The use of FTICR-MS to detect chemical tags from a combinatorial library
Matthew H.
Todd
*a and
Chris
Abell
b
aDepartment of Chemistry, Queen Mary, University of London, Mile End Road, London, UK E1 4NS. E-mail: M.H.Todd@qmul.ac.uk; Fax: 020 7882 7794; Tel: 020 7882 7696
bUniversity Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: ca26@cam.ac.uk; Fax: 01223 336362; Tel: 01223 336405
First published on 18th October 2002
Abstract
The use of tagging in combinatorial chemistry permits tracking of the solid phase as it is taken through iterative split and mix cycles. Several analytical approaches to the identification of tags (and hence the chemical history of the support) have been described. We describe herein a novel chemical tagging strategy for combinatorial solid phase chemistry. The identities of the tags attached to a single bead are discovered by the high resolution, accurate mass technique of Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS).
 Matthew H. Todd Matthew H. Todd | Matthew Todd has been Lecturer in Organic Chemistry at Queen Mary, London since September 2001. He completed his PhD with Chris Abell in 1998, then spent two years as a postdoc with Paul Barlett at the University of California, Berkeley, before returning to Cambridge as Fellow at New Hall College. His research interests are in the areas of organic synthesis, medicinal chemistry and asymmetric catalysis. |
Introduction
Combinatorial chemistry is a broadly applied term, but one which essentially means the simultaneous chemical synthesis of a large number of slightly different molecules (a ‘library’) which are then tested for some property, usually in a high-throughput screen.1 The standard way in which we synthesise libraries containing thousands or even millions of members is via the ‘Split-and-Mix’ methodology first described by Furka.2 The chemistry relies on solid phase organic synthesis, where chemical transformations are carried out on a solid support, consisting usually of a collection of polystyrene beads. By anchoring the members to a solid support in this way, and by employing Furka’s method, we may synthesise a library with far fewer steps than would be required if the members of the library were synthesised via traditional solution phase chemistry. Further, the library members may be purified by simple filtration at the end of the synthesis, which means that the individual reactions in a combinatorial sequence may be driven to completion with the use of excesses of solution-phase reagents. Lastly, since any one bead is only ever exposed to one reagent at a time, a bead will contain multiple copies of only one compound, rather than a mixture, which allows us to remove this compound from a bead, and obtain meaningful data about that molecule’s potency from a screening programme.The role of tagging in combinatorial chemistry
Thus the outcome of a combinatorial synthesis in which a split-and-mix methodology has been followed is a collection of combinatorial beads where a particular synthesised chemical compound (and only that compound) is attached to that support, but crucially we do not of course know the identity of the compound attached to a bead simply by looking at it! Encoding strategies for combinatorial chemistry are those methods designed to allow identification of library members by tracing the chemical history of the support. This does not of course necessarily translate into the identity of the compound, since given the great diversity of chemistry being undertaken in a combinatorial synthesis, we must always guard against unexpected synthetic outcomes arising from side reactions. However, compounds shown to be active from a screen may then be synthesised by more traditional solution phase methods to confirm their identity. Knowledge of the chemical history of the bead is very useful information, and in the majority of cases (e.g. peptide synthesis) this history translates into the compound’s identity with no complications. Let us examine some of the methods that have been developed for the encoding of a library.Physical methods
The chemistry used in a combinatorial synthesis should not interfere with that used for the cleavage of compounds from the support or any other features of the synthesis; in other words the chemistries should be ‘orthogonal.’ Hence a good tagging system would be one where the encoding is entirely non-chemical, and there have been several examples of this.Spatial addressing
The most obvious method of encoding a library is ‘spatial encoding,’ whereby a library member has a known position in a two dimensional matrix.3 It is also possible to encode larger supports by writing on them (sometimes called ‘graphical encoding’), as used for example in Houghten’s tea bag methodology wherein a collection of polystyrene beads are enclosed in a labelled porous bag.4 It has also been shown possible to encode a library by embedding a barcode (etched onto a ceramic surface) into a polystyrene-grafted support which may then be read by pattern recognition software,5 or by using coloured glass beads and polypropylene containers.6 There have been examples of library synthesis on paper where small split-and-mix libraries are possible by literally cutting the support into pieces with scissors and using an indelible marker to write on the surface.7 All these methods, whilst useful for small libraries, are not practical for large split-and-mix libraries. This is simply due to the physical impracticality of handling thousands of large supports in a laboratory. For split-and-mix chemistry to be possible, it is desirable to use small (polystyrene) beads with typical dimensions of ca. 200 micron diameter. This permits the generation of large (multi-thousand member) libraries with no practical difficulties.Radiofrequency methods
Libraries have also been electromagnetically encoded. In the first case,8 transponders similar to those used to identify laboratory mice were used. These consisted of a glass-coated chip that is pre-tuned to emit a binary code when irradiated with electromagnetic radiation, and which was then enhanced so that information could also be written to it. The chip was encased in a polypropylene capsule containing polystyrene resin. A computer program directed the distribution of the capsules in each case to ensure that all possible compounds were actually made. (Statistical variations in a combinatorial synthesis mean that the number of beads used in a standard split-and-mix synthesis has to be larger than the number of members of a library if one wants to be reasonably certain of making all possible library members with a defined set of chemical steps, in much the same way as an opinion poll exhibits fewer statistical anomalies as the number of responses increases.)In the second, similar, case9 an addressable radiofrequency semiconductor unit encased in glass was embedded in polymer beads, surrounded by an inert porous wall. Such devices are passive and acquire their energy from the encoding radiofrequency pulse. A library is made again under the control of the appropriate hard- and software, and using standard split-and-mix techniques. The storage capacity of such units is large, and can record not only the reagents at each step, but also the reaction time and temperature used (the device contains a temperature sensor). These microreactors are capable of withstanding a temperature range of −78 °C to 150 °C, and are inert to most solvents. This technology has been used in the synthesis of taxoid10 and epothilone11 and other libraries,12 and has been commercialised.13
These reactors are quite popular for small to medium sized libraries and are relatively inexpensive. Problems with these strategies in general hinge around the large size of the units (and the corresponding limit on the size of a library that can be made using them). These devices must be at least as large as the wavelength of the radiation used to encode them.
