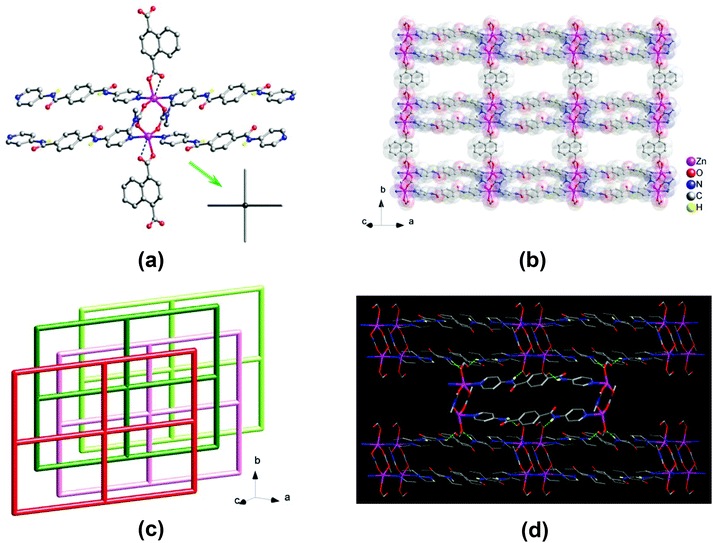 |
| | Fig. 2 Crystal structure of 2: (a) view of a two-blade paddlewheel-like [Zn2(CO2)4N4] cluster as a 4-connected node; (b) view of the 2D layered structure; (c) topologic view of the packing of the 44-sql layers along slight off the crystallographic [101] direction, showing an ABA′B′ offset-stacking fashion; and (d) highlighting the net-to-net N–H⋯O hydrogen-bonding interactions. | |
Two Zn centers are bridged by two dmc− ligands to form a two-blade paddlewheel-like dinuclear [Zn2(CO2)4N4] cluster SBU, with a Zn⋯Zn separation of 3.95 Å (Fig. 2a). These dinuclear SBUs are stringed by the 1,4-ndc2− ligands to generate a neutral 1D chain array of Zn2(1,4-ndc)(dmc)2, which is linked by pairs of bpda ligands showing a trans-conformation to yield a neutral 2D zinc(II)–organic 44-sql layered network (Fig. 2b),15 with a nanosized mesh of 15.06 × 20.06 Å2. These 2D layers are stacked in an offset ABA′B′ fashion along the crystallographic [101] direction (Fig. 2c), causing a loss of potential openings with negligible solvent accessible volume (∼0%).16 This is comparable with the ABAB stacking arrangement in 1 that resulted in the formation of open channels. Among these offset-stacked layers, moderate net-to-net N–H⋯O hydrogen-bonding interactions (N⋯O = 2.88 and 3.03 Å, Table S2†) formed between the amide hydrogens of the bpda ligands and the carboxyl oxygens of the 1,4-ndc2− ligands provide clear support for stabilizing the overall crystal structure of 2 (Fig. 2d and S4†).