Chemical methods
The standard approach to chemical tagging (Scheme 1) involves the attachment of a small amount of a specific molecular tag (TX) to the solid phase before or after each synthetic step in the library synthesis (RX). The combination of tags at the end of the synthesis reveals the synthetic history of the bead. There are fairly evident criteria for any such strategy, namely that the tagging chemistry and the library chemistry must be orthogonal, that the tags be readily identifiable, that the tags and library members may be removed from the support under different conditions (so that screening and compound identification may be carried out separately), and that the tag variation is large enough to code for large libraries. The different approaches to chemical tagging have been reviewed.14 The first reported methods of chemical tagging employed oligonucleotides15 and peptides16 as the tags. Subsequently, small molecules such as chromophores,17 secondary amines18 and haloaromatic alcohols19–22 were employed.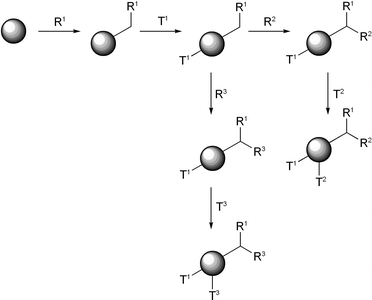 | ||
| Scheme 1 General method of chemical encoding. TX = tag, RX = library component/synthetic step. | ||
Arguably the most important of these approaches is that developed by Still. This methodology has been developed commercially and has been repeatedly shown to be effective in chemically tagging a combinatorial library. The method was first used to encode a peptide library,19 and has been used to probe binding affinities between a receptor and a support-bound library of over 50000 compounds.20 The tags were inert haloaromatic alcohols, where variability in the tag was achieved by variation in the position and nature of the halogen substituents, and the length of the alcohol chain (Fig. 1a). The tags were attached to a small proportion (∼1%) of the available sites of amino-functionalised polystyrene resin by means of a carbonate linkage to a photocleavable linker via its carboxylic acid. The tags were detected as their trimethylsilyl derivatives at less than femtomolar levels by electron capture gas chromatography (ECGC), which is very sensitive to such electrophoric molecules, and formed a binary code by their elution order (i.e. the presence of a tag is a ‘1’ and its absence is a ‘0’, see below). The original scheme allowed for 40 tags differing in chain length and number/position of halide substituents, technically allowing for the encoding of a huge library (240 components). A single ECGC apparatus could at the time decode at least 50 structures per day, which is satisfactory given that one tends to be interested only in the active compounds from a library.
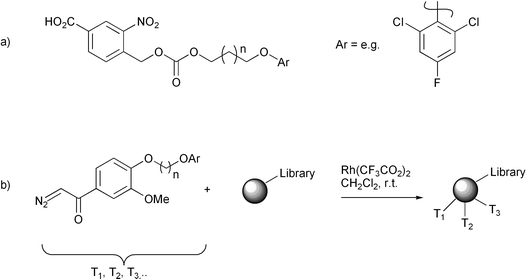 | ||
| Fig. 1 Still’s encoding method. | ||
A limitation of the initial tagging approach was its reliance on the presence of amino functionality on the resin for tag attachment. The scheme was generalised21 to allow for the encoding of virtually any library by attaching the tag via a catechol ether linker introduced by indiscriminate rhodium-catalysed carbene insertion into the solid support (at a level of about 1 pmol per bead, compared to roughly 100 pmol of the library compound, Fig. 1b). The tag is detached by ceric ammonium nitrate oxidative cleavage and detected as before. The uses of Still’s system have been reviewed.22
An approach developed by Affymax shares several characteristics with that devised by Still, but instead uses secondary amines as tags.18 The tags are attached to the resin sequentially to form a tag oligomer using chemistry orthogonal to that of the library synthesis (e.g. the alloc protecting group was used to mediate the tagging chemistry, while the Fmoc group was used in the construction of the library). The tags are detached using 6 M HCl. Identification of the tags involves initial derivatisation to form the dansylates, followed by separation using HPLC on a standard column with fluorescent detection.
Use of mass spectrometry in library synthesis
A report of the use of mass spectrometry in tagging described a number of strategies relying on isotopic variations in tags that are contained within the linker to the library.23 The versatility of the method is compromised by the low number of commercially available isotopically labelled compounds suitable for this task. It has been suggested that mass spectrometric analysis of compounds from beads obviates the need for tagging in the first place.24 However it should be noted that the mass spectrometric technique must be tailored to the library type such that the compounds ‘fly’ well in the mass spectrometer. Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS) was recently used to identify the members of 100-member peptide libraries with no tagging.25 Even in this small library, only 70–80% of the members could be identified with certainty (i.e. within a pre-defined mass error limit).We felt at the outset of our work that FTICR-MS was an ideal analytical technique in a chemical tagging method. We shall now describe our overall approach.
A new chemical tagging method
We have developed a tagging approach distinct from the approaches used by Still and Affymax. We were able to reduce the number of additional steps dedicated to tagging, and to develop a detection technique based on mass spectrometry, that does not require a chromatographic separation step.To achieve this, the specific analytical issues we had to address were:
The mass spectral behaviour of a number of tag candidates, and the detection of such species at picomole levels.
The attachment of these tags to the solid support followed by their release and reliable detection from single beads, either as the tag or some derivative which is trivial to synthesise.
The detection of cleaved library and tag compounds from single beads of a chemical library.
Further, the parallel synthetic achievements were:
The development of a novel set of inert chemical tags suitable for detection at low levels by mass spectrometry.
The attachment of these tags to a solid support in a manner which allowed iterative attachment of the tags.
The use of this orthogonal tagging system to code for a chemical library, requiring the selective release of the library in the presence of the tags, followed by the release of tags from the solid support on demand.
We were able to fulfil all these requirements for the development of our tagging strategy, which is described in full elsewhere.26 We present here a summary of the salient results.
New tagging strategy
The strategy we developed is shown in Scheme 2. The solid support is loaded with an acid-labile linker (4-hydroxybenzyl alcohol, known as the Wang linker), the first phenol tag, and 4-iodophenol to give 1. The linker occupies the great majority of the sites on the resin, and the tag and iodophenol are ‘doped’ in at 1–10% levels. The resin 1 is stable under ambient conditions and can therefore be synthesised in bulk and kept as the starting point of any solid phase synthesis. Attachment of the library to the linker then begins, via the benzyl alcohol of the linker. A variety of substrates could be attached by, for example, Mitsunobu couplings. The first component of the library is already coded for by tag 1.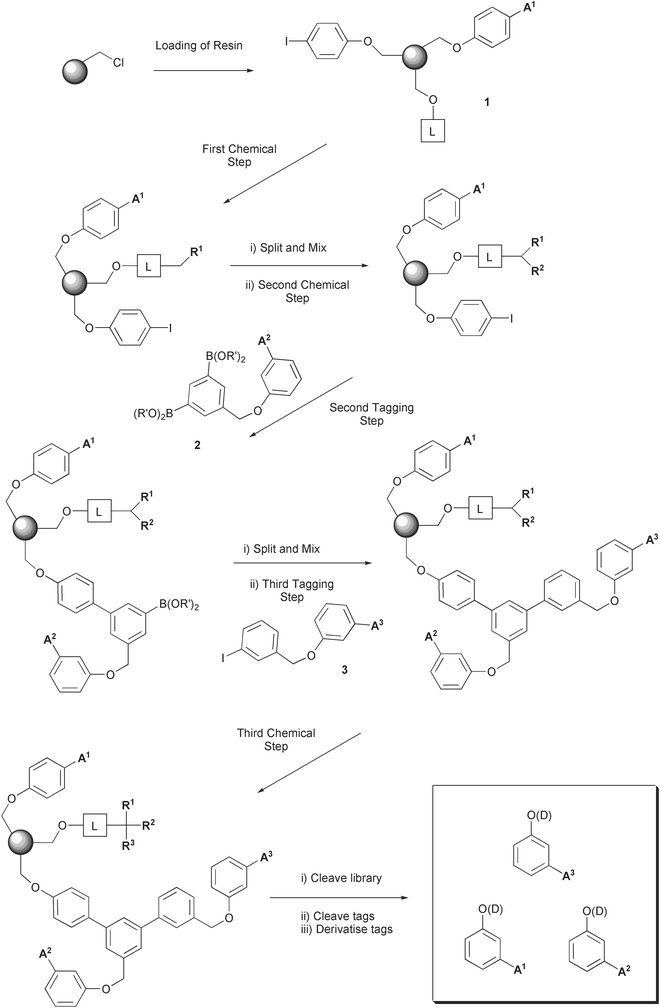 | ||
| Scheme 2 Outline of the tagging strategy. L = Wang linker (4-hydroxybenzyl alcohol), A = alkyl group, R = library element, D = tag derivative. | ||
A split-and-mix step is then performed, followed by attachment of the second element of diversity, or some other chemical step. This is coded for by the second tagging step. This consists in our strategy of an organometallic reaction of the support-bound aryl iodide with a 1,3-diboronate containing tag 2 linked to it via a benzyl ether linkage (2). (The palladium-catalysed reaction between a boronate and a halide to form a carbon–carbon bond is known as a Suzuki reaction.) These diboronates are the key molecules to the whole strategy. After coupling to the aryl iodide, to generate an inert biaryl linkage, the second boronate group is left intact. A second split-and-mix step is performed, after which tag 3 is introduced by a second Suzuki reaction between the pendant boronate and an aryl iodide (3). This iodide is of the same form as the diboronates used previously, in that they contain the tag attached via a benzyl ether bond. (In theory diiodides could be used, setting up a pendant aryl iodide which could be used to couple with another boronate in a further round of tagging.) This third tag codes for the third library step, which is performed subsequently, as the last step in the scheme. In this way a library with three elements of diversity can be constructed, coded for by three sets of tags. The first tag resides in the resin at the outset of the synthesis, the second is introduced pre-attached to a diboronate, and the third is introduced pre-attached to an aryl iodide.