Thermogravimetric properties
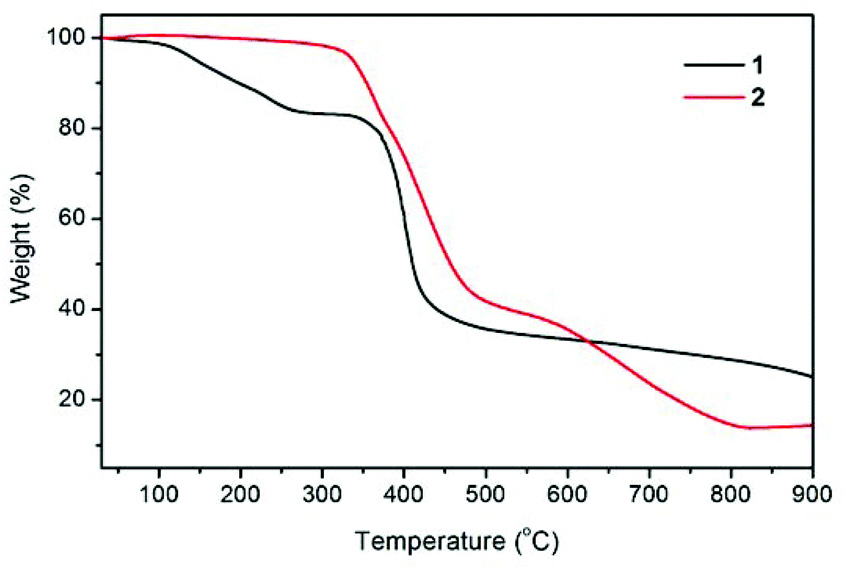
The thermal stabilities of compounds 1 and 2 were examined by thermogravimetric (TG) analysis, which was performed on polycrystalline samples under a nitrogen atmosphere (Fig. 3). The TG curve for 1 reveals a weight loss of 16.7% between 30 and 270 °C, corresponding to the loss of guest H2O and DMF molecules (calcd 17.2%). The de-solvated sample then remained stable at temperatures up to ∼335 °C without any weight loss and then began to decompose. The TG trace of 2 shows no significant weight loss prior to its decomposition at a temperature approaching 320 °C. The decomposition process was complete at a temperature of about 815 °C, leaving a final residue of 14.1% that can reasonably be assumed to be ZnO (calcd 14.1%).
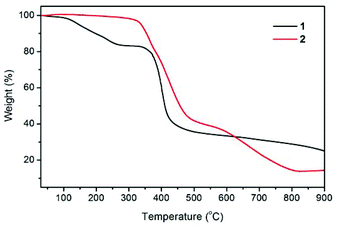 |
| | Fig. 3 Thermogravimetric (TG) curves of compounds 1 (black line) and 2 (red line). | |
Photoluminescence properties
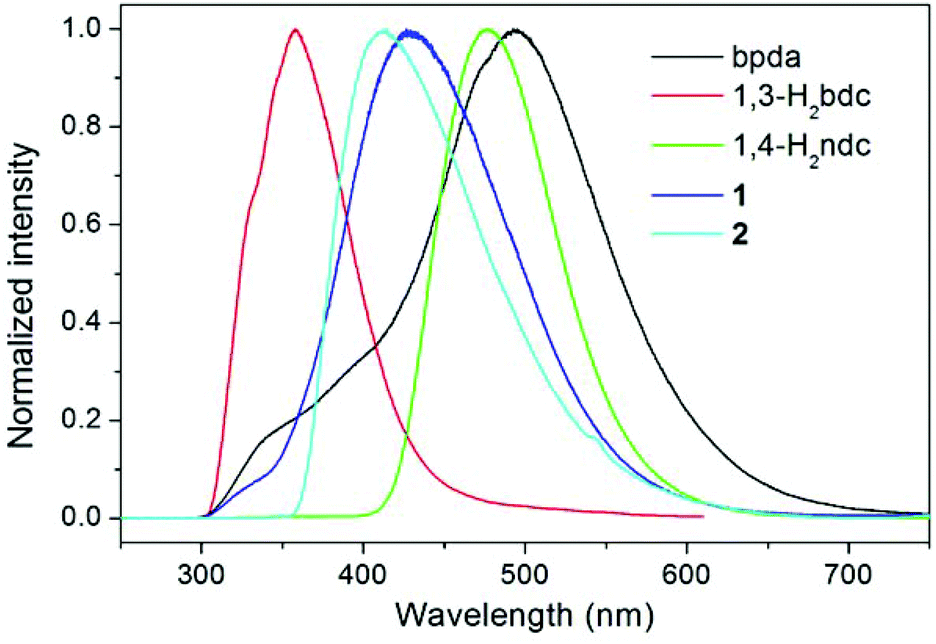
The luminescence properties of compounds 1 and 2 were investigated in the solid state at room temperature, together with the free bpda, 1,3-H2bdc, and 1,4-H2ndc ligands (Fig. 4). Representative photoluminescence emission bands at 493 (λex = 276 nm), 358 (λex = 274 nm), and 476 nm (λex = 274 nm) were observed for the free bpda, 1,3-H2bdc, and 1,4-H2ndc, respectively. The emissions of the organic ligands may be ascribed to π*→n or π*→π transitions.17