The linker chemistry employed allows for library molecules to be cleaved from the solid support under mild conditions and screened; those beads yielding compounds deemed important may then have the tags removed from them under harsher conditions, followed by detection of the tags by mass spectrometry.
There are a number of attractive features of this strategy:
The first tag is loaded when the linker is added to the bead, and the second and third tags are added ‘back to back’ using the sequential reaction of the bisboronate, with an intervening split-and-mix operation, but without work-up of the Suzuki reactions. In effect the second and third tags are introduced in the same reaction, and overall this is the only additional step involved in the library synthesis. No protecting group chemistry is employed.
The Suzuki chemistry employed in the tagging steps is performed under mild conditions and can tolerate a wide variety of chemical functionalities. There is ample precedence of Suzuki coupling on solid support.27
The biaryl and benzylic ether linkages used for the ultimate attachment of the tags to the support are robust, and allow for the facile selective removal of the library. Harsher conditions can then be used to remove the tags.
It is important to understand the power of binary coding in such a strategy. Suppose that the first element of diversity in the library sequence (R1 in Scheme 2) may take one of three forms (e.g. R1 may be one of three amino acids). We require only two chemical tags to code for this. If we translate the presence of a tag in the decoding spectrum as a ‘1’ and its absence as a ‘0’ (we met this idea earlier in the description of the Still encoding method) then we have at our disposal three situations: presence of one tag (‘10’) corresponding to the first amino acid, presence of alternative tag (‘01’) corresponding to the second amino acid, or presence of both tags (‘11’) corresponding to the third amino acid. Absence of both (‘00’) is not employed in the coding owing to the inherent ambiguity of such a result. This method may be applied to each step of library construction, as long as new tags are employed to avoid confusion with earlier steps. If we are able to employ one of three amino acids at each step of our library synthesis, then our complete three-step library as shown in Scheme 2 consists of a 3 × 3 × 3 library, i.e. 27 tripeptides. Yet as we have seen, each stage of library construction required only two tags, allowing this 27-member library to be completely coded for by only 6 tags. The size of library it is possible to encode in this way increases exponentially for a linear increase in the number of tags.
The analytical strategy
The strategy involves detection of the tags using mass spectrometry, specifically Fourier transform ion cyclotron resonance mass spectrometry. FTICR-MS has been used widely in the detection of complex molecules with high sensitivity.28 FTICR-MS was selected primarily because of its great sensitivity (which would allow the identification of tags from single library beads), but also because of its accurate mass capability. Once properly calibrated, all peaks in a FTICR mass spectrum are assigned an accurate mass (by default). This is a very important point. The precision of the mass determination means that the presence of a tag can be confirmed almost regardless of the signal to noise in the mass spectrum. It also identifies a tag even in the presence of something else with the same integral mass. Thus by judicious choice of tags, it is possible to separate the tags from background noise with a high degree of certainty. This has the added benefit that no chromatography is required prior to the detection of the tags, a saving of cost and time were the process to be automated.Chemical tags for FTICR-MS
Alkyl phenols are used in the encoding strategy as they are suitable for MS detection, and chemically relatively inert. Six alkyl phenols used in the tagging studies are shown in Fig. 2. tert-Butylphenol (4a), tert-amylphenol (4b), and tert-octylphenol (4c) are all crystalline solids. The others, heptylphenol (4d), nonylphenol (4e) and dodecylphenol (4f), are gelatinous liquids easily handled as dilute solutions. All are stable except heptylphenol which slowly decomposes over time at room temperature.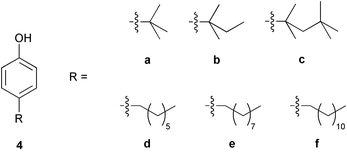 | ||
| Fig. 2 Alkyl phenol tags employed in the tagging strategy. | ||
Electrospray ionisation is used with the FTICR mass spectrometer. For the tags to become charged well they require a protonatable site, or one that can chelate a metal ion. However for the tags to be inert during solid phase chemistry such a functional group ‘handle’ would be undesirable. Consequently the tags are attached to the resin via a benzyl ether linkage during library synthesis, and then derivatised in situ after cleavage to introduce the mass spectral handle on the exposed phenolic oxygen. This derivatisation step is an extra level of complication in the procedure and so had to be as simple as possible. Other tagging techniques use a derivatisation step. In Still’s technique,22 for example, cleaved tags are derivatised with bis-trimethylsilylacetamide prior to analysis, and in the secondary amine method,18 dansylation is used.
We developed a facile in situ dansylation procedure for the derivatisation step by modification and simplification of a literature procedure.29 The conversion of phenols to their dansylates is an attractive strategy because the dansyl group contains a protonatable nitrogen atom which assists in their detection by positive ion ES-MS, and the added mass of the dansyl group moves the region of interest in the ES mass spectrum to one of improved signal-to-noise ratio.
Attachment, cleavage and derivatisation of phenols
The attachment of phenols to chloromethyl polystyrene is a simple reaction involving sodium methoxide as base, and the use of dimethylacetamide (DMA) as solvent.30The phenols are attached to the solid support via a benzyl ether linkage. Removal of the tags prior to mass spectral analysis requires cleavage of this bond. This can be achieved very effectively using thioanisole, in combination with trifluoroacetic acid (TFA) and a Lewis acid such as trimethylsilyl trifluoromethanesulfonate.30 TFA-based reagent mixtures are amenable to dilution with dichloromethane (DCM), providing a solvent system that is frequently used for solid phase work. Dichloromethane ‘swells’ the resin effectively, allowing penetration of reagents into the interior of the polystyrene bead.
It is usual in peptide chemistry to cleave the Wang linker (and hence remove the library compound from the resin) with neat TFA containing traces of triisopropylamine and water. We have shown that the tags remain on the resin under these conditions. Indeed, lower concentrations of TFA in DCM are sufficient to cleave the Wang linker.
To illustrate facile removal of tags using thioanisole after cleavage of the Wang linker, the comparison reaction shown in Scheme 3 was performed. Thus treatment of support-bound phenol 5 with a 10% TFA–DCM solution produced no cleavage of phenol as before, whilst the same reaction mixture containing a drop each of thioanisole and TMS–triflate liberated the phenol within 2 min at room temperature. This method of cleavage is very simple practically. Cleavage of library compounds is therefore carried out in a 10% TFA–DCM solution, to which is then added thioanisole and TMS–triflate for the release of the tags.
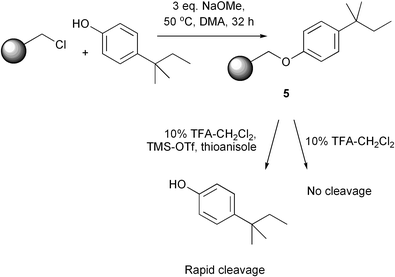 | ||
| Scheme 3 Solid phase loading and cleavage of phenols. | ||
A further advantage of this cleavage system is that all the reagents are volatile. In traditional peptide synthesis, cleavage reactions done with TFA involve the simple removal of excess TFA under a stream of nitrogen. The same method may be used with the thioanisole cleavage mixture, prior to injection of the cleaved tags into the mass spectrometer.
This cleavage–derivatisation procedure is capable of detecting phenol from a single resin bead. For example, a single bead of the tert-amylphenol loaded resin 5 was subjected (Scheme 4) to the thioanisole-based cleavage for 5 min at room temperature, and the supernatant concentrated in vacuo. The phenol was then derivatised with dansyl chloride and analysed by FTICR-MS. The resultant mass spectrum from this single bead experiment is shown in Fig. 3.30 The spectrum has an excellent signal-to-noise ratio, and the mass of the tag peak is only 5 ppm different to the calculated mass. Similar results are seen when mixtures of phenols were attached to the support, and cleaved and analysed in the same way, confirming that the phenol dansylation strategy with mass spectrometric detection is suitable for use in a tagging experiment.
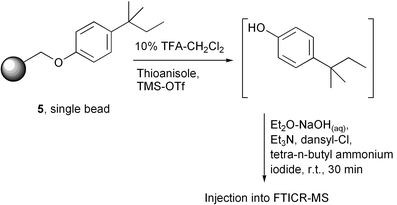 | ||
| Scheme 4 Single-bead cleavage experiment. | ||
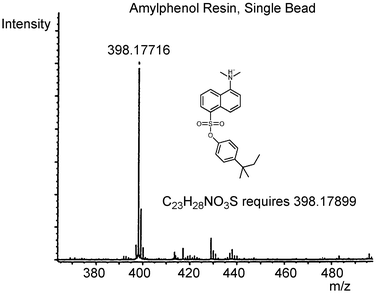 | ||
| Fig. 3 FTICR mass spectrum from single cleaved amylphenol bead. Reprinted from ref. 30, Copyright 2000, with permission from Elsevier Science. | ||
Synthesis of a complete set of chemical tags
An example of the approach to using this strategy involves the synthesis of appropriate chemical tags. For example heptyl- and nonylphenol were attached to the resin as the parent phenols at the beginning of the solid phase synthesis, to code for the first element of diversity (e.g. the first amino acid in a tripeptide). Amyl- and octylphenol were used to code for the second element of diversity, and so were incorporated into the key diboronic acid-based molecules. The final element of diversity was coded for by butyl- and dodecylphenol, and so was attached as part of the aryl iodides. The six molecules comprising the complete novel set of chemical tags are shown in Fig. 4.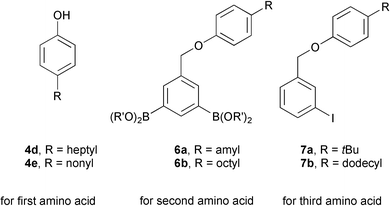 | ||
| Fig. 4 Tagging constructs used in the strategy. | ||
The synthetic route to all the tag constructs has been described in full elsewhere.26 In summary, the benzyl ether bonds in the four molecules 6a–7b were constructed either with Mitsunobu chemistry or via conversion of the benzyl alcohol of the aryl iodide to its trichloroacetimidate derivative 10 (Scheme 5).