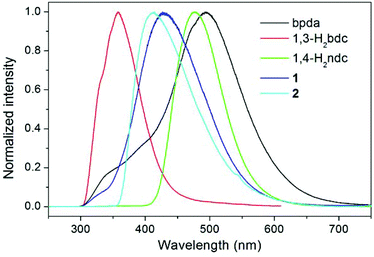 |
| | Fig. 4 Room temperature, solid-state emission spectra of the free organic ligands and compounds 1 and 2. | |
Compound 1 displays an emission band with a maximum at 426 nm upon excitation at 276 nm, whereas compound 2 emits at 414 nm (λex = 345 nm). Based on the band position and shape, it appears that the emissions for both compounds originated from the ligand-to-metal charge transfer (LMCT) transition from carboxylate groups to Zn(II) ions, which may be admixed with a ligand-to-ligand charge transfer (LLCT), as has been observed in other Zn(II) complexes with the d10 valence electron configuration.17,18
CO2 sorption
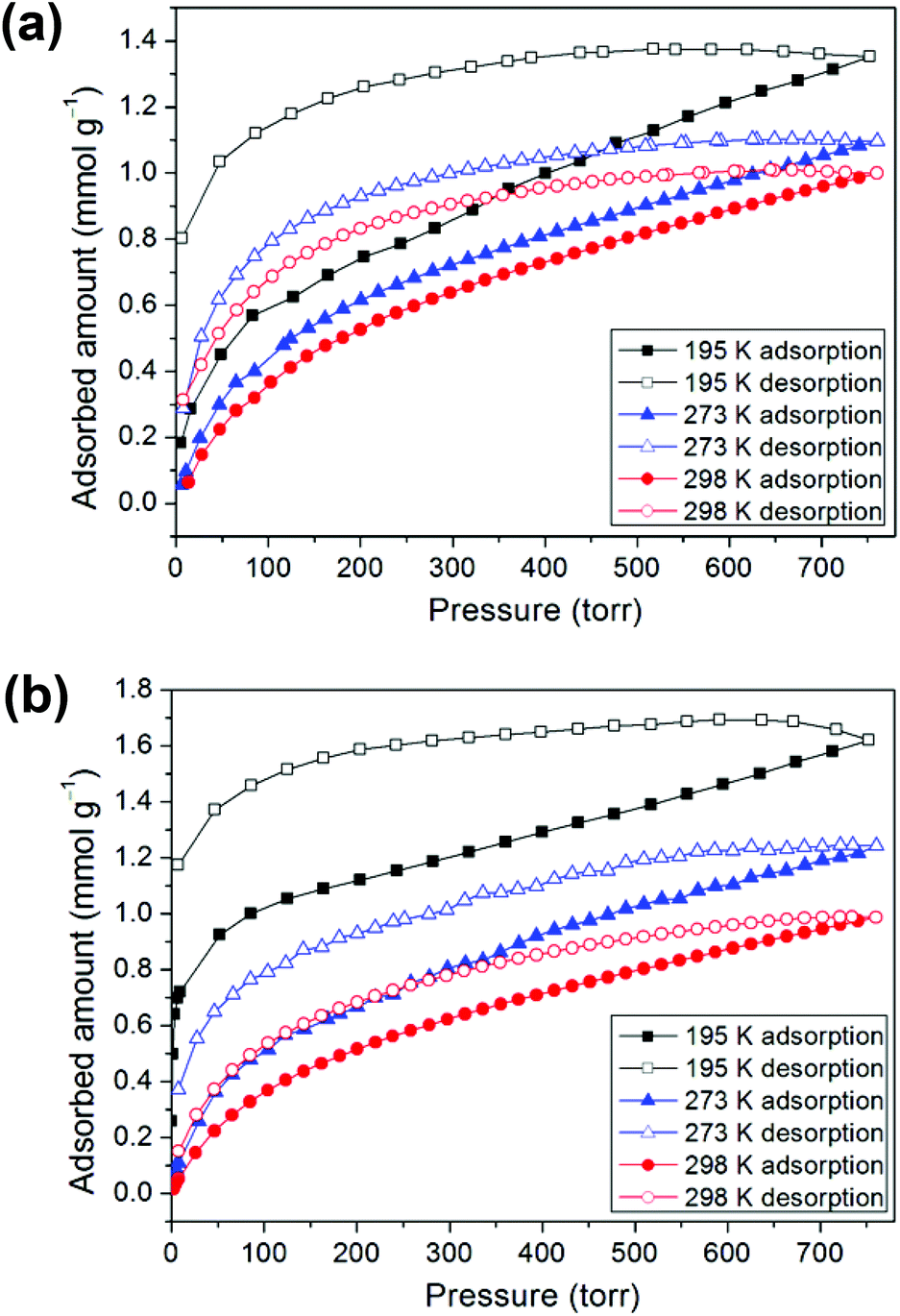
Prior to the gas adsorption experiments, we prepared a desolvated sample of 1, hereafter denoted as 1′, in which guest solvent molecules in the framework had been removed. In addition, compound 2 was thermally activated, labeled as 2′, by heating the as-synthesized sample at 120 °C for 24 h. Sorption studies showed that compounds 1′ and 2′ both exhibit moderate or low CO2 uptake capacities at 1 atm and controlled temperatures (195, 