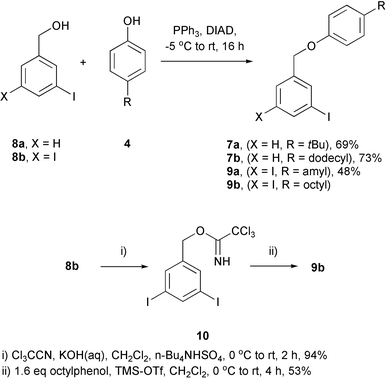 | ||
| Scheme 5 Mitsunobu and trichloroacetimidate chemistry used in the synthesis of the tag constructs. | ||
Synthesis of the diboronates is from the corresponding diiodides via a palladium-catalysed coupling with pinacolatodiboronate (Scheme 6). This is the first demonstration of aromatic diboronate synthesis with this strategy.
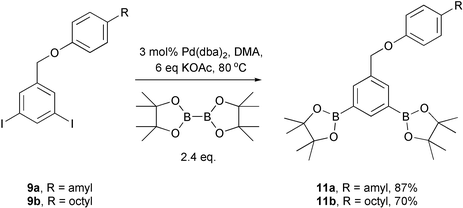 | ||
| Scheme 6 Synthesis of diboronate tag constructs. | ||
Exemplification of the tagging strategy
The tagging method was exemplified in the synthesis of an encoded tripeptide. The sequence of reactions involved is shown in Scheme 7.26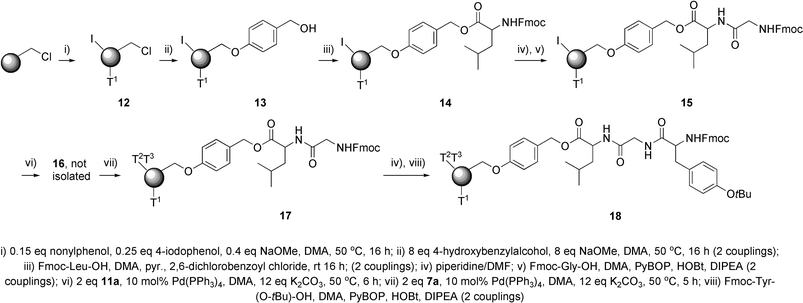 | ||
| Scheme 7 Solid phase synthesis of encoded tripeptide. | ||
The tripeptide leucine–glycine–tyrosine was coded for with nonylphenol–amylphenol–butylphenol. Chloromethyl polystyrene resin was loaded with low levels of nonylphenol (<15% loading) and iodophenol (<25% loading) to give 12. This resin was loaded with the Wang linker to produce 13. The first amino acid was attached to this resin using the standard 2,6-dichlorobenzoyl chloride/pyridine method31 to give 14. The second amino acid was attached to the first via standard peptide coupling chemistry to give 15. This second amino acid was coded for by the attachment of the amylphenol-based diboronate. The final coding step was performed by palladium-catalysed attachment of the butylphenol-based iodide to give 17. Approximately two to three equivalents of diboronate and iodide tags were found to be necessary for these coupling reactions. This final tag coded for tyrosine, which was then attached in the final step to yield 18.
The peptide was readily identifiable with low resolution ES-MS after cleavage of the Wang linker with 10% TFA–DCM at room temperature for 10 min. Cleavage with this mixture can be used to verify that each step in a peptide synthesis has gone to completion.
After peptide removal, single beads were treated using the thioanisole–dansyl chloride cleavage–derivatisation sequence. The FTICR mass spectrum revealed clearly the three expected tags (Fig. 5). The masses of the molecular ions observed for the dansylated tags were all correct to within 4 ppm. While it would be desirable to improve the signal-to-noise ratio observed in these spectra, the encoding strategy is exemplified by this result since the three expected tags are unequivocally identified (at high resolution) whilst those tags not included are absent (asterisks indicate where they would have appeared). No peak on the mass spectrum was within 10 ppm of the expected masses of these tags. This provides the appropriate binary code for this peptide. The other peaks present we could only ascribe to noise that is typically present in a sample of this high dilution. Further, no chromatography was required prior to injection of this mixture into the mass spectrometer.
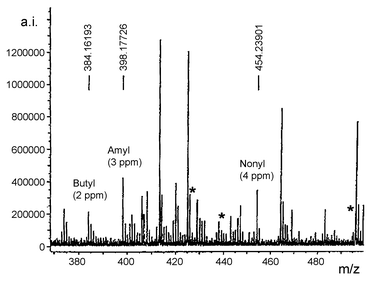 | ||
| Fig. 5 FTICR mass spectrum showing the three required tags obtained from treatment of a single bead of resin 18, after removal of peptide, according to the cleavage–derivatisation method (Scheme 4). Asterisks indicate absence of tag peaks for heptyl (expected at m/z 426.210293), octyl (440.225943) and dodecyl (496.288543). Reprinted with permission from ref. 26. Copyright 2001 American Chemical Society. | ||
Future of chemical tagging
We have described a novel approach for the use of high-resolution mass spectrometry for the decoding of a chemical compound attached to a single resin bead via the use of chemical tags. We met the general requirements of a chemical encoding method for combinatorial chemistry, in that the tags could be easily attached to and removed from the resin, that the tagging chemistry was orthogonal to the library synthesis, and that the tags could be detected at very low levels. However, we showed for the first time that high resolution mass spectrometry is a useful addition to the analytical strategies that may be called upon to solve the problem of identifying chemical tags from an encoded solid phase synthesis sequence. We also diverged from the principal chemical tagging systems described earlier in that we required no chromatography in the identification of the tags.It is likely that large split-and-mix libraries will continue to feature in combinatorial chemistry due to their power to produce so many compounds so rapidly. There will therefore continue to be great analytical demands on the identification of compounds shown to be active from the screening of such libraries, and we look forward to future developments in this field.
Acknowledgement
We thank Zeneca Pharmaceuticals (now AstraZeneca) for a studentship (to M.H.T). We thank Lancaster Synthesis for a gift of pinacolatodiboronate.References
- N. K. Terrett, Combinatorial Chemistry, Oxford University Press, Oxford, 1998 Search PubMed.
- A. Furka, F. Sebestyen, M. Asgedom and G. Dibo, Int. J. Pept. Protein Res., 1991, 37, 487–493 Search PubMed.
- B. A. Bunin and J. A. Ellman, J. Am. Chem. Soc., 1992, 114, 10997–10998 CrossRef CAS; H. M. Geysen, R. N. Meloen and S. J. Barleling, Proc. Natl. Acad. Sci., USA, 1984, 81, 3998–4002 CAS; S. H. DeWitt, J. S. Kiely, C. J. Stankovic, M. C. Schroeder, D. M. R. Cody and M. R. Pavia, Proc. Natl. Acad. Sci., USA, 1993, 90, 6909–6913 CAS; S. P. A. Fodor, J. L. Read, M. C. Pirrung, L. Stryer, A. T. Lu and D. Solas, Science, 1991, 251, 767–773 CAS.
- R. A. Houghten, Proc. Natl. Acad. Sci., USA, 1985, 82, 5131–5135 CAS.
- X. Xiao, C. F. Zhao, H. Potash and M. P. Nova, Angew. Chem. Int. Ed., 1997, 36, 780–782 CrossRef CAS.
- J. W. Guiles, C. L. Lanter and R. A. Rivero, Angew. Chem. Int. Ed., 1998, 37, 926–928 CrossRef CAS.
- F. Dittrich, W. Tegge and R. Frank, Bioorg. Med. Chem. Lett., 1998, 8, 2351–2356 CrossRef CAS.
- E. J. Moran, S. Sarshar, J. F. Cargill, M. M. Shahbaz, A. Lio, A. M. M. Mjalli and R. W. Armstrong, J. Am. Chem. Soc., 1995, 117, 10787–10788 CrossRef CAS.
- K. C. Nicolaou, X.-Y. Xiao, Z. Parandoosh, A. Senyei and M. P. Nova, Angew. Chem. Int. Ed., 1995, 34, 2289–2291 CrossRef CAS.
- X.-Y. Xiao, Z. Parandoosh and M. P. Nova, J. Org. Chem., 1997, 62, 6029–6033 CrossRef CAS.
- K. C. Nicolaou, N. Winssinger, J. Pastor, S. Ninkovic, F. Sarabia, Y. He, D. Vourloumis, Z. Yang, T. Li, P. Giannakakou and E. Hamel, Nature, 1997, 387, 268–272 CrossRef CAS.
- K. C. Nicoloau, J. A. Pfefferkorn, H. J. Mitchell, A. J. Roecker, S. Barluenga, G. Q. Cao, R. L. Affleck and J. E. Lillig, J. Am. Chem. Soc., 2000, 122, 9954–9967 CrossRef.
- http://www.irori.com .
- J. C. Chabala, Curr. Opin. Biotechnol., 1995, 6, 632–639 CrossRef CAS; J. C. Chabala, J. J. Baldwin, J. J. Burbaum, D. Chelsky, L. W. Dillard, I. Henderson, G. Li, M. J. H. Ohlmeyer, T. L. Randle and J. C. Reader, Perspect. Drug Discovery Des., 1995, 2, 305–318 Search PubMed.
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci., USA, 1992, 89, 5381–5383 CAS; M. C. Needles, D. G. Jones, E. H. Tate, G. L. Heinkel, L. M. Kochersperger, W. J. Dower, R. W. Barrett and M. A. Gallop, Proc. Natl. Acad. Sci., USA, 1993, 90, 10700–10704 CAS; J. Nielsen, S. Brenner and K. D. Janda, J. Am. Chem. Soc., 1993, 115, 9812–9813 CrossRef CAS.