273, and 298 K) with a reversible type I isotherm (Fig. 5), which is characteristic of microporous materials. The estimated apparent Langmuir and Brunauer–Emmett–Teller (BET) surface areas were determined to be 85 m2 g−1 and 59 m2 g−1, respectively, for 1′ and 122 m2 g−1 and 82 m2 g−1, respectively, for 2′. Compound 1′ takes up only a small amount (1.35 mmol g−1) of CO2 at 195 K, 1.10 mmol g−1 of CO2 at 273 K and 1.00 mmol g−1 of CO2 at 298 K, whereas 2′ takes up 1.62, 1.24, and 0.99 mmol g−1 of CO2 at 195 K, 273 K and 298 K, respectively. In comparison with some 2D layered networks, such as [Zn(5NO2-ip)(bpy)] (5NO2-ip = 5-nitroisophthalate, bpy = 4,4′-bipyridine; 2.54 mmol g−1 of CO2 at 273 K),19 and [{NiL1}3(BTC)2] (L1 = 3,10-bis(4-fluorobenzyl)-1,3,5,8,10,12-hexaazacyclotetradecane, BTC = 1,3,5-benzenetricarboxylate; 2.66 mmol g−1 of CO2 at 273 K),20 both 1′ and 2′ showed low CO2 adsorption, which may be due to the poor porosity of both frameworks and a decrease in the amount of appropriate intermolecular interactions. Furthermore, the framework of 2, after thermal activation, may be transformed into a new phase, which is likely to be porous due to a layer slip from off-stacking, as evidenced by the PXRD pattern (Fig. S2, ESI†) and the moderate CO2 adsorption capability (Table 1).
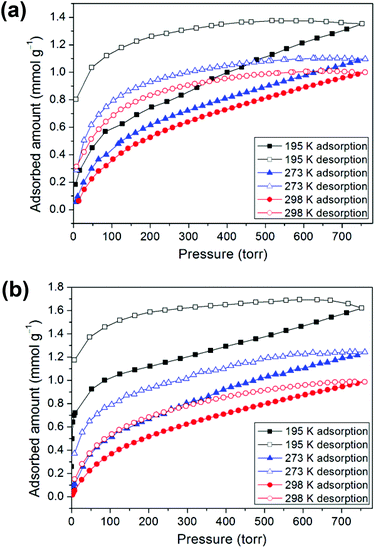 |
| | Fig. 5 Low-pressure CO2 isotherms of (a) 1′ and (b) 2′ measured at 195 K, 273 K, and 298 K. | |
Table 1 Comparison of the structural characteristics and CO2 adsorption properties of compounds 1–3
| |
1
|
2
|
3
|
| Dicarboxylate ligand |
1,3-bdc |
1,4-ndc |
1,4-bdc |
| Frameworks |
2D sql layer with ABAB stacking |
2D sql layer with ABA′B′ stacking |
3D 2-fold interpenetrated pillared-layered framework |
| Amide groups exposed to channels |
No |
No |
Yes |
| Solvent accessible voids (%) |
28 |
∼0 |
29 |
| Langmuir surface area (m2 g−1) |
85 |
122 |
468 |
| BET surface area (m2 g−1) |
59 |
82 |
331 |
| CO2 adsorption amount (195, 273, 298 K) (mmol g−1) |
1.35, 1.10, 1.00 |
1.62, 1.24, 0.99 |
5.54, 2.83, 1.87 |
|
Q
st (kJ mol−1) |
15.9–3.3 |
26.3–13.2 |
30.2–37.2 |
| Amide:CO2 interaction |
No |
No |
Yes |
| Amide:CO2:CO2 interaction |
No |
No |
Yes |
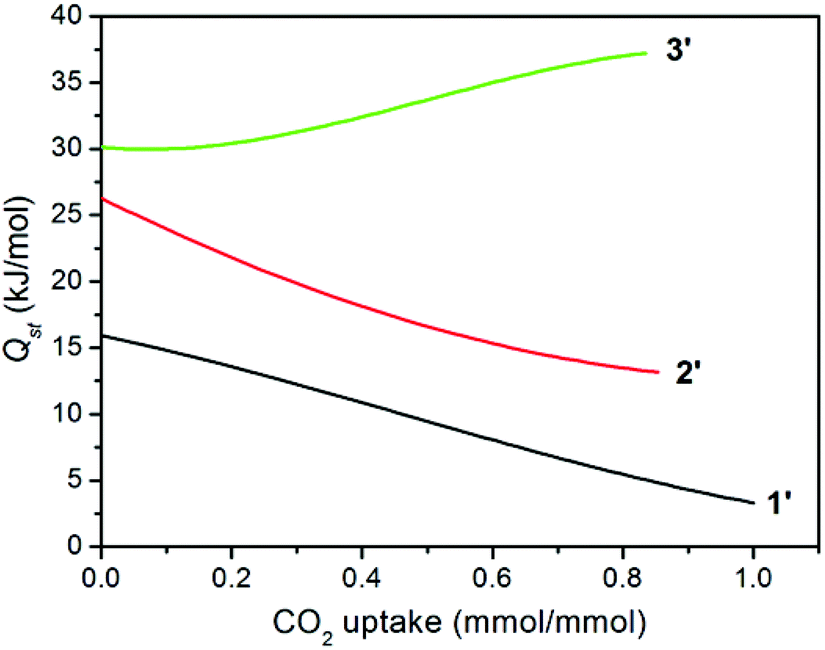
The isosteric heat of CO2 adsorption was calculated by the virial method,7a,21 which displays commonly decreased Qst traces for 1′ and 2′ with increasing CO2 uptake (Fig. 6), depending on the amount of adsorbed CO2 molecules. This is much different from a related report for compound {[Zn4(1,4-bdc)4(bpda)4]·5DMF·3H2O}n (3), the desolvated sample (3′) of which displayed an abnormally increased trace arising (30.2–37.2 kJ mol−1) from the significant cooperative effect of amide groups in capturing CO2 (Table 1).11 Surprisingly, compound 1′ at zero coverage exhibits a weak (15.9 kJ mol−1) CO2 binding affinity, which is close to the enthalpy of liquefaction of CO2 (17 kJ mol−1), indicating that the framework surface does not provide an improvement over the affinity of the gas for itself.8e For 2′, although the Qst value of 26.2 kJ mol−1 is much lower than 30.2 kJ mol−1 for 3′ and 36.5 kJ mol−1 for [Cu3L2(H2O)5]·Guest (where H3L = 5-(4-carboxybenzoylamino)isophthalic acid),10b,11 it is still comparable to that of [Cu24(TPBTM6−)8(H2O)24] (26.3 kJ mol−1),10c Cu3(BTB6−) (24.1 kJ mol−1), and Cu3(TATB6−) (24.4 kJ mol−1).10a
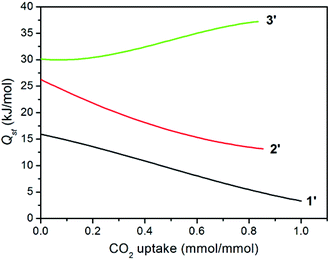 |
| | Fig. 6 Isosteric heat (Qst) of CO2 adsorption. | |
Structure–CO2 capture relationship
Understanding the interrelationship between the structural characteristics of porous hosts and their CO2 adsorption capabilities is essential for the development of new materials for use in the capture and storage of CO2. In addition to amine-mediated MOFs, amide-mediated MOFs have also emerged as potential candidates for effectively capturing CO2.10,11,22 In this work, the most interesting observation is that the CO2 adsorption behavior of the amide-containing Zn(II)-based MOFs 1 and 2 is completely different from that of a related species 3. A detailed comparison was therefore made to illustrate their interrelationship. As can be seen from Scheme 1, the aromatic dicarboxylate ligands (1,3-bdc, 1,4-ndc, and 1,4-bdc, respectively) of 1–3 are quite similar. Interestingly, because of a minor variation of the ligand structures from 1,4-bdc (smaller spacer, linear ligating orientation) to 1,3-bdc (smaller spacer, angular ligating orientation) and 1,4-ndc (larger bulky spacer, linear ligating orientation), the crystal structures of 1–3 were drastically changed from a two-fold interpenetrated pillared-layer framework (3) to non-interpenetrated 2D sql layers (1 and 2), even though they were constructed from very similar two-blade paddlewheel SBUs. More important, this change results in a significant effect on the CO2 adsorption ability of the frameworks (Table 1). For 3, the amide groups are exposed to the large channel openings of a two-fold interpenetrated framework, permitting a significant level of cooperative binding of CO2 in an amide:CO2 manner and even in an amide:CO2:CO2 manner.11 These findings help to explain the observed increase in Qst from a low to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CO2 uptake. On the other hand, both layered structures 1 and 2 show compact stacking, in which all of the amide groups are sheltered and contribute to hydrogen bonds. Therefore, it is reasonable that no meaningful cooperative binding, even primary amide:CO2 interactions, between the amide groups and CO2 molecules occurs in the condensed 2D structures. Weak Qst values that would not increase with increasing CO2 uptake are estimated. As a consequence, the three amide-mediated zinc(II) MOFs 1–3 represent an excellent example to demonstrate the intimate interrelationships between CO2 adsorption capability and the structural characteristics of their host materials.
:1 CO2 uptake. On the other hand, both layered structures 1 and 2 show compact stacking, in which all of the amide groups are sheltered and contribute to hydrogen bonds. Therefore, it is reasonable that no meaningful cooperative binding, even primary amide:CO2 interactions, between the amide groups and CO2 molecules occurs in the condensed 2D structures. Weak Qst values that would not increase with increasing CO2 uptake are estimated. As a consequence, the three amide-mediated zinc(II) MOFs 1–3 represent an excellent example to demonstrate the intimate interrelationships between CO2 adsorption capability and the structural characteristics of their host materials.
Conclusion
Three amide-mediated Zn(II) MOFs were synthesized from related aromatic dicarboxylate bridging ligands, i.e. from 1,4-bdc to 1,3-bdc and 1,4-ndc. An intimate interrelationship between their structural characteristics and CO2 adsorption behavior is unambiguously demonstrated. The arrangement of the amide group in the framework determines whether or not the amide:CO2 (cooperative) interaction occurs which plays a key role in influencing the extent of CO2 sorption. This unique case indeed adds to our understanding of CO2 capture and the design of the porous host materials for achieving this.
Acknowledgements
We are grateful to the Academia Sinica and the Ministry of Science and Technology, Taiwan, for financial support.
Notes and references
-
(a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed;
(b) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed;
(c) S. Ma and H. C. Zhou, Chem. Commun., 2010, 46, 44–53 RSC;
(d) A. Shigematsu, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 13145–13147 CrossRef CAS PubMed;
(e) P. Thanasekaran, T.-T. Luo, J.-Y. Wu and K.-L. Lu, Dalton Trans., 2012, 41, 5437–5453 RSC.
-
(a) X. M. Liu, R. B. Lin, J. P. Zhang and X. M. Chen, Inorg. Chem., 2012, 51, 5686–5692 CrossRef CAS PubMed;
(b) Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 RSC;
(c) M. R. Kishan, J. Tian, P. K. Thallapally, C. A. Fernandez, S. J. Dalgarno, J. E. Warren, B. P. McGrail and J. L. Atwood, Chem. Commun., 2010, 46, 538–540 RSC;
(d) R. Zou, A. I. Abdel-Fattah, H. Xu, Y. Zhao and D. D. Hickmott, CrystEngComm, 2010, 12, 1337–1353 RSC.
-
(a) N. Yanai, K. Kitayama, Y. Hijikata, H. Sato, R. Matsuda, Y. Kubota, M. Takata, M. Mizuno, T. Uemura and S. Kitagawa, Nat. Mater., 2011, 10, 787–793 CrossRef CAS PubMed;
(b) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed.
-
(a) K. S. Jeong, Y. B. Go, S. M. Shin, S. J. Lee, J. Kim, O. M. Yaghi and N. Jeong, Chem. Sci., 2011, 2, 877–882 RSC;
(b) H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899–1909 RSC;
(c) L. Chen, Y. Yang, Z. Guo and D. Jiang, Adv. Mater., 2011, 23, 3149–3154 CrossRef CAS PubMed;
(d) L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed.
-
(a) R. C. Huxford, J. Della Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef CAS PubMed;
(b) P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
-
(a) T. Devic, P. Horcajada, C. Serre, F. Salles, G. Maurin, B. Moulin, D. Heurtaux, G. Clet, A. Vimont, J. M. Grenèche, B. Le Ouay, F. Moreau, E. Magnier, Y. Filinchuk, J. Marrot, J. C. Lavalley, M. Daturi and G. Férey, J. Am. Chem. Soc., 2010, 132, 1127–1136 CrossRef CAS PubMed;
(b) R. M. P. Colodrero, P. Olivera-Pastor, E. R. Losilla, D. Hernandez-Alonso, M. A. G. Aranda, L. Leon-Reina, J. Rius, K. D. Demadis, B. Moreau, D. Villemin, M. Palomino, F. Rey and A. Cabeza, Inorg. Chem., 2012, 51, 7689–7698 CrossRef CAS PubMed.
-
(a) Y.-S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed;
(b) D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed;
(c) T. Düren, Y.-S. Bae and R. Q. Snurr, Chem. Soc. Rev., 2009, 38, 1237–1247 RSC;
(d) A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed.
-
(a) A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gándara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef CAS PubMed;
(b) R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed;
(c) R. Vaidhyanathan, S. S. Iremonger, K. W. Dawson and G. K. H. Shimizu, Chem. Commun., 2009, 5230–5232 RSC;
(d) J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 38–39 CrossRef CAS PubMed;
(e) A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS PubMed;
(f) S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed;
(g) B. Arstad, H. Fjellvåg, K. O. Kongshaug, O. Swang and R. Blom, Adsorption, 2008, 14, 755–762 CrossRef CAS.
-
(a) S. S. Mondal, A. Bhunia, I. A. Baburin, C. Jäger, A. Kelling, U. Schilde, G. Seifert, C. Janiak and H.-J. Holdt, Chem. Commun., 2013, 49, 7599–7601 RSC;
(b) F. Debatin, A. Thomas, A. Kelling, N. Hedin, Z. Bacsik, I. Senkovska, S. Kaskel, M. Junginger, H. Müller, U. Schilde, C. Jäger, A. Friedrich and H.-J. Holdt, Angew. Chem., Int. Ed., 2010, 49, 1258–1262 CrossRef CAS PubMed;
(c) Y. Zhao, H. Wu, T. J. Emge, Q. Gong, N. Nijem, Y. J. Chabal, L. Kong, D. C. Langreth, H. Liu, H. Zeng and J. Li, Chem. – Eur. J., 2011, 17, 5101–5109 CrossRef CAS PubMed;
(d) E. Neofotistou, C. D. Malliakas and P. N. Trikalitis, Chem. – Eur. J., 2009, 15, 4523–4527 CrossRef CAS PubMed.
-
(a) B. Zheng, Z. Yang, J. Bai, Y. Li and S. Li, Chem. Commun., 2012, 48, 7025–7027 RSC;
(b) J. Duan, Z. Yang, J. Bai, B. Zheng, Y. Li and S. Li, Chem. Commun., 2012, 48, 3058–3060 RSC;
(c) B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748–751 CrossRef CAS PubMed;
(d) B. Zheng, H. Liu, Z. Wang, X. Yu, P. Yi and J. Bai, CrystEngComm, 2013, 15, 3517–3520 RSC;
(e) C. Hou, Q. Liu, T. Okamura, P. Wang and W.-Y. Sun, CrystEngComm, 2012, 14, 8569–8576 RSC;
(f) M.-J. Sie, Y.-J. Chang, P.-W. Cheng, P.-T. Kuo, C.-W. Yeh, C.-F. Cheng, J.-D. Chen and J.-C. Wang, CrystEngComm, 2012, 14, 5505–5516 RSC.
- C.-H. Lee, H.-Y. Huang, Y.-H. Liu, T.-T. Luo, G.-H. Lee, S.-M. Peng, J.-C. Jiang, I. Chao and K.-L. Lu, Inorg. Chem., 2013, 52, 3962–3968 CrossRef CAS PubMed.
- C.-H. Lee, J.-Y. Wu, G.-H. Lee, S.-M. Peng, J.-C. Jiang and K.-L. Lu, Cryst. Growth Des., 2014, 14, 5608–5616 CAS.
-
G. M. Sheldrick, SHELX-97 (including SHELXS and SHELXL), University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- For the three-letter net codes, see
(a) M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef PubMed;
(b) N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS PubMed;
(c) The associated website, http://rcsr.anu.edu.au/home.
- PLATON: A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- W. Chen, J.-Y. Wang, C. Chen, Q. Yue, Y.-M. Yuan, J.-S. Chen and S.-N. Wang, Inorg. Chem., 2003, 42, 944–946 CrossRef CAS PubMed.
-
(a) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC;
(b) J.-Y. Wu, P.-T. Yuan and C.-C. Hsiao, CrystEngComm, 2014, 16, 3128–3140 RSC;
(c) C. Y. Xu, Q. Q. Guo, X. J. Wang, H. W. Hou and Y. T. Fan, Cryst. Growth Des., 2011, 11, 1869–1879 CrossRef CAS;
(d) M. Du, X.-J. Jiang and X.-J. Zhao, Inorg. Chem., 2007, 46, 3984–3995 CrossRef CAS PubMed;
(e) Q. Chu, G.-X. Liu, Y.-Q. Huang, X.-F. Wang and W.-Y. Sun, Dalton Trans., 2007, 4302–4311 RSC;
(f) S.-L. Zheng, J.-H. Yang, X.-L. Yu, X.-M. Chen and W.-T. Wong, Inorg. Chem., 2004, 43, 830–838 CrossRef CAS PubMed.
- S. Horike, Y. Inubushi, T. Hori, T. Fukushima and S. Kitagawa, Chem. Sci., 2012, 3, 116–120 RSC.
- Y. Inubushi, S. Horike, T. Fukushima, G. Akiyama, R. Matsuda and S. Kitagawa, Chem. Commun., 2010, 46, 9229–9231 RSC.
- J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304–1315 CrossRef CAS PubMed.
-
(a) B.-C. Tzeng, T.-H. Chiu, B.-S. Chen and G.-H. Lee, Chem. – Eur. J., 2008, 14, 5237–5245 CrossRef CAS PubMed;
(b) S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS PubMed;
(c) N. N. Adarsh, D. K. Kumar and P. Dastidar, CrystEngComm, 2009, 11, 796–802 RSC;
(d) X. Song, Y. Zou, X. Liu, M. Oha and M. S. Lah, New J. Chem., 2010, 34, 2396–2399 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns, additional structure diagrams, CO2 adsorption isotherms and hydrogen-bond parameters. CCDC 1029622 and 1029623. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00241e |
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.