- M. Ptek and M. Lebl, Tetrahedron Lett., 1991, 32, 3891–3894 CrossRef; J. M. Kerr, S. C. Banville and R. N. Zuckermann, J. Am. Chem. Soc., 1993, 115, 2529–2531 CrossRef CAS; V. Nikolaiev, V. Stierandova, V. Krchnak, B. Seligmann, K. S. Lam, S. E. Salmon and M. Lebl, Pept. Res., 1993, 6, 161–170 Search PubMed.
- B. J. Egner, S. Rana, H. Smith, N. Bouloc, J. Frey, W. S. Brocklesby and M. Bradley, Chem. Commun., 1997, 735–736 RSC; E. Campian, F. Sebestyen and A. Furka, in Innovation and Perspectives in Solid Phase Synthesis, ed. R. Epton, Mayflower, Birmingham, 1994, pp. 469–472 Search PubMed.
- Z. J. Ni, D. Maclean, C. P. Holmes, M. M. Murphy, B. Ruhland, J. W. Jacobs, E. M. Gordon and M. A. Gallop, J. Med. Chem., 1996, 39, 1601–1608 CrossRef CAS; A. Atuegbu, D. Maclean, C. Nguyen, E. M. Gordon and J. W. Jacobs, Bioorg. Med. Chem., 1996, 4, 1097–1106 CrossRef CAS; D. Maclean, J. R. Schullek, M. M. Murphy, Z. J. Ni, E. M. Gordon and M. A. Gallop, Proc. Natl. Acad. Sci., USA, 1997, 94, 2805–2810 CrossRef CAS.
- M. H. J. Ohlmeyer, R. N. Swanson, L. W. Dillard, J. C. Reader, G. Asouline, R. Kobayashi, M. Wigler and W. C. Still, Proc. Natl. Acad. Sci., USA, 1993, 90, 10922–10926 CAS.
- A. Borchardt and W. C. Still, J. Am. Chem. Soc., 1994, 116, 373–374 CrossRef CAS.
- H. P. Nestler, P. A. Bartlett and W. C. Still, J. Org. Chem., 1994, 59, 4723–4724 CrossRef CAS.
- W. C. Still, Acc. Chem. Res., 1996, 29, 155–163 CrossRef CAS.
- H. M. Geysen, C. D. Wagner, W. M. Bodnar, C. J. Markworth, G. J. Parke, F. J. Schoenen, D. S. Wanger and D. S. Kinder, Chem. Biol., 1996, 3, 679–688 CrossRef CAS.
- C. L. Brummel, J. C. Vickerman, S. A. Carr, M. E. Hemling, G. D. Roberts, W. Johnson, J. Weinstock, D. Gaitanopoulos, S. J. Benkovic and N. Winograd, Anal. Chem., 1996, 68, 237–242 CrossRef CAS; M. C. Fitzgerald, K. Harris, C. G. Shevlin and G. Siuzdak, Bioorg. Med. Chem. Lett., 1996, 6, 979–982 CrossRef CAS; B. B. Brown, D. S. Wagner and H. M. Geysen, Mol. Div., 1995, 1, 4–12 Search PubMed.
- A. S. Fang, P. Vouros, C. C. Stacey, G. H. Kruppa, F. H. Laukien, E. A. Wintner, T. Carell and J. Rebek, Comb. Chem. High Throughput Screening, 1998, 1, 23–33 Search PubMed.
- M. H. Todd and C. Abell, J. Comb. Chem., 2001, 3, 319–327 CrossRef CAS.
- M. H. Todd and C. Abell, in Solid Phase Synthesis, ed. K. Burgess, Wiley-Interscience, New York, 2000, pp. 25–79. Search PubMed.
- D. F. Hunt, J. Shabanowitz, J. R. Yates, R. T. McIver, R. L. Hunter, J. E. P. Syka and J. Amy, Anal. Chem., 1985, 57, 2728–2733 CrossRef CAS; R. E. Shomo, A. G. Marshall and R. G. Weisenberger, Anal. Chem., 1985, 57, 2940–2944 CrossRef CAS; B. E. Winger, S. A. Hofstadler, J. E. Bruce, H. R. Udseth and R. D. Smith, J. Am. Soc. Mass Spectrom., 1993, 4, 566–577 CrossRef CAS; S. D. H. Shi, C. L. Hendrickson and A. G. Marshall, Proc. Natl. Acad. Sci., USA, 1998, 95, 11532–11537 CrossRef CAS; J. S. Pyrek, Synlett, 1999, 249–266 CAS.
- P. J. M. Kwakman, D. A. Kamminga, U. A. T. Brinkman and G. J. DeJong, J. Chromatogr., 1991, 553, 345–356 CrossRef CAS.
- M. H. Todd and C. Abell, Tetrahedron Lett., 2000, 41, 8183–8187 CrossRef CAS.
- P. Sieber, Tetrahedron Lett., 1987, 28, 6147–6150 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |